

Abstract
Toluate dioxygenase (TADO) of Pseudomonas putida mt-2 catalyzes the dihydroxylation of a broad range of substituted benzoates. The two components of this enzyme were hyperexpressed and anaerobically purified. Reconstituted TADO had a specific activity of 3.8 U/mg with m-toluate, and each component had a full complement of their respective Fe2S2 centers. Steady-state kinetics data obtained by using an oxygraph assay and by varying the toluate and dioxygen concentrations were analyzed by a compulsory order ternary complex mechanism. TADO had greatest specificity for m-toluate, displaying apparent parameters of KmA = 9 ± 1 μM, kcat = 3.9 ± 0.2 s−1, and KmO2 = 16 ± 2 μM (100 mM sodium phosphate, pH 7.0; 25°C), where KmO2 represents the Km for O2 and KmA represents the Km for the aromatic substrate. The enzyme utilized benzoates in the following order of specificity: m-toluate > benzoate ≃ 3-chlorobenzoate > p-toluate ≃ 4-chlorobenzoate ≫ o-toluate ≃ 2-chlorobenzoate. The transformation of each of the first five compounds was well coupled to O2 utilization and yielded the corresponding 1,2-cis-dihydrodiol. In contrast, the transformation of ortho-substituted benzoates was poorly coupled to O2 utilization, with >10 times more O2 being consumed than benzoate. However, the apparent Km of TADO for these benzoates was >100 μM, indicating that they do not effectively inhibit the turnover of good substrates.
Ring-hydroxylating dioxygenases catalyze the dihydroxylation of aromatic compounds. These multicomponent enzymes are key players in the microbial degradation of aromatic compounds and thus constitute an essential link in the global carbon cycle (9, 18). Moreover, ring-hydroxylating dioxygenases are important biocatalysts in a growing number of applications: the cis-dihydrodiols typically produced by these enzymes are useful chiral synthons, and these enzymes catalyze a range of other reactions of use to green chemistry (19, 28). Their versatility has also been exploited in a variety of bioremediation applications (47). Despite recent advances in the understanding of these dioxygenases, including structures of naphthalene dioxygenase (NDO) (5, 31), many important aspects of their function remain unknown.
Toluate 1,2-dioxygenase (TADO) of Pseudomonas putida mt-2 (16, 39, 49) transforms meta- and para-substituted benzoates to the corresponding cis-1,2-dihydroxycyclohexadienes (Fig. 1). TADO is encoded by the xylXYZ genes, which are part of the xyl regulon found on the pWW0 plasmid involved in the degradation of xylenes and substituted toluenes (24). Transcription of the xylXYZ genes is under control of the Pm promoter, which is activated by the XylS regulator and various benzoates that act as positive effectors (37). The nucleotide sequence of xylXYZ (25) indicates that TADO is a group II dioxygenase, a group that includes anthranilate, benzoate, and 2-halobenzoate dioxygenases (ANDO, BADO, and 2-HBADO, respectively) and other enzymes that were classified as class IB dioxygenases according to a previous classification scheme (19, 40). Accordingly, TADO comprises a two-subunit oxygenase (ISPTADO), presumably of α3β3 constitution, and a reductase (REDTADO). Sequence analyses indicate that the xylX-encoded α-subunit of ISPTADO contains a Rieske-type Fe2S2 cluster and a mononuclear iron center that is thought to be the site of toluate transformation. Similarly, REDTADO contains an FAD and a plant-type Fe2S2 cluster and presumably transfers electrons from NADH to ISPTADO.
FIG. 1.

Reaction catalyzed by toluate 1,2-dioxygenase of P. putida mt-2. TADO initiates the catabolism of substituted benzoates, catalyzing their transformation to the corresponding cis-1,2-dihydroxycyclohexadiene. TADO consists of two components: a two-subunit oxygenase, encoded by xylXY, and an oxidoreductase encoded by xylZ.
TADO elicited attention many years ago due to its substrate specificity, which is generally held to be broader than that of BADO (38, 42). Accordingly, TADO has been studied at a genetic level (26) and has been used to engineer improved pollutant-degrading characteristics in a microorganism (43). Other class IB enzymes possessing different activities were subsequently discovered. For example, ANDO and 2-HBADO preferentially transform ortho-substituted benzoates, although the latter has a preference for benzoates with electron-withdrawing substituents and catalyzes the dehalogenation of ortho-chlorinated substrates (14, 17). The availability of these related enzymes and the relative solubility of their substrates facilitate a range of studies to investigate structure-function relationships of ring-hydroxylating dioxygenases. However, TADO is relatively poorly characterized on the biochemical level. Indeed, the substrate specificity of this enzyme has never been reported despite statements concerning the nature of this specificity.
In the present study, we developed an efficient expression system for each of the TADO components, based in part on native transcriptional regulatory machinery of the xyl genes. Each component of TADO was purified in a highly active form. An oxygraph assay was used to investigate the specificity of TADO and the steady-state utilization of O2 in the presence of different substituted benzoates. Finally, the transformation products of these benzoates were identified.
MATERIALS AND METHODS
Reagents.
The following reagents were from Sigma-Aldrich (percent purity): benzoate (99.5%), o-toluate (99.5%), m-toluate (99%), p-toluate (98%), 2-Cl benzoate (98%), 3-Cl benzoate (99%), 4-Cl benzoate (97%), catechol, 3-methyl catechol, 4-methyl catechol, and catalase. 3-Cl Catechol was a gift from Victor Snieckus. Restriction enzymes, PfuI polymerase, and T4 ligase were from Amersham Pharmacia Biotech and Stratagene. Nickel-NTA Superflow resin was purchased from Qiagen. Other resins were purchased from Amersham Pharmacia Biotech. The oligonucleotides were purchased from Gibco-BRL Life Technologies. All other chemicals were of analytical grade.
Strains and plasmids.
Strains and plasmids used in this work are listed in Table 1. Escherichia coli and P. putida strains were grown at 37 and 30°C, respectively. Strains used in the propagation of DNA were grown in Luria-Bertani broth. Strains used in the expression of protein were grown in Terrific Broth (1) supplemented at 10 ml/liter with an HCl-solubilized mineral solution (48). For protein expression, 1 liter of medium in a 2-liter flask was inoculated with 10 ml of an overnight culture. Media were supplemented with 100 μg of ampicillin and/or 10 μg of tetracycline/ml as needed. ISPTADO was expressed in strain P. putida CL01 containing pVLTXYZ1. When the culture reached an optical density at 600 nm (OD600) of 0.6, IPTG (isopropyl-β-d-thiogalactopyranoside) and m-toluate were added to final respective concentrations of 0.1 and 1 mM. This amount of m-toluate was added a second time 3 h after the initial induction. REDTADO was expressed in P. putida KT2442 containing pVLTZ1. When the culture reached an OD600 of 0.6, IPTG was added to a final concentration of 0.5 mM. The culture was incubated for an additional 20 h before being harvested.
TABLE 1.
Strains and plasmids used in this studya
| Strain or plasmid | Relevant genotype/properties | Source or reference |
|---|---|---|
| Strains | ||
| E. coli DH5α | 22 | |
| E. coli GJ1158 | T7 RNAP under the control of proU | 3 |
| E. coli S17-1λpir | λpir lysogen, Smr | 10 |
| E. coli BL21(DE3)/pLysS | Stratagene | |
| P. putida KT2442 | Prototrophic, hsdR Rifr | 27 |
| P. putida CL01 | P. putida KT2442 carrying xylS, Rifr Pipr | This study |
| Plasmids | ||
| pPL392 | pBR322 containing the xyl meta operon, Apr | 23 |
| pT7-7 | T7 promoter, ColE1 origin, Apr | 46 |
| pVLT31 | Broad-host-range expression vector, Ptac promoter, RSF1010-lacIq, Tcr | 10 |
| pCNB5 | pUT mini-Tn5 delivery system, Apr Kmr | 10, 11 |
| pLEHP20 | Plac promoter, His tag expression vector, Apr | 15 |
| pBKT7-1 | T7 promoter, Apr | 32 |
| pEMBL18 | Plac promoter, Apr | 13 |
| pBKT7-S | T7 promoter, pBKT7-1 carrying xylS, Apr | 32 |
| pEMXYZ1 | pEMBL18 carrying xylXYZ, Apr | This study |
| pCNB5-S | pCNB5 carrying xylS, Apr Kmr | This study |
| pFVZ8 | pLEHP20 carrying xylZ with His tag, Apr | This study |
| pFVZ11 | pT7-7 carrying xylZ with His tag, Apr | This study |
| pVLTXYZ1 | pVLT31 carrying xylXYZ, Tcr | This study |
| pVLTZ1 | pVLT31 carrying xylZ with His tag, Tcr | This study |
Apr, ampicillin resistance; Kmr, kanamycin resistance; Pipr, piperacillin resistance; Rifr, rifampin resistance; Smr, streptomycin resistance; Tcr, tetracycline resistance.
DNA manipulation.
DNA was purified, digested, and ligated by standard protocols (44). Cells were transformed with plasmids via electroporation by using a Gene Pulser Transfection Apparatus and a Pulse Controller (Bio-Rad, Hercules, Calif.) according to the instructions of the manufacturer. Conjugal mating was performed by using standard protocols (44). PCR amplification was performed by using a Thermolyne Model DB66P25 thermocycler (Barnstead, San Diego, Calif.). Oligonucleotide-directed mutagenesis was performed by a strategy based on the elimination of a restriction site (12) and with derivatives of pEMBL18 as the template DNA. Plasmid DNA was sequenced by using an ABI model 373 Stretch DNA sequencer at the Nucleic Acid Analysis Unit at the Université Laval. Sequencing reactions were performed according to the ABI dyedeoxy terminator protocol.
General handling of proteins.
Anaerobic protein samples were handled in a glovebox maintained at <2 ppm O2 or in stoppered glass vials flushed with argon. Buffers were prepared with water purified on a Barnstead NANOpure UV apparatus to a resistivity of >17 MΩ · cm. Buffers for anaerobic procedures were filtered, bubbled vigorously with argon for 20 min, and then equilibrated in the glovebox for 24 h prior to use. Protein-containing samples were concentrated by ultrafiltration by using an Amicon stirred cell equipped with a YM10 filter. Small samples of protein were “desalted” by passage over a 1- by 8-cm column of Bio-Gel P-6 DG (Bio-Rad) equilibrated with the appropriate buffer. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was performed by using a Bio-Rad MiniPROTEAN II apparatus and staining with Coomassie blue according to established procedures (33). Protein concentrations were determined by the Bradford method (4) with bovine serum albumin as a standard. The concentration of ISPTADO was determined by using an extinction coefficient of 83.4 mM−1 cm−1 at 323 nm, calculated as described in Results.
Purification of proteins.
Cell pellets were resuspended in buffer containing 0.01 mg of DNase (Boehringer Mannheim)/ml and disrupted by a single passage through a French press (Aminco) at an operating pressure of 18,000 lb/in2. The cell debris was removed by ultracentrifugation at 170,000 × g for 75 min. The clear supernatant fluid was filtered through a 0.45-μm (pore-size) filter and transferred to a glove box. All subsequent procedures were performed anaerobically as described previously (48). High-resolution chromatographic techniques were performed by using an ÄKTA Explorer (Amersham Pharmacia Biotech).
In purifying ISPTADO, buffer A was 25 mM HEPES (pH 7.3) and contained 10% glycerol, 0.25 mM Fe(NH4)2(SO4)2, and 2 mM dithiothreitol to maximize the specific activity of the preparation. Column eluates were monitored at 280, 323, and 455 nm. The supernatant, prepared from 24 g of cells resuspended in 30 ml of 25 mM HEPES (pH 7.3)-10% glycerol, was divided into two portions, each of which was loaded onto a 2- by 9-cm column of SourceQ anion-exchange resin equilibrated with buffer A and operated at a flow rate of 20 ml/min. The oxygenase was eluted by using a linear gradient of 0.2 to 0.4 M NaCl in 20 column volumes of buffer A. Brown ISPTADO-containing fractions, which eluted at ca. 0.3 M NaCl, were pooled and concentrated to 2.0 ml. The sample was loaded onto a Superdex 200HR 26/60 column equilibrated with buffer A containing 0.15 M NaCl and operated at a flow rate of 2 ml/min. ISPTADO-containing fractions were pooled and concentrated. This sample was combined with two volumes of buffer A containing 2 M ammonium sulfate, filtered, and loaded onto a Phenyl-Sepharose column (1 by 9 cm) equilibrated with buffer A containing 1.26 M ammonium sulfate. ISPTADO was eluted by using a linear gradient of 1.26 to 0 M ammonium sulfate in 10 column volumes. Oxygenase-containing fractions, which eluted at 0.6 M ammonium sulfate, were pooled, concentrated, and exchanged into buffer A by ultrafiltration.
REDTADO was purified as a His-tagged protein in a single step by using immobilized metal affinity chromatography (IMAC). Columns of Nickel-NTA resin were prepared by using glass wool-plugged Pasteur pipettes. The volume of resin utilized was calculated to minimize the nonspecific binding of other proteins. The resin was equilibrated with 20 mM morpholinepropanesulfonic acid (MOPS; pH 8.0)-300 mM NaCl. The supernatant, prepared from 20 g of cell resuspended in 30 ml of 20 mM MOPS (pH 8.0)-300 mM NaCl was loaded onto the column. The column was washed with the same buffer containing 20 mM imidazole to remove nonspecifically bound proteins. The reductase was eluted with buffer containing 150 mM imidazole. Ht-REDTADO was concentrated and exchanged into 50 mM Tris (pH 8.0) containing 100 mM NaCl and 1 mM CaCl2 by using ultrafiltration.
XylL was overexpressed and purified as described elsewhere (Y. Ge and L. D. Eltis, unpublished data).
Determination of iron and sulfur content.
Iron and sulfur concentrations were determined colorimetrically by using Ferene S (20) and N,N-dimethyl-p-phenylenediamine (6), respectively. Sulfur assays were performed with gas-tight cuvettes. All assays were performed in duplicate. The correlation coefficients of the standard curves were at least 0.98.
Steady-state kinetic studies.
The TADO-catalyzed reaction was monitored polarographically after the consumption of O2 using a microcomputer-interfaced Clarke-type oxygen electrode essentially as described previously (48). The oxygen electrode was calibrated by using either catechol and excess catechol 2,3-dioxygenase or 2,3-dihydroxybiphenyl and excess 2,3-dihydroxybiphenyl dioxygenase. The full scale was established with buffer equilibrated with 20, 50, or 100% O2 (see below) according to the experiment. The instrument was zeroed by adding excess sodium hydrosulfite to the assay mixture. Data were recorded every 0.1 s. Initial velocities were determined from progress curves by analyzing the data by using Microsoft Excel (Redmond, Wash.).
Reaction buffers containing different concentrations of dissolved O2 were prepared by vigorously bubbling them with humidified mixtures of O2 and N2 gases for at least 15 min prior to the experiment as described previously (48). The reaction chamber was flushed continuously with the humidified gas mixture, the equilibrated buffer was transferred to the reaction chamber by using a gas-tight syringe, and the stopper was inserted into the reaction chamber.
The standard activity assay was performed in a total volume of 1.4 ml of air-saturated 100 mM phosphate buffer (pH 7.0) at 25 ± 1°C containing 430 μM NADH and 100 μM m-toluate. REDTADO (4 μl) was added to a final concentration of 2.0 μM, and the background was recorded. The reaction was initiated by injecting ISPTADO (∼4 μl) to a final concentration of 0.37 μM into the reaction chamber. One unit of enzymatic activity (U) was defined as the amount of enzyme required to consume 1 μmol of O2/min.
Analysis of steady-state data.
Steady-state kinetic data were analyzed by using an equation that describes a compulsory order ternary complex mechanism in which the binding of the aromatic substrate, A, precedes that of O2 (50) as shown below:
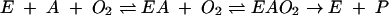 |
The steady-state rate equation derived from this mechanism is given in the following equation (7):
 |
where KmA represents the Km for the aromatic substrate, KmO2 represents the Km for O2, and KdA represents the dissociation constant for the aromatic substrate. The equation was fit to the data by using the least-squares and dynamic-weighting options of LEONORA (8). The parameters determined in this study are apparent since they depend on the concentration of REDTADO.
Coupling measurements.
Coupling experiments for all substrates were carried out by using an O2 electrode and the same conditions as the standard activity assay, except that the REDTADO, ISPTADO and substrate concentrations were 4.0, 0.74, and 215 μM, respectively. Reactions were initiated by adding ISPTADO and quenched by diluting 200 μl of the reaction mixture with 400 μl of methanol. The reaction was quenched 3 min after the initiation of the reaction or when O2 consumption stopped. The amount of aromatic substrate remaining in these assays was determined by using high-pressure liquid chromatography (HPLC) measurements, and standard curves were constructed with known amounts of substrate prepared in a 1:2 mixture of reaction buffer and methanol. The amount of hydrogen peroxide generated in coupling experiments was determined by adding 800 U of catalase to the reaction mixture at the time corresponding to the methanol quench and measuring the amount of O2 produced. Measurements were corrected for background O2 consumption and H2O2 formation.
HPLC measurements.
HPLC measurements were performed by using a Waters Millennium32 system (Waters Corp., Milford, Mass.) equipped with a Waters 2996 Photodiode Array Detector, an Alliance Waters 2695 Separations Module, and a C18 reversed-phase, Hewlett-Packard ODS hypersil column (4 by 125 mm, 5 μm). This system was interfaced to a Milliennium32 Client/Server. Samples of 10 μl were injected and eluted with 80% H2O and 20% acetonitrile at a flow rate of 1 ml/min. To identify the cis-diol transformation products of substituted benzoates, the former were further transformed to catechols by adding 1.2 μM XylL and 128 μM NAD+ to the reaction mixture. The reactions were initiated and quenched as described for the coupling assay. The retention times of transformation products were compared to those of authentic catechols.
UV/visible absorption spectroscopy.
Absorption spectra were recorded by using a Varian Cary 3 spectrophotometer equipped with a thermojacketed cuvette holder. The spectrophotometer was interfaced to a microcomputer and controlled by Cary OS/2 multitasking software. Spectra were recorded by using 25 mM HEPES buffer (pH 7.3) at 25°C and a 3-ml gas-tight cuvette (Hellma, Concord, Ontario, Canada). All measurements were performed in duplicate.
RESULTS
Construction of expression systems.
A host cell for TADO expression was constructed by inserting an inducible copy of xylS into the chromosome of P. putida KT2442. Briefly, the xylS gene was excised from pBKT7-S (32) as a ∼1.5-kb NotI fragment and was ligated into the NotI site of pCNB5 (10, 11). The resulting construct, pCNB5-S, places xylS under the control of the Ptrc promoter on a mini-Tn5 transposon. The constructed mini-Tn5 was inserted into the chromosome of P. putida KT2442 by biparental mating with E. coli S17-1 λpir containing pCNB5-S as the donor, thereby generating P. putida CL01.
The genes encoding TADO, xylXYZ, together with the Pm promoter which lies upstream of xylX, were subcloned from pPL392 (23) into pEMBL18 (13) and pVLT31 (10) as a SacI-BamHI fragment, generating constructs pEMXYZ1 and pVLTXYZ1, respectively. An NheI site was introduced at the beginning of xylZ by directed mutagenesis by using a synthetic oligonucleotide with the sequence 5′-GGCAACCTTGTGGCTAGCGGCGGCACCTCA-3′. The xylZ gene was then cloned into pLEHP20 (15) as an NheI-PstI fragment, generating pFVZ8, which contains a gene encoding an N-terminal His-tagged REDTADO (ht-REDTADO). The gene encoding ht-REDTADO was cloned into pT7-7 and pVLT31 as an XbaI-PstI fragment, generating pFVZ11 and pVLTZ1, respectively.
Expression and purification of TADO.
Among the E. coli- and Pseudomonas-based systems tested for the expression of ISPTADO, P. putida CL01 containing pVLTXYZ1 yielded the highest levels of soluble protein (results not shown). In contrast, E. coli DH5α containing pEMXYZ1 yielded approximately one-third the amount of ISPTADO, much of which was insoluble. Optimal expression of ISPTADO in P. putida was achieved by growing the cells to an OD600 of 0.6, adding 0.1 mM IPTG and 1 mM m-toluate, and then adding a further 1 mM m-toluate 3 h later. In cells harvested 24 h after the initial induction, ISPTADO constituted ca. 20% of the total cellular protein.
ISPTADO was anaerobically purified from 4 liters of P. putida CL01 containing pVLTXYZ1 by using anion-exchange, gel filtration, and hydrophobic interaction chromatographies as summarized in Table 2. A total of 39 mg of ISPTADO was purified from 24 g of cells (wet weight). The final preparation of ISPTADO was judged to be >95% pure based on a Coomassie blue-stained denaturing gel (Fig. 2). Elution of ISPTADO from the gel filtration column was consistent with it having a molecular mass of ca. 215 kDa and, therefore, an α3β3 constitution (results not shown). Purified protein flash frozen in liquid nitrogen and stored as beads at −80°C exhibited no loss of activity over 6 months.
TABLE 2.
Purification of the oxygenase component of toluate dioxygenasea
| Purification step | Total protein (mg) | Total activity (U) | Sp act (U/mg) | Yield (%) |
|---|---|---|---|---|
| Crude extract | 1,840 | 883 | 0.48 | 100 |
| SourceQ | 175 | 385 | 2.2 | 44 |
| Superdex 200HR | 104 | 260 | 2.5 | 29 |
| Phenyl-Sepharose | 39 | 148 | 3.8 | 17 |
Activity units (U) are as defined in Materials and Methods.
FIG. 2.

Denaturing gel of purified preparations of P. putida mt-2 TADO components. The lanes were loaded with 5 μg each of molecular mass standards (lanes 1), purified ISPTADO (lane 2), and purified REDTADO (lane 3). Arrows indicate the α and β subunits and the reductase.
Among the systems tested, P. putida KT2442/pVLTZ1 produced the largest quantity of ht-REDTADO (results not shown). Cells grown to an OD600 of 0.6 and incubated for a further 20 h in the presence of 0.5 mM IPTG expressed ht-REDTADO to 20% of the total cellular protein, essentially all of which was soluble. High levels of ht-REDTADO were also produced in both E. coli BL21(DE3)/pLysS and E. coli GJ1158/pFVZ11. However, under all expression protocols tested, most of the fusion protein was present in inclusion bodies in these strains.
During aerobic purification, brown solutions of ht-REDTADO turned yellow, a finding consistent with loss of the Fe2S2 cluster and oxidation of the FAD. During subsequent concentration of the ht-REDTADO-containing solution, the yellow color passed through the ultrafiltration membrane, indicating that the FAD had dissociated from the reductase. To minimize the loss of cofactors during the IMAC-based purification of ht-REDTADO, manipulations were therefore performed anaerobically as described in Materials and Methods. A total of 60 mg of fusion protein was obtained from 4 liters of culture. This was judged to be >99% pure by SDS-PAGE (Fig. 2). Ht-REDTADO was not stable in the presence of imidazole, even under anaerobic conditions; thus, it was important to rapidly remove the imidazole after IMAC. The specific activity of ht-REDTADO in the TADO oxygraph assay was essentially identical to that of a preparation of ht-REDTADO in which the His tag had been proteolytically removed. Therefore, ht-REDTADO was used in subsequent TADO assays. Purified ht-REDTADO flash frozen in liquid nitrogen and stored as beads at −80°C exhibited no detectable loss of activity over 6 months.
Characterization of the Fe2S2 clusters.
Purified ISPTADO contained 16 ± 3 mol of iron and 5.8 ± 0.4 mol of sulfur per mol of α3β3 hexamer. Passage of the ISPTADO preparation over a small desalting column equilibrated with buffer A containing no added iron did not affect these values. Preparations of REDTADO contained 2.1 ± 0.3 mol of iron and 1.9 ± 0.5 mol of sulfur per mol of reductase.
As purified here, ISPTADO absorbed maximally at 280, 323, and 455 nm, a finding typical of reduced Rieske-type Fe2S2 clusters (Fig. 3). The R value (ratio of A280/A323) was 6.7 and was unaffected by the anaerobic addition of sodium hydrosulfite, indicating that the Rieske-type Fe2S2 cluster of ISPTADO remained fully reduced during purification. ISPTADO could be oxidized by treating it with a slight excess of potassium ferricyanide or by exposing the sample to air for 20 min. The R value of oxidized ISPTADO was 4.5 (Fig. 3). These spectra are similar to those of the oxygenases of other ring-hydroxylating dioxygenases (17, 29). The extinction coefficient of ISPTADO was 83.4 mM−1 cm−1 at 323 nm (25 mM HEPES, pH 7.3; 25°C) based on its sulfur content.
FIG. 3.

Spectra of reduced (solid line) and oxidized (dotted line) purified ISPTADO. The sample cuvette contained 1.2 μM protein in 25 mM HEPES buffer (pH 7.3) at 25°C.
The absorption spectra of REDTADO was typical of class IB reductases (Fig. 4) (14, 17). As isolated here, the REDTADO absorbed maximally at 270, 340, and 455 nm in anaerobic buffer. Addition of NADH did not alter the spectrum. However, exposure of the sample to air resulted in a spectrum typical of oxidized reductases. The addition of 1 eq of NADH to the air-oxidized sample reduced the protein, as indicated by the spectrum. This indicates that the loss of the Fe2S2 cluster from the oxidized REDTADO was not immediate.
FIG. 4.

Spectra of reduced (solid line) and oxidized (dotted line) REDTADO. The sample cuvette contained 1 μM protein in 50 mM Tris, 100 mM NaCl, and 1 mM CaCl2 (pH 8.0) at 25°C.
In vitro reconstitution of TADO activity.
TADO was reconstituted in vitro, and its activity was monitored by using an oxygraph assay. At concentrations of ISPTADO ranging from 0.1 to 0.6 μM, the oxygenase could not be saturated by REDTADO, even in the presence of a 50-fold excess of the latter. Moreover, higher molar ratios of REDTADO to ISPTADO increased the rate of O2 consumption observed in the absence of aromatic substrate. Therefore, in the standard assay, the respective concentrations of REDTADO and ISPTADO were fixed at 2.0 and 0.37 μM (i.e., a molar ratio of ca. 5.4:1). Under these conditions, the activity of TADO was proportional to the concentration of ISPTADO over an order of magnitude. With 0.1 M ionic-strength phosphate buffers at 25°C, the TADO activity was highest at pH 7.0 (results not shown). The addition of iron, FAD, and/or reducing agents did not affect the activity of TADO. Under the standard assay conditions, the specific activity of TADO was 3.8 U/mg.
Steady-state kinetic studies.
To investigate the specificity of TADO, the initial rate of O2 utilization by the enzyme was determined as a function of substituted benzoate and O2 concentrations. For the substituted benzoates that were efficiently transformed by TADO, the steady-state rate equation describing a compulsory order, ternary complex mechanism (see equation, Materials and Methods) fits the initial rate data (representative data in air-saturated buffer are shown in Fig. 5), a finding consistent with the proposed mechanism of ring-hydroxylating dioxygenases (50). However, the data could not be used to exclude other steady-state mechanisms. Of the benzoates tested, TADO's preferred substrate was m-toluate, which it utilized with an apparent specificity three times higher than benzoate and five times higher than p-toluate (Table 3). The ortho-substituted benzoates were even poorer substrates for TADO. Finally, the enzyme was more specific for methylated benzoates than for the corresponding chlorinated analogues. Interestingly, the enzyme's affinity for the various substrates (reflected in the calculated values of KdA [data not shown]) varied to a much greater extent than the turnover numbers.
FIG. 5.

Steady-state dihydroxylation of m-toluate by reconstituted TADO. The dependence of the initial rate of O2 consumption on concentrations of m-toluate in air-saturated buffer (100 mM phosphate buffer, pH 7.0; 25°C). Additional experimental details are provided in Materials and Methods. The line represents the best fit of the Michaelis-Menten equation to the data. The fitted parameters were an apparent KmA of 5.3 ± 0.3 μM and apparent Vmax of 55 ± 1 μM/min.
TABLE 3.
Apparent steady-state kinetic parameters of TADO for selected substituted benzoatesa
| Substrate | KmA (μM) | KmO2 (μM) | kcat (s−1) | kcat/KmA (106 M−1 s−1) | kcat/KmO2 (106 M−1 s−1) |
|---|---|---|---|---|---|
| Benzoate | 19 (4) | 13 (2) | 2.8 (0.1) | 0.15 (0.03) | 0.22 (0.02) |
| o-Toluate | 250 (65) | 46 (10) | 2.3 (0.13)b | 0.009 (0.002)b | 0.049 (0.009) |
| m-Toluate | 9.1 (1.3) | 16 (2) | 3.9 (0.2) | 0.43 (0.04) | 0.25 (0.02) |
| p-Toluate | 28 (6) | 8.4 (1.5) | 2.5 (0.1) | 0.09 (0.02) | 0.30 (0.04) |
| 2-Cl benzoate | 144 (90) | 92 (22) | 1.9 (0.1)b | 0.013 (0.008)b | 0.021 (0.004) |
| 3-Cl benzoate | 26 (3) | 19 (6) | 3.1 (0.1) | 0.15 (0.01) | 0.21 (0.01) |
| 4-Cl benzoate | 41 (9) | 12 (2) | 3.0 (0.1) | 0.08 (0.01) | 0.25 (0.03) |
Experiments were performed with 100 mM sodium phosphate (pH 7.0) at 25°C containing 430 μM NADH. Standard errors are given in parentheses. The data sets used to calculate the parameters for different substrates contain 78 to 110 points.
Values were based on O2 consumption to be consistent with the other values in the table. Values calculated based on cis-dihydrodiol production are at least an order of magnitude lower.
The ability of TADO to utilize O2 also varied markedly with the aromatic substrate (Table 3). Thus, the dioxygenase was equally reactive with O2 in the presence of meta-substituted, para-substituted, and unsubstituted benzoates, as determined by kcat/KmO2 and, in the presence of these substrates, was essentially saturated with O2 in air-saturated buffer (KmO2 ≃ 15 μM). In contrast, the specificity of TADO for O2 was approximately five times lower in the presence of ortho-substituted benzoates. As few studies have investigated the specificity of ring-hydroxylating dioxygenases as a function of O2 concentration, the steady-state parameters of TADO for substituted benzoates in air-saturated buffers are presented in Table 4 to facilitate comparison with other enzymes.
TABLE 4.
Apparent steady-state kinetic and coupling parameters of TADO for selected substituted benzoates in air-saturated buffera
| Substrate | Km (μM) | kcat (s−1) | kcat/Km (106 M−1 s−1) | Substrate/O2 | H2O2/O2 |
|---|---|---|---|---|---|
| Benzoate | 15 (1) | 2.4 (0.1) | 0.16 (0.01) | 0.96 (0.10) | 0.03 (0.006) |
| o-Toluate | 590 (60) | 1.6 (0.1)b | 0.003 (0.0002)b | 0.10 (0.10) | 0.27 (0.02) |
| m-Toluate | 5.3 (0.3) | 2.9 (0.04) | 0.46 (0.03) | 0.92 (0.13) | 0.01 (0.003) |
| p-Toluate | 13 (1) | 1.95 (0.06) | 0.14 (0.02) | 0.90 (0.10) | 0.03 (0.005) |
| 2-Cl benzoate | 1200 (240) | 1.4 (0.1)b | 0.001 (0.0001)b | 0.10 (0.09) | 0.2 (0.05) |
| 3-Cl benzoate | 29 (2) | 1.03 (0.01) | 0.035 (0.005) | 0.94 (0.10) | 0.002 (0.0005) |
| 4-Cl benzoate | 83 (7) | 0.86 (0.03) | 0.01 (0.001) | 0.93 (0.10) | 0.03 (0.01) |
Experiments were performed with air-saturated 100 mM sodium phosphate (pH 7.0) at 25°C containing 430 μM NADH. Standard errors are given in parentheses.
Values were based on O2 consumption to be consistent with the other values in the table. Values calculated based on cis-dihydrodiol production were at least an order of magnitude lower.
Coupling of O2 consumption to toluate transformation.
To investigate whether substrate utilization is coupled in TADO, the stoichiometry of aromatic substrate and O2 consumption in the dioxygenase-catalyzed reaction was investigated. In these experiments, the amount of aromatic substrate consumed in the reaction was determined by HPLC. In the presence of saturating quantities of good substrates (i.e., benzoate and meta- and para-substituted benzoates), the amount of substrate consumed corresponded to the amount of O2 consumed (Table 4). In contrast, in the presence of the ortho-substituted benzoates, substrate utilization was significantly uncoupled: at least 10 times more O2 was consumed than benzoate. Moreover, H2O2 accounted for between 20 and 30% of the total O2 consumed (Table 4).
Identification of reaction products.
For each of the well-coupled substrates (i.e., benzoate and meta- and para-substituted benzoates), a single reaction product was detected. XylL-treated transformation products coeluted with the respective substituted catechol standards in HPLC analyses, indicating that the TADO transformation products were exclusively 1,2-dihydrodiols. No dehalogenation or demethylation products were detected. In each case, the amount of catechol detected corresponded to the amount of benzoate transformed within experimental error.
A single reaction product was also detected in the case of o-toluate. This product coeluted with the cis-dihydrodiol produced from benzoate (2.152 min) and had the same absorption spectrum as the latter (λmax = 261.5 nm). In contrast, the cis-dihydrodiols produced from m- and p-toluates had different elution times (2.168 and 2.086 min) and spectra (262.7 and 265.0 nm). Moreover, the TADO transformation product of o-toluate was quantitatively transformed by XylL to a compound that coeluted with catechol and could be further transformed by catechol 2,3-dioxygenase to yield a spectrum identical to that of 2-hydroxymuconic semialdehyde (λmax = 376 nm). The amount of catechol detected corresponded to ca. 1% of the O2 consumed. The detected cis-dihydrodiol did not arise from contaminating benzoate, since HPLC analysis indicated that a 1 mM solution of o-toluate contained <10 nM benzoate. In the case of 2-chlorobenzoate, no transformation product was detected, even when an initial concentration of 1 mM 2-chlorobenzoate was used.
DISCUSSION
Production of TADO components in suitably engineered pseudomonad strains greatly increased the amount of expressed, soluble protein, as was observed for biphenyl dioxygenase (BPDO [29]). Thus, the yield of ca. 10 mg of purified ISPTADO per liter of culture of P. putida CL01 represents a much higher yield of oxygenase than obtained from native strains grown on appropriate carbon sources or from recombinant strains of E. coli. For example, Fetzner et al. (17) obtained less than 0.5 mg of 2-HBADO per liter of culture of P. cepacia 2CBS. Although the oxygenase components of some ring-hydroxylating enzymes, including ANDO (14), have been overproduced in E. coli, the production of others appears to be limited by the formation of inclusion bodies (36, 45), as was observed in expressing xylXY in E. coli. This may reflect the inability of E. coli strains to effectively incorporate the Fe2S2 cluster into the dioxygenase components as they are translated.
Anaerobic purification is critical to obtaining highly active preparations of ISPTADO and REDTADO, as was necessary in purifying BPDO (29). It is difficult to compare the specific activity of reconstituted TADO to those of other class IB dioxygenases due to differences in assay procedures and the intrinsic enzyme activities. However, elemental analyses indicate that preparations of ISPTADO and REDTADO contained full complements of prosthetic groups. Moreover, the addition of exogenous iron or FAD did not increase the preparation's specific activity, in contrast to what has been reported for some aerobically purified enzyme preparations (14, 17, 34). The high iron content of ISPTADO may be due to nonspecific binding of iron to the oxygenase, as has been observed in 2,3-dihydroxybiphenyl dioxygenase (21) and BPDO (29). Finally, anaerobically purified preparations of ISPTADO have an R value that is up to half that of aerobic preparations of BADO and 2-HBADO (17, 52), indicating that the ISPTADO preparation contains a higher proportion of Fe2S2 clusters.
The inability to saturate ISPTADO with REDTADO, even at a 48-fold molar excess of the latter, is consistent with what has been observed for other dioxygenases, including phthalate dioxygenase and BPDO (2, 29). For BADO and ANDO, the Km values of oxygenase for the reductase were 26 and 1 μM, respectively (14, 52). It is impractical to perform steady-state kinetics at high concentrations of reductase due to the amount of protein required and the background consumption of O2. However, at the limiting concentrations of REDTADO used, TADO activity is linearly dependent on ISP concentration, and the apparent kinetic parameters obtained provide a useful means for evaluating the catalytic properties of the enzyme.
The current studies demonstrate that the transformation of ortho-substituted benzoates by TADO is severely uncoupled from O2 utilization. The degree of uncoupling indicates that the steady-state rate constants determined on the basis of O2 consumption overestimate the true values by at least 1 order of magnitude. Uncoupling has been observed in other ring-hydroxylating dioxygenases, including naphthalene dioxygenase (34) and BPDO (29), and may reflect the inability of ortho-substituted benzoates to displace solvent molecules from the active site of TADO. In principle, ortho-substituted benzoates would competitively inhibit the transformation of meta- and para-substituted benzoates. However, the relatively high Km of ISPTADO for ortho-substituted benzoates indicates that high concentrations would be required, a finding consistent with the previous finding that o-toluate does not significantly inhibit TADO activity (51). The similar elution times and spectra of the cis-dihydrodiols obtained from o-toluate and benzoate, respectively, as well as the transformation of the former to catechol by XylL, suggest that TADO catalyzes the concomitant dihydroxylation and demethylation of o-toluate. Interestingly, XylL has been reported to catalyze the demethylation of toluene cis-dihydrodiol. However, this reaction required adenosylcobalamin (35). Further characterization of these demethylation reactions is warranted.
The lack of reported specificity data for TADO or BADO precludes direct comparison of the current results with other studies. However, the maximal activity of TADO for substituted benzoates (meta > unsubstituted > para > ortho; from apparent kcat [Table 4]) differs from the relative rates reported for BADO (unsubstituted > meta > ortho > para [52]). Interestingly, the maximal activity of TADO for substituted benzoates corresponds to the relative rates of transformation of these compounds by E. coli cells containing TADO (53) but not by m-toluate-grown cells of P. putida mt-2 (unsubstituted > meta > para > ortho [38, 42, 53]). It is possible that these differences reflect differences in substrate uptake by the cells, as previously noted (42). However, it seems more likely that the differences reflect the presence of a second benzoate-transforming enzyme in m-toluate-grown cells of P. putida mt-2 (30, 39, 49). The specificity of TADO contrasts markedly to those of 2-HBADO and ANDO, which show strong preferences for ortho-substituted benzoates (14, 17). Moreover, TADO differs from 2-HBADO in that the former has no apparent ability to dehalogenate ortho-chlorinated benzoates. Highly active preparations of TADO, together with the availability of closely related enzymes with markedly different substrate specificities, should facilitate the elucidation of the structural determinants of reactivity, including specificity, in this important class of enzymes.
The specificity of TADO for methyl benzoates differs significantly from the effector preference of XylS, which is strongly activated by both meta- and ortho-substituted benzoates (meta ≃ ortho > unsubstituted ≫ para [41]). This may reflect a vestigial activity of XylS that predates its recruitment to regulate the TOL meta operon, particularly since high concentrations of o-methylated and o-chlorinated benzoates uncouple TADO. Interestingly, XylS appears to also activate the transcription of the chromosomally located BADO genes of P. putida mt-2 (30). It is thus possible that the specificity of XylS reflects a dual regulatory role. In this respect, it would be interesting to determine the substrate specificity of the chromosomally encoded BADO and the effector specificity of BenR, the primary transcriptional regulator of the chromosomal genes.
Acknowledgments
This work was supported by Natural Sciences and Engineering Research Council of Canada Operating grant 171359-99 to L.D.E.
We thank Claude Lévesque and Christian Blouin for technical assistance and Stephen Seah for useful discussions.
REFERENCES
- 1.Ausubel, F. M., R. Brent, R. E. Kinston, D. D. Moore, J. G. Seidman, J. A. Smith, and K. Struhl. 2000. Current protocols in molecular biology. J. Wiley & Sons, Inc., New York, N.Y.
- 2.Batie, J. C., E. LaHaie, and P. D. Ballou. 1987. Purification and characterization of phthalate oxygenase and phthalate oxygenase reductase from Pseudomonas cepacia. J. Biol. Chem. 262:1510-1518. [PubMed] [Google Scholar]
- 3.Bhandari, P., and J. Gowrishankar. 1997. An Escherichia coli host strain useful for efficient overproduction of cloned gene products with NaCl as the inducer. J. Bacteriol. 179:4403-4406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bradford, M. M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248-254. [DOI] [PubMed] [Google Scholar]
- 5.Carredano, E., A Karlsson, B. Kauppi, D. Choudhury, R. E. Parales, J. V. Parales, K. Lee, D. T. Gibson, H. Eklund, and S. Ramaswamy. 2000. Substrate binding site of naphthalene 1,2-dioxygenase: functional implications of indole binding. J. Mol. Biol. 296:701-712. [DOI] [PubMed] [Google Scholar]
- 6.Chen, J. S., and L. E. Mortenson. 1977. Inhibition of methylene blue formation during determination of the acid-labile sulfide of iron-sulfur protein samples containing dithionite. Anal. Biochem. 79:157-165. [DOI] [PubMed] [Google Scholar]
- 7.Cleland, W. W. 1963. The kinetics of enzyme-catalyzed reactions with two or more substrates or products. Biochim. Biophys. Acta 67:104-137. [DOI] [PubMed] [Google Scholar]
- 8.Cornish-Bowden, A. 1995. Analysis of enzyme kinetics data, Oxford University Press, New York, N.Y.
- 9.Dagley, S. 1978. Determinants of biodegradability. Q. Rev. Biophys. 11:577-602. [DOI] [PubMed] [Google Scholar]
- 10.de Lorenzo, V., L. D. Eltis, B. Kessler, and K. N. Timmis. 1993. Analysis of Pseudomonas gene products using lacIq/Ptrp-lac plasmids and transposons that confer conditional phenotypes. Gene 123:17-24. [DOI] [PubMed] [Google Scholar]
- 11.de Lorenzo, V., S. Fernandez, M. Herrero, U. Jakubzik, and K. N. Timmis. 1993. Engineering of alkyl- and haloaromatic-responsive gene expression with mini-transposons containing regulated promoters of biodegradative pathways of Pseudomonas. Gene 130:41-46. [DOI] [PubMed] [Google Scholar]
- 12.Deng, W. P., and J. A. Nickoloff. 1992. Site-directed mutagenesis of virtually any plasmid by eliminating a unique site. Anal. Biochem. 200:81-88. [DOI] [PubMed] [Google Scholar]
- 13.Dente, L., and R. Cortese. 1987. pEMBL: a new family of single-stranded plasmids for sequencing DNA. Methods Enzymol. 155:111-119. [DOI] [PubMed] [Google Scholar]
- 14.Eby, D. M., Z. M. Beharry, E. D. Coulter, D. M. Kurtz, Jr., and E. L. Neidle. 2001. Characterization and evolution of anthranilate 1,2-dioxygenase from Acinetobacter sp. strain ADP1. J. Bacteriol. 183:109-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eltis, L. D., S. Iwagami, and M. Smith. 1994. Hyperexpression of a synthetic gene encoding a high potential iron sulfur protein. Protein Eng. 7:1145-1150. [DOI] [PubMed] [Google Scholar]
- 16.Feist, C. F., and G. D. Hegeman. 1969. Phenol and benzoate metabolism by Pseudomonas putida: regulation of tangential pathways. J. Bacteriol. 100:869-877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fetzner, S., R. Muller, and F. Lingens. 1992. Purification and some properties of 2-halobenzoate 1,2-dioxygenase, a two-component enzyme system from Pseudomonas cepacia 2CBS. J. Bacteriol. 174:279-290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gibson, D. T. 1987. Microbial metabolism of aromatic hydrocarbons and the carbon cycle, p. 33-58. In S. R. Hagedorn, R. S. Hanson, and D. A. Kunz (ed.), Microbial metabolism and the carbon cycle. Harwood Academic Press, Chur, Switzerland.
- 19.Gibson, D. T., and R. E. Parales. 2000. Aromatic hydrocarbon dioxygenases in environmental biotechnology. Curr. Opin. Biotechnol. 11:236-243. [DOI] [PubMed] [Google Scholar]
- 20.Haigler, B. E., and D. T. Gibson. 1990. Purification and properties of NADH-ferredoxinNAP reductase, a component of naphthalene dioxygenase from Pseudomonas sp. strain NCIB 9816. J. Bacteriol. 172:457-464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Han, S., L. D. Eltis, K. N. Timmis, S. W. Muchmore, and J. T. Bolin. 1995. Crystal structure of the biphenyl-cleaving extradiol dioxygenase from a PCB-degrading pseudomonad. Science 270:976-980. [DOI] [PubMed] [Google Scholar]
- 22.Hanahan, D. 1983. Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol. 166:557-580. [DOI] [PubMed] [Google Scholar]
- 23.Harayama, S., P. R. Lehrbach, and K. N. Timmis. 1984. Transposon mutagenesis analysis of meta-cleavage pathway operon genes of the TOL plasmid of Pseudomonas putida mt-2. J. Bacteriol. 160:251-255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harayama, S., and M. Rekik. 1990. The meta cleavage operon of TOL degradative plasmid pWW0 comprises 13 genes. Mol. Gen. Genet. 221:113-120. [DOI] [PubMed] [Google Scholar]
- 25.Harayama, S., M. Rekik, A. Bairoch, E. L. Neidle, and L. N. Ornston. 1991. Potential DNA slippage structures acquired during evolutionary divergence of Acinetobacter calcoaceticus chromosomal benABC and Pseudomonas putida TOL pWW0 plasmid xylXYZ genes, encoding benzoate dioxygenases. J. Bacteriol. 173:7540-7548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harayama, S., M. Rekik, and K. N. Timmis. 1986. Genetic analysis of a relaxed substrate specificity aromatic ring dioxygenase, toluate 1,2-dioxygenase, encoded by TOL plasmid pWW0 of Pseudomonas putida. Mol. Gen. Genet. 202:226-234. [DOI] [PubMed] [Google Scholar]
- 27.Herrero, M., V. V. de Lorenzo, and K. N. Timmis. 1990. Transposon vectors containing non-antibiotic resistance selection markers for cloning and stable chromosomal insertion of foreign genes in gram-negative bacteria. J. Bacteriol. 172:6557-6567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hudlicky, T., D. Gonzalez, and D. T. Gibson. 1999. Enzymatic hydroxylation of aromatics in enantioselective synthesis: expanding asymmetric methodology. Aldrichim. Acta 32:35-62. [Google Scholar]
- 29.Imbeault, N. Y. R., J. B. Powlowski, C. L. Colbert, J. T. Bolin, and L. D. Eltis. 2000. Steady-state kinetic characterization and crystallization of a polychlorinated biphenyl-transforming dioxygenase. J. Biol. Chem. 275:12430-12437. [DOI] [PubMed] [Google Scholar]
- 30.Jeffrey, W. H., S. M. Cuskey, P. J. Chapman, S. Resnick, and R. H. Olsen. 1992. Characterization of Pseudomonas putida mutants unable to catabolize benzoate: cloning and characterization of Pseudomonas genes involved in benzoate catabolism and isolation of a chromosomal DNA fragment able to substitute for xylS in activation of the TOL lower-pathway promoter. J. Bacteriol. 174:4986-4996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kauppi, B., K. Lee, E. Carredano, E. R. Parales, D. T. Gibson, H. Eklund, and S. Samaswamy. 1998. Structure of an aromatic-ring-hydroxylating dioxygenase-naphthalene 1,2-dioxygenase. Structure 6:571-586. [DOI] [PubMed] [Google Scholar]
- 32.Kessler, B., K. N. Timmis, and V. de Lorenzo. 1994. The organization of the Pm promoter of the TOL plasmid reflects the structure of its cognate activator protein XylS. Mol. Gen. Genet. 244:596-605. [DOI] [PubMed] [Google Scholar]
- 33.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680-685. [DOI] [PubMed] [Google Scholar]
- 34.Lee, K. 1999. Benzene-induced uncoupling of naphthalene dioxygenase activity and enzyme inactivation by production of hydrogen peroxide. J. Bacteriol. 181:2719-2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee, J.-Y., H.-S. Park, and H. S. Kim. 1999. Adenosylcobalamin-mediated methyl transfer by toluate cis-dihydrodiol dehydrogenase of the TOL plasmid pWW0. J. Bacteriol. 181:2953-2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maeda, T., Y. Takahashi, H. Suenage, A. Suyama, M. Goto, and K. Furukawa. 2001. Functional analyses of Bph-Tod hybrid dioxygenase, which exhibits high degradation activity toward trichloroethylene. J. Biol. Chem. 276:29833-29838. [DOI] [PubMed] [Google Scholar]
- 37.Mermod, N., J. L. Ramos, A. Bairoch, and K. N. Timmis. 1987. The xylS gene positive regulator of TOL plasmid pWW0: identification, sequence analysis, and overproduction leading to constitutive expression of meta cleavage operon. Mol. Gen. Genet. 207:349-354. [DOI] [PubMed] [Google Scholar]
- 38.Murray, K., C. J. Duggleby, J. M. Sala-Trepat, and P. A. Williams. 1972. The metabolism of benzoate and methybenzoates via the meta-cleavage pathway by Pseudomonas arvilla mt-2. Eur. J. Biochem. 28:301-310. [DOI] [PubMed] [Google Scholar]
- 39.Nakazawa, T., and T. Yokota. 1973. Benzoate metabolism in Pseudomonas putida(arvilla) mt-2: demonstration of two benzoate pathways. J. Bacteriol. 115:262-267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nam, J. W., H. Nojiri, T. Yoshida, H. Habe, H. Yamane, and T. Omori. 2001. New classification system for oxygenase components involved in ring-hydroxylating oxygenations. Biosci. Biotechnol. Biochem. 65:254-263. [DOI] [PubMed] [Google Scholar]
- 41.Ramos, J. L., A. Stolz, W. Reineke, and K. N. Timmis. 1986. Altered effector specificities in regulators of gene expression: TOL plasmid xylS mutants and their use to engineer expansion of the range of aromatics degraded by bacteria. Proc. Natl. Acad. Sci. USA 83:8467-8471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reineke, W., and H. J. Knackmuss. 1978. Chemical structure and biodegradability of halogenate aromatic compounds: substituent effects on 1,2-dioxygenation of benzoic acid. Biochim. Biophys. Acta 542:412-423. [DOI] [PubMed] [Google Scholar]
- 43.Rojo, F., D. H. Pieper, K. H. Engesser, H. J. Knackmuss, and K. N. Timmis. 1987. Assemblage of ortho cleavage route for simultaneous degradation of chloro- and methylaromatics. Science 238:1395-1398. [DOI] [PubMed] [Google Scholar]
- 44.Sambrook, J., E. F. Fristch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 45.Suen, W. C., and D. T. Gibson. 1994. Recombinant Escherichia coli strains synthesize active forms of naphthalene dioxygenase and its individual alpha and beta subunits. Gene 143:67-71. [DOI] [PubMed] [Google Scholar]
- 46.Tabor, S., and C. C. Richardson. 1985. A bacteriophage T7 RNA polymerase/promoter system for controlled exclusive expression of specific genes. Proc. Natl. Acad. Sci. USA 82:1074-1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Timmis, K. N., and D. H. Pieper. 1999. Bacteria designed for bioremediation. Trends Biotechnol. 17:201-204. [DOI] [PubMed] [Google Scholar]
- 48.Vaillancourt, F. H., S. Han, P. D. Fortin, J. T. Bolin, and L. D. Eltis. 1998. Molecular basis for the stabilization and inhibition of 2, 3-dihydroxybiphenyl 1,2-dioxygenase by t-butanol. J. Biol. Chem. 273:34887-34895. [DOI] [PubMed] [Google Scholar]
- 49.Williams, P. A., and K. Murray. 1974. Metabolism of benzoate and the methylbenzoates by Pseudomonas putida (arvilla) mt-2: evidence for the existence of a TOL plasmid. J. Bacteriol. 120:416-423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wolfe, M. D., J. V. Parales, D. T. Gibson, and J. D. Lipscomb. 2001. Single turnover chemistry and regulation of O2 activation by the oxygenase component of naphthalene 1,2-dioxygenase. J. Biol. Chem. 276:1945-1953. [DOI] [PubMed] [Google Scholar]
- 51.Wubbolts, M. G., and K. N. Timmis. 1990. Biotransformation of substituted benzoates to the corresponding cis-diols by an engineered strain of Pseudomonas oleovorans producing the TOL plasmid-specified enzyme toluate-1,2-dioxygenase. Appl. Environ. Microbiol. 56:569-571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yamaguchi, M., and H. Fujisawa. 1980. Purification and characterization of an oxygenase component in benzoate 1,2-dioxygenase system from Pseudomonas arvilla C-1. J. Biol. Chem. 255:5058-5063. [PubMed] [Google Scholar]
- 53.Zeyer, J., P. R. Lehrbach, and K. N. Timmis. 1985. Use of cloned genes of Pseudononas TOL plasmid to effect biotransformation of benzoates to cis-dihydrodiols and catechols by Escherichia coli cells. Appl. Environ. Microbiol. 50:1409-1413. [DOI] [PMC free article] [PubMed] [Google Scholar]