

Abstract
The 2-aminoethylphosphonate transaminase (AEPT; the phnW gene product) of the Salmonella enterica serovar Typhimurium 2-aminoethylphosphonate (AEP) degradation pathway catalyzes the reversible reaction of AEP and pyruvate to form phosphonoacetaldehyde (P-Ald) and l-alanine (l-Ala). Here, we describe the purification and characterization of recombinant AEPT. pH rate profiles (log Vm and log Vm/Km versus pH) revealed a pH optimum of 8.5. At pH 8.5, Keq is equal to 0.5 and the kcat values of the forward and reverse reactions are 7 and 9 s−1, respectively. The Km for AEP is 1.11 ± 0.03 mM; for pyruvate it is 0.15 ± 0.02 mM, for P-Ald it is 0.09 ± 0.01 mM, and for l-Ala it is 1.4 ± 0.03 mM. Substrate specificity tests revealed a high degree of discrimination, indicating a singular physiological role for the transaminase in AEP degradation. The 40-kDa subunit of the homodimeric enzyme is homologous to other members of the pyridoxalphosphate-dependent amino acid transaminase superfamily. Catalytic residues conserved within well-characterized members are also conserved within the seven known AEPT sequences. Site-directed mutagenesis demonstrated the importance of three selected residues (Asp168, Lys194, and Arg340) in AEPT catalysis.
2-Aminoethylphosphonate (AEP) and its N-alkylated derivatives are the most abundant and ubiquitous of naturally occurring phosphonates (16). These are typically found as conjugates of glycans (7), lipids (3, 19, 31), and proteins (15), which in turn perform essential biochemical functions in specialized lower organisms. In pathogens, AEP conjugates are used for host infection and persistence. Thus, the enzymes responsible for AEP metabolism are prime targets for inhibitor development.
AEP is synthesized by a variety of organisms according to the pathway shown in Fig. 1 (6, 24, 36). Because of its natural abundance and resistance to acid-, base-, and phosphotransferase-catalyzed hydrolysis (16), soil-dwelling bacteria have acquired a unique pathway for the degradation of AEP to usable forms of carbon, nitrogen, and phosphorus (Fig. 1) (8, 18, 22, 28, 33).
FIG. 1.

Pathway of AEP synthesis. PEP, phosphoenolpyruvate.
Cloning and sequencing of genes for the Salmonella enterica serovar Typhimurium AEP pathway operon revealed a cluster of seven genes (phnR to phnX) that are activated by the Pho regulon under conditions of phosphate deprivation (18, 23; W. W. Metcalf, W. Jiang, and B. L. Wanner, unpublished data). Based on sequence similarities at the protein level, PhnR is thought to act as a transcriptional regulator (S.-K. Kim, W. Jiang, K. A. Datsenko, K.-S. Lee, and B. L. Wanner, unpublished data), PhnS is thought to act as a periplasmic binding protein, PhnT is thought to act as an ABC family traffic ATPase, and PhnU and PhnV function as the integral membrane channel proteins. PhnW and PhnX are the AEP pathway enzymes AEP aminotransferase (AEPT) (EC 2.6.1.37) and a phosphonoacetaldehyde (P-Ald) hydrolase (trivial name, phosphonatase), respectively (35). Phosphonatase has been isolated from several bacterial sources, including Salmonella serovar Typhimurium, and both its three-dimensional structure (26) and mechanism of action are well characterized (5, 13, 22, 26, 27). AEPT was not as well understood.
Previous studies of AEPT structure and catalysis have focused on the Pseudomonas aeruginosa enzyme (12, 13). In agreement with the mass predicted by its gene sequence (encoding a 147-residue protein; GenBank accession number U61982), the P. aeruginosa AEPT was reported to be a homotetramer of 16.5-kDa subunits (12). The enzyme requires the cofactor pyridoxalphosphate (PLP), has a pH optimum of 8.5 to 9.0, and is specific for pyruvate as the amino group acceptor and AEP as the amino group donor. During catalysis, the pro-S proton of AEP is abstracted (21).
Based on the phnW sequence, Salmonella serovar Typhimurium AEPT is predicted to be 367 amino acids in length, about twice the reported size of the P. aeruginosa AEPT. Sequence analysis of the Bacillus cereus AEPT gene (GenBank accession number AY077635) (4) identified a 355-amino-acid protein, similar in size to the Salmonella serovar Typhimurium AEPT. Sequence alignments revealed 40 to 52% sequence identity among different AEPT sequences. The P. aeruginosa AEPT sequence aligns with the C-terminal halves of the Salmonella serovar Typhimurium and B. cereus AEPT sequences, suggesting that they possess an N-terminal domain in addition to the C-terminal catalytic domain that they have in common. In this paper, we describe the isolation and kinetic properties of recombinant Salmonella serovar Typhimurium AEPT and provide evidence that it is a homodimer of 40-kDa subunits. Through sequence comparisons with amino acid transaminase homologues, we identified probable substrate and PLP binding residues. By using site-directed mutagenesis, we confirmed that these residues are important in catalytic functioning. Since two of the residues (D168 and K194) are located within its N-terminal half, the full-length protein is required for catalytic activity. Based on the P. aeruginosa genome sequence (30) (GenBank accession number NP 250001), the AEPT gene encodes a 371-residue protein homologous to the Salmonella serovar Typhimurium AEPT, thus providing a consistent picture of AEPT structure and catalysis. The initial sequence of the P. aeruginosa AEPT has an internal stop codon absent from the genome sequence. As in Salmonella serovar Typhimurium, the P. aeruginosa phnW and phnX genes are juxtaposed.
MATERIALS AND METHODS
AEPT activity assay.
P-Ald formation was monitored at 340 nm (ɛ = 6.2 mM−1 cm−1) using 1-ml reaction mixtures containing 20 mM AEP, 5 mM pyruvate, 0.5 mM β-NADH, 100 μM PLP, 10 U of alcohol dehydrogenase, 2 U of phosphonatase, and 5 mM MgCl2 in a 50 mM potassium salt of N-tris(hydroxymethyl)methylglycine (K+TRICINE) (pH 8.5; 25°C). AEP formation was monitored at 340 nm (ɛ = 6.2 mM−1 cm−1) using 1-ml reaction mixtures containing 2.5 mM P-Ald, 20 mM l-Ala, 0.5 mM β-NADH, 100 μM PLP, 10 U of lactate dehydrogenase, and 5 mM MgCl2 in 50 mM K+TRICINE (pH 8.5; 25°C).
AEPT preparation.
Salmonella serovar Typhimurium phnW was PCR amplified using pWM67 (18) as a template and cloned into pET3a to generate pTAS. The cloned gene was verified by DNA sequencing. A pTAS transformant of Escherichia coli BL21(DE3) was grown aerobically at 37°C in Luria-Bertani medium containing 50 μg of ampicillin/ml. Protein production was induced with 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside) ca. 13 h postinoculation when the cell density reached an optical density at 600 nm of 0.93. The cells were harvested after 4.5 h by centrifugation at 18,000 × g for 15 min and then resuspended in 140 ml of buffer (10 mM KH2PO4, 1 mM dithiothreitol, 5 μM PLP; pH adjusted to 7.5 with KOH) at 0°C. The cell suspension was passed twice through a French press at 16,000 lb/in2 and clarified by centrifugation at 18,000 × g for 30 min at 4°C. Powdered ammonium sulfate was slowly added to the supernatant with gentle stirring at 0°C to 45% saturation. The mixture was centrifuged at 18,000 × g for 30 min (4°C). The pellet was dissolved in 50 ml of buffer and dialyzed against the same buffer at 4°C. The dialysate was chromatographed on a preequilibrated 3.5- by 40-cm DEAE-cellulose column. The column was eluted with 2 liters of a linear gradient of NaCl (0 to 0.5 M) in buffer. The AEPT eluted at approximately 0.2 M NaCl. The enzyme was concentrated using an Amicon YM30 membrane and dialyzed against buffer to yield 13 mg of AEPT/g of wet cells. The sample was stored in buffer at −80°C for 2 months without significant activity loss.
N-terminal amino acid sequence determination.
Salmonella serovar Typhimurium AEPT was transferred from an unstained sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gel to a polyvinylidene difluoride membrane using the protocol provided by Novex. The N-terminal sequence was determined by Edman degradation by using an Applied Biosystems 470 gas phase protein sequencer. The N-terminal amino acid sequence is TSRNYLLLTPGP, indicating that Met1 was removed by posttranslational modification.
AEPT molecular mass determination.
The molecular mass of the AEPT monomer has a calculated value of 40,117.68 Da based on its predicted amino acid sequence (assuming loss of Met1 and determined using the program Compute pI/Mw on the ExPASy Molecular Biology Server [2]). The AEPT subunit mass was measured by SDS-PAGE (4% stacking gel and 12% separating gel) analysis carried out with commercial protein molecular weight (MW) standards. From the plot of log MW versus distance traveled, the AEPT subunit mass of 42 kDa was determined. The mass of native AEPT was estimated by using fast protein liquid chromatography gel filtration chromatography (Sephacryl S-200; buffer, 10 mM KH2PO4, 1 mM dithiothreitol, 5 μM PLP; pH adjusted to 7.5 with KOH) carried out with commercial protein MW standards. From a plot of log MW versus elution volume, the molecular mass was determined to be ∼100 kDa.
Steady-state kinetic-constant determination.
The steady-state kinetic catalytic constants Vmax and Km were measured at pH 8.5 and 25°C using the two spectrophotometric coupled assays described above. Reactions were carried out at 5 mM pyruvate and various concentrations (1 to 10 mM) of AEP, at 20 mM AEP and various concentrations (0.1 to 5 mM) of pyruvate, at 20 mM l-Ala and various concentrations (0.04 to 1 mM) of P-Ald, or at 2.5 mM P-Ald and various concentrations (0.2 to 10 mM) of l-Ala. The initial-velocity data were fitted to the Michaelis-Menten equation (9).
Initial-velocity data were also measured at various P-Ald concentrations (2.5 to 25 mM) and l-Ala concentration (5 to 40 mM). The data were plotted in double-reciprocal form to yield the parallel pattern of a ping-pong reaction. The equilibrium constant for the reaction was calculated from the Vmax and Km derived from the initial-velocity data by using the Haldane equation (equation 1) for a Bi-Bi ping-pong mechanism:
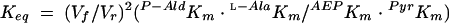 |
(1) |
where Vf is maximum velocity in the P-Ald-forming direction, Vr is maximum velocity in the AEP-forming direction, and Km is the Michaelis constant for the given substrate.
pH rate profile determination.
Initial-velocity data were measured as a function of the reaction pH by using the following buffers at the indicated pHs: 50 mM potassium salt of 2-(N-morpholino)ethanesulfonic acid (pH 6.0 and 6.5), 50 mM potassium salt of N-(2-hydroxyethyl)piperazine-N′-2-ethanesulfonic acid (pH 7.0 and 7.5), 50 mM K+TRICINE (pH 8.0 and 8.5), 50 mM potassium salt of 3-(cyclohexylamino)-2-hydroxy-1-propanesulfonic acid (pH 9.0 and 9.5), and 50 mM potassium salt of 3-(cyclohexylamino)-1-butanesulfonic acid (pH 10.0 and 10.5). The Vmax and Vmax/Km values were determined as described earlier and fitted to equation 2, 3, or 4:
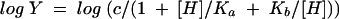 |
(2) |
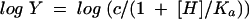 |
(3) |
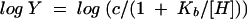 |
(4) |
where Y is Vmax or Vmax/Km, [H] is the hydrogen ion concentration, Ka is the acid dissociation constant, and Kb is the base dissociation constant.
Construction of site-directed AEPT mutants.
Mutagenesis was done using a PCR strategy (14) based on pTAS as the template, commercial primers, the PCR kit supplied by Stratagene, and the Power Block IITM System thermal cycler manufactured by ERICOMP. PCR-amplified DNAs were cloned into pET3a (Stratagene) for expression in E. coli BL21(DE3). The mutated genes were verified by DNA sequencing. The mutant proteins were purified as described above for the wild-type AEPT and were shown to be homogeneous on the basis of SDS-PAGE analysis. The yields of the pure proteins were as follows: 3.6 mg/g of cells for D168A, 10.3 mg/g of cells for K194R, 11.8 mg/g of cells for K194L, 19.1 mg/g of cells for R340K, and 4.3 mg/g of cells for R340A.
RESULTS AND DISCUSSION
Purification.
The Salmonella serovar Typhimurium LT-2 phnW gene was PCR amplified using pWM67 (18) as a template and cloned into the pET3a expression vector to generate the pTAS clone. Following induction, a pTAS transformant of E. coli BL21(DE3) yielded AEPT at 13 mg/g of wet cells. The identity of AEPT and the posttranslation removal of Met1 were confirmed by N-terminal sequencing. The enzyme was purified in two steps: ammonium sulfate precipitation followed by DEAE-cellulose chromatography (Fig. 2). The mass of the monomer was estimated to be 42 kDa (40117.68-Da theoretical mass), while the mass of the native enzyme was found to be ca. 100 kDa. These results suggest a homodimeric quaternary structure, which has been observed for structurally related amino acid aminotransferases.
FIG. 2.

Coomassie blue-stained SDS-PAGE gels of the AEP transaminase isolated at each stage of the purification procedure. Lane 1, protein standards; lane 2, total soluble protein; lane 3, AEPT fraction following the ammonium sulfate precipitation step; lane 4, combined AEPT fractions from DEAE-cellulose column.
Kinetic properties.
The reaction catalyzed by AEPT takes place in two partial reactions (Fig. 3). In the first partial reaction, a Schiff base is formed between AEP and PLP, which then undergoes hydrolysis to P-Ald and pyridoxamine. During the second partial reaction, pyruvate displaces P-Ald at the substrate binding site, where it forms a Schiff base with the pyridoxamine. The Schiff base is then hydrolyzed to form l-Ala and PLP. The plot of the reciprocal velocity versus the reciprocal P-Ald concentration measured at changing fixed l-Ala concentrations is parallel (data not shown), consistent with a Bi-Bi ping-pong kinetic mechanism. The steady-state kinetic constants measured at pH 8.5 and 25°C for homogeneous enzyme (>95% pure) for the P-Ald-forming direction are kcat, 7.4 s−1; AEP Km, 1.11 ± 0.03 mM; and pyruvate Km, 0.15 ± 0.02; and for the AEP-forming direction, they are kcat, 9.3 s−1; P-Ald Km, 0.09 ± 0.01 mM; and l-Ala Km, 1.4 ± 0.03 mM. The value Keq = [P-Ald] [l-Ala]/[AEP] [pyruvate] = 0.5 was calculated using the Haldane relationship.
FIG. 3.

Two partial reactions catalyzed by AEPT.
pH optimum.
To determine the optimal pH range for AEPT catalysis, the pH rate profiles of the AEPT-catalyzed P-Ald formation (Fig. 4A) and of AEP formation (Fig. 4B) were measured using initial-velocity techniques. For the P-Ald-forming reaction, the Vmax was constant between pH 6.5 and 9.5. The Vmax/Km value for AEP was constant between pH 8 and 9.5 but decreased with decreasing pH below pH 8. The computer fit of the Vmax/Km data gave an apparent pKa of 7.0 ± 0.2. For the AEP-forming reaction, the Vmax was highest between pH 7.5 and 8.5. The Vmax decreased below pH 7.5 and above pH 8.5. The Vmax/Km value for l-Ala reflected a narrow pH optimum, dropping both above and below pH 8. The computer fit of the Vmax data gave an apparent pKa of 6.9 ± 0.3 for the break on the acid side and an apparent pKa of 8.9 ± 0.3 for the break on the base side. From the Vmax/Km profile, these values are 8 ± 1 and 9 ± 1, respectively (however, note that pKa values that are not separated by >1 pH unit are not accurately defined).
FIG. 4.

(A) Plot of log Vmax (Vm) or log Vmax/Km AEP measured for the Salmonella serovar Typhimurium AEPT-catalyzed conversion of AEP and pyruvate to P-Ald and l-Ala at 25°C versus the reaction solution pH. (B) Plot of log Vmax and log Vmax/Km l-Ala measured for the Salmonella serovar Typhimurium AEPT-catalyzed conversion of P-Ald and l-Ala to AEP and pyruvate at 25°C versus the reaction solution pH.
The pH dependencies observed for the forward and reverse directions of the transaminase-catalyzed reactions are clearly different. This difference shows that the pH dependence arises from the requirement for specific protonation states of binding and catalytic groups rather than from a pH-induced conformational change leading to loss of activity. The differences in the pH profiles observed for the forward and reverse directions reflect the differences in ionization requirements for enzyme-substrate complexes as well as the positions of proton transfer steps relative to rate-limiting steps. Owing to the numerous proton transfer steps that are likely to occur and to the lack of structural data, it is not possible at this time to assign the measured pKa values to specific active-site groups.
Substrate specificity.
Transaminases function in amino acid metabolism, where one amino acid (the NH3 donor) is deaminated to form an α-ketoacid while a second ketoacid accepts the NH3 group to form the corresponding amino acid (29). The predominant NH3 acceptor in the cell is α-ketoglutarate (which forms glutamate). In the reaction catalyzed by the Salmonella serovar Typhimurium (this study) or P. aeruginosa (10) AEPT, pyruvate serves as the NH3 acceptor from AEP, thus forming l-Ala and P-Ald. The specificity of the Salmonella serovar Typhimurium AEPT transaminase towards other potential amino group acceptors was examined to determine if AEPT functions in a metabolic pathway in addition to the AEP degradation pathway. In particular, we were interested in the possible role of AEPT in phosphonoalanine (P-Ala) metabolism. Like AEP, P-Ala is a ubiquitous natural aminophosphonate (32). P-Ala can be synthesized from phosphonopyruvate (the phosphonate common to all phosphonate biosynthetic pathways characterized to date) via transamination and converted to phosphonopyruvate by the reverse process. Catalysis of this transamination reaction was tested using phosphonopyruvate-l-Ala and phosphonopyruvate-l-Asp reactant pairs. (A convenient assay to test the reverse direction was not available.) No detectable activity was observed (the kcat detection limit was 10−4 s−1).
To further examine the substrate specificity of AEPT, the common NH3 acceptors α-ketoglutarate and oxaloacetate were tested in the AEPT-catalyzed deamination of AEP, l-Ala, or l-Asp (Table 1). No activity was detected with oxaloacetate as the NH3 acceptor (the kcat detection limit was 10−4 s−1). While α-ketoglutarate was converted to l-glutamate with l-Ala or l-Asp serving as the NH3 donor, this occurred at a very low rate: 0.25 and 0.5% of the kcat observed with P-Ald. Thus, the Salmonella serovar Typhimurium AEPT appears to be a highly specialized transaminase functioning only in AEP metabolism.
TABLE 1.
Steady-state kinetic constants measured for Salmonella serovar Typhimurium AEPT at pH 8.5 and 25°C using alternate substrates
| NH3 acceptor | NH3 donor | Km of NH3 acceptor (mM) | Km of NH3 donor (mM) | kcat (s−1) | kcat/Km of NH3 donor (M−1 s−1) |
|---|---|---|---|---|---|
| P-Ald | l-Alaa | 0.09 ± 0.01c | 1.4 ± 0.3d | 9.3 | 6,600 |
| α-Ketoglutaratei | l-Alaa | 120e | 0.0165 ± 0.0002f | 0.025 | 1,500 |
| P-Pyrj | l-Alaa | No activity | No activity | ≤10−4 | |
| Oxaloacetate | l-Alaa | No activity | No activity | ≤10−4 | |
| P-Ald | l-Aspb | 0.66 ± 0.07g | 9 ± 2h | 0.029 | 3.2 |
| α-Ketoglutaratek | l-Aspb | 0.9 ± 0.4g | 1.6 ± 0.2f | 0.050 | 31 |
| P-Pyr | l-Aspb | No activity | No activity | ≤10−4 | |
| α-Ketoglutaratel | AEP | No activity | No activity | ≤10−4 |
The LDH/NADH coupled assay was used.
The MDH/NADH coupled assay was used.
The Km of P-Ald and was measured with 20 mM l-Ala.
The Km of l-Ala was measured with 2.5 mM P-Ald.
The estimated Km of α-ketoglutaric acid was measured with 1 mM l-Ala.
The Kms of l-Ala and l-Asp were measured with 20 mM α-ketoglutaric acid.
The Kms of P-Ald and α-ketoglutarate were measured with 20 mM l-Asp.
The Km of l-Asp was measured with 5.0 mM P-Ald.
Glutamate-pyruvate transaminase activity (EC 2.6.1.2.).
P-Pyr, phosphonopyruvate.
Aspartate transaminase activity (l-aspartate-2-oxoglutarate transaminase; l-aspartate-2-oxoglutarate transaminase; EC 2.6.1.1).
The activity was measured in the direction of P-Ald formation as described in Materials and Methods.
The stereospecificity of the pyruvate-AEP transamination reaction was examined by comparing the relative reactivity of l-Ala and d-Ala as NH3 donors in the amination of P-Ald. The kcat and Km with d-Ala as the NH3 donor were measured at various concentrations (3 to 20 mM) of d-Ala in the presence of saturating concentration (3 mM) of P-Ald (Km = 0.028 ± 0.005 mM). For the reaction of d-Ala, kcat was 0.04 s−1 and Km was 11 ± 3 mM, whereas the kinetic constants measured for l-Ala are kcat, 9.3 s−1, and l-Ala Km, 1.4 ± 0.03 mM. These results indicate a preference, but not an absolute requirement, for the l isomer.
Identification of potential catalytic groups.
A recent search of the GenBank database, using the advanced BLAST search tool (1), revealed seven probable AEPT gene sequences, including six of bacterial origin and one from Leishmania major. In L. major, as well as in at least one bacterium (Bacteroides fragilis), AEPT functions in the biosynthetic pathway where phosphoenolpyruvate is converted to AEP as shown in Fig. 1. In five other bacteria (Salmonella serovar Typhimurium, B. cereus, Vibrio cholerae, Sinorhizobium meliloti, and P. aeruginosa), AEPT probably functions in AEP degradation. Pairwise alignments between sequences revealed 30 to 64% identities. Residues conserved among all seven sequences constitute 12% identity. Among the conserved residues are the polar residues S65, N89, Y92, H140, E142, T143, D168, S171, K194, S221, Q229, T243, Y329, and R340.
Homologues that have activities different from that of AEPT include serine-pyruvate transaminases, phosphoserine transaminases, alanine-glyoxylate transaminase, aspartate transaminase from Methanobacterium thermoformicicum, isopenicillin N epimerase (gene, cefD), cystathionine synthase, and cyanobacterial soluble hydrogenase. All of these transaminases belong to a class of aminotransferase folds called subgroup IV (25). A search of the SCOP protein database using the 3D-PSSM search tool (20) identified the structural homologues phosphoserine aminotransferase and cystathionine synthase with 95% certainty. An alignment generated by ClustalW (34) of AEPT, serine-pyruvate transaminase, and phosphoserine transaminase sequences identified nine common residues, three of which (D168, K194, and R340) correspond to catalytic groups found in members of the amino acid transaminase superfamily. By analogy to the roles played by these residues in members of the transaminase superfamily, K194 may function in AEPT to bind the PLP cofactor as the Schiff base, D168 may function in H bonding to the PLP N(1)H, and R340 may function in binding the l-Ala carboxylate group (Fig. 3).
To evaluate D168, K194, and R340 as possible catalytic residues in AEPT, they were replaced by site-directed mutagenesis. The site-directed mutants, K194R, K194L, R340A, R340K, and D168A, were prepared using PCR techniques, and the mutant proteins were purified to homogeneity. The steady-state kinetic properties of the AEPT mutants are shown in Table 2. Catalytic activity in the mutant enzymes D168A, K194R, and K194L was undetectable, even when large amounts of enzyme (0.2 mg/ml) were used in the assays. Under these conditions, the detection limit for activity was ca. 10−4 s−1. The R340K and R340A AEPT mutants were partially active.
TABLE 2.
Steady-state kinetic constants for Salmonella serovar Typhimurium AEPT mutants
| AEPTa | Km of AEP (mM) | Km of pyruvate (mM) | Km of P-Ald (mM) | Km of l-Ala (mM) | kcat (s−1)→P-Aldb | kcat (s−1)→AEPc |
|---|---|---|---|---|---|---|
| Wild type | 1.11 ± 0.03 | 0.150 ± 0.02 | 0.09 ± 0.01 | 1.4 ± 0.3 | 7.4 | 9.3 |
| D168A | NAd | NA | NA | NA | NA | ≤1 × 10−4 |
| K194R | NA | NA | NA | NA | NA | ≤1 × 10−4 |
| K194L | NA | NA | NA | NA | NA | ≤1 × 10−4 |
| R340K | −e | 0.50 ± 0.05 | 2.9 ± 0.4 | 20 ± 1 | 0.2 | 0.6 |
| R340A | 26 ± 1 | 6.1 ± 0.3 | 0.19 ± 0.01 | 140 ± 70 | 0.2 | 0.2 |
The 1-ml assay solution for the direction of P-Ald formation contains 0.5 mM β-NADH, 100 μM PLP, 10 U of ADH, 2 U of phosphonatase, and 5 mM MgCl2 in 50 mM K+TRICINE (pH 8.5) at various concentrations of AEP (5 to 40 mM) with 5 mM pyruvate or various concentrations of pyruvate (0.4 to 7 mM) with 20 mM AEP. The 1-ml assay solution for the direction of AEP formation contains 0.5 mM β-NADH, 100 μM PLP, 10 U of LDH, and 5 mM MgCl2 in 50 mM K+TRICINE (pH 8.5) at various concentrations of P-Ald (0.25 to 10 mM) with 20 mM l-Ala or various concentrations of l-Ala (3 to 20 mM) with 5 mM P-Ald.
→P-Ald, in the direction of P-Ald formation.
→AEP, in the direction of AEP formation.
NA, no activity.
The Km was too large to measure.
Mutagenesis studies of the conserved Asp, Lys, and Arg have been carried out on several amino acid transaminases. The aspartate transaminase is, however, the most thoroughly investigated of the transaminases (11, 17). We compared the kinetic properties of the D222, K258, and R386 mutants of the E. coli aspartate transaminase with the kinetic properties of the corresponding D168, K194, and R340 AEPT mutants. The D222A mutant of aspartate transaminase is active, but 3,000-fold less active than the wild-type enzyme. No activity was detected (detection limit, 10−4 s−1) for the D168A AEPT mutant, indicating that the kcat is reduced by at least 100,000-fold. The K258A and K258R mutants of aspartate transaminase were found to be inactive, as were the AEPT K194L and K194R mutants. The kcat of the R386K mutant of aspartate transaminase was reduced by 55-fold, while the Asp Km increased 18-fold. The kcat of the R340K mutant was reduced 35-fold, and the AEP Km was increased 20-fold. The aspartate transaminase R386A mutant proved to be inactive, whereas for the R340A AEPT mutant, the kcat was ca. 40-fold lower than that of the wild-type enzyme and each of the substrate Km values was elevated.
Since it is has been postulated that the conserved Arg in the amino acid transaminase superfamily (R386 in the Asp transaminase) has a role as a docking site for the substrate carboxylate substituent, we surmised that in the AEPT, R340 may function in binding the carboxylate of the l-Ala. As mentioned earlier, AEPT displays stereopreference for the l-Ala enantiomers. To test the hypothesis that stereoisomer discrimination derives from the interaction of the l-Ala carboxylate with R340, the stereospecificities of the R194 mutants were measured and compared to that of the wild-type AEPT. The steady-state kinetic constants measured with l-Ala and d-Ala are listed in Table 3. As predicted, the ability of AEPT to discriminate between the l and d forms of Ala is lost in the R194 mutants.
TABLE 3.
Kinetic constants of wild-type, R340K, and R340A AEPT measured with d-Ala as the amino donor
| AEPTA | Km of d-Ala (mM)a | kcat (s−1) | kcat/Km of d-Ala (M−1 s−1) | kcat/Km of l-Ala (M−1 s−1) |
|---|---|---|---|---|
| Wild type | 11 ± 3 | 0.040 | 3.8 | 1,700 |
| R340K | 2.8 ± 0.5 | 0.011 | 4.0 | 38 |
| R340A | 3.7 ± 0.4 | 0.010 | 2.5 | 1.3 |
The Km of d-Ala was measured with various concentrations of d-Ala (3 to 20 mM) in the presence of a saturating concentration of 3 mM P-Ald (Km = 0.028 ± 0.005 mM).
Conclusions.
AEP synthesis and degradation are dependent on AEPT. We have shown that AEPT is homologous both in structure and catalytic mechanism to members of the aminotransferase family. Through demonstration of stringent AEPT substrate specificity, its singular function in AEP pathways has been shown. Thus, AEPT is a suitable target for drug development aimed at AEP-dependent microbial pathogens.
Acknowledgments
This work was supported by NIH grants GM28688 and GM57695 to D.D.-M. and B.L.W., respectively.
REFERENCES
- 1.Altschul, S. F., T. L. Madden, A. A. Schäffer, J. H. Zhang, Z. Zhang, W. Miller, and D. J. Lipman. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Appel, R. D., A. Bairoch, and D. F. Hochstrasser. 1994. A new generation of information retrieval tools for biologists: the example of the ExPASy WWW server. Trends Biochem. Sci. 19:258-260. [DOI] [PubMed] [Google Scholar]
- 3.Araki, S., S. Abe, S. Yamada, M. Satake, N. Fujiwara, K. Kon, and S. Ando. 1992. Characterization of two novel pyruvylated glycosphingolipids containing 2′-aminoethylphosphoryl(→6)-galactose from the nervous system of Aplysia kurodai. J. Biochem. (Tokyo) 112:461-469. [DOI] [PubMed] [Google Scholar]
- 4.Baker, A. S. 1996. Investigation of the enzymes of the 2-aminophosphonate degradation pathway of Salmonella typhimurium LT2 and Bacillus cereus AI-2. Ph.D. thesis. University of Maryland, College Park.
- 5.Baker, A. S., M. J. Ciocci, W. W. Metcalf, J. Kim, P. C. Babbitt, B. L. Wanner, B. M. Martin, and D. Dunaway-Mariano. 1998. Insights into the mechanism of catalysis by the P-C bond cleaving enzyme phosphonoacetaldehyde hydrolase derived from gene sequence analysis and mutagenesis. Biochemistry 37:9305-9315. [DOI] [PubMed] [Google Scholar]
- 6.Barry, R. J., E. Bowman, M. McQueney, and D. Dunaway-Mariano. 1988. Elucidation of the 2-aminoethylphosphonate biosynthetic pathway in Tetrahymena pyriformis. Biochem. Biophys. Res. Commun. 153:177-182. [DOI] [PubMed] [Google Scholar]
- 7.Baumann, H., A. O. Tzianabos, J.-R. Brisson, D. L. Kasper, and H. J. Jennings. 1992. Structural elucidation of two capsular polysaccharides from one strain of Bacteroides fragilis using high-resolution NMR spectroscopy. Biochemistry 31:4081-4089. [DOI] [PubMed] [Google Scholar]
- 8.Cassaigne, A., A. M. Lacoste, and E. Neuzil. 1976. Recherches sur le catabolisme des acids phosphoniques biodégradation de la liaison C-P par Pseudomonas aeruginosa. C. R. Acad. Sci. Ser. D. 282:1637-1639. [PubMed] [Google Scholar]
- 9.Cleland, W. W. 1979. Statistical analysis of enzyme kinetic data. Methods Enzymol. 63:103-138. [DOI] [PubMed] [Google Scholar]
- 10.Cole, S. T., and N. Honore. 1989. Transcription of the sulA-ompA region of Escherichia coli during the SOS response and the role of an antisense RNA molecule. Mol. Microbiol. 3:715-722. [DOI] [PubMed] [Google Scholar]
- 11.Cronin, C. N., and J. F. Kirsch. 1988. Role of arginine-292 in the substrate specificity of aspartate aminotransferase as examined by site-directed mutagenesis. Biochemistry 27:4572-4579. [DOI] [PubMed] [Google Scholar]
- 12.Dumora, C., A.-M. Lacoste, and A. Cassaigne. 1983. Purification and properties of 2-aminoethylphosphonate:pyruvate aminotransferase from Pseudomonas aeruginosa. Eur. J. Biochem. 133:119-125. [DOI] [PubMed] [Google Scholar]
- 13.Dumora, C., A.-M. Lacoste, and A. Cassaigne. 1989. Phosphonoacetaldehyde hydrolase from Pseudomonas aeruginosa: purification properties and comparison with Bacillus cereus enzyme. Biochim. Biophys. Acta 997:193-198. [DOI] [PubMed] [Google Scholar]
- 14.Erlich, H. A., and N. Arnheim. 1992. Genetic analysis using the polymerase chain reaction. Annu. Rev. Genet. 26:479-506. [DOI] [PubMed] [Google Scholar]
- 15.Hard, K., J. M. Van Doorn, J. E. Thomas-Oates, J. P. Kamerling, and D. J. Van der Horst. 1993. Structure of the asn-linked oligosaccharides of apolipophorin III from the insect Locusta migratoria. Carbohydrate-linked 2-aminoethylphosphonate as a constituent of a glycoprotein. Biochemistry 32:766-775. [DOI] [PubMed] [Google Scholar]
- 16.Hildebrand, R. L. 1983. The effects of synthetic phosphonates on living systems, p. 139-169. In R. L. Hildebrand (ed.), The role of phosphonates in living systems. CRC Press, Inc., Boca Raton, Fla.
- 17.Inoue, Y., S. Kuramitsu, K. Inoue, H. Kagamiyama, K. Hiromi, S. Tanase, and Y. Morino. 1989. Substitution of a lysyl residue for arginine 386 of Escherichia coli aspartate aminotransferase. J. Biol. Chem. 264:9673-9681. [PubMed] [Google Scholar]
- 18.Jiang, W., W. W. Metcalf, K.-S. Lee, and B. L. Wanner. 1995. Molecular cloning, mapping, and regulation of Pho regulon genes for phosphonate breakdown by the phosphonatase pathway of Salmonella typhimurium LT2. J. Bacteriol. 177:6411-6421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kariotoglou, D. M., and S. K. Mastronicolis. 2001. Sphingophosphonolipids, phospholipids, and fatty acids from Aegean jellyfish Aurelia aurita. Lipids 36:1255-1264. [DOI] [PubMed] [Google Scholar]
- 20.Kelley, L. A., R. M. MacCallum, and M. J. Sternberg. 2000. Enhanced genome annotation using structural profiles in the program 3D-PSSM. J. Mol. Biol. 299:499-520. [DOI] [PubMed] [Google Scholar]
- 21.Lacoste, A.-M., C. Dumora, L. Balas, F. Hammerschmidt, and J. Vercauteren. 1993. Stereochemistry of the reaction catalysed by 2-aminoethylphosphonate aminotransferase—a 1H-NMR study. Eur. J. Biochem. 215:841-844. [DOI] [PubMed] [Google Scholar]
- 22.La Nauze, J. M., H. Rosenberg, and D. C. Shaw. 1970. The enzymic cleavage of the carbon-phosphorus bond: purification and properties of phosphonatase. Biochim. Biophys. Acta 212:332-350. [DOI] [PubMed] [Google Scholar]
- 23.Lee, K.-S., W. W. Metcalf, and B. L. Wanner. 1992. Evidence for two phosphonate degradative pathways in Enterobacter aerogenes. J. Bacteriol. 174:2501-2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liang, C. R., and H. Rosenberg. 1968. The biosynthesis of the carbon-phosphorus bond in Tetrahymena. Biochim. Biophys. Acta 156:437-439. [DOI] [PubMed] [Google Scholar]
- 25.Mehta, P. K., T. I. Hale, and P. Christen. 1993. Aminotransferases: demonstration of homology and division into evolutionary subgroups. Eur. J. Biochem. 214:549-561. [DOI] [PubMed] [Google Scholar]
- 26.Morais, M. C., W. Zhang, A. S. Baker, G. Zhang, D. Dunaway-Mariano, and K. N. Allen. 2000. The crystal structure of Bacillus cereus phosphonoacetaldehyde hydrolase: insight into catalysis of phosphorus bond cleavage and catalytic diversification within the HAD enzyme superfamily. Biochemistry 39:10385-10396. [DOI] [PubMed] [Google Scholar]
- 27.Olsen, D. B., T. W. Hepburn, S. Lee, B. M. Martin, P. S. Mariano, and D. Dunaway-Mariano. 1992. Investigation of the substrate binding and catalytic groups of the P-C bond cleaving enzyme, phosphonoacetaldehyde hydrolase. Arch. Biochem. Biophys. 296:144-151. [DOI] [PubMed] [Google Scholar]
- 28.Parker, G. F., T. P. Higgins, T. Hawkes, and R. L. Robson. 1999. Rhizobium (Sinorhizobium) meliloti phn genes: characterization and identification of their protein products. J. Bacteriol. 181:389-395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schneider, G., H. Kack, and Y. Lindqvist. 2000. The manifold of vitamin B6 dependent enzymes. Structure Fold. Des. 8:R1-R6. [DOI] [PubMed] [Google Scholar]
- 30.Stover, C. K., X. Q. Pham, A. L. Erwin, S. D. Mizoguchi, P. Warrener, M. J. Hickey, F. S. L. Brinkman, W. O. Hufnagle, D. J. Kowalik, M. Lagrou, R. L. Garber, L. Goltry, E. Tolentino, S. Westbrock-Wadman, Y. Yuan, L. L. Brody, S. N. Coulter, K. R. Folger, A. Kas, K. Larbig, R. Lim, K. Smith, D. Spencer, G. K. S. Wong, Z. Wu, I. T. Paulsen, J. Reizer, M. H. Saier, R. E. W. Hancock, S. Lory, and M. V. Olson. 2000. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 406:959-964. [DOI] [PubMed] [Google Scholar]
- 31.Tamari, M. 1984. C-P compounds associated with peptide, carbohydrate and steroid, p. 195-200. In T. Hori, M. Horiguchi, and A. Hayashi (ed.), Biochemistry of natural C-P compounds. Maruzen, Ltd., Kyoto, Japan.
- 32.Tan, S. A., and L. G. Tan. 1989. Distribution of ciliatine (2-aminoethylphosphonic acid) and phosphonoalanine (2-amino-3-phosphonopropionic acid) in human tissues. Clin. Physiol. Biochem. 7:303-309. [PubMed] [Google Scholar]
- 33.Ternan, N. G., and J. P. Quinn. 1998. Phosphate starvation-independent 2-aminoethylphosphonic acid biodegradation in a newly isolated strain of Pseudomonas putida, NG2. Syst. Appl. Microbiol. 21:346-352. [DOI] [PubMed] [Google Scholar]
- 34.Thompson, J. D., D. G. Higgins, and T. J. Gibson. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673-4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wanner, B. L. 1996. Phosphorus assimilation and control of the phosphate regulon, p. 1357-1381. In F. C. Neidhardt, R. Curtiss III, J. L. Ingraham, E. C. C. Lin, K. B. Low, Jr., B. Magasanik, W. S. Reznikoff, M. Riley, M. Schaechter, and H. E. Umbarger (ed.), Escherichia coli and Salmonella: cellular and molecular biology. ASM Press, Washington, D.C.
- 36.Warren, W. A. 1968. Biosynthesis of phosphonic acids in tetrahymena. Biochim. Biophys. Acta 156:340-346. [DOI] [PubMed] [Google Scholar]