

Abstract
Tristetraprolin (TTP) is an mRNA-binding protein, but studies of this interaction have been difficult due to problems with the purification of recombinant TTP. In the present study, we expressed human and mouse TTP as glutathione S-transferase and maltose-binding protein (MBP) fusion proteins in Escherichia coli, and purified them by affinity resins and Mono Q chromatography. TTP cleaved from the fusion protein was identified by immunoblotting, MALDI-MS, and protein sequencing, and was further purified to homogeneity by continuous-elution SDS–gel electrophoresis. Purified recombinant TTP bound to the AU-rich element of tumor necrosis factor-α (TNFα) mRNA and this binding was dependent on Zn2+. Results from sizing columns suggested that the active species might be in the form of an oligomer of MBP-TTP. Recombinant TTP was phosphorylated by three members of the mitogen-activated protein (MAP) kinase family, p42, p38, and JNK, with half-maximal phosphorylation occurring at approximately 0.5, 0.25, and 0.25 μM protein, respectively. Phosphorylation by these kinases did not appear to affect the ability of TTP to bind to TNFα mRNA under the assay conditions. This study describes a procedure for purifying nonfusion protein TTP to homogeneity, demonstrates that TTP's RNA-binding activity is zinc dependent, and that TTP can be phosphorylated by JNK as well as by the other members of the greater MAP kinase family.
Keywords: Tristetraprolin, Tumor necrosis factor-α, Protein expression and purification, Protein phosphorylation, mRNA ARE-binding protein, Mitogen-activated protein kinase
Tristetraprolin (TTP)1, also known as ZFP36, Nup475, TIS11, and G0S24, is the prototype of a small family of mammalian proteins with two CCCH (CX8CX5CX3H) zinc-finger motifs separated by 18 amino acid residues [1-3]. Similar zinc-finger sequences are found in the GenBank database in at least 14 species, including human, bovine, mouse, rat, Xenopus, carp, zebrafish, Drosophila, C. elegans, baker's yeast, fission yeast, oyster, rice, and Arabidopsis ([4], and data not shown). TTP mRNA could be detected by Northern blotting in a number of tissues including spleen, thymus, lymph node, lung, liver, and intestine [2]. The expression level of TTP mRNA in cells is increased in response to several kinds of stimuli, such as insulin and other growth factors, as well as stimulators of innate immunity such as lipopolysaccharide.
TTP has been documented to be an mRNA-binding protein based on evidence from transfected cells. TTP and its related proteins can bind to the 3′-untranslated AU-rich elements (ARE) of certain clinically important mRNAs, such as those encoding tumor necrosis factor-α (TNFα), granulocyte-macrophage colony-stimulating factor (GM-CSF), and interleukin 3 (IL3) [4-8]. TTP and its related proteins can bind to these mRNAs in a cell-free assay, and can destabilize them in transfected 293 cells [4-8]. Mice deficient in TTP develop a severe inflammatory syndrome, including polyarticular arthritis, myeloid hyperplasia, autoimmunity, and cachexia [9]. This syndrome is largely due to the increased stability of mRNAs for TNFα and GM-CSF, and the resulting increased secretion of these cytokines [5,6].
To understand the structure and function of TTP and its related proteins, ZFP36L1 and ZFP36L2, in mammals, and to produce high-quality antibodies against them, it was first necessary to produce sufficient amounts of highly purified proteins. However, progress on these proteins has been slow due to the difficulty of expressing and purifying TTP from various expression systems. In earlier studies, TTP was expressed as a His-tagged protein in Escherichia coli for antibody production and for zinc-finger-binding characterization [1,10]. TTP fused to glutathione S-transferase (GST) was expressed in E. coli and used in an in vitro phosphorylation study [11]. Those fusion proteins were described as extensively degraded and largely precipitated [11,12]. In fact, to our knowledge there is no report to date that TTP has been purified to homogeneity from any source.
Fusion proteins with the E. coli maltose-binding protein (MBP) and N-utilization substance protein A (NusA) have exhibited excellent solubility in several cases [13,14], and the MBP fusion proteins have been used to characterize a variety of proteins otherwise difficult to purify. Examples of these fusion proteins include human and murine single-chain antibodies [15], the globular head region of the human C1q B-chain [16], human phenylalanine hydroxylase [17], the SarR protein from Staphylococcus aureus [18], hen steroid receptor-binding factor [19], an archaeal geranylgeranyl diphosphate synthase [20], and pea NADPH:protochlorophyllide oxidoreductase [21].
We compared the GST, MBP, and NusA systems for the expression of soluble TTP in E. coli. We report here the expression of soluble GST-TTP, MBP-TTP, and NusA-TTP, the partial purification of the fusion proteins by affinity resins and Mono Q columns, and the final purification of TTP after digestion of the MBP-TTP fusion protein by continuous-elution gel electrophoresis (CEGE). To our knowledge, this is the first report that nonfusion TTP can be purified to homogeneity with retention of RNA-binding activity. The purified GST-TTP and MBP-TTP fusion proteins, as well as CEGE-purified TTP itself, were shown to be competent for RNA binding. MBP-TTP, but not MBP, could be phosphorylated by p42, p38, and c-jun-N-terminal kinase (JNK) in vitro. We also demonstrated directly, using purified TTP, that TTP is a zinc-dependent mRNA-binding protein and that it can be phosphorylated by JNK as well as other mitogen-activated protein (MAP) kinases.
Experimental procedures
Plasmid construction
Plasmids pGST-hTTP, pMBP-hTTP, pNusA-hTTP, and pMBP-mTTP were designed to express human TTP (hTTP) and mouse TTP (mTTP) as fusion proteins in E. coli protein expression systems. The fusion proteins contained GST, MBP, or NusA at the amino terminus and hTTP residues 2 to 326 (GenBank Accession No. NP_003398) or mTTP residues 2 to 319 (GenBank Accession No. NP_035886) at the carboxyl terminus. The plasmid pGST-hTTP was constructed by cloning the PCR-amplified DNA fragment using human TTP cDNA sequence as a template (GenBank Accession No. NM_003407) [22] into expression vector pGEX4T-1 (Amersham Pharmacia Biotech, Uppsala, Sweden) at the BamHI and XhoI sites with a forward primer containing sequence for a PreScission protease (Amersham Pharmacia Biotech) cleavage site. The plasmids pMBP-hTTP and pNusA-hTTP were constructed by cloning the human TTP cDNA sequence (GenBank Accession No. NM_003407) [22] with a PreScission protease cleavage site and E. coli MBP or E. coli NusA DNA sequences into expression vector pT7#3.3 (AstraZeneca Pharmaceuticals, Mereside, UK), respectively. The plasmid pMBP-mTTP was constructed by exchanging the hTTP fragment in plasmid pMBP-hTTP with the PCR-amplified mTTP fragment at the SspI and SpeI sites. Plasmid A1 (GenBank Accession No. NM_011756) 2] was used as a template and the forward primer contained sequence for a PreScission protease cleavage site. The plasmid constructions were confirmed with restriction enzyme digestion and DNA sequencing using the dRhodamine Terminator Cycle Sequencing kit (Perkin Elmer Life Sciences, Gaithersburg, MD).
Expression of TTP fusion proteins in E. coli
Plasmids were transformed into the E. coli BL21(DE3) strain by the heat-shock procedure [23]. The optimum conditions for GST-TTP, MBP-TTP, and NusA-TTP expression were determined with several isopropylthio-β-d-galactoside (IPTG) induction conditions including various temperatures and induction times [24,25]. The optimum conditions for MBP-TTP expression were as follows: A single colony was inoculated into Luria–Bertani (LB)-tetracycline (15μg/ml) medium and grown overnight at 37 °C. The overnight culture was inoculated at a 1 to 20 dilution into fresh medium and grown for about 2 h at 37 °C to reach a cell density of about 0.6–1 at A600. Then IPTG was added to the culture medium (0.4 mM final concentration) and protein expression was induced at 28 °C for about 3 h before harvesting. Cells were homogenized by sonication in homogenization buffer containing 20 mM Tris–HCl, pH 7.4, 200 mM NaCl, 10 mM β-mercaptoethanol, 1 mM EDTA, 1 mM phenylmethylsulfonyl fluoride (PMSF), 2μM leupeptin, 2 μM pepstatin, and 2 μg/ml aprotinin. The homogenate was centrifuged at 10,000g for 10 min. The supernatant and the pellet were used to evaluate the expression level and solubility of TTP fusion proteins.
Purification of TTP fusion proteins from E. coli
MBP-TTP fusion proteins were purified by fast protein liquid chromatography (Amersham Pharmacia Biotech) from the 10,000g supernatant with two chromatographic procedures, including amylose resin affinity and Mono Q anion-exchange chromatography in buffers with (MBP-mTTP) or without (MBP-hTTP) zinc chloride. The procedures for amylose resin and Mono Q chromatography were similar to those previously described [26,27]. Briefly, the bacterial cell supernatant containing MBP-hTTP was loaded onto an amylose resin (New England BioLab, Beverly, MA) column (2:5 × 4:5cm). The column was washed with 150 ml of amylose resin washing buffer (20 mM Tris–HCl, pH 7.4, 200 mM NaCl, 10 mM β-mercaptoethanol) and then eluted with 150 ml of amylose resin elution buffer (10 mM maltose in amylose resin washing buffer). MBP-mTTP was affinity-purified by the amylose resin column by similar procedures except that the sonication buffer contained 2 mM ZnCl2 and the amylose washing and elution buffers contained 1 mM ZnCl2.
Fractions containing MBP-hTTP from the amylose resin affinity column were centrifuged at 10,000g for 10 min before being loaded onto a Mono Q HR 5/5 column (Amersham Pharmacia Biotech). The column was washed with 5 bed vol of Mono Q buffer A (20 mM ethanolamine, 5 mM EDTA, 10 mM b-mercaptoethanol, pH 9.0) and eluted with 20 bed vol of a linear gradient from 0 to 100% of Mono Q buffer B (1 M NaCl in Mono Q buffer A). MBP-TTP fractions with the highest purity were pooled and concentrated with Amicon Centricon-10 (Amicon, Beverly, MA). The concentrated MBP-TTP fusion protein was adjusted to 20% (v/v) glycerol and stored at )70 °C. MBP-mTTP also was purified by Mono Q column in the presence of 0.1 mM ZnCl2, without EDTA, in both Mono Q buffers A and B.
MBP-TTP was separated also by size-exclusion chromatography for the determination of protein size. Fractions containing MBP-hTTP from the amylose resin affinity chromatography were centrifuged at 10,000g for 10 min before being loaded onto a Superose 12 HR 10/30 column (Amersham Pharmacia Biotech) or a Sephacryl S200 column (2:5 × 112cm) [28]. Proteins were eluted with Mono Q buffer A and analyzed by SDS–PAGE, immunoblotting using anti-MBP serum (New England BioLab), and RNA gel mobility-shift assay (GMSA) [29] using 10 μl of the fractions. The molecular masses of MBP-hTTP were determined against standard curves generated from protein standards on the same column. The protein standards (Amersham Pharmacia Biotech) used were bovine pancreas ribonuclease A (13.7 kDa), bovine pancreas chymotrypsinogen (25 kDa), hen egg ovalbumin (43 kDa), bovine serum albumin (67 kDa), rabbit muscle aldolase (158 kDa), and bovine liver catalase (232 kDa).
GST-hTTP was partially purified by glutathione Sepharose 4B beads (Amersham Pharmacia Biotech) using a procedure similar to that described previously [24]. GST-hTTP was eluted with 20 mM reduced glutathione, 120 mM NaCl, 100 mM Tris–HCl at pH 6.8, plus 20% (v/v) of glycerol. MBP was affinity-purified also from E. coli-expressing plasmid pMALc2x (New England Biolabs) by amylose resin chromatography and used as a control in the experiments.
Purification of TTP from MBP-TTP
MBP-TTP partially purified from amylose resin affinity chromatography was used for PreScission protease digestion and CEGE separation. For a small-scale digestion, MBP-hTTP (70 μg) was digested with 4 units of GST fusion PreScission protease (Amersham Pharmacia Biotech) in a total volume of 50 μl of digestion buffer containing 50 mM Tris–HCl, pH 7.0, 150 mM NaCl, 1 mM EDTA, and 1 mM dithiothreitol (DTT). The digestion reaction was incubated at 5 °C or room temperature for various times, and 10 μl of the digestion mixture was removed at each time point. The digestion reaction was terminated with 5X SDS–PAGE sample buffer (51.25% sucrose, 0.006% Pyronin Y, 6% SDS, 7.7% DTT, 60 mM EDTA) and analyzed by SDS–PAGE. The digested hTTP was identified by immuno-blotting with affinity-purified anti-GST-hTTP antibodies and by matrix-assisted laser desorption ionization (MALDI) mass spectrometry (MS) analysis (see below).
For a large-scale digestion, MBP-hTTP (2 ml of 1 mg/ ml protein in 20% glycerol) eluted from an amylose column was digested directly with 30 μl (60 units) of PreScission protease at room temperature for 17 h. The digestion reaction was terminated with 500 μl of 5X SDS–PAGE sample buffer. The digestion mixture was loaded onto a tubular SDS–PAGE gel consisting of 40 ml of 12% separating gel and 4 ml of 5% stacking gel. Human TTP was separated from other proteins by CEGE at 250 V overnight at 1 ml/min using the Model 491 Prep Cell (Bio-Rad Laboratories, Hercules, CA). The eluted proteins were collected with a fraction collector, analyzed by SDS–PAGE, and detected with silver staining [30], or immunoblotting using anti-MBP serum and affinity-purified anti-GST-hTTP antibodies. The purified hTTP protein was further identified with direct amino-terminal Edman sequencing [31].
Protein determination, SDS–PAGE, and immunoblotting
Determination of protein concentrations, SDS–PAGE, and immunoblotting followed previously described procedures [32] except that the nitrocellulose membranes were washed only with TTBS, and proteins were detected using Western Blot Chemiluminescence Reagent (NEN Life Science, Boston, MA) or Super-Signal West Pico Chemiluminescent Substrate (Pierce, Rockford, IL).
The primary antibodies used were anti-GST serum (Sigma Chemical Co., St. Louis, MO), anti-MBP serum (New England BioLab), and affinity-purified anti-GST-hTTP antibodies. Anti-GST-hTTP (DU88) was raised in rabbits against partially purified GST-hTTP eluted from glutathione beads as described before [7]. The crude antiserum was affinity-purified against GST-hTTP. Briefly, E. coli strain BL21(DE3) was transformed with expression plasmid pGST-hTTP. GST-hTTP expression was induced by IPTG. The cells were homogenized by sonication. Following centrifugation at 10,000g for 10 min, proteins in the pellet were separated by preparative SDS–PAGE and transferred onto nitrocellulose membranes. Membrane strips corresponding to GST-hTTP were excised and used to affinity purify anti-GST-hTTP from the crude antiserum following the procedures as described [33]. The secondary antibodies used were affinity-purified goat anti-rabbit IgG (H+L) horseradish peroxidase conjugate with human IgG absorbed (Bio-Rad Laboratory, using 1:5000 or 1:10,000 dilutions in TTBS).
MALDI mass spectrometry analysis and amino-terminal sequencing
For MALDI analysis, MBP-hTTP was partially purified by amylose resin affinity chromatography as described above. Following digestion with GST fusion protein PreScission protease, hTTP (calculated molecular mass: 34 kDa) was separated by 12% SDS–PAGE from the MBP fusion partner (calculated molecular mass: 41 kDa), the GST-PreScission protease (calculated molecular mass: 46 kDa), and other minor contaminating proteins in the digestion mixture. After the gel was stained with Coomassie blue, the band corresponding to the lower molecular mass band recognized by the affinity-purified anti-GST-hTTP antibodies was excised from the gel and processed by an in-gel digestion with trypsin using the Investigator ProGest Protein Digestion Station (Genomic Solutions, Ann Arbor, MI). The peptide mixtures were freeze-dried and suspended in 5 μl of acetonitrile:H2O:formic acid (45:45:10), followed by cocrystallization with a saturated MALDI matrix solution containing α-cyano-4-hydroxycinnamic acid: water: formic acid (45:45:10, v/v). The crystals were analyzed with a Voyager-DE-STR MALDI/TOF mass spectrometer (Perseptive Biosystems, Framingham, MA) as described before [34]. The ionized tryptic fragments generated from the MS analysis were used to search the NCBI NR database using Protein Prospector.
For protein sequencing, nonfusion human TTP was purified by amylose resin affinity chromatography of MBP-hTTP, PreScission protease digestion, and CEGE as described above. About 4 μg of MBP-hTTP and 0.13 μg of hTTP were directly spotted onto a square piece of PolyScreen PVDF transfer membrane (NEN Life Products). The membrane was air-dried for 10 min before being extensively washed with water for 1 h and then dried at room temperature for 10 min. The membrane containing the protein sample was loaded onto a reaction cartridge and sequenced with the amino-terminal protein sequencer (Applied Biosystems Procise Model 49X cLC Protein Sequencing System, Foster City, CA) and/or the carboxyl-terminal protein sequencer (Applied Biosystems Procise C). The sequencing information was analyzed with ABI 610A 2.1 data analysis software and FASTA programs.
RNA-binding activity and MAP kinase activity assays
For the RNA-binding activity assay, GMSA was performed following previously reported procedures [5]. Briefly, an RNA probe (about 1––200,000 cpm/reaction) was prepared from [α-32P]UTP and T7 RNA polymerase using Promega's RiboProbe in vitro Transcription System (Promega Corp., Madison, WI), with a mouse TNFα mRNA ARE region (nucleotides 128–1350 of GenBank Accession No. X02611) as the template [35]. Various amounts of GST-TTP, MBP-TTP, or TTP as indicated in the figure legends were incubated with the ARE probe at room temperature for 20 min in 1X RNA gel-shift buffer (10 mM Hepes, pH 7.6, 3 mM MgCl2, 40 mM KCl, 5% (v/v) glycerol, 0.5% (v/v) NP-40, and 2 mM DTT). Following addition of 50 μg of heparin and 1.2 μg of yeast tRNA and incubation at room temperature for 10 min, unprotected RNA was digested with 100 units of RNase T1 at 37 °C for 15 min. The reaction mixtures were separated at 250 V for 60–80 min through a 6% nondenaturing acrylamide gel in 0.4X TBE buffer (1X, 10.8% Tris, 5.5% boric acid, 20 mM EDTA, pH 8.0). The gel was dried at 80 °C for 90 min before imaging using autoradiography or phosphorimaging (Molecular Dynamics, Sunnyvale, CA) for quantitation.
For the MAP kinase phosphorylation assays, MBPTTP or MBP alone was purified from E. coil by amylose resin columns and then used for phosphorylation reactions with recombinant His-tagged rat p42 (ERK2) MAP kinase, GST-tagged mouse p38 MAP kinase, or calmodulin-binding peptide (24 amino acid residues)-tagged rat JNK purified from E. coli (Calbiochem, La Jolla, CA). The p42 MAP kinase protocol was similar to a previous procedure [11]. The phosphorylation reaction mixture contained 25 mM Tris–HCl, pH 7.4, 10 mM MgCl2, 1 mM DTT, 0.1 mM EGTA, 0.1 mM Na3VO4, and 10–100 μM [γ-32P]ATP or [γ-33P]ATP (specific activity was about 2000 cpm/pmol), variable amounts of the fusion protein, and 0.01–0.1 lg of p42 MAP kinase. The protocols for p38 MAP kinase and JNK were similar to the one described before [29]. The phosphorylation reaction mixture contained 25 mM Hepes, pH 7.6, 10 mM Mg acetate, 10–50 μM [γ-32P]ATP, or [γ-33P]ATP (specific activity was about 5–10,000 cpm/ pmol), variable amounts of the fusion protein, and 0.5 μg of p38 MAP kinase or 0.2 μg of JNK. The phosphorylation reactions were initiated by the addition of labeled ATP, incubated at 30 °C for various times, and terminated by the addition of 1/5 vol of 5X SDS–PAGE sample buffer. The labeled protein was separated from free ATP by 10% SDS–PAGE and detected with X-ray film and Phosphorimager for quantitation.
Results
Expression of recombinant TTP in E. coli
TTP expressed in E. coli as a GST fusion protein was shown in previous studies to be extensively degraded and mostly insoluble [11,12]. We found that significant amounts of full-length GST-hTTP (calculated Mr 61,324) accumulated in the soluble fraction of E. coli extracts by lowering the expression temperature to 22, 25, or 30 °C and by controlling the IPTG induction time; nonetheless, the majority of GST-hTTP was found in the 10,000g pellet at 22 and 25 °C (Fig. 1A). More soluble GST-hTTP was obtained under the induction conditions of 16 h at 22 °C, 4 and 16 h at 25 °C, and 3 and 4 h at 30 °C than under other conditions (Fig. 1A).
Fig. 1.
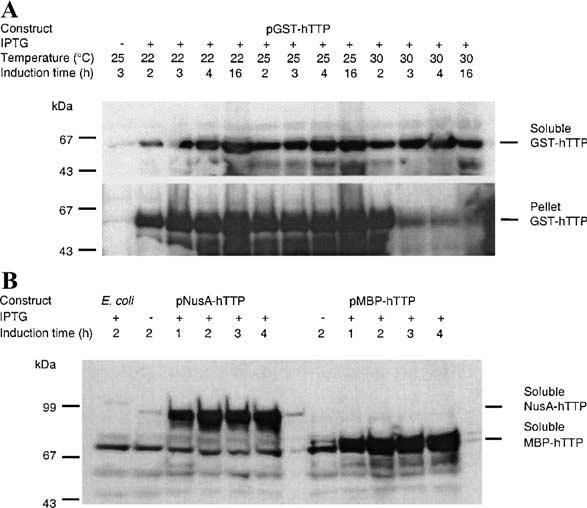
Expression of recombinant hTTP in E. coli. E. coli BL21(DE3) cells were transformed with expression plasmids pGST-hTTP, pMBP-hTTP, and pNusA-hTTP. The fusion proteins were induced by 0.1, 0.4, and 0.4 mM IPTG, respectively, at 22, 25, 30 °C (for pGST-hTTP), and 28 °C (for pMBP-hTTP and pNusA-hTTP) for various times as indicated. (A) GST-hTTP in the 10,000g supernatant and the pellet from the same volume of homogenate were analyzed by immunoblotting using anti-GST serum. Immunoreactive GST-hTTP is indicated. (B) MBP-hTTP and NusA-hTTP in the 10,000g supernatant were detected by immunoblotting using affinity-purified anti-GST-hTTP, as indicated. Similar results were observed using plasmid pMBP-mTTP (data not shown). Lanes labeled E. coli and no IPTG(−) as two negative controls were soluble extracts from E. coli cells without plasmids and E. coli cells with plasmids but without IPTG induction, respectively.
To increase the yield of soluble TTP fusion proteins, we used the MBP and NusA fusion systems, which have been reported to promote the solubility of polypeptides to which they are fused [13,14]. A protein band with the predicted molecular mass of MBP-hTTP (calculated Mr 75,265) could be readily detected on Coomassie blue-stained gels in the 10,000g supernatant of IPTG-induced cells (data not shown). The presence of MBP-hTTP was confirmed by immunoblotting using affinity-purified anti-GST-hTTP antibodies (Fig. 1B). The expression and solubility of MBP-mTTP (calculated Mr 74,875) were similar to MBP-hTTP (data not shown). NusA-hTTP (calculated Mr 90,482) also was expressed well in E. coli with significant solubility (Fig. 1B).
Purification of recombinant TTP from E. coli
We partially purified GST-hTTP from the 10,000g supernatant using glutathione Sepharose 4B beads (Fig. 2A), although considerable GST-hTTP was still associated with the beads following eight successive elutions (data not shown). However, GST-hTTP eluted from the beads (Fig. 2A) rapidly precipitated, and was not used in further purification steps (data not shown).
Fig. 2.
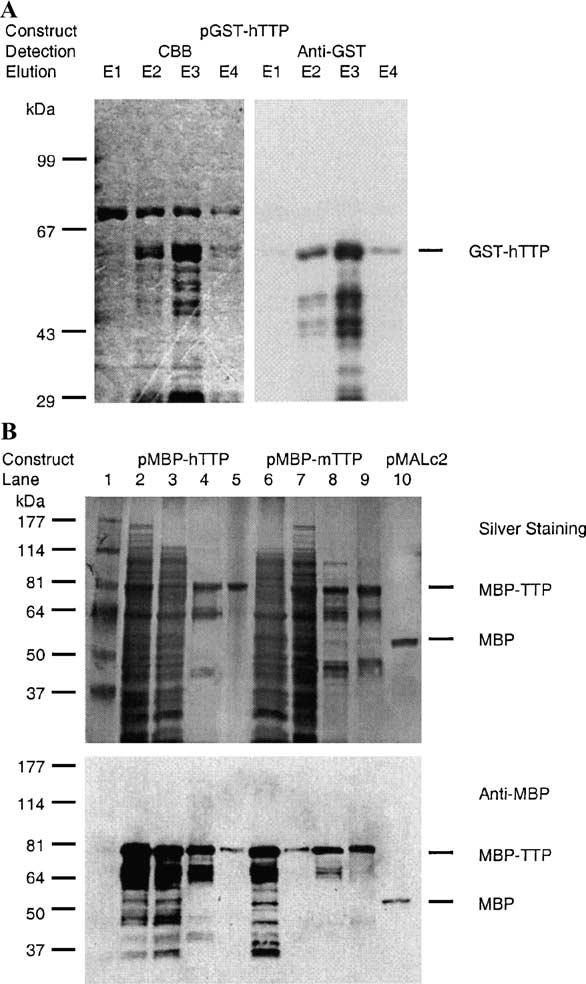
Purification of recombinant TTP fusion proteins and MBP from E. coli. GST-hTTP, MBP-hTTP, MBP-mTTP, and MBP were induced by IPTG in E. coli as described in the legend for Fig. 1. (A) GST-hTTP was affinity-purified with glutathione Sepharose 4B beads and eluted from the beads four times successively, designating elutions 1, 2, 3, and 4 as E1, E2, E3, and E4. The eluted proteins were separated with 8% SDS–PAGE and detected with Commassie brilliant blue (CBB) staining (left panel) or immunoblotting with anti-GST serum (right panel). Immunoreactive GST-hTTP is indicated. (B) MBP-hTTP, MBP-mTTP, and MBP were initially affinity-purified from the 10,000g supernatant by an amylose resin affinity column and eluted with 10 mM maltose. MBP-hTTP (left panel) and MBP-mTTP (right panel) from the amylose resin column were further purified using a Mono Q column. Top panel: 10% SDS–PAGE separation and silver staining. Lane 1, protein molecular size standards; lane 2, total E. coli homogenate expressing MBP-hTTP (50 μg of protein); lane 3, S10,000g supernatant (50 μg); lane 4, peak fraction eluted from the amylose resin column (5 μg); lane 5, peak fraction eluted from the Mono Q column (1 μg). Lane 6, total E. coli homogenate expressing MBP-mTTP (50 μg); lane 7, S10,000g supernatant (50 μg); lane 8, peak fraction eluted from the amylose resin column (5 μg); lane 9, peak fraction eluted from the Mono Q column (5 μg); and lane 10, the peak fraction of MBP from amylose resin column (5 μg). The positions of purified MBP-TTP (both hTTP and mTTP) and MBP are indicated. Bottom panel: Immunological detection of MBP-hTTP, MBP-mTTP, and MBP by anti-MBP serum. The samples were identical to those used in the upper panel except that 10% of the amount of protein used in the upper panel was used.
The MBP-TTP protein was chosen for further purification because of the availability of the amylose resin for affinity purification of MBP fusion proteins as well as its higher level of expression compared to NusA-TTP. About 30 mg of total protein was eluted from the amylose resin affinity column using a cell extract from 1 liter of E. coli transformed with pMBP-hTTP. The majority of the eluted protein was MBP-hTTP, although the eluted fractions also contained other proteins (Fig. 2B, lane 4). MBP-hTTP was further purified by Mono Q anion-exchange column (Fig. 2B, lane 5), although the protein eluted in a broad range of fractions corresponding to 0.15 to 0.60 M NaCl (data not shown). The identity of MBP-hTTP in the eluted fractions was confirmed by immunoblotting using anti-MBP serum (Fig. 2B). This purification procedure was also used for MBP-mTTP purification with buffers containing zinc chloride and yielded similar results (Fig. 2B, lanes 6–9). It appears that MBP-TTP was purified to a higher level of purity in buffers without zinc than with zinc (Fig. 2B, lanes 5 vs 9). MBP (calculated Mr 51,929 from pMALc2) could be purified also from E. coli by a single step using the amylose resin column (Fig. 2B, lane 10).
Purification of TTP from the recombinant MBP fusion protein
MBP-hTTP eluted from the amylose resin affinity column was digested with PreScission protease in a time-dependent manner; more than 80% of the full-length MBP-hTTP was digested after an 18-h incubation at either 5 or 24 °C (H. Cao and P.J. Blackshear, unpublished results). The degree of protease digestion was similar in buffers with or without protease inhibitors including 1 mM PMSF and 2 μM leupeptin (Fig. 3, left panel).
Fig. 3.
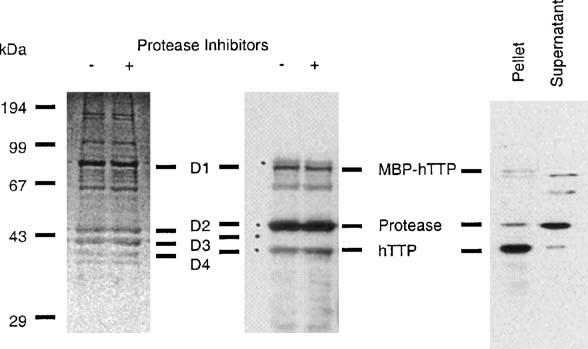
PreScission protease digestion of MBP-hTTP and precipitation of hTTP following the digestion. Left panel: MBP-hTTP from the amylose resin column was digested at 5 °C for 18 h in the digestion buffer with or without 1 mM PMSF and 2 μM leupeptin (protease inhibitors), separated by 12% SDS–PAGE, and stained with CBB. Middle panel: Proteins were digested, separated by 12% SDS–PAGE, transferred onto nitrocellulose membranes, and detected with anti-GST-hTTP antibodies. The four major protein bands (D1–D4) detected by Ponceau S staining before immunoblotting are indicated. The bands corresponding to MBP-hTTP, the PreScission protease, and hTTP are indicated. Right panel: MBP-hTTP was purified by amylose resin affinity chromatography and digested with PreScission protease overnight. The digestion mixture was centrifuged at 20,000g for 5 min. Proteins in the supernatant and pellet fractions were separated by 12% SDS–PAGE and immunoblotted with anti-GST-hTTP antibodies. The bands corresponding to MBP-hTTP, the PreScission protease, and hTTP are indicated.
Previous reports have shown that TTP migrates with an apparent molecular weight of about 43 kDa on SDS gels [5]. It was therefore important to determine which band on SDS–PAGE was hTTP before further purification. The identities of hTTP (calculated molecular mass: 34 kDa), MBP (calculated molecular mass from pMBP-TTP: 41 kDa), and GST fusion PreScission protease (calculated molecular mass: 46 kDa) were determined by immunoblotting with a combination of anti-MBP serum, affinity-purified anti-GST-hTTP antibodies, and MALDI mass spectrometry. Anti-GST-hTTP recognized protein bands D1 (undigested MBP-hTTP), D2, and D4 (Fig. 3, middle panel) and anti-MBP recognized D1 and D3, MBP (data not shown). The protein band D4 was further identified as hTTP by MALDI mass spectrometry. Nine tryptic fragments of protein band D4 had masses (m/z) of 915.5, 1218.4, 1230.4, 1235.4, 1522.6, 1748.5, 1835.5, 2083.5, and 2306.5 Da. These fragments corresponded to the tryptic fragments of hTTP 315–321, 183–194, 124–134, 183–194, 256–270, 66–82, 135–149, 83–103, and 162–182, respectively (GenBank Accession No. NP_003398). The tryptic peptide identified by MALDI MS corresponding to amino acid residues 315–321 indicated that hTTP was intact at the carboxyl-terminus. However, we were unable to purify the released hTTP from the digestion mixture by conventional chromatographic procedures, including Mono Q anion-exchange, Superdex 75 size-exclusion, Superose 12 size-exclusion, and Sephasil C4 reverse-phase columns, due to its extensive precipitation following the protease digestion (Fig. 3, right panel).
We further purified hTTP from the PreScission pro-tease-digested mixture using CEGE (Fig. 4). Human TTP was purified to homogeneity in fractions 40 to 46 (Fig. 4A). When a second similar purification was analyzed (Fig. 4B), anti-GST-hTTP antibodies recognized the undigested MBP-hTTP (lane L), the GST PreScission protease (lanes L and 82), and hTTP (lanes L, 30, and 37). A mixture of anti-GST-hTTP and anti-MBP antibodies recognized an additional band between the GST PreScission protease and hTTP bands (lane 52). The identity of the purified hTTP was confirmed by amino-terminal sequencing. The first 13 amino acid residues of hTTP were determined to be GPDLTAIYE SLLS, with the first two amino acid residues from the PreScission protease site in the vector and residues 3–13 represented the 2–12 residues of human TTP (GenBank Accession No. NP_003398).
Fig. 4.
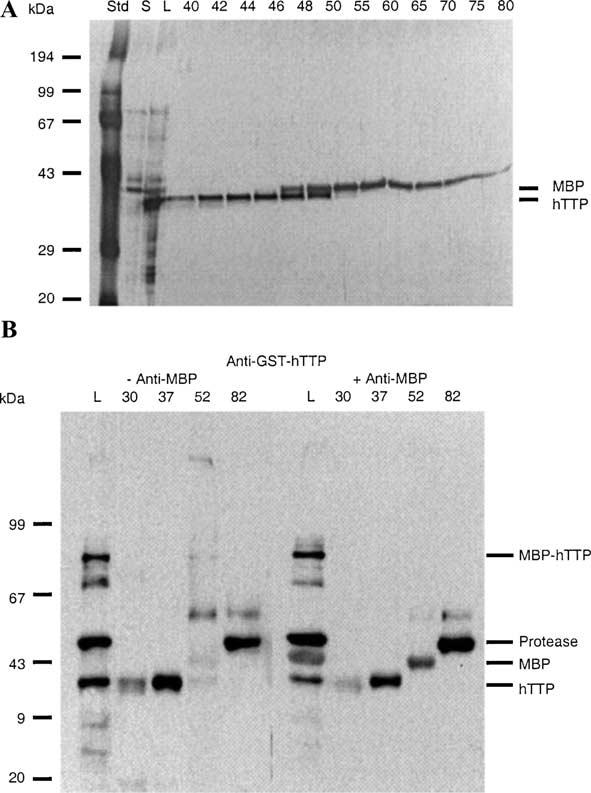
Purification of hTTP by continuous-elution gel electrophoresis following PreScission protease digestion. Proteins (2 mg in 2 ml) from an overnight digestion as described in the legend of Fig. 3 were separated by 12% SDS–PAGE and eluted continuously with SDS–PAGE running buffer at 1 ml/min. Eluted proteins were collected in 5-ml fractions. (A) Proteins in the digestion mixture (L; 1 μl) and selected fractions between 40 and 80 (20 μl) were separated by 12% SDS–PAGE and visualized by silver staining. The positions of purified hTTP and MBP are indicated. S, the 10,000g supernatant of the protease digestion mixture. (B) Similar fractions from another experiment were concentrated and proteins (1/3 of the concentrated protein per lane) were used for immunoblotting with anti-GST-hTTP antibodies (left panel) or a mixture of anti-GST-hTTP plus anti-MBP antibodies (right panel). Note that the anti-GST-hTTP antibodies recognized both the bottom hTTP band and the top GST fusion protease band whereas the anti-MBP antibody recognized the middle MBP band. L, the protease digestion mixture.
RNA-binding activity of recombinant TTP
TTP has been shown to bind to the AREs of TNFα, GM-CSF, and IL3 mRNAs in an assay using soluble extracts from transfected human cells [5-8]. We therefore investigated the ARE-binding activity of the purified recombinant TTP using the labeled TNFα mRNA ARE as a probe. Partially purified GST-hTTP was active at binding the mRNA probe, resulting in complexes similar to those formed with HA-tagged hTTP expressed in human 293 cells (Fig. 5A); these cells do not normally express detectable amounts of TTP mRNA [7]. MBP-hTTP from the amylose resin affinity column also was able to bind to the ARE probe (Fig. 5B). Similar results were obtained using MBP-mTTP (data not shown). However, the highly purified MBP-hTTP from the Mono Q column purification was unable to bind to the probe (data not shown, also refer to Fig. 7), probably due to the high concentration of EDTA (5 mM) in the Mono Q buffers, which we show below inhibited ARE-binding activity (refer to Fig. 7).
Fig. 5.
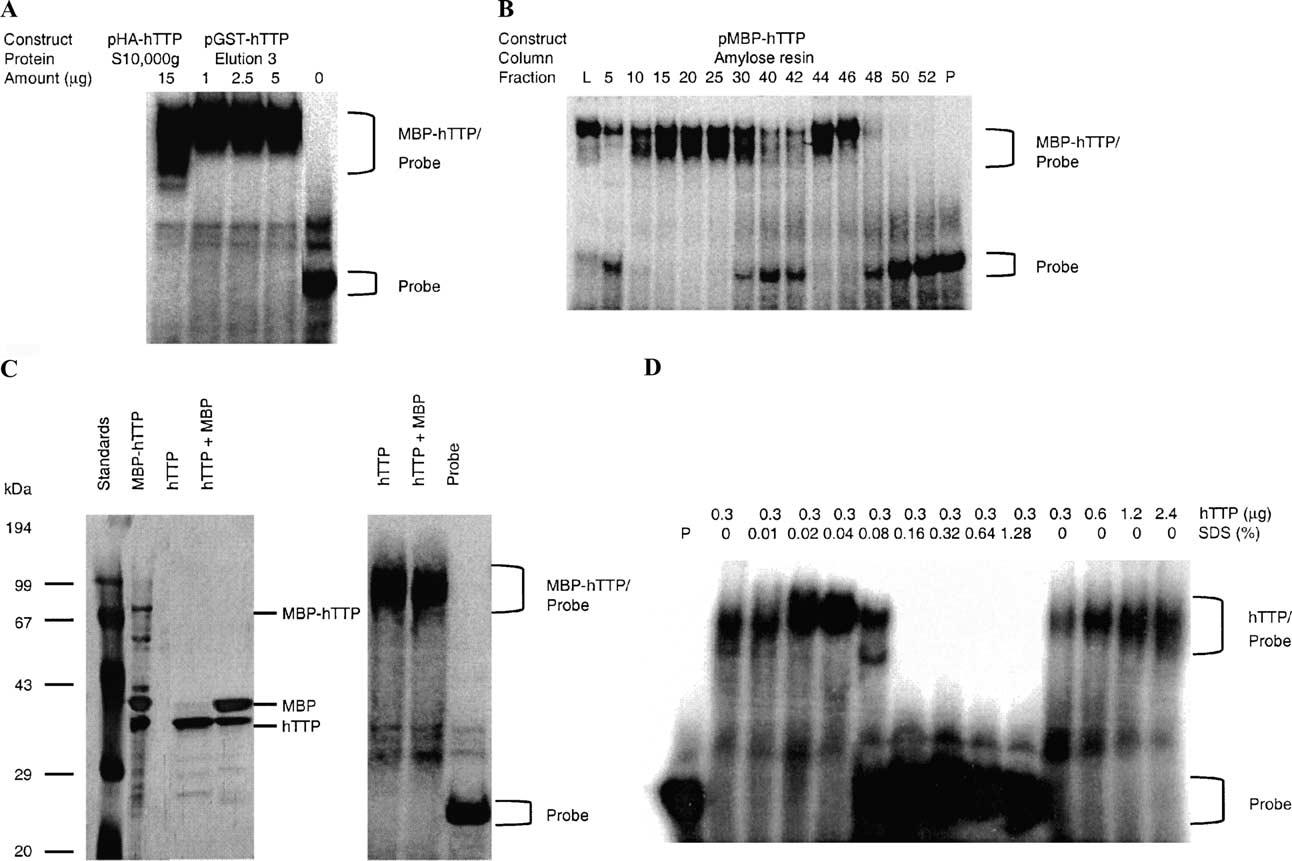
ARE-binding activity of GST-hTTP, MBP-hTTP, and hTTP. (A) GST-hTTP-binding activity. Various amounts (0, 1, 2.5, and 5 μg) of GST-hTTP in the third elution from glutathione-Sepharose 4B beads (E3 in Fig. 2A) were used for TNFα mRNA ARE-binding activity assays. As a comparison, HA-hTTP (15 μg of total protein in the 10,000g supernatant) from transfected human HEK 293 cells was used. (B) MBP-hTTP-binding activity. The ARE-binding activity of MBP-hTTP was determined using the TNFα mRNA ARE probe and 1 μl of amylose resin fractions 5 to 25 (unbound), fractions 30 to 42 (wash), and fractions 44 to 52 (elution). L, load (the 10,000g supernatant); P, probe control. Similar results were obtained using MBP-mTTP (data not shown). No activity was observed with fractions from E. coli cells transformed with pMALc2 (gel not shown). (C) Purified hTTP-binding activity. Human TTP was cleaved from MBP-hTTP by PreScission protease and separated from other proteins by CEGE. The CEGE-purified hTTP and the digested MBP-hTTP were separated by 10% SDS–PAGE, silver-stained (left panel), and used in the ARE-binding assay using the TNFα mRNA probe (right panel). (D) Effect of SDS on hTTP's ARE-binding activity. Human TTP purified by CEGE was used for ARE-binding activity assay in buffers containing various amounts of hTTP without SDS (right panel) or various concentrations of SDS with the same amount of hTTP (left panel) as indicated. P, probe control.
Fig. 7.
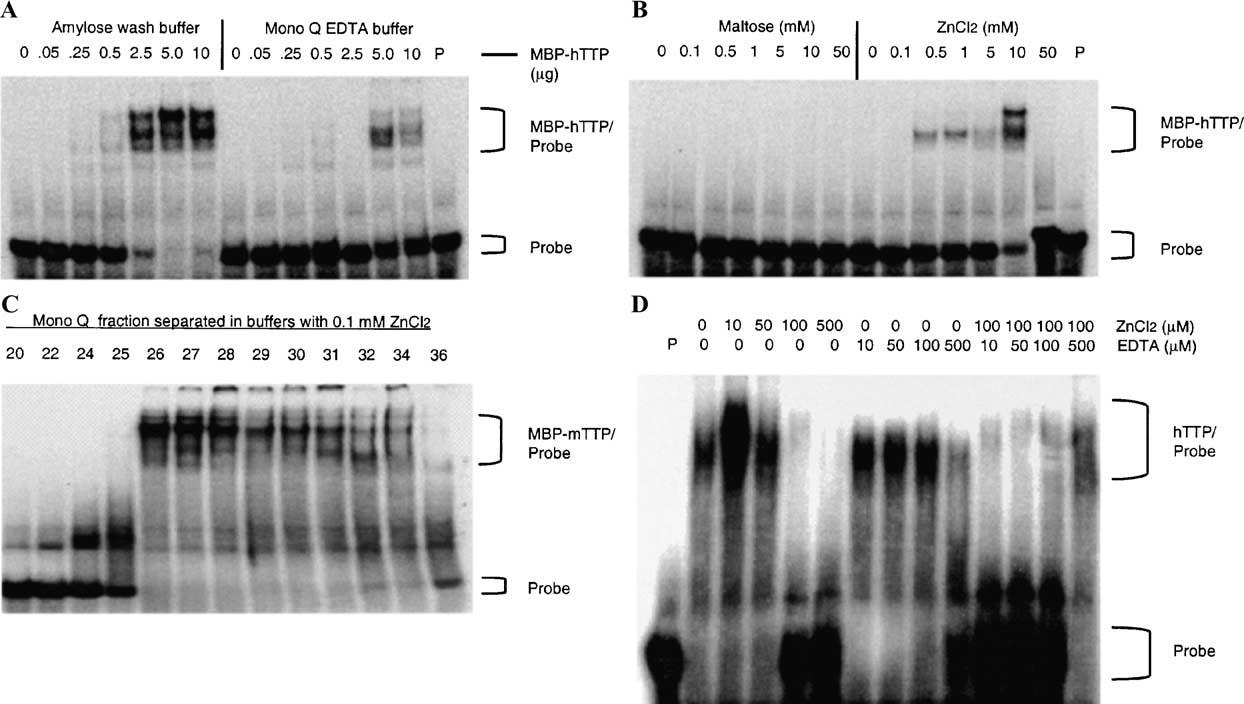
Effect of zinc on the ARE-binding activity of MBP-TTP and hTTP. (A) Effect of EDTA on the ARE-binding activity of MBP-TTP. ARE-binding activity was determined using various amounts of MBP-hTTP (as indicated in the figure) eluted from the amylose resin column and diluted with either amylose wash buffer with 10 mM maltose or Mono Q buffer A with 5 mM EDTA. P, probe alone. (B) Effect of zinc on the ARE-binding activity of inactive MBP-TTP. The ARE-binding activity was determined by using the same amount of inactive MBP-hTTP (10 μl) from the Mono Q fractions in Mono Q buffer that were supplemented with various amounts of maltose or ZnCl2, as indicated. P, probe alone. (C) Purification of active MBP-TTP by Mono Q column with zinc-containing buffer. MBP-mTTP from amylose resin was separated by Mono Q column in buffers containing 0.1 mM ZnCl2 and no EDTA. MBP-mTTP ARE-binding activity was determined with GMSA using the TNFα mRNA ARE probe. (D) Effect of zinc and EDTA on ARE-binding activity of hTTP. CEGE-purified hTTP (0.3 μg) was used for ARE-binding activity assay in buffers containing various concentrations of ZnCl2, EDTA, and mixtures of ZnCl2 and EDTA as indicated. P, probe control.
The ARE-binding activity of hTTP after PreScission protease cleavage, SDS-denaturation, and CEGE purification was also investigated. Human TTP could be separated from MBP and the other proteins in the digestion mixture by CEGE (Fig. 5C). Somewhat surprisingly, this SDS-denatured and purified hTTP was able to bind to the ARE probe in assay buffers containing a final concentration of 0.025% SDS (Fig. 5C). Therefore, we did more extensive GMSA using the purified hTTP, and summarized the results in Fig. 5D. Our results showed that the ARE-binding activity of hTTP purified by CEGE was dependent on the amount of hTTP used in the binding assay (Fig. 5D, right panel). Furthermore, this ARE-binding activity was actually increased by lower concentrations of SDS (0.02–0.04%) but was completely inhibited by higher concentrations of SDS (0.16–1.28%) in the assay buffer (Fig. 5D, left panel).
Size of active recombinant TTP
Most of the MBP-TTP fusion protein was recovered in the void volume of Superose 12 size-exclusion column (data not shown), suggesting that MBP-TTP may exist as a larger complex under these conditions. When MBP-hTTP (50 mg) eluted from amylose resin was subjected to chromatography on Sephacryl S-200, it separated into one sharp peak (Peak 1) of apparent Mr 400,000 and one broad protein peak (Peak 2) of apparent Mr 70,000–160,000 (Fig. 6A). Both peaks contained full-length MBP-hTTP, as determined by SDS–PAGE and immunoblotting with anti-MBP antibodies (Figs. 6B and C). However, ARE-binding activity was only detected in fractions from Peak 1 (Mr about 400,000) and in the earlier portion of Peak 2 (Peak 2a, Fig. 6A) (Mr about 150,000); no activity was detected in the later portion of Peak 2 (Peak 2b, Fig. 6A) (Mr about 70, 000), a size close to that of MBP-hTTP monomer (Fig. 6D).
Fig. 6.
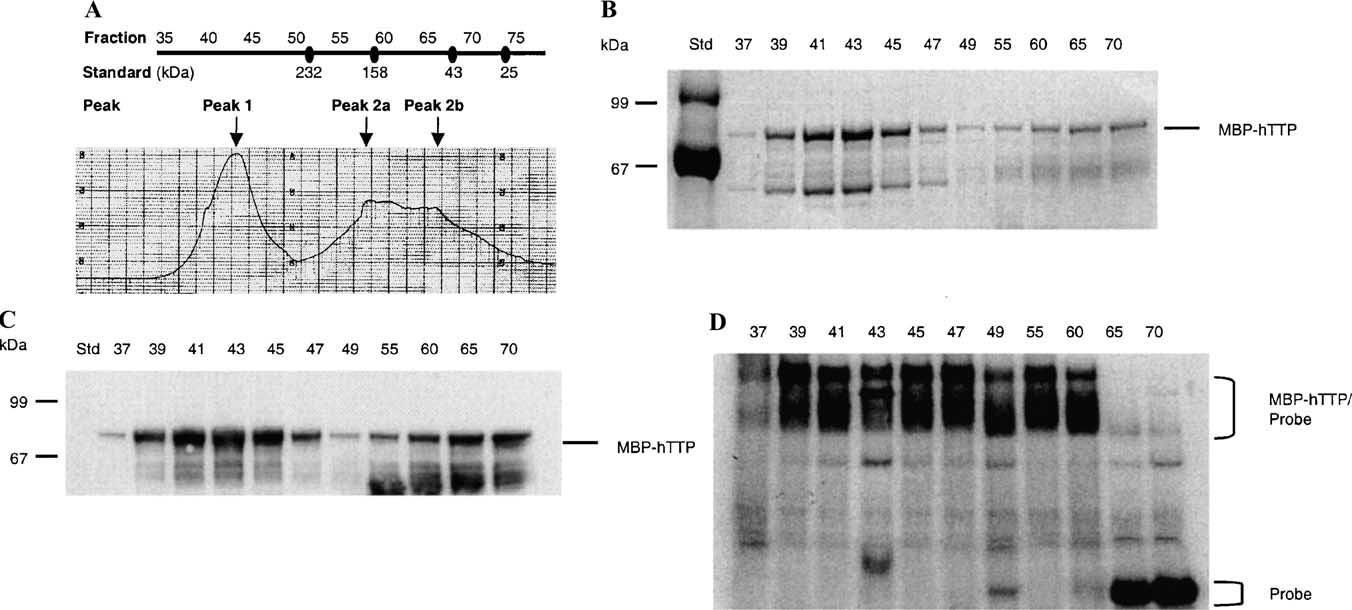
ARE-binding activity of MBP-hTTP peaks from gel-filtration chromatography. (A) Chromatogram. MBP-hTTP (50 mg) was partially purified by amylose resin chromatography and applied to a Sephacryl S200 column (2:5 × 112 cm). Fraction numbers are indicated on the top. The protein peaks (at 280 nm) were labeled as Peak 1, Peak 2a, and Peak 2b. The approximate molecular masses of proteins were determined using a standard curve generated on the same column. The standard protein sizes are indicated below the fraction numbers and include bovine pancreas ribonuclease A (13.7 kDa), bovine pancreas chymotrypsinogen (25 kDa), hen egg ovalbumin (43 kDa), bovine serum albumin (67 kDa), rabbit muscle aldolase (158 kDa), and bovine liver catalase (232 kDa). (B) SDS–PAGE. Selected protein fractions from the column were separated with 10% SDS–PAGE and stained with CBB. The position of MBP-hTTP is indicated. (C) Immunoblotting. MBP-hTTP in the column fractions was identified by immunoblotting using anti-MBP serum. (D) ARE-binding activity. ARE-binding activity of the column fractions indicated was assayed using the TNFa mRNA probe.
Requirement of zinc for ARE-binding activity of recombinant TTP
Previous studies have indicated that recombinant TTP can bind zinc [1]. We therefore investigated whether zinc is required for TTP's mRNA ARE-binding activity. We found that the ARE-binding activity of MBP-hTTP eluted from the amylose affinity column was much higher in buffers containing 10 mM maltose but no EDTA than in buffers containing 5 mM EDTA but no maltose (Fig. 7A). This result suggested that the ARE-binding activity of MBP-hTTP was decreased either by removing maltose or zinc or some other divalent cation from MBP-hTTP in the assay buffer. The RNA-binding activity of the inactive MBP-hTTP purified by Mono Q column chromatography could be partially recovered by addition of 0.5–10 mM ZnCl2; but not maltose, to the assay mixtures (Fig. 7B). In contrast to the purification of MBP-hTTP in buffers without ZnCl2, MBP-mTTP eluted from the Mono Q column in buffers containing 0.1 mM ZnCl2 was recovered in 8 fractions instead of 40 fractions in the absence of zinc (data not shown). Under these conditions, the mTTP fusion protein was able to bind to the ARE probe in the gel-shift assay (Fig. 7C). Finally, we tested the effect of zinc and EDTA on TTP's ARE-binding activity using the CEGE-purified hTTP free of the MBP fusion partner. The ARE-binding activity of the purified hTTP was significantly increased by 10 μM ZnCl2 but was decreased by higher concentrations of zinc in the binding buffer (Fig. 7D, left panel). Furthermore, this binding activity was almost completely inhibited by 500 μM EDTA in the binding buffer (Fig. 7D, middle panel). In addition, the inhibition of hTTP's mRNA ARE-binding activity by 100 lM ZnCl2 was neutralized by 500 μM EDTA in the same assay buffers (Fig. 7D, right panel). Finally, our recent results showed that the effect of zinc on TTP's mRNA ARE-binding activity was also observed when using 6 His-tagged hTTP partially purified from human HEK 293 cells (H. Cao and P.J. Blackshear, unpublished results).
Phosphorylation of recombinant TTP by MAP kinases in vitro
MBP-TTP (both MBP-hTTP and MBP-mTTP) partially purified from E. coli by amylose resin affinity columns was used to investigate the phosphorylation of TTP by MAP kinases. MBP-TTP could be phosphorylated by p42, p38, and JNK MAP kinases (Fig. 8A). However, MBP was not phosphorylated by the MAP kinases, and MBP-TTP was not autophosphorylated under these conditions (Fig. 8A). Apparent autophosphorylation of p38 and JNK protein occurred (Fig. 8A), as described previously on p38 autophosphorylation [29] and on JNK autophosphorylation from the manufacturer's information (Calbiochem). As an example, we tested the stoichiometry of phosphorylation of MBP-hTTP and MBP-mTTP by p42 MAP kinase under these in vitro assay conditions. The gel slices corresponding to the bands of the phosphorylated MBP-hTTP and MBP-mTTP were cut out from the dried gels and the radioactivity was counted. According to this calculation, following background correction using gel slices from lanes with MBP as a substrate, approximately 1.5 to 2 pmol of phosphate was incorporated into each pmol of the fusion proteins under these assay conditions.
Fig. 8.

Phosphorylation of MBP-TTP (both hTTP and mTTP) by MAP kinases. (A) MBP-TTP (1 μg) was partially purified by amylose resin column and used as a substrate for p42 (0.1 μg), p38 (0.5 μg), or JNK (0.2 μg). The reactions in 20 μl were performed at 30 °C for 30 min. (B) Time course (left panel, plots 1–6) and substrate concentration dependence (right panel, plots 7–12) of the phosphorylation reactions. The reactions were performed at 30 °C for various times using 1 μM MBP-TTP as the substrate in 150 μl (10 μl per time point) (left panel), or at 30 °C for 15 min using various amounts of MBP-TTP in 20 μl (right panel). The results were quantified by Phosphorimager, analyzed by ImageQuant 5.1, and plotted by SigmaPlot 8.0. The data presented show the results of single determinations for plots 1–4 and 9, the means of results of two determinations for plots 5–7, 11, and 12, and the means of results of four determinations for plots 8 and 10. The standard deviations for the means were within 20% of the means. The MBP-TTP concentration was estimated by designating the purity of the MBP-TTP purified from the amylose resin affinity column (Fig. 2B, lanes 4 and 8) as 70%. The ATP used in the reactions for p42, p38, and JNK was [γc-32P]ATP, [γ-32P]ATP, and [γ-33P]ATP, respectively.
When 1 μM MBP-TTP was used as a substrate, half-maximal phosphorylation was achieved within 15 min and phosphorylation reached maxima at about 30 min at 30 °C for p42, p38, and JNK. When various amounts of MBP-TTP were used as a substrate in the reactions, performed in the linear portion of the time courses (15 min), half-maximal phosphorylation occurred at approximately 0.5, 0.25, and 0.25 μM for p42, p38, and JNK kinases under these assay conditions (Fig. 8B). The amount of MBP-TTP phosphorylation by the three kinds of MAP kinases decreased with extended reaction times (Fig. 8B, left panel) as well as with high concentrations of protein substrates (Fig. 8B, right panel). Similar findings have been reported with other protein kinases, and we did not pursue further the cause(s) for the decreased phosphorylation in this study.
A previous study from our group determined that dephosphorylated TTP bound an ARE probe with greater affinity than the protein phosphorylated in intact 293 cells [29]. To test if the phosphorylation of recombinant TTP by MAP kinases had any effect on the ability of MBP-TTP to bind to the TNFα mRNA ARE, we performed GMSA following in vitro phosphorylation. The ARE-binding activity of MBP-mTTP was shown first to be dependent on the amount of MBP-mTTP used in the binding assay (Fig. 9A). Using three concentrations of phosphorylated MBP-mTTP, based on the above results, we found no major difference in RNA binding between MBP-mTTP phosphorylated with p42, p38, or JNK MAP kinases and the unphosphorylated controls under these assay conditions (Fig. 9B and data not shown). However, relatively minor quantitative changes in affinity upon phosphorylation cannot be ruled out, and await the development of more accurate assays.
Fig. 9.
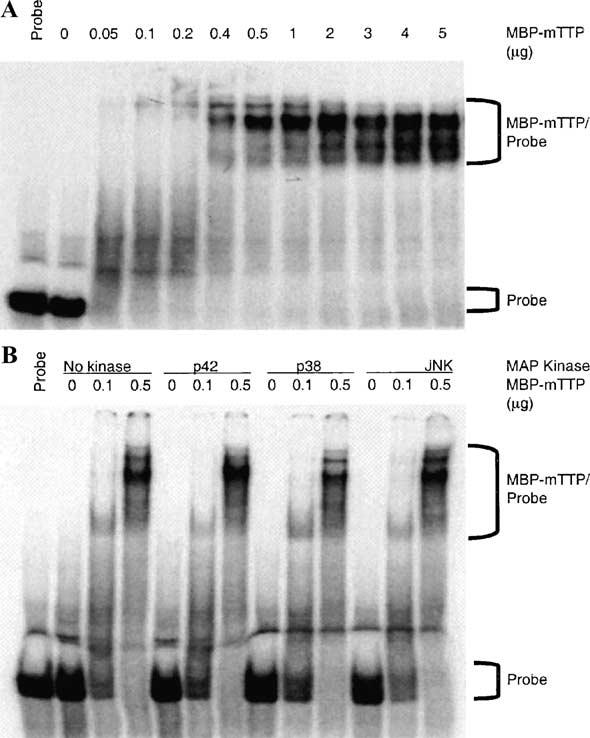
Effect of phosphorylation of MBP-mTTP by MAP kinases on its ARE-binding activity. MBP-mTTP partially purified from the amylose resin column was used for in vitro phosphorylation and GMSA. (A) The binding activity of various amounts of MBP-mTTP is shown. (B) Based on the results in A, two concentrations of MBP-mTTP and the control without MBP-mTTP were selected for phosphorylation by p42, p38, or JNK kinases, under the same conditions as described in Fig. 8. The reaction mixtures were then used for ARE-binding assays under conditions identical to those used in A.
Discussion
In this study we describe a purification procedure for recombinant TTP that exhibits zinc-dependent RNA-binding activity. Although this purification procedure requires SDS denaturation, it results in TTP free from fusion partners that nonetheless can be used to study TTP's RNA-binding activity. This has proven difficult in the past because of TTP's propensity to precipitate in both the presence and the absence of fusion partners [11,12].
In attempts to produce recombinant TTP for biochemical studies, we tried several different fusion protein systems and purification schemes including GST, MBP, and NusA fusions, as well as S-tag and His-tag proteins. Previous studies demonstrated that TTP expressed in E. coli as a GST fusion protein was degraded and precipitated extensively [11,12]. We found that the insolubility of GST-hTTP could be improved by lowering the expression temperature and optimizing the IPTG treatment. Very recently, it was reported that the insolubility of GST-TTP also could be improved by cotransforming E. coli cells with a second plasmid encoding the E. coli GroES/EL chaperone complex [36]. However, the partially purified GST-hTTP in our study became insoluble and was not digested to a significant extent by PreScission protease or thrombin (data not shown), making it unsuitable for further studies.
Human TTP fused at its carboxyl-terminus to E. coli MBP and NusA was more soluble, and both fusion proteins were expressed well in E. coli. We chose to pursue further studies on the purification of active MBP-TTP because of the availability of amylose resin affinity columns. The MBP-TTP fusion protein could be purified by a combination of affinity and conventional chromatographic procedures and could be digested readily with PreScission protease. The released TTP precipitated quickly, but could be resolubilized by SDS denaturation and then purified to homogeneity by CEGE. Both the CEGE-purified TTP and the purified MBP-TTP fusion protein were active in that they could bind to a TNFα mRNA ARE probe in a gel mobility-shift assay. These results suggested that fusion with MBP promoted the solubility of both human and mouse TTP. Similar results were obtained using a related protein, mouse ZFP36L1 (data not shown). These data are in agreement with earlier observations that MBP can act as a “molecular chaperone” that promotes the solubility and stability of proteins that are fused to it [13,15].
The partially purified GST-TTP and MBP-TTP and the highly purified hTTP were able to bind to the TNFα mRNA ARE probe, forming protein/RNA complexes similar to those observed in extracts from human HEK 293 cells expressing TTP. Somewhat surprisingly, the SDS-denatured TTP that was purified away from its fusion partner, MBP, was still capable of binding to the same probe. This result is in consistent with previous findings that SDS-denatured protein/enzymes can sometimes recover activity following SDS–PAGE, either by washing the gel with a buffer [24] or by transferring the SDS-denatured protein onto nitrocellulose membranes [37,38]. In addition, we found that the ARE-binding activity of hTTP was somewhat stimulated by low concentrations of SDS but was completely destroyed by high concentrations of SDS in the binding buffer. SDS stimulation of TTP's ARE-binding activity at low concentrations also was seen in experiments using 6 His-tagged hTTP partially purified from transfected human HEK 293 cells (H. Cao and P.J. Blackshear, unpublished results). The reason for the SDS stimulation is unknown at the present. However, we speculate that some SDS might help in folding correctly those hTTP molecules that might have been precipitated following purification and concentration.
It is not known whether TTP binds to mRNA as a monomer or oligomer. MBP-TTP purified from the amylose affinity column was resolved into one sharp peak, corresponding in size to large oligomers (Mr about 400,000), and one broad protein peak, corresponding in size to dimers and monomers. Unexpectedly, the smallest size of MBP-TTP detected in the second peak was unable to bind to the ARE probe, whereas MBP-TTP in the first peak and in the early portion of the second peak was able to bind to the same probe. One possible interpretation of these results is that MBP-TTP monomer is not able to bind to the TNFα mRNA ARE, and that dimerization or polymerization is necessary for mRNA-binding activity. The fact that the recombinant TTP/probe complexes were detected by GMSA as multiple bands on native gels also may indicate the existence of multiple forms of active recombinant TTP. A number of MBP fusion proteins have been shown to be dimers, tetramers, or larger forms [16,17,20,21]. Although MBP alone has been reported to exist in solution as a monomer [16], studies in which dimerization of MBP fusion protein occurred attributed the dimer formation to the protein moiety rather than the MBP moiety [16]. Finally, it was shown that MBP did not affect the dimerization of the fusion protein SarR-MBP [18]. Further studies will be required to determine which molecular architecture is essential for TTP binding to its target mRNA.
We also found that zinc was essential for the ARE-binding activity of TTP. First, the binding activity of MBP-hTTP was decreased in buffers containing EDTA. Second, the addition of zinc converted the nonbinding MBP-hTTP to an active binding protein. Third, active (in terms of ARE binding) MBP-mTTP could be purified by Mono Q chromatography in buffers containing zinc ions but not in buffers containing EDTA. Finally, the ARE-binding activity of the CEGE-purified hTTP free of MBP moiety was increased by low concentration but was inhibited by high concentration of ZnCl2 in the binding assay buffer. The effect of zinc on TTP's mRNA ARE-binding activity was obtained also when using 6 His-tagged hTTP partially purified from transfected human HEK 293 cells (H. Cao and P.J. Blackshear, unpublished results). These results are consistent with previous data on zinc binding. The primary amino acid sequence of TTP contains two tandem Cys3His repeats [4]. Recombinant TTP was previously shown to bind zinc [1], and each Cys3His finger in synthetic or recombinant peptides can bind to one zinc ion with an affinity similar to that of other classical zinc-finger peptides [10]. Mutagenesis studies have showed that mutation of any one of the cysteine residues, or the histidine residue, of either of the TTP Cys3His repeats results in a mutant protein that is unable to bind to mRNA probes [7]. In studies of other types of zinc-binding proteins, it was reported that EDTA could extract zinc ion from those proteins [39], and that zinc was important for the correct folding and structural stability of various zinc finger-binding proteins [39-41]. The fact that inactive MBP-TTP was eluted from a Mono Q column in an EDTA-containing buffer as very broad peaks suggests that zinc may be required to maintain the structural integrity of TTP. The inhibitory effects of high concentrations of ZnCl2 on ARE-binding activity are in agreement with recent results using a GST-TTP fusion protein [36].
We have shown here that recombinant MBP-TTP from both human and mouse could be phosphorylated directly by JNK, p42, and p38 MAP kinases with similar time courses and with half-maximal phosphorylation occurring at approximately 0.5, 0.25, and 0.25 μM protein, respectively. The Km values of ERK2 (p42) for the His-tagged transcription factor Ets-1 [42], p38 for GST-activating transcription factor 2 [43], and JNK for c-Jun [44] have been reported to be about 19, 6.2, and 8.3–11 μM, respectively; these Km values are within the ranges observed for other protein serine/threonine kinases [45]. However, the Km values for the recombinant commercial protein kinases used in this study toward physiological substrates are not known. We showed previously that TTP could be phosphorylated in intact cells and in cell-free systems by p42 MAP kinase [11]. More recently, we showed that TTP could be used as a substrate for p38 MAP kinase in a cell-free system and suggested that TTP could be involved in signal transduction pathways downstream of p38 MAP kinase [29]. His-tagged TTP was also shown to be highly phosphorylated by p38 MAP kinase [46]; however, another group suggested that p38 MAP kinase did not phosphorylate GST-TTP [12], leading to the suggestion that TTP is unlikely to be a direct substrate of p38 MAP kinase in cells [12,47]. At least under the assay conditions used here, recombinant MBP-TTP from both human and mouse could be directly phosphorylated by p42 and p38 MAP kinases with reasonable affinities, supporting previous findings that TTP can be phosphorylated by p42 and p38 MAP kinases [11,29,46]. More complete correlation of the cell-free and intact cell phosphorylation sites will be necessary to establish whether TTP is a physiological substrate for these kinases.
Our finding that TTP can be phosphorylated by JNK is interesting in view of earlier studies showing that JNK is involved in the stabilization of IL3 mRNA in PB-3c mast cells [48] and IL2 mRNA in activated T cells [49]. However, other studies suggested that JNK was not involved in the stabilization of IL3 mRNA in NIH 3T3 cells [47], or in the stabilization of IL6 and IL8 mRNAs in Hela cells [50]. Further work will be required to determine whether JNK phosphorylation of TTP occurs in intact cells and whether this is important in modulating TTP-dependent mRNA stability.
In the present study, recombinant TTP expressed in E. coli could bind to the TNFα mRNA ARE in a gel mobility-shift assay. It is possible that TTP expressed in E. coli may represent a completely nonphosphorylated protein. We recently showed that TTP expressed in 293 cells and then dephosphorylated by calf intestinal alkaline phosphatase was able to bind more tightly an ARE probe than phosphorylated TTP [29]. Down-regulation of RNA-binding activity by phosphorylation has also been seen with the potato virus A coat protein [51] and the RNA-dependent protein kinase [52]. However, in the present study, we did not observe a major effect of TTP phosphorylation by p42, p38, or JNK MAP kinases on TTP's mRNA-binding activity under our in vitro assay conditions. We need to rigorously investigate the possibility that the ARE-binding affinity might be altered by the in vitro phosphorylation of MBP-TTP fusion protein using a more quantitative assay in the future. Since TTP appears to be extensively phosphorylated in cells, it may be that phosphorylation by one or more additional protein kinases is required to affect mRNA binding. The availability of recombinant TTP should help in the identification of the protein kinases responsible for the phosphorylation of TTP in vivo, as well as the future elucidation of TTP structure-function relationships.
Acknowledgments
We thank April Greene (AstraZeneca Pharmaceuticals) for providing us the MBP-hTTP and NusA-hTTP constructs, Wi S. Lai for providing us the mouse cDNAs A19 and TNFa, Ester Carballo-Jane for her help with the initial GMSA and p38 MAPK assays, Elizabeth A. Kennington for providing us the anti-GST-hTTP serum and technical support, Christopher H. Borchers for the MALDI-TOF MS analysis, and William C. Copeland and Kenneth B. Tomer for critical reading of the manuscript. This work was supported in part by a Cooperative Research and Development Award from Astra Zeneca Pharmaceuticals.
Footnotes
Abbreviations used: ARE, AU-rich element; CBB, Coomassie brilliant blue; CEGE, continuous-elution gel electrophoresis; DTT, dithiothreitol; GM-CSF, granulocyte-macrophage colony-stimulating factor; GMSA, gel mobility-shift assay; GST, glutathione S-transferase; IL, interleukin; IPTG, isopropylthio-β-d-galactoside; JNK, c-jun-N-terminal kinase; m/z, mass-to-charge ratio; MALDI, matrix-assisted laser desorption ionization; MS, mass spectrometry; MAP, mitogen-activated protein; MBP, maltose-binding protein; NP-40, Nonidet P-40; NusA, N utilization substance protein A; PAGE, polyacrylamide gel electrophoresis; PBS, phosphate-buffered saline; PCR, polymerase chain reaction; PMSF, phenylmethylsulfonyl fluoride; TNFα, tumor necrosis factor-α; TTBS, Tween (0.05%) in Tris-buffered saline; TTP, tristetraprolin; hTTP, human TTP; mTTP, mouse TTP.
References
- 1.DuBois RN, McLane MW, Ryder K, Lau LF, Nathans D. J. Biol. Chem. 1990;265:19185–19191. [PubMed] [Google Scholar]
- 2.Lai WS, Stumpo DJ, Blackshear PJ. J. Biol. Chem. 1990;265:16556–16563. [PubMed] [Google Scholar]
- 3.Varnum BC, Ma QF, Chi TH, Fletcher B, Herschman HR. Mol. Cell. Biol. 1991;11:1754–1758. doi: 10.1128/mcb.11.3.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lai WS, Carballo E, Thorn JM, Kennington EA, Blackshear PJ. J. Biol. Chem. 2000;275:17827–17837. doi: 10.1074/jbc.M001696200. [DOI] [PubMed] [Google Scholar]
- 5.Carballo E, Lai WS, Blackshear PJ. Science. 1998;281:1001–1005. doi: 10.1126/science.281.5379.1001. [DOI] [PubMed] [Google Scholar]
- 6.Carballo E, Lai WS, Blackshear PJ. Blood. 2000;95:1891–1899. [PubMed] [Google Scholar]
- 7.Lai WS, Carballo E, Strum JR, Kennington EA, Phillips RS, Blackshear PJ. Mol. Cell. Biol. 1999;19:4311–4323. doi: 10.1128/mcb.19.6.4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lai WS, Blackshear PJ. J. Biol. Chem. 2001;276:23144–23154. doi: 10.1074/jbc.M100680200. [DOI] [PubMed] [Google Scholar]
- 9.Taylor GA, Carballo E, Lee DM, Lai WS, Thompson MJ, Patel DD, Schenkman DI, Gilkeson GS, Broxmeyer HE, Haynes BF, Blackshear PJ. Immunity. 1996;4:445–454. doi: 10.1016/s1074-7613(00)80411-2. [DOI] [PubMed] [Google Scholar]
- 10.Worthington MT, Amann BT, Nathans D, Berg JM. Proc. Natl. Acad. Sci. USA. 1996;93:13754–13759. doi: 10.1073/pnas.93.24.13754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Taylor GA, Thompson MJ, Lai WS, Blackshear PJ. J. Biol. Chem. 1995;270:13341–13347. doi: 10.1074/jbc.270.22.13341. [DOI] [PubMed] [Google Scholar]
- 12.Mahtani KR, Brook M, Dean JL, Sully G, Saklatvala J, Clark AR. Mol. Cell. Biol. 2001;21:6461–6469. doi: 10.1128/MCB.21.9.6461-6469.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kapust RB, Waugh DS. Protein Sci. 1999;8:1668–1674. doi: 10.1110/ps.8.8.1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Davis GD, Elisee C, Newham DM, Harrison RG. Biotechnol. Bioeng. 1999;65:382–388. [PubMed] [Google Scholar]
- 15.Bach H, Mazor Y, Shaky S, Shoham-Lev A, Berdichevsky Y, Gutnick DL, Benhar I. J. Mol. Biol. 3122001:79–93. doi: 10.1006/jmbi.2001.4914. [DOI] [PubMed] [Google Scholar]
- 16.Kishore U, Leigh LE, Eggleton P, Strong P, Perdikoulis MV, Willis AC, Reid KB. Biochem. J. 1998;333:27–32. doi: 10.1042/bj3330027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martinez A, Knappskog PM, Olafsdottir S, Doskeland AP, Eiken HG, Svebak RM, Bozzini M, Apold J, Flatmark T. Biochem. J. 1995;306:589–597. doi: 10.1042/bj3060589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu Y, Manna A, Li R, Martin WE, Murphy RC, Cheung AL, Zhang G. Proc. Natl. Acad. Sci. USA. 2001;98:6877–6882. doi: 10.1073/pnas.121013398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barrett TJ, Sandhu NP, Tomlinson AJ, Benson LM, Subramaniam M, Naylor S, Spelsberg TC. Biochemistry. 2000;39:753–762. doi: 10.1021/bi991809v. [DOI] [PubMed] [Google Scholar]
- 20.Ohto C, Nakane H, Hemmi H, Ohnuma S, Obata S, Nishino T. Biosci. Biotechnol. Biochem. 1998;62:1243–1246. doi: 10.1271/bbb.62.1243. [DOI] [PubMed] [Google Scholar]
- 21.Martin GE, Timko MP, Wilks HM. Biochem. J. 1997;325:139–145. doi: 10.1042/bj3250139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Taylor GA, Lai WS, Oakey RJ, Seldin MF, Shows TB, Eddy RL, Jr., Blackshear PJ. Nucleic Acids Res. 1991;19:3454. doi: 10.1093/nar/19.12.3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ausubel FM, Brent R, Kingston RE, Moore DD, Smith JA, Seidman JG, Struhl K. Current Protocols in Molecular Biology. Wiley; New York: 1989. [Google Scholar]
- 24.Cao H, Imparl-Radosevich J, Guan H, Keeling PL, James MG, Myers AM. Plant Physiol. 1999;120:205–216. doi: 10.1104/pp.120.1.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cao H, Preiss J. J. Protein Chem. 1999;18:379–386. doi: 10.1023/a:1021003915810. [DOI] [PubMed] [Google Scholar]
- 26.Cao H, James MG, Myers AM. Arch. Biochem. Biophys. 2000;373:135–146. doi: 10.1006/abbi.1999.1547. [DOI] [PubMed] [Google Scholar]
- 27.Cao H, Preiss J. J. Protein Chem. 1996;15:291–304. doi: 10.1007/BF01887118. [DOI] [PubMed] [Google Scholar]
- 28.Beatty MK, Rahman A, Cao H, Woodman W, Lee M, Myers AM, James MG. Plant Physiol. 1999;119:255–266. doi: 10.1104/pp.119.1.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carballo E, Cao H, Lai WS, Kennington EA, Campbell D, Blackshear PJ. J. Biol. Chem. 2001;276:42580–42587. doi: 10.1074/jbc.M104953200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shevchenko A, Wilm M, Vorm O, Mann M. Anal. Chem. 1996;68:850–858. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- 31.Matsudaira P. J. Biol. Chem. 1987;262:10035–10038. [PubMed] [Google Scholar]
- 32.Cao H, Shannon JC. Physiol. Plant. 1997;100:400–406. [Google Scholar]
- 33.Sambrook J, Fritsch EF, Naniatis T. Molecular Cloning: A Laboratory Manual. second Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 1989. [Google Scholar]
- 34.Borchers C, Peter JF, Hall MC, Kunkel TA, Tomer KB. Anal. Chem. 2000;72:1163–1168. doi: 10.1021/ac990937m. [DOI] [PubMed] [Google Scholar]
- 35.Fransen L, Muller R, Marmenout A, Tavernier J, Van der Heyden J, Kawashima E, Chollet A, Tizard R, Van Heuverswyn H, Van Vliet A, et al. Nucleic Acids Res. 1985;13:4417–4429. doi: 10.1093/nar/13.12.4417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Worthington MT, Pelo JW, Sachedina MA, Applegate JL, Arseneau KO, Pizarro TT. J. Biol. Chem. 2002;277:48558–48564. doi: 10.1074/jbc.M206505200. [DOI] [PubMed] [Google Scholar]
- 37.van der Meer J, Dorssers L, Zabel P. EMBO J. 1983;2:233–237. doi: 10.1002/j.1460-2075.1983.tb01411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oblong JE, Lamppa GK. EMBO J. 1992;11:4401–4409. doi: 10.1002/j.1460-2075.1992.tb05540.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Day ES, Wen D, Garber EA, Hong J, Avedissian LS, Rayhorn P, Shen W, Zeng C, Bailey VR, Reilly JO, Roden JA, Moore CB, Williams KP, Galdes A, Whitty A, Baker DP. Biochemistry. 1999;38:14868–14880. doi: 10.1021/bi9910068. [DOI] [PubMed] [Google Scholar]
- 40.Cox EH, McLendon GL. Curr. Opin. Chem. Biol. 2000;4:162–165. doi: 10.1016/s1367-5931(99)00070-8. [DOI] [PubMed] [Google Scholar]
- 41.Zamble DB, McClure CP, Penner-Hahn JE, Walsh CT. Biochemistry. 2000;39:16190–16199. doi: 10.1021/bi001398e. [DOI] [PubMed] [Google Scholar]
- 42.Waas WF, Dalby KN. Protein Expression Purif. 2001;23:191–197. doi: 10.1006/prep.2001.1491. [DOI] [PubMed] [Google Scholar]
- 43.LoGrasso PV, Frantz B, Rolando AM, O'Keefe SJ, Hermes JD, O'Neill EA. Biochemistry. 1997;36:10422–10427. doi: 10.1021/bi9706778. [DOI] [PubMed] [Google Scholar]
- 44.Murray BW, Bennett BL, Sasaki DT. Methods Enzymol. 2001;332:432–452. doi: 10.1016/s0076-6879(01)32220-6. [DOI] [PubMed] [Google Scholar]
- 45.Edelman AM, Blumenthal DK, Krebs EG. Annu. Rev. Biochem. 1987;56:567–613. doi: 10.1146/annurev.bi.56.070187.003031. [DOI] [PubMed] [Google Scholar]
- 46.Zhu W, Brauchle MA, Di Padova F, Gram H, New L, Ono K, Downey JS, Han J. Am. J. Physiol. 2001;281:L499–L508. doi: 10.1152/ajplung.2001.281.2.L499. [DOI] [PubMed] [Google Scholar]
- 47.Ming XF, Stoecklin G, Lu M, Looser R, Moroni C. Mol. Cell Biol. 2001;21:5778–5789. doi: 10.1128/MCB.21.17.5778-5789.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ming XF, Kaiser M, Moroni C. EMBO J. 1998;17:6039–6048. doi: 10.1093/emboj/17.20.6039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen CY, Del Gatto-Konczak F, Wu Z, Karin M. Science. 1998;280:1945–1949. doi: 10.1126/science.280.5371.1945. [DOI] [PubMed] [Google Scholar]
- 50.Winzen R, Kracht M, Ritter B, Wilhelm A, Chen CY, Shyu AB, Muller M, Gaestel M, Resch K, Holtmann H. EMBO J. 1999;18:4969–4980. doi: 10.1093/emboj/18.18.4969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ivanov KI, Puustinen P, Merits A, Saarma M, Makinen K. J. Biol. Chem. 2001;276:13530–13540. doi: 10.1074/jbc.M009551200. [DOI] [PubMed] [Google Scholar]
- 52.Jammi NV, Beal PA. Nucleic Acids Res. 2001;29:3020–3029. doi: 10.1093/nar/29.14.3020. [DOI] [PMC free article] [PubMed] [Google Scholar]