

Abstract
CaBP1–8 are neuronal Ca2+-binding proteins with similarity to calmodulin (CaM). Here we show that CaBP4 is specifically expressed in photoreceptors, where it is localized to synaptic terminals. The outer plexiform layer, which contains the photoreceptor synapses with secondary neurons, was thinner in the Cabp4−/− mice than in control mice. Cabp4−/− retinas also had ectopic synapses originating from rod bipolar and horizontal cells that extended into the outer nuclear layer. Responses of Cabp4−/− rod bipolars were reduced in sensitivity about 100-fold. Electroretinograms (ERGs) indicated a reduction in cone and rod synaptic function. The phenotype of Cabp4−/− mice shares similarities with that of incomplete congenital stationary night blindness (CSNB2) patients. CaBP4 directly associated with the C-terminal domain of the Cav1.4 α1-subunit and shifted the activation of Cav1.4 to hyperpolarized voltages in transfected cells. These observations indicate that CaBP4 is important for normal synaptic function, probably through regulation of Ca2+ influx and neurotransmitter release in photoreceptor synaptic terminals.
L-type Ca2+ channels are involved in neuronal differentiation and outgrowth and in synaptic plasticity1,2. At many ribbon synapses, Ca2+ influx through L-type Ca2+ channels triggers neurotransmitter release3–5. The α1-subunit of the L-type Cav1.4 channel (Cav1.4α1) is specific to photoreceptors and is present at highest density in the synaptic terminals5,6. Compared with other L-type Ca2+ channels, Cav1.4 channels are activated at relatively more negative voltages and show slow inactivation7–9, important properties for the ability of photoreceptors to sustain continual glutamate release in the dark4,10. Null mutations in Cav1.4α1 are responsible for an X-linked disorder, CSNB2 (refs. 11,12). ERGs of these patients indicate that a deficit may occur in transmission of signals from rod photoreceptors to bipolar cells. In mice, deletion of the β2-subunit, another component of the photoreceptor L-type channel, alters the expression of Cav1.4 and produces a phenotype similar to that seen in CSNB2 patients13.
CaBPs, a subfamily of calmodulin (CaM)-like neuronal Ca2+-binding proteins14, modulate voltage-dependent Ca2+ channels (VDCCs) and inositol triphosphate receptors15–17. Here we show that CaBP4, which has only been partially characterized in silico14, is found specifically in photoreceptor synaptic terminals, is important for the normal function of photoreceptor synapses and colocalizes with and modulates the activity of expressed Cav1.4 channels. These results indicate that CaBP4 may be an important regulator of Ca2+ influx and transmitter release in photoreceptor synaptic terminals.
RESULTS
CaBP4 is a CaM-related CaBP
CaBP4 was identified by polymerase chain reaction (PCR) cloning using primers based on the sequence of its closest relative, CaBP2 (ref. 14). The full-length cDNA of human CaBP4 contains an open reading frame of 825 bp, predicting a protein of 275 amino acids and a calculated molecular mass of 30,432 Da. Human, bovine, mouse and rat CaBP4 are highly homologous in their C-terminal regions (~90%) and are less conserved in their N-terminal regions (~60%) with inter-species differences occurring in the first exon (Fig. 1a). CaBP4 contains four EF-hand motifs, although the second motif cannot coordinate Ca2+ because the Lys residue in position 1 is not suitable for Ca2+ coordination (Fig. 1a).
Figure 1.
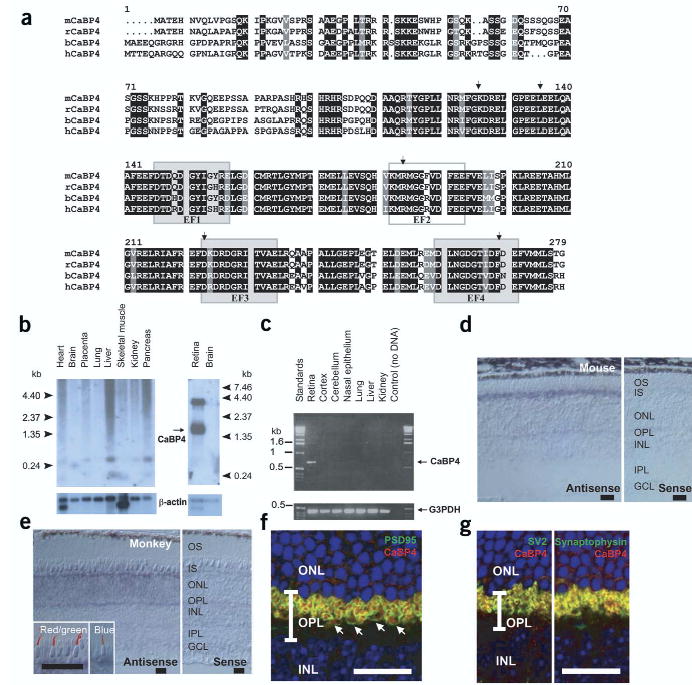
CaBP4 protein sequence, tissue distribution and immunolocalization. (a) Primary structure of mouse (m), rat (r), bovine (b) and human (h) CaBP4 (accession numbers AY039218, XM_344981, AY048883 and AY039271). The identical residues in all sequences are in white on a black background. Conservative substitutions are in white on a gray background. Functional EF-hand motifs are shown as shaded boxes and nonfunctional EF-hand motif as an open box. Arrows indicate the intronexon junction of the CABP4 gene. (b) Northern blot analysis of CaBP4 mRNA from various human (left) or rat (right) tissues. Control hybridization with a 32P-labeled β-actin mRNA is also shown. (c) PCR analysis of CaBP4 transcript in various mouse tissues. A positive control was carried out with primers specific for glyceraldehyde-3-phosphate dehydrogenase (G3PDH). (d) In situ hybridization of CaBP4 transcripts in mouse retina using antisense (left) and sense (right) RNA. (e) In situ hybridization of CaBP4 transcripts in monkey retina using antisense (left) and sense (right) RNA. Inset, immunoreactivity with antibodies to red and green opsin (left) and blue opsin (right) was covisualized with CaBP4 transcripts (blue signal). (f) Presynaptic localization of mouse CaBP4 (red) demonstrated by covisualization with a presynaptic protein (PSD95; green). Arrows indicate rod spherules and cone pedicles. (g) Colocalization of synaptic vesicle proteins SV2 and synaptophysin (green) with mouse CaBP4 (red). In f and g, yellow indicates overlap of two immunoreactivities and blue indicates cell nuclei (Hoechst 33342 staining). INL, inner nuclear layer; ONL, outer nuclear layer; OPL, outer plexiform layer. (d–f) Scale bars, 20 μm.
The Cabp4 gene is short (~4 kb) with a six-exon structure similar to CaM (see Supplementary Fig. 1 online). The human genome data at the NCBI predict that Cabp4 is in the opposite orientation and is separated by four genes and ~60 kb from Cabp2 on chromosome 11 at q13.1, a region with conserved synteny with mouse chromosome 19.
CaBP4 is localized in rod and cone photoreceptor synapses
Northern blot analyses with the CaBP4 cDNA probe showed two transcripts of 1.6 kb and 3.8 kb in the retina (Fig. 1b). CaBP4 products from PCR with reverse transcription (RT-PCR) were observed in the retina but not in other locations (Fig. 1c). The CaBP4 antisense RNA probe hybridized to the mouse photoreceptor inner segments (Fig. 1d). CaBP4 was expressed in monkey rods and cones; in situ hybridization coupled with immunocytochemistry confirmed the expression of monkey CaBP4 in all cone types (Fig. 1e). CaBP4 was not detected in retinal cells other than photoreceptors (Fig. 1d,e).
Subcellular localization of CaBP4 in the mouse retina was carried out using specific antibodies to CaBP4. The staining pattern with these antibodies was compared with the localization of PSD95 (Fig. 1f), a presynaptic protein that is found in terminals from both types of photoreceptor18. CaBP4 was observed in both rod spherules and cone pedicles (Fig. 1f). The presynaptic localization of CaBP4 was confirmed using double-labeling studies with antibodies to synaptic vesicle protein SV2 and synaptophysin (Fig. 1g).
Cabp4−/− mice have abnormal outer plexiform layers
CaBP4-deficient mice were generated by replacing exon 1 and part of exon 2 with the PGK-neo cassette (Supplementary Fig. 1). A PCR-based assay confirmed the disruption of Cabp4 in Cabp4−/− mice (Supplementary Fig. 1). CaBP4 proteins were not detected in Cabp4−/− retinas by immunoblotting or immunocytochemistry (Fig. 2a,b).
Figure 2.
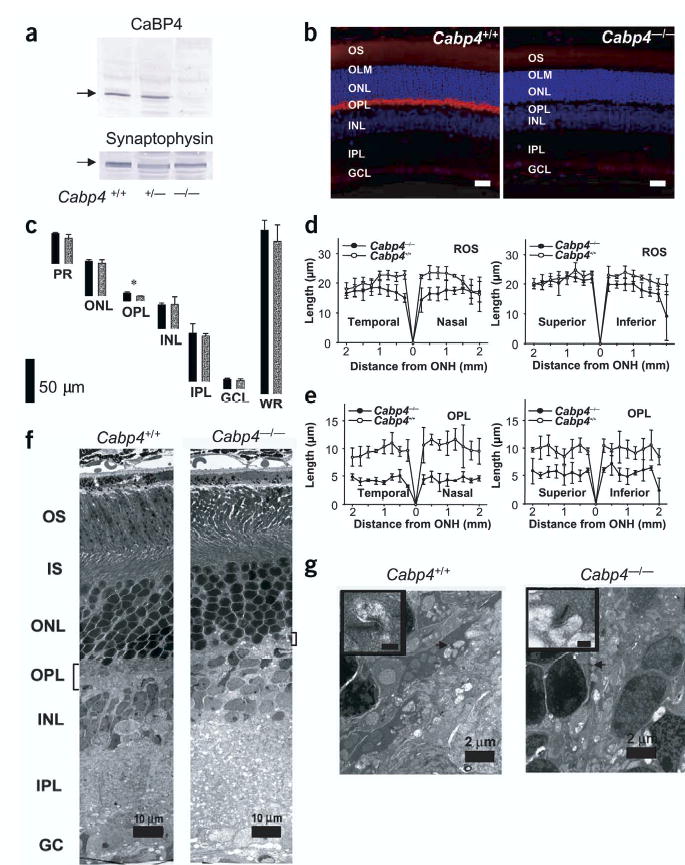
Characterization of CaBP4 knockout mice. (a) Immunoblotting of retinal extracts from Cabp4+/+, Cabp4+/− and Cabp4−/− mice probed with anti-CaBP4 or anti-synaptophysin (control). (b) Immunolocalization of CaBP4 in the retina of Cabp4+/+ mouse (left). Lack of CaBP4 immunoreactivity in the retina of Cabp4−/− mouse (right). Scale bars, 20 μm. Nuclei are visualized by staining with Hoechst 33342 dye (blue). (c) The thickness of individual retinal layers from 8-to 10-week-old Cabp4+/+ (black bars) and Cabp4−/− mice (gray bars) measured at 1.25 mm inferior from the optic nerve head. *P < 0.01. GCL, ganglion cell layer; INL, inner nuclear layer; IPL, inner plexiform layer; ONL, outer nuclear layer; OPL, outer plexiform layer; PR, photoreceptor outer and inner segments; WR, whole retina. (d,e) Rod outer segment (ROS; d) and outer plexiform layer (ORPL; e) thickness (in micrometers) plotted as a function of the retinal location (in millimeters) from the optic nerve head. (f) Montage of cross-sections through the retinas of 2-month-old mice analyzed by transmission electron microscopy. The outer plexiform layer is thinner in Cabp4−/− mice than it is in Cabp4+/+ mice. IS, inner segment; OS, outer segment. (g) Higher magnification of a cross-section through the outer plexiform layer. Photoreceptor terminals are present in the outer plexiform layer of Cabp4+/+ mice (arrow), but fewer and altered terminals are observed in Cabp4−/− mice. A synaptic ribbon is shown at higher magnification (inset; scale bars, 0.2 μm).
Retinal morphologies of 2-month-old Cabp4−/− and Cabp4+/+ mice were compared using light and electron microscopy. Structural changes were observed in the outer segment and outer nuclear layers: rod outer segments were shorter, the disk density was lower and the outer nuclear layer was missing one or two layers of cell bodies. The outer segments of Cabp4−/− mice were 15% shorter than those of Cabp4+/+ mice (Fig. 2d; averages of rod outer-segment thickness at 250–2,000 μm from the optic nerve were compared). This observation is consistent with a similar drop in rhodopsin content as measured directly by UV-visible spectroscopy and retinoid analysis (data not shown)19.
Marked changes were observed in the outer plexiform layer, which showed ~51% reduction in thickness in Cabp4−/− mice as compared with Cabp4+/+ mice (P < 0.001; Fig. 2e; averages of outer-plexiform-layer thickness at 250–2,000 μm from the optic nerve were compared). Quantitative calculations of the thickness of other retinal layers showed no significant differences between these two genetic backgrounds (P > 0.05; Fig. 2c). Electron micrographs showed a paucity of photoreceptor terminals and synaptic ribbons in the outer plexiform layer of Cabp4−/− mice (Fig. 2f,g and Supplementary Fig. 2). Quantitative analysis of the electron micrographs showed that the number of photoreceptor terminals was ~33% lower in Cabp4−/−mice (251 ± 59 photoreceptor terminals, n = 3 mice) as compared with Cabp4+/+ mice (372 ± 17, n = 3 mice), and the number of synaptic ribbons was 37 ± 14% (n = 3 mice) lower in Cabp4−/− mice. These data indicate that the photoreceptor terminals are both less numerous and defective. Changes in the outer plexiform layer were more pronounced in 6- to 8-month-old Cabp4−/− mice than in 2-month-old mice (Supplementary Figs. 2 and 3).
To investigate the consequence of CaBP4 deficiency on synapses of the outer plexiform layer in more detail, we analyzed synaptic morphology in Cabp4−/− mice using antibodies that label specific synaptic proteins or cell types. In Cabp4+/+ and Cabp4−/− mice, the ionotropic glutamate receptor 1 was highly concentrated at postsynaptic sites(Fig. 3a). Photoreceptor synaptic terminals were visualized using antibody to PSD95 (anti-PSD95; ref. 18; Fig. 3a,b). In Cabp4+/+ mice, rod spherules and cone pedicles were inflated, whereas those in Cabp4−/− mice were disorganized and flat (also see Supplementary Fig. 3). mGluR6 immunoreactivity21 was aggregated in hot spots at the tips of dendritic processes in both Cabp4+/+ and Cabp4−/− mice (Fig. 3b). Fewer hot spots were observed, however, in Cabp4−/− mice. mGluR6 localized proximally to PSD95 (Fig. 3b) in both cases.
Figure 3.
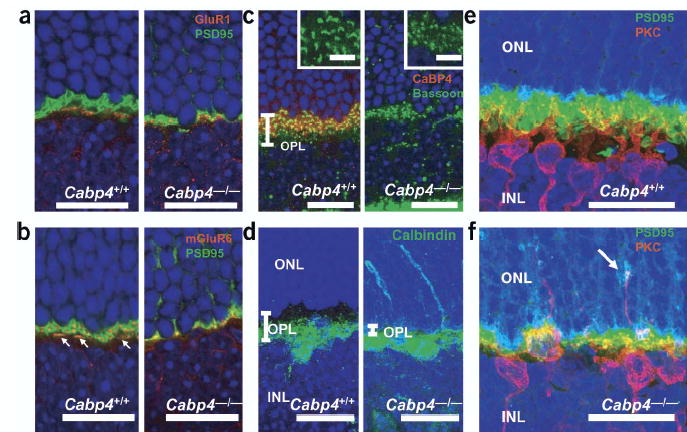
Synaptic connection between photoreceptors and bipolar cells. (a) Immunolocalization of GluR1 (red) and PSD95 (green). In Cabp4+/+ (left) and Cabp4−/− (right) mice, GluR1 is expressed in off-cone bipolar cell dendrites20 proximal to cone pedicles. PSD95 is localized proximal to the plasma membrane of the photoreceptor synaptic terminals. (b) Immunolocalization of mGluR6 (red) and PSD95 (green). A photoreceptor presynapse in a Cabp4+/+ mouse shows morphologically distinguishable rod spherules and cone pedicles (arrows indicate cone pedicles). Cabp4−/− mice have disorganized synaptic processes.(c) Immunolocalization of CaBP4 (red) and the presynaptic marker bassoon (green). Insets show higher-magnification images. (d) Immunolocalization of calbindin, a marker for H1 horizontal cells. Each picture is a three-dimensional projection made from multiple stacks of confocal images. (e,f) Three-dimensional projections of PKCα (red, a marker for rod bipolar cells) and PSD95 (green) immunolocalization in (e) Cabp4+/+ and (f) Cabp4−/− mice. Arrow indicates ectopic synapse in the outer nuclear layer between rod photoreceptor and bipolar cells. In a–f, nuclei were visualized by staining with Hoechst 33342 dye. Scale bars, 20 μm (in inset, 5 μm).
An antibody to bassoon (anti-bassoon), which is a presynaptic cytomatrix protein22, labeled horseshoe-shaped structures in Cabp4+/+ mice but produced only punctuate staining in Cabp4−/−mice (Fig. 3c). In addition, some labeled puncta in Cabp4−/− retinas were located in the outer nuclear layer. The number of bassoon-positive presynaptic terminals in the outer plexiform layer and outer nuclear layer were compared between Cabp4−/− and Cabp4+/+ mice. A decrease of 43.7 ± 13.7% (mean ± s.d.; n = 3) in the number of presynaptic terminals was observed (in a cross-section of the retina, ~1,250 μm from the optic nerve head).
Dendrites of rod bipolar cells that were visualized using an antibody to PKCα were poorly branched and underdeveloped in Cabp4−/− mice as compared with Cabp4+/+ mice (Fig. 3e,f). In some instances, bipolar and horizontal cell dendrites extended into the outer nuclear layer of Cabp4−/− mice and formed ectopic synapses with photoreceptors (Fig. 3d,f); such branching was never observed in Cabp4+/+ mice, in which dendritic processes of rod bipolar and horizontal cells ended at the outer plexiform layer (Fig. 3d,e).
Visual responses in Cabp4−/− mice are abnormal
The disruption of normal synaptic morphology in Cabp4−/− mice indicates that corresponding deficits in synaptic function may be present. Indeed, as described below, transmission from rods and cones to bipolar cells was severely attenuated in Cabp4−/− mice, whereas photoreceptor responses were only modestly affected.
We compared responses of rod photoreceptors from Cabp4+/+ and Cabp4−/− mice using suction electrode recordings23. At all flash strengths, the responses of Cabp4−/− rods were faster and less sensitive than the corresponding Cabp4+/+ responses (Fig. 4a,b). The difference in sensitivity was quantified by fitting each cell’s stimulus-response relationship to determine the flash that produced a half-maximal response (Fig. 4c). The half-saturating flash strength was 9.4 ± 0.5 Rh* for Cabp4+/+ rods (mean ± s.e.m.; n = 22) and 13.4 ± 0.6 Rh* for Cabp4−/− rods (n = 38). The difference in kinetics was characterized by measuring the time to peak and integration time of responses to dim flashes. Cabp4−/− rods had a shorter time to peak (171 ± 5 ms in 31 Cabp4−/− rods versus 220 ± 11 ms in 25 Cabp4+/+ rods; mean ± s.e.m.) and smaller integration time (210 ± 17 ms versus 340 ± 22 ms). We do not know what causes the difference between light responses in Cabp4−/− and Cabp4+/+ rods; changes in outer-segment Ca2+ dynamics, as measured from exchange currents24, were not significantly different (data not shown).
Figure 4.
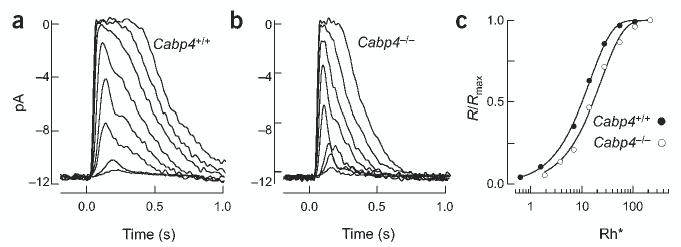
Current responses of Cabp4+/+ and Cabp4−/− rod outer segments. (a) Flash family measured from a Cabp4+/+ rod. Average responses (5–100 trials at each flash strength) are superimposed for flashes producing 0.65, 1.3, 7, 14, 28, 56, 112 and 224 Rh*. (b) Flash family measured from a Cabp4−/− rod as in a. Flash strengths are 1.9, 3.8, 7.6, 14, 26, 56, 112 and 224 Rh*. (c) Stimulus-response relationship for the Cabp4+/+ (•) and Cabp4−/− (○) rod in a and b. Response amplitudes were normalized to the maximal response and were plotted against flash strength. Saturating exponential fits to the data were used to estimate the half-saturating flash strength.
To determine whether signal transfer from rods to rod bipolar cells was altered in Cabp4−/− mice, we measured light responses of rod bipolar cells in a retinal slice preparation25. Cabp4+/+ rod bipolars had maximal responses of up to 400 pA (n = 47), whereas Cabp4−/− rod bipolars had maximal responses of at most 12 pA (n = 52; Fig. 5a,b). Furthermore, the half-saturating flash strength was higher for Cabp4−/− rod bipolars than for Cabp4+/+ cells (Fig. 5d). The half-saturating flash strength was 2.8 ± 0.04 Rh*/rod (mean ± s.e.m.; n = 30) in Cabp4+/+ rod bipolars and 25 ± 3 Rh*/rod in Cabp4−/− rod bipolars (n = 16). The increased half-saturating flash strength and decreased maximal response cause the sensitivity of Cabp4−/− rod bipolars to be at least 100-fold less than that of Cabp4+/+ cells (Fig. 5c). Because the rod responses in the two mouse strains differed by a factor of close to two, this large difference in the rod bipolar responses indicates a deficit in synaptic transmission.
Figure 5.
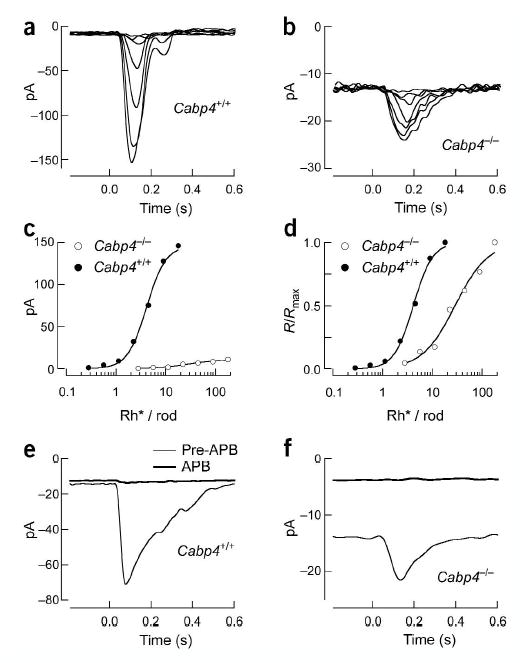
Voltage-clamp responses of Cabp4+/+ and Cabp4−/− rod bipolars. (a) Flash family measured from a Cabp4+/+ rod bipolar held at −60 mV. Average responses are superimposed for flashes producing 0.28, 0.55, 1.1, 2.2, 4.5, 9 and 18 Rh*/rod. (b) Flash family measured from a Cabp4−/− rod bipolar as in a. Flash strengths are 2.8, 5.6, 11, 22, 44, 90 and 180 Rh*/rod. (c) Stimulus-response relationship for the Cabp4+/+ (•) and Cabp4−/− (○) rod bipolar cells in a and b. (d) Stimulus-response relationship as in c with the response amplitudes normalized to the maximal response. Hill curves that were fit to the data were used to estimate the half-saturating flash strength. (e) Effect of APB on the dark current and saturating response of a Cabp4+/+ rod bipolar. Saturating flashes producing 36 Rh*/rod were delivered every 3 s. The two traces shown are responses measured before and after adding 8 μM APB to the superfusion solution. (f) Effect of APB on the dark current and saturating response of a Cabp4−/− rod bipolar, as in e. Flash strength was 180 Rh*/rod.
Rod bipolar cells use mGluR6 metabotropic glutamate receptors to detect glutamate released from the rods21. Activation of these receptors leads to closure of nonselective cation channels. To determine what fraction of these channels was closed in darkness, we exposed rod bipolars to a saturating concentration of the mGluR6 agonist APB26, which creates a high level of activity in the transduction cascade that links receptors and channels and closes most or all of the open channels27.
As expected, APB eliminated the light response in a Cabp4+/+ rod bipolar cells by activating mGluR6 receptors and rendering them insensitive to changes in glutamate (Fig. 5e). APB also produced a minimal change in dark current, as previously reported27. APB again eliminated the light response in a Cabp4−/− rod bipolar cell but also produced a large decrease in the inward dark current (Fig. 5f). In Cabp4+/+ rod bipolar cells, the change in saturating response was more than 50 times the change in dark current27. In Cabp4−/− rod bipolar cells, the change in dark current produced by APB was similar in magnitude to the saturating light response (n = 18). Thus, the number of additional channels open at the peak of the saturating light response was comparable to the number that were open in darkness in Cabp4−/− rod bipolars. This indicates that the glutamate concentration present at the mGluR6 receptors is substantially lower in Cabp4−/− rod bipolars as compared with that in Cabp4+/+ cells.
Disruption of cone and rod signaling
ERG responses in Cabp4−/− mice and Cabp4+/+ mice confirmed the differences described above for rod signaling and indicated a similar disruption of signaling between cones and cone bipolar cells. The a-wave (generated by photoreceptors) of dark-adapted Cabp4−/− mice was about half the amplitude of that of Cabp4+/+ mice (Fig. 6a,b and Supplementary Fig. 4). The b-wave (the bipolar cell component) was dramatically reduced in Cabp4−/− mice. In some cases, a small positive voltage change remained in the normal location of the b-wave, indicating that some transmission remained between photoreceptor and bipolar cells.
Figure 6.
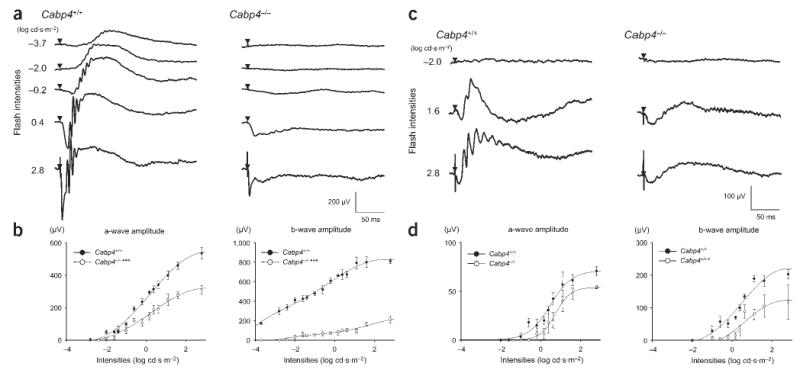
Single-flash ERG responses of increasing intensity for Cabp4−/− and Cabp4+/+ mice. (a,c) Serial ERG responses to increasing flash stimuli obtained from Cabp4−/− and Cabp4+/+ mice under (a) dark-adapted and (c) light-adapted conditions. Single-flash timing is indicated by the filled triangle. (b) Plotted ERG a-wave and b-wave amplitudes in response to increasing stimuli in Cabp4−/− mice showed significantly lower responses as compared with those in Cabp4+/+ mice in dark-adapted conditions (***P < 0.0001; n = 8). (d) The b-wave amplitudes in Cabp4−/− mice were also lower as compared with those in Cabp4+/+ mice in light-adapted conditions (*P < 0.01; n = 8) but the a-wave amplitudes showed smaller differences. Light-adapted responses were examined after bleaching at 1.4 log cd·m−2 for 15 min.
Under light-adapted conditions, the a-waves of Cabp4−/− and Cabp4+/+ mice differed by ~20%, suggesting that cone function was near normal (Fig. 6c,d). The b-wave was significantly reduced at all stimulus intensities in Cabp4−/− mice as compared with Cabp4+/+ mice. Thus, the absence of CaBP4 disrupted normal transmission of signals from rods and cones to bipolar cells.
CaBP4 interacts with and modulates Cav1.4
L-type VDCCs are modulated by CaM, which binds to multiple sites in the C-terminal domain of the α1-subunit28–30. CaBP1 interacts with and modulates Cav2.1 channels15 and Cav1.2 channels31. To test whether CaBP4 might have a similar role, we monitored direct interactions between CaBP4 and a cytoplasmic fragment of Cav1.4α1. We coupled the Cav1.4α1 fragment (amino acids 1445–1983) to a resin and loaded purified CaBP4 in the presence of Ca2+. CaBP4 bound to the Cav1.4α1 C terminus in a Ca2+-dependent manner as shown by EGTA elution, but a significant fraction of the protein bound to the column in a Ca2+-independent manner as shown by subsequent acid elution (Fig. 7a).
Figure 7.
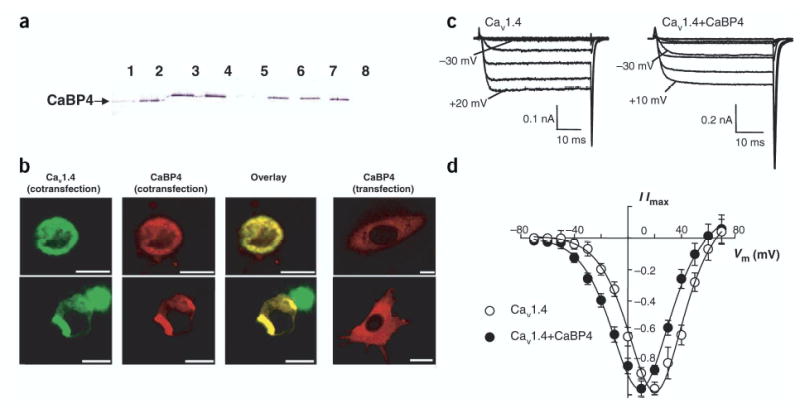
CaBP4 interacts with and modulates Cav1.4. (a) Affinity chromatography of purified recombinant mCaBP4 on Ca1.4vα1 column. The eluted fractions were probed with anti-CaBP4. Lanes 1–4, elution with 3 mM EGTA; lane 5, final elution with 3 mM EGTA; lanes 6–8: further elution with 0.1 M glycine buffer, pH 2.1. (b) Colocalization of CaBP4-DsRed2 with Cav1.4-GFP in HEK293 cells. Confocal images of HEK293 cells transfected with mCaBP4-DsRed2 and Cav1.4–GFP (left three panels) or transfected with mCaBP4-DsRed2 alone (right panel). The yellow color indicates colocalization of the overexpressed fusion proteins. Scale bars, 12.5 μ) m. (c) Modulation of Cav1.4 activation by CaBP4 in transfected HEK293T cells. Whole-cell Ca2+ currents recorded in cells transfected with Cav1.4 subunits (α1 1.4, β2A, α2δ) alone (left) or cotransfected with CaBP4 (right). Shown are representative traces of Ca2+ currents evoked by 50-ms steps from a holding voltage of −80 mV to various test voltages. Extracellular recording solution contained 20 mM Ca2+ and intracellular solution contained 5 mM EGTA. CaBP4 enhanced activation of Ca2+ currents at negative voltages as indicated. (d) Current-voltage relationship from cells transfected with Cav1.4 alone (○) or cotransfected with CaBP4 (•). For each test voltage (Vm), I/Imax represents the current amplitude measured at 45 ms normalized to the maximal current amplitude obtained in the series (mean ± s.e.m.; n = 7–10).
To further investigate if CaBP4 interacts with Cav1.4 in a more physiological setting, we analyzed the colocalization of these proteins in transfected HEK293 cells. To visualize the proteins, we constructed enhanced green fluorescent protein (EGFP) and DsRed2 fusion proteins. When HEK293 cells were transfected with mCaBP4-DsRed2, the red fluorescence was detected throughout the cytoplasm (Fig. 7b). Expression of Cav1.4-GFP with mCaBP4-DsRed2 resulted in localization of mCaBP4-DsRed2 with Cav1.4-GFP to the plasma membrane (Fig. 7b), indicating that these two proteins may interact with each other in HEK293 cells.
To test whether CaBP4 altered the function of Cav1.4 channels, we analyzed whole-cell patch-clamp recordings of Ca2+ currents in transfected HEK293T cells. In cells transfected with Cav1.4 alone, inward Ca2+ currents were evoked from a holding voltage of −80 mV to a range of voltages from −30 to +60 mV (Fig. 7c,d). Cav1.4 showed virtually no inactivation during 50-ms step depolarizations, as described previously7–9. In cotransfected cells, CaBP4 enhanced activation of Cav1.4 by shifting the I-V relationship to hyperpolarized voltages (Fig. 7c,d). Maximal inward current was evoked at significantly more negative voltages in cells cotransfected with CaBP4 (Vhalf = −15.4 ± 1.2, Vmax = 7.8 ± 1.6 mV, n = 7) as compared with cells transfected with Cav1.4 alone (Vhalf = −5.3 ± 1.9, Vmax = 17.2 ± 1.3 mV, n = 10; P < 0.05).
DISCUSSION
CaBP4 is a photoreceptor-specific protein that belongs to a subfamily of at least eight CaM-related proteins14–17. The specific functions of these proteins in vivo remain unclear, although in vitro CaBP1 interacts with inositol triphosphate receptors and Cav2.1 channels15–17 and with Cav1.2 channels31. The work presented here shows that CaBP4 is essential for the development and/or maintenance of the photoreceptor output synapse, probably through modulation of photoreceptor Ca2+ channels and transmitter release.
CaBP4 is important for synapse formation and function
Photoreceptor synapses in CaBP4-deficient mice were severely disrupted both anatomically and functionally. In situ hybridization and confocal microscopy showed that CaBP4 was located specifically in photoreceptors, and primarily in their synaptic terminals. Light and electron microscopy showed dramatic changes in photoreceptor synapses, including a thinner outer plexiform layer, a reduction in the number of synaptic ribbons and photoreceptor terminals and deflation of rod spherules and cone pedicles. The absence of CaBP4 also resulted in the formation of ectopic synapses between rods and rod bipolar or horizontal cells in the outer nuclear layer. Ectopic synapses are also present in mice that are deficient in the bassoon protein, which is required for the anchoring of the photoreceptor ribbon to the active zone22. Although the morphology of the retina and outer plexiform layer are preserved in Bassoon-deficient mice, ERG recordings show that the transfer of signals from photoreceptors to postsynaptic cells is attenuated22.
Light responses of CaBP4-deficient mice were also altered as compared with those of control mice. Single-cell recordings and ERG measurements showed a disruption in the transmission of photoreceptor signals to postsynaptic cells. In darkness, a significant fraction of the cation channels that were coupled to mGluR6 receptors were open in rod bipolar cells of CaBP4-deficient mice, unlike those in control rod bipolars27. Thus, the glutamate concentration that was present at the bipolar mGluR6 receptors was reduced in the absence of CaBP4.
CaBP4 modulates Cav1.4
CaBP4 colocalized and interacted with expressed Cav1.4 VDCCs and modulated their functional properties. Thus, like CaM15, CaBPs may be physiological regulators of VDCCs.
The photoreceptor Ca2+ currents activate at voltages of 10–15 mV more negative than currents obtained after heterologous expression of Cav1.4 (ref. 8). The voltage dependence of heterologously expressed Cav1.4 channels would be poorly suited to control transmitter release at physiological voltages for photoreceptors. The absence of a cytosolic modulator might explain the difference between native and recombinant channels. Indeed, CaBP4 shifted the activation range of expressed L-type VDCCs to more hyperpolarized voltages, suggesting that CaBP4 is a modulator of Cav1.4 in vivo. As a consequence of this voltage shift, at the photoreceptor resting potential of approximately −40 mV32, Ca2+ influx through expressed L-type VDCCs increased about fivefold in the presence of CaBP4 as compared with its absence. This change in Ca2+ influx should produce a corresponding change in transmitter release, which is consistent with the lowered activity of mGluR6 receptors observed in Cabp4−/− rod bipolar cells.
The IQ motifs and the regions known to bind CaM and to be necessary for Ca2+-dependent inactivation28–30,33,34 are conserved between the α1-subunits of the L-type VDCCs, suggesting that Cav1.4 might also be modulated by CaM or by a CaM-like protein. Apo-CaM is tethered to L-type VDCCs in its resting state. The tethering is to two peptides located upstream from the IQ motif in the cytoplasmic C-terminal part of the channel α1-subunit28. CaBP4 also bound to Cav1.4 in the absence of Ca2+, although less effectively than in the presence of Ca2+ (Fig. 7a). It is possible that CaBP4 is also tethered to the channel in its resting state.
Interaction of CaBP4 with L-type VDCCs could lead to a variety of effects that are necessary for proper formation of the photoreceptor synapse. L-type VDCCs are essential in the development of neuritic outgrowth in several neuronal cell types2. In rods, L-type VDCCs are the primary Ca2+ channels required for structural plasticity and upregulation of synaptic vesicle synthesis1,2. A splice variant of CaBP1, caldendrin, colocalizes with synaptophysin in the inner plexiform layer and might be involved in the development of amacrine cell processes before functional synapses have formed35. L-type VDCCs also activate signaling pathways that lead to transcriptional activation36. The cAMP response element–binding proteins, which drive the expression of a number of genes that regulate neuronal survival and plasticity37,38, and MEF-2, a MADS transcription factor that mediates neuronal survival39, depend on Ca2+ flux through L-type VDCCs.
Although the molecular identity of the cone Ca2+ channels is not fully elucidated, some cone channels contain the Cav1.3 subunit5,40,41. The disruption of cone signaling in CaBP4-deficient mice indicates that CaBP4 is also critical for the normal development of cone synaptic terminals. The peptide sequences of Cav1.4 that bind CaM are conserved in the Cav1.4 and the Cav1.3 α1-subunits. Cav1.4 is also expressed in retinal bipolar cells42. CaBP1, 2 and 5, which are members of the same neuronal CaBP subfamily as CaBP4, are expressed in other neurons including the cone and rod bipolar cells14. A similar mode of regulation of calcium channels by CaBPs might also occur in other neurons.
The phenotype of Cabp4−/− mice resembles that observed in CSNB2 patients carrying mutations in the gene CACNA1F12,43 and in mice deficient for the β2-subunit of the Cav1.4 channel13. CSNB2 patients show reduced b-wave and impaired rod and cone function. The β2-subunit-deficient mice show downregulation of Cav1.4 α1-subunit and have a normal a-wave, a highly reduced b-wave and a thinner outer plexiform layer. Both of these channel disruptions may lead to a reduction in Ca2+ influx and transmitter release from photoreceptor synaptic terminals, which in turn disrupts normal synaptic development and function.
METHODS
Cloning of CaBP4 and Cav1.4 α1-subunit.
A partial cDNA clone encoding CaBP4 was amplified from a human retina cDNA library using primers based on CaBP2. The full-length cDNA sequence was isolated as two overlapping fragments by nested PCR from retinal cDNA libraries using primers that hybridized to the arm of the vector and CaBP4 sequence. The PCR products were cloned in pCRII-TOPO vector (Invitrogen) and were sequenced using the DyeDeoxyTerminator method (ABI-Prism, Perkin Elmer).
After removal of the stop codon by PCR, the mouse CaBP4 was cloned with a fragment encoding a (Gly)7-Leu-(Gly)7 fused to Ds-Red in pFastCMV44. Recombinant baculovirus carrying this fusion protein under the control of the CMV promoter was prepared as described previously44.
The mouse Cav1.4α1 gene was cloned in four fragments from mouse retina RNA by RT-PCR using primers based on published sequences; it encodes a protein of 1,984 amino acids that are identical to sequence for AJ579852 (ref. 8). The full-length coding sequence was put together into pcDNA3.1(−). For fusion to EGFP, mouse Cav1.4α1 was cloned in pEGFP-C2 (Clontech). The β2A- and α2δ-subunits were cloned into pcDNA3.1(+)45,46.
Tissue distribution of CaBP4.
Rat retina and brain mRNAs, and tissue northern blot containing 2 μg of poly(A)+ RNA from various human tissues were purchased from Clontech Laboratories. Northern blots were probed with 32P-labeled CABP4, Cabp4 or ACTB DNAs (encoding human and mouse CaBP4 and human β-actin, respectively)47. In addition, the tissue distribution was analyzed by RT-PCR with primers designed to amplify the complete mouse CaBP4 coding sequence as described previously14.
Expression of CaBP4 and the C-terminal fragment of Cav1.4α1 in bacteria.
His-tagged CaBP4 and Cav1.4α1 partial peptides were generated by cloning appropriate PCR fragments into pET30b (Novagen). The proteins were expressed in BL21 bacteria after induction with 0.2 mM IPTG and were purified on Ni2+-NTA columns according to the manufacturer’s protocol (Qiagen).
Transfection of Cav1.4-EGFP and CaBP4-DsRed2 in HEK293 cells.
HEK293 cells were transfected with Cav1.4-EGFP using Geneporter 2 reagent (Gene Therapy Systems) according to the manufacturer’s instructions. At 24 h after transfection, the cells were incubated with pFastCMV-mCaBP4-DsRed2 baculovirus for 5 h. The cells were incubated for an additional 2 d before fixation in 4% paraformaldehyde and analysis on a Zeiss LSM510 laser-scanning microscope (Carl Zeiss).
Antibody production.
Rabbit polyclonal antibodies against bacterially expressed full-length mouse CaBP4 (sera UW145 and UW146) were raised in New Zealand white rabbits as described previously14. Monoclonal mouse anti-PSD95 (clone K28/43) was purchased from Upstate Biotechnology. Monoclonal mouse anti–protein kinase C (clone MC5) was purchased from Sigma. Anti-synaptophysin was purchased from Sigma. Monoclonal mouse anti-bassoon antibody was purchased from Stressgen. Rabbit anti-GluR1 was purchased from Chemicon. Monoclonal mouse anti-calbindin (clone CL-300) was purchased from Sigma Immunochemicals.
CaBP4 knockout mice.
The targeting vector was constructed by replacing exon 1 and part of exon 2 with the neo gene cassette (Supplementary Fig. 1). The mice were generated as described in the legend to Supplementary Figure 1b. The Cabp4-targeted allele was maintained in a C57Bl/6J background. All procedures using mice were approved by the University of Washington Institutional Animal Care and Use Committee.
Histology, immunocytochemistry and in situ hybridization.
For histology, we fixed eyecups in 2% glutaraldehyde/2% paraformaldehyde for 18 h. For immunocytochemistry and in situ hybridization, we fixed eyecups in 4% paraformaldehyde in 0.1 M phosphate buffer (PB) for 8 h. After fixation, tissues were infiltrated with 20% sucrose in PB and then were embedded in 33% OCT compound (Miles) diluted with 20% sucrose in PB. We cut 10-μm sections for immunocytochemistry and in situ hybridization.
For histology, we cut sections at 5 μm, stained them with hematoxylin (Harris modified hematoxylin solution; Sigma) and viewed them with Nomarski optics (labophot-2, Nikon). Images were captured with a digital CCD (charge-coupled device) camera (Diagnostic Instruments). Probe synthesis and in situ hybridization for retinal sections were as previously described48.
Immunocytochemistry was carried out as described previously48. For double staining, a mixture of Cy3-conjugated goat anti–rabbit IgG and Alexa488–conjugated goat anti–mouse IgG was used. Sections were occasionally stained with Hoechst 33342 dye (Molecular Probes). Sections were analyzed under a confocal microscope (Zeiss LSM510, Carl Zeiss). Three-dimensional reconstructions (Fig. 3) were made using LSM510 software 3.0.
Transmission electron microscopy.
Tissue preparation and electron microscopy were carried out as described previously49. Photoreceptor terminals and synaptic ribbons were counted from working prints of sections obtained from three mice and at two different positions (~20 μm apart). Sections of ~800 μm were analyzed from one end to the other.
Affinity chromatographies.
Purified His-tagged mCaBP4 was loaded onto a Cav1.4α1 C terminus (amino acid 1445–1983)-Sepharose column that was equilibrated with 10 mM 1,3-bis[tris(hydroxymethy)methylamino] propane (BTP) (pH 7.5), 2 mM benzamidine and 0.1 mM CaCl2. The column was then washed successively with the same buffer containing 150 mM NaCl. The elution was done with 3 mM EGTA followed by 0.1 M Gly, pH 2.5. Fractions (1 ml) were collected and aliquots were analyzed by SDS-PAGE.
ERGs.
ERGs were recorded from anesthetized mice as described previously50. Leading edges of the ERG responses were fitted (as an ensemble) with a model of rod phototransduction activation as previously described19. C57Bl/6NCrlBr mice were used as controls.
Recordings from rods and rod bipolar cells.
Suction electrode recordings from rod photoreceptors and patch-clamp recordings from rod bipolar cells followed published procedures27. C57Bl/6J mice were used as controls. Rod and rod bipolar responses were each measured from four mice that were dark adapted for at least 12 h. For rod bipolar experiments, three or four slices were examined from each mouse. Photon densities measured at the preparation were converted to photoisomerizations per rod (Rh*/rod) assuming a collecting area of 0.5 μm2 (ref. 25). All experiments were at 35–37 °C.
Whole-cell patch-clamp recordings.
HEK293T cells were grown to ~70–80% confluence and were transfected with Gene Porter reagent (Gene Therapy Systems) according to manufacturer’s protocols. Cells were plated on 35-mm dishes and were transfected with a total of 5 μg DNA (α11.4, β2A, α2δ ± CaBP4) including 0.3 μg of pEGFP–N1 for fluorescence detection of transfected cells. At least 48 h after transfection, we acquired whole-cell patch-clamp recordings of transfected cells with a HEKA EPC-9 patch-clamp amplifier (HEKA Instruments). Data acquisition and leak subtraction using a P/−4 protocol were done with Pulse software (HEKA Instruments). Extracellular recording solutions contained 140 mM Tris, 2 mM MgCl2 and 20 mM CaCl2. Intracellular solutions consisted of 140 mM N-methyl-d-glucamine, 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, 2 mM MgCl2, 2 mM Mg-ATP and 5 mM EGTA. The pH of recording solutions was adjusted to 7.3 with methanesulfonic acid. I-V curves were fitted according to the following relationship: I = Gmax(V−Vrev)/(1+exp{(Vhalf−V)/kact}), where Vrev is the extrapolated reversal potential for ICa, V is the test voltage, I is the peak current, Gmax is the maximum conductance, Vhalf is the voltage for half-maximal activation and kact is the slope factor. Data were analyzed using Igor software (Wavemetrics) and graphs and statistical analyses were done with Sigma Plot (SPSS).
Statistical analyses.
A one-way analysis of variance was used for statistical analyses.
Supplementary Material
Acknowledgments
We thank W. Baehr, P. Detwiler, T. Doan, S. Bajjalieh and A. Polans for comments on the manuscript. We thank Y. Kim, M. Batten, A. Alekseev and M. Kalnoky for technical assistance. Anti-mGluR6 and anti-SV2 antibodies were gifts from S. Nakanishi and S.M. Bajjalieh, respectively. This research was supported by National Institutes of Health grants EY09339 to K.P., EY014561 to F.H., EY11850 to F.R. and NS044922 to A.L., the Whitehall Foundation to A.L., a grant from Research to Prevent Blindness (RPB), Inc. to the Department of Ophthalmology at the University of Washington and a grant from the E.K. Bishop Foundation. K.P. is a RPB Senior Investigator. D.P. was supported by a Vision Core Grant EY01730.
Footnotes
Note: Supplementary information is available on the Nature Neuroscience website.
COMPETING INTERESTS STATEMENT
The authors declare that they have no competing financial interests.
References
- 1.Zhang N, Townes-Anderson E. Regulation of structural plasticity by different channel types in rod and cone photoreceptors. J Neurosci. 2002;22:7065–7079. doi: 10.1523/JNEUROSCI.22-16-07065.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nachman-Clewner M, St Jules R, Townes-Anderson E. L-type calcium channels in the photoreceptor ribbon synapse: localization and role in plasticity. J Comp Neurol. 1999;415:1–16. [PubMed] [Google Scholar]
- 3.Schmitz F, Konigstorfer A, Sudhof TC. RIBEYE, a component of synaptic ribbons: a protein’s journey through evolution provides insight into synaptic ribbon function. Neuron. 2000;28:857–872. doi: 10.1016/s0896-6273(00)00159-8. [DOI] [PubMed] [Google Scholar]
- 4.Schmitz Y, Witkovsky P. Dependence of photoreceptor glutamate release on a dihydropyridine-sensitive calcium channel. Neuroscience. 1997;78:1209–1216. doi: 10.1016/s0306-4522(96)00678-1. [DOI] [PubMed] [Google Scholar]
- 5.Barnes S, Kelly MEM. Calcium channels at the photoreceptor synapse. in. Photorecept Calcium. 2002;514:465–476. doi: 10.1007/978-1-4615-0121-3_28. [DOI] [PubMed] [Google Scholar]
- 6.Morgans CW. Localization of the α1F calcium channel subunit in the rat retina. Invest Ophthalmol Vis Sci. 2001;42:2414–2418. [PubMed] [Google Scholar]
- 7.McRory JE, et al. The CACNA1F gene encodes an L-type calcium channel with unique biophysical properties and tissue distribution. J Neurosci. 2004;24:1707–1718. doi: 10.1523/JNEUROSCI.4846-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baumann L, Gerstner A, Zong X, Biel M, Wahl-Schott C. Functional characterization of the L-type Ca2+ channel Cav1.4α1 from mouse retina. Invest Ophthalmol Vis Sci. 2004;45:708–713. doi: 10.1167/iovs.03-0937. [DOI] [PubMed] [Google Scholar]
- 9.Koschak A, et al. Cav1.4α1 subunits can form slowly inactivating dihydropyridine-sensitive L-type Ca2+ channels lacking Ca2+-dependent inactivation. J Neurosci. 2003;23:6041–6049. doi: 10.1523/JNEUROSCI.23-14-06041.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rieke F, Schwartz EA. A cGMP-gated current can control exocytosis at cone synapses. Neuron. 1994;13:863–873. doi: 10.1016/0896-6273(94)90252-6. [DOI] [PubMed] [Google Scholar]
- 11.Bech-Hansen NT, et al. Mutations in NYX, encoding the leucine-rich proteoglycan nyctalopin, cause X-linked complete congenital stationary night blindness. Nat Genet. 2000;26:319–323. doi: 10.1038/81619. [DOI] [PubMed] [Google Scholar]
- 12.Strom TM, et al. An L-type calcium-channel gene mutated in incomplete X-linked congenital stationary night blindness. Nat Genet. 1998;19:260–263. doi: 10.1038/940. [DOI] [PubMed] [Google Scholar]
- 13.Ball SL, et al. Role of the β2 subunit of voltage-dependent calcium channels in the retinal outer plexiform layer. Invest Ophthalmol Vis Sci. 2002;43:1595–1603. [PubMed] [Google Scholar]
- 14.Haeseleer F, et al. Five members of a novel Ca2+-binding protein (CABP) subfamily with similarity to calmodulin. J Biol Chem. 2000;275:1247–1260. doi: 10.1074/jbc.275.2.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee A, et al. Differential modulation of Cav2.1 channels by calmodulin and Ca2+-binding protein 1. Nat Neurosci. 2002;5:210–217. doi: 10.1038/nn805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang J, et al. Identification of a family of calcium sensors as protein ligands of inositol trisphosphate receptor Ca2+ release channels. Proc Natl Acad Sci USA. 2002;99:7711–7716. doi: 10.1073/pnas.102006299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haynes LP, Tepikin AV, Burgoyne RD. Calcium-binding protein 1 is an inhibitor of agonist-evoked, inositol 1,4,5-trisphosphate-mediated calcium signaling. J Biol Chem. 2004;279:547–555. doi: 10.1074/jbc.M309617200. [DOI] [PubMed] [Google Scholar]
- 18.Koulen P, Fletcher EL, Craven SE, Bredt DS, Wassle H. Immunocytochemical localization of the postsynaptic density protein PSD-95 in the mammalian retina. J Neurosci. 1998;18:10136–10149. doi: 10.1523/JNEUROSCI.18-23-10136.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Van Hooser JP, et al. Rapid restoration of visual pigment and function with oral retinoid in a mouse model of childhood blindness. Proc Natl Acad Sci USA. 2000;97:8623–8628. doi: 10.1073/pnas.150236297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grunert U, Haverkamp S, Fletcher EL, Wassle H. Synaptic distribution of ionotropic glutamate receptors in the inner plexiform layer of the primate retina. J Comp Neurol. 2002;447:138–151. doi: 10.1002/cne.10220. [DOI] [PubMed] [Google Scholar]
- 21.Nomura A, et al. Developmentally regulated postsynaptic localization of a metabotropic glutamate receptor in rat rod bipolar cells. Cell. 1994;77:361–369. doi: 10.1016/0092-8674(94)90151-1. [DOI] [PubMed] [Google Scholar]
- 22.Dick O, et al. The presynaptic active zone protein bassoon is essential for photoreceptor ribbon synapse formation in the retina. Neuron. 2003;37:775–786. doi: 10.1016/s0896-6273(03)00086-2. [DOI] [PubMed] [Google Scholar]
- 23.Baylor DA, Lamb TD, Yau KW. Responses of retinal rods to single photons. J Physiol (Lond) 1979;288:613–634. [PMC free article] [PubMed] [Google Scholar]
- 24.Nakatani K, Yau KW. Calcium and magnesium fluxes across the plasma membrane of the toad rod outer segment. J Physiol (Lond) 1988;395:695–729. doi: 10.1113/jphysiol.1988.sp016942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Field GD, Rieke F. Nonlinear signal transfer from mouse rods to bipolar cells and implications for visual sensitivity. Neuron. 2002;34:773–785. doi: 10.1016/s0896-6273(02)00700-6. [DOI] [PubMed] [Google Scholar]
- 26.Slaughter MM, Miller RF. 2-amino-4-phosphonobutyric acid: a new pharmacological tool for retina research. Science. 1981;211:182–185. doi: 10.1126/science.6255566. [DOI] [PubMed] [Google Scholar]
- 27.Sampath AP, Rieke F. Selective transmission of single photon responses by saturation at the rod-to-rod bipolar synapse. Neuron. 2004;41:431–443. doi: 10.1016/s0896-6273(04)00005-4. [DOI] [PubMed] [Google Scholar]
- 28.Pitt GS, et al. Molecular basis of calmodulin tethering and Ca2+-dependent inactivation of L-type Ca2+ channels. J Biol Chem. 2001;276:30794–30802. doi: 10.1074/jbc.M104959200. [DOI] [PubMed] [Google Scholar]
- 29.Erickson MG, Alseikhan BA, Peterson BZ, Yue DT. Preassociation of calmodulin with voltage-gated Ca2+ channels revealed by FRET in single living cells. Neuron. 2001;31:973–985. doi: 10.1016/s0896-6273(01)00438-x. [DOI] [PubMed] [Google Scholar]
- 30.Zuhlke RD, Pitt GS, Deisseroth K, Tsien RW, Reuter H. Calmodulin supports both inactivation and facilitation of L-type calcium channels. Nature. 1999;399:159–162. doi: 10.1038/20200. [DOI] [PubMed] [Google Scholar]
- 31.Zhou H, et al. Ca2+-binding protein-1 facilitates and forms a postsynaptic complex with Cav1.2 (L-type) Ca2+ channels. J Neurosci. 2004;24:4698–4708. doi: 10.1523/JNEUROSCI.5523-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baylor DA, Fuortes MG. Electrical responses of single cones in the retina of the turtle. J Physiol (Lond) 1970;207:77–92. doi: 10.1113/jphysiol.1970.sp009049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pate P, et al. Determinants for calmodulin binding on voltage-dependent Ca2+ channels. J Biol Chem. 2000;275:39786–39792. doi: 10.1074/jbc.M007158200. [DOI] [PubMed] [Google Scholar]
- 34.Peterson BZ, DeMaria CD, Yue DT. Calmodulin is the Ca2+ sensor for Ca2+-dependent inactivation of 1-type calcium channels. Neuron. 1999;22:549–558. doi: 10.1016/s0896-6273(00)80709-6. [DOI] [PubMed] [Google Scholar]
- 35.Seidenbecher CI, Reissner C, Kreutz MR. Caldendrins in the inner retina. in. Photorecept Calcium. 2002;514:451–463. doi: 10.1007/978-1-4615-0121-3_27. [DOI] [PubMed] [Google Scholar]
- 36.Dolmetsch R. Excitation-transcription coupling: signaling by ion channels to the nucleus. Sci STKE. 2003;2003:PE4 . doi: 10.1126/stke.2003.166.pe4. [DOI] [PubMed] [Google Scholar]
- 37.Deisseroth K, Heist EK, Tsien RW. Translocation of calmodulin to the nucleus supports CREB phosphorylation in hippocampal neurons. Nature. 1998;392:198–202. doi: 10.1038/32448. [DOI] [PubMed] [Google Scholar]
- 38.Bading H, Ginty DD, Greenberg ME. Regulation of gene expression in hippocampal neurons by distinct calcium signaling pathways. Science. 1993;260:181–186. doi: 10.1126/science.8097060. [DOI] [PubMed] [Google Scholar]
- 39.Mao ZX, Bonni A, Xia F, Nadal-Vicens M, Greenberg ME. Neuronal activity-dependent cell survival mediated by transcription factor MEF2. Science. 1999;286:785–790. doi: 10.1126/science.286.5440.785. [DOI] [PubMed] [Google Scholar]
- 40.Morgans CW, El Far O, Berntson A, Wassle H, Taylor WR. Calcium extrusion from mammalian photoreceptor terminals. J Neurosci. 1998;18:2467–2474. doi: 10.1523/JNEUROSCI.18-07-02467.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morgans CW. Calcium channel heterogeneity among cone photoreceptors in the tree shrew retina. Eur J Neurosci. 1999;11:2989–2993. doi: 10.1046/j.1460-9568.1999.00719.x. [DOI] [PubMed] [Google Scholar]
- 42.Berntson A, Taylor WR, Morgans CW. Molecular identity, synaptic localization, and physiology oOf calcium channels in retinal bipolar cells. J Neurosci Res. 2003;71:146–151. doi: 10.1002/jnr.10459. [DOI] [PubMed] [Google Scholar]
- 43.Bech-Hansen NT, et al. Loss-of-function mutations in a calcium-channel α1-subunit gene in Xp11.23 cause incomplete X-linked congenital stationary night blindness. Nat Genet. 1998;19:264–267. doi: 10.1038/947. [DOI] [PubMed] [Google Scholar]
- 44.Haeseleer F, Imanishi Y, Saperstein DA, Palczewski K. Gene transfer mediated by recombinant baculovirus into mouse eye. Invest Ophthalmol Vis Sci. 2001;42:3294–3300. [PMC free article] [PubMed] [Google Scholar]
- 45.Ellis SB, et al. Sequence and expression of messenger RNAs encoding the α1-subunit and α2-subunit of a Dhp-sensitive calcium channel. Science. 1988;241:1661–1664. doi: 10.1126/science.2458626. [DOI] [PubMed] [Google Scholar]
- 46.Perezreyes E, et al. Cloning and expression of a cardiac brain β-subunit of the L-type calcium channel. J Biol Chem. 1992;267:1792–1797. [PubMed] [Google Scholar]
- 47.Haeseleer F, et al. Molecular characterization of a third member of the guanylyl cyclase–activating protein subfamily. J Biol Chem. 1999;274:6526–6535. doi: 10.1074/jbc.274.10.6526. [DOI] [PubMed] [Google Scholar]
- 48.Imanishi Y, et al. Characterization of retinal guanylate cyclase–activating protein 3 (GCAP3) from zebrafish to man. Eur J Neurosci. 2002;15:63–78. doi: 10.1046/j.0953-816x.2001.01835.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Batten ML, et al. Lecithin-retinol acyltransferase is essential for accumulation of all-trans-retinyl esters in the eye and in the liver. J Biol Chem. 2004;279:10422–10432. doi: 10.1074/jbc.M312410200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Maeda T. et al. Evaluation of the role of the retinal G protein–coupled receptor (RGR) in the vertebrate retina in vivo. J Neurochem. 2003;85:944–956. doi: 10.1046/j.1471-4159.2003.01741.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.