

Abstract
Leptosphaeria maculans is the most ubiquitous fungal pathogen of Brassica crops and causes the devastating stem canker disease of oilseed rape worldwide. We used minisatellite markers to determine the genetic structure of L. maculans in four field populations from France. Isolates were collected at three different spatial scales (leaf, 2-m2 field plot, and field) enabling the evaluation of spatial distribution of the mating type alleles and of genetic variability within and among field populations. Within each field population, no gametic disequilibrium between the minisatellite loci was detected and the mating type alleles were present at equal frequencies. Both sexual and asexual reproduction occur in the field, but the genetic structure of these populations is consistent with annual cycles of randomly mating sexual reproduction. All L. maculans field populations had a high level of gene diversity (H = 0.68 to 0.75) and genotypic diversity. Within each field population, the number of genotypes often was very close to the number of isolates. Analysis of molecular variance indicated that >99.5% of the total genetic variability was distributed at a small spatial scale, i.e., within 2-m2 field plots. Population differentiation among the four field populations was low (GST < 0.02), suggesting a high degree of gene exchange between these populations. The high gene flow evidenced here in French populations of L. maculans suggests a rapid countrywide diffusion of novel virulence alleles whenever novel resistance sources are used.
Stem canker of crucifers, also termed blackleg disease, is caused by Leptosphaeria maculans (Desm.) Ces. & de Not. (anamorph Phoma lingam Tode ex Fr.), a heterothallic haploid dothideomycete fungus. Stem canker of crucifers is the most damaging disease of oilseed Brassica crops, including oilseed rape (OSR; winter cultivars of Brassica napus) and canola (spring cultivars of B. napus and B. rapa) (39). This disease is widespread in Europe, Canada, and Australia and causes average OSR yield losses of 5 to 20% in France due to lodging of the crop (31).
The epidemiology of the stem canker disease has been extensively investigated (19, 39) since the first descriptions of the causal fungus by Tode in 1751 and Desmazières in 1849 (34). In Europe, OSR is grown as a winter crop and L. maculans produces pseudothecia in the autumn following sexual outcrosses on infected debris from the previous growing season. Airborne ascospores discharged from these pseudothecia infect leaves of the recently emerged crop, resulting in the development of leaf lesions (phoma leaf spots) that contain the asexual fruiting bodies, pycnidia. Asexual pycnidiospores can cause secondary infections, which are thought to be important in Australia but rare in Canada and Europe, where the disease is thought to be monocyclic (39). From the initial necrotrophic phase when leaf lesions occur, the fungus enters a symptomless biotrophic phase when, during winter and early spring, the fungus grows systemically in intercellular spaces down the petiole toward the crown at the stem base. During late spring and early summer in Europe, L. maculans again becomes necrotrophic and causes necrosis of the crown tissues that may result in the plant lodging prior to harvest (20, 21).
Strategies for stem canker disease management include cultural practices such as crop rotation, isolation of the crops from infected stubble of the previous growing season crops, and stubble management. Some control is achieved through the use of fungicides but, at present, disease control relies mainly on the use of disease-resistant cultivars. There are two types of disease resistance in B. napus: polygenic quantitative resistance (29, 30) or specific resistance, which involves major resistance gene(s) (19, 39). Nine avirulence genes (AvrLm genes), namely, AvrLm1 to AvrLm9, have been identified in L. maculans (2, 6-8), and the corresponding nine major resistance genes (Rlm1 to Rlm9) have been described in Brassica sp. (13), as expected in a host-pathogen system with gene-for-gene interactions (17). The Rlm genes effectively control the disease as long as the corresponding avirulent allele (AvrLm) dominates the pathogen population (3, 9, 31, 35). However, large-scale cropping of cultivars with a single effective Rlm gene results in the rapid breakdown of major gene resistance within a few years after commercial release of the cultivars due to the adaptation of the L. maculans field populations. Such breakdown of resistance was documented in France for Rlm1 (35) and in Australia for the breakdown of the “Surpass 400” resistance (23).
Our knowledge of genetic variation in L. maculans populations and the role of the evolutionary processes such as mutation, migration, genetic drift, selection, and/or recombination in these rapid shifts in the field population is still very limited. Previous studies of genetic variation in L. maculans populations from Canada and Australia found high levels of genetic variation in field populations of L. maculans (10, 11, 24), which is consistent with the hypothesis that sexual reproduction is an important part of the life cycle of L. maculans. However, reports of the distribution of genetic variation among and within field populations have been inconsistent (10, 11, 24). To date, there have been no studies of the genetic structure of L. maculans field populations in France and other countries of Western Europe. In these regions, previous work has focused mainly on field population race structure and has used virulence markers to assess genetic diversity in the fungus (5, 9, 22, 35).
Our objective here was to assess genetic variation in French populations of L. maculans with minisatellite loci (15), mating type, and virulence markers (8). We tested the hypotheses that the field populations were randomly mating, that populations from different fields were genetically separable, and that epidemics are initiated by ascospores. The present study provides the first estimates of genetic structure of field populations of L. maculans from France and suggests that genetic structure of populations from Western Europe is similar to that in Australia.
MATERIALS AND METHODS
Sampling of L. maculans field populations.
A total of 401 isolates were obtained from pycnidiospores oozing from individual pycnidia of leaf lesions. The resulting cultures were subcultured as hyphal tips for further purification (38). All fungal cultures were maintained on V8 juice agar, and conidia were produced on V8 juice medium as previously described (2). Isolates of L. maculans were collected from four OSR fields in France as soon as the first leaf lesions were observed in Le Rheu (western France), Oucques (central France), and Grignon (near Paris) in autumn 2000 and in Versailles (near Paris) in autumn 2003. In each field, cultivar Drakkar, a cultivar that lacks the nine known resistance alleles (Rlm genes), was grown, and samples were taken from four arbitrarily selected plots (2 by 1 m). From each 2-m2 plot, 25 naturally infected plants were arbitrarily selected, and one leaf with phoma leaf lesions was collected from each plant. One lesion per leaf was arbitrarily selected, and the causal organism was isolated. In addition, isolations were made from each of 50 leaf lesions occurring on one leaf from Oucques (autumn 2000) and from every lesion occurring on five leaves (43 total lesions) collected at Versailles in December 2003.
Pathogenicity tests.
Races of the L. maculans isolates were determined by inoculating the 401 isolates onto a B. napus differential set, comprising fixed lines or commercial cultivars with as few Rlm alleles as possible (8). Cotyledons of the fully susceptible cv. Westar (no known Rlm allele), the cvs. Columbus (Rlm1 and Rlm3), Bristol (Rlm2 and Rlm9), 22-1-1 (Rlm3), Jet Neuf (Rlm4), 23-2-1 (Rlm7), Falcon-MX (Rlm4 and Rlm6), and Samourai-MX (Rlm1 and Rlm6) or Darmor-MX (Rlm6) were inoculated according to established protocols (2, 7). At 14 to 27 days after inoculation, symptoms were scored from 10 to 12 plants by using the IMASCORE rating scale comprising six infection classes (IC1 to IC6), where IC1 to IC3 corresponded to avirulent isolates (AvrLm) and IC4 to IC6 corresponded to virulent isolates (avrLm) (7).
Mating type determination.
A multiplex PCR, developed to rapidly characterize the mating type of L. maculans isolates (12), was used to determine the distribution of the Mat1-1 and Mat1-2 alleles within the 401 field isolates collected. The distribution of mating type alleles was analyzed with a χ2 test.
Minisatellite analyses.
We evaluated six polymorphic minisatellite loci (MinLm1 to MinLm6), all on different chromosomes in L. maculans (4, 15). New specific primers based on the published sequences (accession numbers AJ621802 and AJ621805, respectively) were designed for MinLm3 and MinLm5 by using Oligo v.5.0 software (Molecular Biology Insights, Inc., Cascade, Iowa). Primers MinLm3-ULG (5′-GGCTCGGTCGGTTAGTTA-3′) and MinLm3-LLG (5′-AATGATGTACAGGACGGGATTT-3′) or primers MinLm5-ULG (5′-GCCGCCCGCCGCCTTACC-3′) and MinLm5-LLG (5′-GAGCTCCTGCGCCACAGTG-3′) hybridized to the flanks of the minisatellite locus and allow amplification of the repeated motifs only. MinLm2, MinLm4, and MinLm6 were amplified by using primers that had been previously described (15).
For PCR, genomic DNA was extracted from pycnidiospores by using the DNeasy 96 plant kit (QIAGEN S.A., Courtaboeuf, France) as previously described (4). PCR amplification was done in a total volume of 15 μl containing 0.2 μM of each deoxynucleotide triphosphate, 1.2 μM of each primer, 0.6 U of Taq DNA polymerase (Qbiogen, Illkirch, France), 1.5 μl of a 10× reaction buffer supplied with the enzyme, and 10 to 30 ng of genomic DNA. PCR amplifications were done in an Eppendorf Mastercycler EP Gradient thermocycler (Eppendorf, Le Pecq, France), with 30 cycles of 94°C for 30 s, 60°C for 30 s, and 72°C for 80 s, with a final extension at 72°C for 5 min. The cycling conditions were the same for the amplification of all minisatellite loci. The PCR products were separated by electrophoresis on a 2.5% SeaKem LE agarose gel (FMC, Rockland, Maine) and visualized by staining with ethidium bromide and UV illumination. Allele sizes were determined by using Quantity One 1-D Analysis software (Bio-Rad, Marnes-la-Coquette, France) and comparing the bands for the alleles with those of the 1-kb ladder (Invitrogen, Cergy Pontoise, France) and with PCR products obtained after amplification of alleles of known size for each minisatellite. Each allele identified at the six minisatellite loci was previously sequenced from at least two isolates to determine the exact allele size and to confirm that the size polymorphism observed after electrophoresis was due to variability in the number of tandemly repeated core motifs (4, 15). Sequencing of the same allele amplified from at least three different individuals also confirmed that sequences of the same size are homologous and size homoplasy did not occur (4, 15). The data were scored as the number of repeat units for each minisatellite.
Statistical analysis.
For statistical analyses, the 401 isolates were divided into a “leaf-scale group” comprising 50 isolates collected from one leaf in Oucques and 43 isolates collected from five leaves in Versailles and a “field-scale group” comprising 308 isolates. The field scale group was structured into four field populations, each of which was further subdivided into four subpopulations corresponding to four 2-m2 plots.
Genetic diversity, which was studied separately for each field population and for all fields sampled, was estimated from genotype and gene diversity. Genotypes of isolates in both the field- and leaf-scale groups were determined by combining information from the alleles at minisatellite loci and at the mating type locus. Genotype diversity (G) was then estimated as the percentage of unique genotypes obtained in each sample unit, in each field population, and in the overall sample units. A discriminatory index (DI), based on Simpson's index of diversity, was used to estimate the discriminatory power of the six minisatellites. DI measures the probability that two individuals randomly selected from a small sample will have the same haplotype. DI is determined by the number and relative frequencies of the different haplotypes and is calculated as follows:
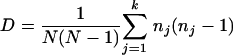 |
where N is the total number of isolates, k is the total number of haplotypes, and nj is the number of isolates sharing the jth multilocus haplotype (14).
POPGENE version 1.31 software (40) was used to compute allele frequency, Nei's gene diversity (h) (27) for each population and Nei's measures of genetic identity (I) between populations over all loci (28). The mean gene diversity (H) was the mean of h over loci within each population.
Gametic linkage disequilibria were computed by using GENEPOP version 3.3 software (32) for each pair of loci within each of the four field populations and in the overall field samples. GENEPOP performs the Fisher exact test using a Markov chain to test for gametic disequilibrium between two loci. The null hypothesis H0 was: “genotypes at one locus are independent from genotypes at the other locus.” The significance of departure from linkage equilibrium was assessed by using Bonferroni adjusted P values (33). Linkage disequilibrium also was estimated by pairwise comparison of haplotypes using the index of multilocus gametic disequilibrium 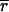 d (1), which tests to what extent individuals that are identical (or different) at one locus are more likely to be identical (or different) at the other loci. To determine whether an observation deviates significantly from the null hypothesis of random mating (no linkage disequilibrium among loci), the observed value was compared to the results of 500 randomized data sets. The program MULTILOCUS version 1.3 (1) was used for both estimation and randomization.
d (1), which tests to what extent individuals that are identical (or different) at one locus are more likely to be identical (or different) at the other loci. To determine whether an observation deviates significantly from the null hypothesis of random mating (no linkage disequilibrium among loci), the observed value was compared to the results of 500 randomized data sets. The program MULTILOCUS version 1.3 (1) was used for both estimation and randomization.
Heterogeneity of allele frequencies among field populations was investigated for each locus with contingency tables analysis and the Fisher exact test as implemented in the GENEPOP program. The null hypothesis tested was H0: “allele frequencies are homogeneous across populations.” Genetic structure was analyzed with Nei's coefficient of population differentiation (GST), which indicates the proportion of the total genetic variation attributable to population differentiation. Analyses of molecular variance (AMOVA) were computed with Arlequin version 2.000 software (37) and used to estimate how genetic variability is partitioned among fields, among plots within fields, and within plots. Φ statistics associated with these components of variance (ΦCT, ΦSC, and ΦST, respectively) were estimated and their significance tested by using a nonparametric permutation approach (16).
RESULTS
Diversity of virulence.
Six avirulence loci (AvrLm1, AvrLm2, AvrLm3, AvrLm4, AvrLm6, and AvrLm7) were characterized in four field-scale populations. All isolates were avirulent at the AvrLm6 and AvrLm7 loci and were virulent at the AvrLm2 and AvrLm3 loci. Isolates were polymorphic only at the AvrLm1 and AvrLm4 loci where virulent isolates (avrLm1 and avrLm4 isolates, respectively) were more frequent. The frequency of avrLm1 varied significantly by population, ranging from 0.60 in the Le Rheu population to 0.97 in the Versailles population, whereas avrLm4 frequencies were close to 0.90 at all locations (Table 1).
TABLE 1.
Allelic frequencies and gene diversities at two avirulence loci of Leptosphaeria maculans in French field populations
| Locusb | Allele frequencya in population at:
|
Pc | |||
|---|---|---|---|---|---|
| Grignon (n = 82) | Le Rheu (n = 81) | Oucques (n = 82) | Versailles (n = 63) | ||
| AvrLm1 | 0.05 | 0.40 | 0.22 | 0.03 | <0.05 |
| h | 0.09 | 0.48 | 0.34 | 0.06 | |
| AvrLm4 | 0.10 | 0.07 | 0.11 | 0.06 | 0.77 |
| h | 0.18 | 0.14 | 0.20 | 0.12 | |
| H | 0.13 | 0.31 | 0.27 | 0.09 | |
Frequency of avirulence allele; n, number of isolates analyzed.
h, gene diversity per locus; H, gene diversity averaged over both loci.
Probability for the Fisher exact test for frequency heterogeneity across the populations.
Minisatellite variation.
Five of the six minisatellite loci tested in the present study were polymorphic at a 5% level in the populations analyzed (Table 2). The data from the MinLm4 marker were not evaluated further because one of the three alleles at this locus was found in >95% of the isolates (Table 2). The remaining five minisatellite loci—MinLm1, MinLm2, MinLm3, MinLm5, and MinLm6—all had a high level of allelic variation in the four field populations studied with 7, 16, 12, 6, and 7 alleles, respectively (Table 3). Based on the discriminatory index DI, the probability that two isolates randomly selected from the 401 isolates would have the same combination of alleles over these five loci was 8.5 × 10−4 (Table 4).
TABLE 2.
Frequencies of the most common minisatellite alleles, genic differentiation, and gene diversityd for minisatellite loci across four field populations of Leptosphaeria maculans
| Locus | Allelea | Allele frequency in population at:
|
Pb | |||
|---|---|---|---|---|---|---|
| Grignon | Le Rheu | Oucques | Versailles | |||
| MinLm1 | 2× | 0.20 | 0.25 | 0.24 | 0.16 | <0.05 |
| 5× | 0.72 | 0.52 | 0.61 | 0.70 | ||
| h | 0.44 | 0.65 | 0.57 | 0.48 | ||
| MinLm2 | 6× | 0.23 | 0.20 | 0.07 | 0.25 | 0.07 |
| 7× | 0.09 | 0.12 | 0.15 | 0.18 | ||
| 9× | 0.21 | 0.21 | 0.20 | 0.16 | ||
| h | 0.86 | 0.86 | 0.88 | 0.86 | ||
| MinLm3 | 3× | 0.56 | 0.35 | 0.40 | 0.57 | 0.12 |
| 9× | 0.13 | 0.21 | 0.16 | 0.10 | ||
| 15× | 0.04 | 0.17 | 0.12 | 0.11 | ||
| h | 0.65 | 0.78 | 0.78 | 0.64 | ||
| MinLm4 | 3× | 0.99 | 0.96 | 0.96 | 0.98 | 0.86 |
| h | NDc | ND | ND | ND | ||
| MinLm5 | 4× | 0.52 | 0.59 | 0.45 | 0.61 | 0.59 |
| 5× | 0.33 | 0.28 | 0.38 | 0.32 | ||
| h | 0.61 | 0.56 | 0.64 | 0.51 | ||
| MinLm6 | 4× | 0.43 | 0.51 | 0.39 | 0.40 | 0.18 |
| 5× | 0.31 | 0.37 | 0.37 | 0.35 | ||
| h | 0.69 | 0.60 | 0.69 | 0.70 | ||
| H | 0.68 | 0.71 | 0.75 | 0.71 | ||
2×, 5×, etc., alleles are scored as the number of repeat units; h, gene diversity per locus; H, gene diversity averaged over all four loci (MinLm2, MinLm3, MinLm5, and MinLm6).
Probability for the Fisher exact test for frequency heterogeneity across the populations.
ND, not determined.
Gene diversity was computed only for polymorphic loci at a 5% level.
TABLE 3.
Number of alleles at the six minisatellite loci in field populations of Leptosphaeria maculans
| Locus | No. of allelesa
|
|||
|---|---|---|---|---|
| Grignon | Le Rheu | Oucques | Versailles | |
| MinLm1 | 5 | 7 (1) | 6 | 6 |
| MinLm2 | 13 (1) | 12 | 15 (1) | 12 |
| MinLm3 | 12 (2) | 9 | 11 (3) | 9 |
| MinLm4 | 3* | 3* | 3* | 3* |
| MinLm5 | 5 | 6 | 5 | 4 |
| MinLm6 | 7 | 6 | 6 | 6 |
| Total | 45 | 43 | 46 | 40 |
The number of low-frequency alleles present in only one field population is indicated in parentheses.
, not polymorphic at a 5% level.
TABLE 4.
Genotype diversity at three spatial scales in field populations of Leptosphaeria maculans in France
| Scale group and geographic origin | Subpopulation | nc | No. of leaves | No. of genotypesa | Gb |
|---|---|---|---|---|---|
| Field scale | |||||
| Grignon | P1 | 20 | 20 | 20 | 100 |
| P2 | 24 | 24 | 23 | 96 | |
| P3 | 18 | 18 | 17 | 94 | |
| P4 | 20 | 20 | 20 | 100 | |
| Le Rheu | P1 | 22 | 22 | 22 | 100 |
| P2 | 22 | 22 | 22 | 100 | |
| P3 | 18 | 18 | 18 | 100 | |
| P4 | 19 | 19 | 19 | 100 | |
| Oucques | P1 | 18 | 18 | 17 | 94 |
| P2 | 21 | 21 | 20 | 95 | |
| P3 | 21 | 21 | 21 | 100 | |
| P4 | 22 | 22 | 21 | 95 | |
| Versailles | M5 | 13 | 13 | 13 | 100 |
| M6 | 17 | 17 | 17 | 100 | |
| M7 | 17 | 17 | 15 | 88 | |
| M9 | 16 | 16 | 16 | 100 | |
| Leaf scale | |||||
| Oucques | FI | 50 | 1 | 49 | 98 |
| Versailles | FI | 8 | 1 | 8 | 100 |
| FII | 10 | 1 | 10 | 100 | |
| FIII | 7 | 1 | 7 | 100 | |
| FIV | 10 | 1 | 10 | 100 | |
| FV | 8 | 1 | 8 | 100 |
Genotypes are based on multilocus haplotypes determined by combining information from the alleles at MinLm1, MinLm2, MinLm3, MinLm5, MinLm6, and Mat1.
Genotypic diversity is expressed as the percentage of unique genotypes.
n, number of isolates analyzed.
Genetic diversity in field populations.
Overall, 349 different multilocus genotypes were obtained with the mating type and five minisatellite loci among the 401 analyzed isolates (Table 4). The mean number of isolates per genotype was low (1.15) and 309 isolates (78%) had unique genotypes. A few putatively clonal genotypes were distributed within and among the four populations of L. maculans. These isolates are unlikely to be true clones since the addition of the two polymorphic virulence loci, AvrLm1 and AvrLm4, enabled us to distinguish 12 additional genotypes among the 92 isolates with “clonal” genotypes (data not shown). The most common genotype identified with the mating type locus and the five minisatellite loci was shared by five isolates, of which four originated from the four field plots sampled in Le Rheu. One genotype, shared by three isolates, was found in three different populations, and 17 genotypes occurring twice were found in two populations. The genotype distribution exhibited no variation between the different geographic scales. The genotypic diversity reached its maximum value within 2-m2 field plots or at the leaf scale (Table 4). Even at the smallest spatial scale, i.e., the leaf scale, the number of genotypes often was very close to the number of individuals collected (Table 4). Only 2 of 50 isolates collected from the same leaf had the same genotype. These two isolates originated from lesions in close proximity to each other, and we think that they originate from a single strain that either had caused two separate lesions or was sampled twice.
The Grignon and Oucques populations contained the most alleles over the six loci (Table 3). These two populations also had alleles at the MinLm2 and MinLm3 loci that occurred at low frequency and that were not present in the other field populations (Table 3). Two or three alleles at MinLm2, MinLm3, MinLm5, and MinLm6 were more frequent and were present at similar frequencies in all of the analyzed populations (Table 2). MinLm1 is physically close to the AvrLm1 avirulence gene, and the allele frequencies at these two loci were significantly different across the four populations (Tables 1 and 2). MinLm1 was excluded from analysis of gene diversity and distribution of genetic diversity because this locus was in linkage disequilibrium with AvrLm1 (4, 18). Nei's measures of gene diversity averaged over the four loci MinLm2, MinLm3, MinLm5, and MinLm6 were similar among the four field populations and ranged from 0.68 in the Grignon population to 0.75 in the Oucques population (Table 2).
Mating type distribution and linkage disequilibria in field populations.
PCR products corresponding to one of the two mating type idiomorphs could be amplified from all 401 isolates. Both mating types were present in the populations at all of the tested spatial scales, including different lesions on the same leaf, different plots within a field, and different fields within the country. No significant departures from the 1:1 ratio between the two mating types were observed at the field scale (Table 5). Linkage disequilibria were assessed with MinLm1, MinLm2, MinLm3, MinLm5, and MinLm6 and using two different approaches. Within field populations, only two pairwise tests of linkage disequilibria were significant in the Le Rheu and Versailles populations, but none of them were significant after Bonferroni's corrections for multiple comparisons were made. When the complete field-scale data set was analyzed as a single group, none of the pairwise comparisons were significant. The multilocus linkage disequilibrium values ( d) obtained for the four field populations were close to zero and not significant (P > 0.05). No significant deviation from random association of alleles was detected within the total population (
d) obtained for the four field populations were close to zero and not significant (P > 0.05). No significant deviation from random association of alleles was detected within the total population (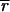 d < 0.01, P = 0.40), suggesting that recombination regularly generates new genotypes at the field level. Linkage disequilibria between the minisatellite loci and the polymorphic avirulence loci (AvrLm1 and AvrLm4) also were assessed in field populations. Overall, only one pairwise test of linkage disequilibria, which corresponded to the MinLm1 and AvrLm1 loci, was significant after Bonferroni's corrections. These two loci were in significant linkage disequilibria only within the Le Rheu and Oucques populations.
d < 0.01, P = 0.40), suggesting that recombination regularly generates new genotypes at the field level. Linkage disequilibria between the minisatellite loci and the polymorphic avirulence loci (AvrLm1 and AvrLm4) also were assessed in field populations. Overall, only one pairwise test of linkage disequilibria, which corresponded to the MinLm1 and AvrLm1 loci, was significant after Bonferroni's corrections. These two loci were in significant linkage disequilibria only within the Le Rheu and Oucques populations.
TABLE 5.
Distribution of the mating type alleles of Leptosphaeria maculans in field populations in France
| Scale group and geographic origin | Subpopulation(s) | nb | No. of alleles for mating typea:
|
|
|---|---|---|---|---|
| Mat1-1 | Mat1-2 | |||
| Field scale | ||||
| Grignon | P1-P4 | 82 | 49 | 33 |
| Le Rheu | P1-P4 | 81 | 34 | 47 |
| Oucques | P1-P4 | 82 | 48 | 34 |
| Versailles | P1-P4 | 63 | 34 | 29 |
| Leaf scale | ||||
| Oucques | FI | 50 | 22 | 28 |
| Versailles | FI | 8 | 2 | 6 |
| FII | 10 | 5 | 5 | |
| FIII | 7 | 4 | 3 | |
| FIV | 10 | 4 | 6 | |
| FV | 8 | 1 | 7 | |
| FI-FV | 43 | 16 | 27 | |
Whenever sample sizes were large enough to perform a χ2 test, mating type frequencies were not significantly different at P = 0.05.
n, number of isolates analyzed.
Distribution of genetic diversity within and among populations.
Distribution of genetic diversity was estimated using MinLm2, MinLm3, MinLm5, and MinLm6. Pairwise estimates of Nei's coefficient of population differentiation (GST) were relatively low and ranged from 0.004 to 0.011 (Table 6). These estimates are consistent with the hypothesis that there is no genetic differentiation between these French field populations. Similar results were obtained from the Fisher exact test for heterogeneity of allele frequencies among field populations (Table 2) and from an AMOVA performed with the four field populations as a whole (ΦST = 0.0007, P = 0.14; data not shown). The results of the AMOVA, which partitions the genetic variation hierarchically from field plot level to an among-field level, revealed that most of the allelic variability (>99.5%) was found in the 2-m2 field plots and that the genetic variation among field populations accounted for only 0.11% of the total variability (Table 7). Nei's measure of genetic identity was high for each pairwise comparison of field populations (Table 6). The highest estimate of genetic identity was obtained from a pairwise comparison of the Versailles and Grignon populations, which also were the geographically closest locations (Table 6).
TABLE 6.
Pairwise comparisons of population differentiation (GST) and Nei's unbiased measure of gene identity (I) in Leptosphaeria maculans field populations from France
| Population |
GST (I) values for population:
|
||
|---|---|---|---|
| Le Rheu | Oucques | Versailles | |
| Grignon | 0.011 (0.95) | 0.008 (0.96) | 0.004 (0.98) |
| Le Rheu | 0.010 (0.95) | 0.011 (0.95) | |
| Oucques | 0.011 (0.95) | ||
TABLE 7.
Analysis of molecular variance of the multilocus haplotypes for four Leptosphaeria maculans populations isolated from OSR fields in France
| Source of variation | df | Variance components | % of variation | Pa | Φ statistics |
|---|---|---|---|---|---|
| Among field populations | 3 | 0.00053 | 0.11 | 0.062 | ΦCT = 0.001 |
| Among field plots, but within field populations | 12 | 0.00165 | 0.33 | 0.017 | ΦSC = 0.003 |
| Within field plots | 292 | 0.49864 | 99.56 | 0.001 | ΦST = 0.004 |
Probability of having values larger than those observed based on 1,000 randomizations of the treatments.
DISCUSSION
The four minisatellite loci examined had a large number of alleles at each locus, which enabled a high level of discrimination between field isolates. Most of the isolates could be distinguished by using this set of minisatellite loci, and adding one or two more polymorphic minisatellite loci (currently unavailable) to this set probably would differentiate all of the isolates except those that are true clones. The most common genotype among the 401 isolates was shared by only five isolates and corresponds to the combination of the most frequent alleles at each locus. Furthermore, these five isolates were polymorphic at the virulence loci and correspond to at least two genotypes (data not shown), suggesting that some of these isolates are genetically distinct individuals and that they do not represent a widely distributed clone.
The 1:1 distribution of mating type alleles we observed also has been observed in field populations of L. maculans in Australia (10) and is expected in randomly mating populations (26). Milgroom (26) proposed several tests for random mating in natural populations of fungal pathogens. Among these, the frequent occurrence of fungal sexual structures, a high level of genotypic diversity, and gametic linkage equilibrium, all provide evidence that is consistent with sexual reproduction. In L. maculans, the frequent occurrence of pseudothecia has been documented in many instances (19, 39). The present study further supports the importance of sexual reproduction by showing that minisatellite loci were in linkage equilibrium and that genotypic diversity was very high even at small spatial scales in field populations of L. maculans in France.
Linkage disequilibrium was detected between the MinLm1 and AvrLm1 loci, which are physically close and genetically linked. The genetic distance between these loci ranges from 0 to >30 cM depending on the L. maculans genetic map considered (L. Gout, unpublished data). The suppression of recombination observed in some genetic maps may be related to nonhomologous sequences present in this region in the avirulent and virulent parental isolates used (L. Gout, unpublished data). The nonhomologous genome organization of the AvrLm1 region in avirulent and virulent isolates has been conserved in field populations from Western Europe based on the lack of apparent recombination between markers spanning this region (5). In the Le Rheu and Oucques populations, the presence of avirulent and virulent isolates could have reduced recombination near AvrLm1 and have led to the linkage disequilibrium between MinLm1 and AvrLm1 via “hitchhiking” to AvrLm1, which would be selected for when cultivars carrying Rlm1 were planted. MinLm1 and AvrLm1 were in linkage equilibrium in the Grignon and Versailles populations, but >95% of the isolates in these field populations were virulent. The lack of disequilibrium in this genomic region in these populations could result because the recombination rate in the region is not the same when populations are mainly virulent as it is when both avirulent and virulent isolates coexist in the population.
L. maculans has a mixed reproductive system (39). Ascospores are thought to be the major source of inoculum for the annual epidemics. Our results are consistent with this hypothesis and similar to those of studies of field populations from Canada (24) or Australia (10). Pycnidia are produced throughout the growing season, but the role of the asexual pycnidiospores in the epidemiology of the disease remains unclear, and secondary infections via pycnidiospores are thought to be rare in Western Europe (39). Our collections were mainly made at a time corresponding to the initiation of epidemics, and the variation we observed is consistent with the hypothesis that ascospores are the primary inoculum. Additional samples collected throughout the growing season are needed to determine the role of asexual spores in the epidemiology of L. maculans in France.
Most of the total genetic variability in these populations of L. maculans (>99.5%) was distributed at a small spatial scale, i.e., within 2-m2 field plots. These populations also had a very high level of gene (0.68 ≤ H ≤ 0.75) and genotypic (88% ≤ G ≤ 100%) diversity even over small spatial scales. The four field populations, despite being collected in two different years and from sites more than 500 km apart, may be coevolving parts of a large panmictic population. Ascospores are airborne, can potentially be dispersed over distances of several kilometers, can survive up to 6 weeks (19, 39), and could enable this genetic homogenization.
In contrast to the minisatellite loci, the level of diversity at the virulence loci was low, with only two of the six characterized avirulence loci (AvrLm1 and AvrLm4) being polymorphic. Polymorphism was similar across the four field populations for AvrLm4 but not for AvrLm1. The two corresponding resistance genes, Rlm1 and Rlm4, have been widely deployed in France, with cultivars carrying Rlm4 in use since the 1970s (36) and cultivars carrying Rlm1 in use since the early 1990s (36). Thus, subpopulations of L. maculans may be differentiated based on selectable markers, e.g., the AvrLm1 locus, as a result of spatial heterogeneity in the use of Rlm1 cultivars (35). This genic differentiation of subpopulations may be transitory, and the genetic exchange occurring in French populations of L. maculans should homogenize the frequencies of these virulence alleles. Increased use of cultivars carrying Rlm gene(s) across all OSR growing regions could give the same result, but this scenario is less likely since the commercial success of cultivars with a given Rlm gene should be inversely proportional to the frequency of the corresponding virulence allele in the pathogen populations. Indeed, the frequencies of the avrLm4 virulence allele are very similar across all of the field populations we studied, and all of the isolates characterized in the present study carried the avrLm2 virulence allele. The corresponding resistance gene, Rlm2, was used in France prior to Rlm4 and is still present in many cultivars (36).
A low level of differentiation between field populations of L. maculans also was observed in Australia, where the high level of genetic variability revealed by amplified fragment length polymorphism analyses was distributed mainly within field populations (10). In contrast, AMOVA done on RAPD [random(ly) amplified polymorphic DNA] data attributed only 55% of the total variability to differences within field populations in Canada. Simultaneous analysis of field populations from these continents together with some European field populations is now needed (i) to evaluate the genetic structure of worldwide populations, (ii) to identify potential subpopulations, and (iii) to estimate the level of intercontinental genetic exchange. The amount of intercontinental genetic exchange is of particular interest since gene flow, due to natural long distance dispersal of propagules or linked to human activities, may lead to genetic exchange between populations located on different continents. Thus, the durability of a resistance gene on one continent could be altered by the durability of the same resistance gene on another. Resistance gene durability is important because only a few Rlm genes are known (13) and maintaining the effectiveness of each resistance gene is critical for the sustainable control of stem canker. The minisatellite markers we used in the present study were useful in determining the amount of genetic variability within and between field populations in France and may be useful for a global population study.
Our study is the first to evaluate the genetic variability of L. maculans field populations in France. Based on the level of gametic linkage equilibria observed and the frequency-dependent selection of mating type alleles, we showed that sexual recombination is an important part of this fungus's life cycle and occurs regularly in L. maculans field populations in France. Indeed, we showed that ascospores could be the primary source of inoculum and could be responsible for the leaf-spotting phase of the disease in both autumn and winter. High levels of gene flow also were inferred from the low levels of population differentiation. McDonald and Linde (25) recently proposed a framework to predict the evolutionary potential of pathogen populations based on analysis of their genetic structure. According to their model, pathogens such as L. maculans with high gene flow and mixed reproduction system have moderate to high evolutionary potential. However, the recent breakdown of the Rlm1 resistance occurred in a time frame (only 3 years) that is similar to that predicted for pathogens with high evolutionary potential, which pose the greatest “risk” of breaking down novel specific resistance genes. Fungal pathogens such as rusts or mildews assigned to this highest risk category (25) often have mixed reproductive systems and high levels of migration (genotype flow) through long-distance dispersal. Thus, the rapid breakdown of Rlm1 now questions the level of migration among L. maculans field populations.
Acknowledgments
This study was supported by EU contract QLK5-CT-2002-01813 SECURE and a grant from the French Ministry of Agriculture (Contrat de Branche Résistance du Colza au Phoma: Nouvelles Resistances et Suivi Dynamique des Races-2000-2003). M.E. was funded by the European Union (Marie Curie Fellowship HPMT-CT-2001-00395), the Biotechnology and Biological Sciences Research Council, and DuPont.
We thank L. Bousset (INRA Bio3P, Le Rheu, France), E. Fournier (INRA, Versailles, France), and J. Enjalbert (INRA, Grignon, France) for helpful discussions during the preparation of the manuscript; anonymous reviewers for comments and suggestions; K. Louvard (INRA PMDV, Versailles, France) for isolate collections and DNA extractions; M. Chabirand and L. Coudard (INRA PMDV, Versailles, France) for plant management; J. P. Narcy and J. Roux (INRA-PMDV) for technical assistance; and C. Cogoluènhes and F. Le Coz, from Institut National Agronomique Paris-Grignon, for their contributions to this study.
REFERENCES
- 1.Agapow, P.-M., and A. Burt. 2001. Indices of multilocus linkage disequilibrium. Mol. Ecol. Notes 1:101-102. [Google Scholar]
- 2.Ansan-Melayah, D., M. H. Balesdent, M. Buée, and T. Rouxel. 1995. Genetic characterization of AvrLm1, the first avirulence gene of Leptosphaeria maculans. Phytopathology 85:1525-1529. [Google Scholar]
- 3.Ansan-Melayah, D., T. Rouxel, J. Bertrandy, B. Letarnec, E. Mendes-Pereira, and M. H. Balesdent. 1997. Field efficiency of Brassica napus specific resistance correlates with Leptosphaeria maculans population structure. Eur. J. Plant Pathol. 103:835-841. [Google Scholar]
- 4.Attard, A., M. Gourgues, L. Gout, J. Schmit, J. Roux, J. P. Narcy, M. H. Balesdent, and T. Rouxel. 2001. Molecular characterisation and polymorphism of MinLm1, a minisatellite from the phytopathogenic ascomycete Leptosphaeria maculans. Curr. Genet. 40:54-64. [DOI] [PubMed] [Google Scholar]
- 5.Attard, A., L. Gout, M. Gourgues, M. L. Kuhn, J. Schmit, S. Laroche, D. Ansan-Melayah, A. Billault, L. Cattolico, M. H. Balesdent, and T. Rouxel. 2002. Analysis of molecular markers genetically linked to the Leptosphaeria maculans avirulence gene AvrLm1 in field populations indicates a highly conserved event leading to virulence on Rlm1 genotypes. Mol. Plant-Microbe Interact. 15:672-682. [DOI] [PubMed] [Google Scholar]
- 6.Balesdent, M. H., A. Attard, D. Ansan-Melayah, R. Delourme, M. Renard, and T. Rouxel. 2001. Genetic control and host range of avirulence toward Brassica napus cultivars Quinta and Jet Neuf in Leptosphaeria maculans. Phytopathology 91:70-76. [DOI] [PubMed] [Google Scholar]
- 7.Balesdent, M. H., A. Attard, M. L. Kuhn, and T. Rouxel. 2002. New avirulence genes in the phytopathogenic fungus Leptosphaeria maculans. Phytopathology 92:1122-1133. [DOI] [PubMed] [Google Scholar]
- 8.Balesdent, M. H., M. J. Barbetti, H. Li, K. Sivasithamparam, L. Gout, and T. Rouxel. 2005. Analysis of Leptosphaeria maculans race structure in a worldwide collection of isolates. Phytopathology 95:1061-1071. [DOI] [PubMed] [Google Scholar]
- 9.Balesdent, M. H., K. Louvard, X. Pinochet, and T. Rouxel. 2006. A large-scale survey of Leptosphaeria maculans occurring on oilseed rape in France. Eur. J. Plant Pathol. 114:53-65. [Google Scholar]
- 10.Barrins, J. M., P. K. Ades, P. A. Salisbury, and B. J. Howlett. 2004. Genetic diversity of Australian isolates of Leptosphaeria maculans, the fungus that causes blackleg of canola (Brassica napus). Aust. Plant Pathol. 33:529-536. [Google Scholar]
- 11.Barrins, J. M., A. Purwantara, P. K. Ades, P. A. Salisbury, and B. J. Howlett. 2002. Genetic diversity of isolates of Leptosphaeria maculans from a canola (Brassica napus) paddock in Australia. Aust. Plant Pathol. 31:129-135. [Google Scholar]
- 12.Cozijnsen, A. J., and B. J. Howlett. 2003. Characterisation of the mating-type locus of the plant pathogenic ascomycete Leptosphaeria maculans. Curr. Genet. 43:351-357. [DOI] [PubMed] [Google Scholar]
- 13.Delourme, R., M. L. Pilet-Nayel, M. Archipiano, R. Horvais, X. Tanguy, T. Rouxel, H. Brun, A. Renard, and M. H. Balesdent. 2004. A cluster of major specific resistance genes to Leptosphaeria maculans in Brassica napus. Phytopathology 94:578-583. [DOI] [PubMed] [Google Scholar]
- 14.Dillon, J. R., M. Rahman, and K. Yeung. 1993. Discriminatory power of typing schemes based on Simpson's index of diversity for Neisseria gonorrhoeae. J. Clin. Microbiol. 31:2831-2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eckert, M., L. Gout, T. Rouxel, F. Blaise, M. Jedryczka, B. D. L. Fitt, and M. H. Balesdent. 2005. Identification and characterisation of polymorphic minisatellites in the phytopathogenic ascomycete Leptosphaeria maculans. Curr. Genet. 47:37-48. [DOI] [PubMed] [Google Scholar]
- 16.Excoffier, L., P. E. Smouse, and J. M. Quattro. 1992. Analysis of molecular variance inferred from metric distances among DNA haplotypes-application to human mitochondrial-DNA restriction data. Genetics 131:479-491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Flor, H. H. 1971. Current status of the sene-for-fene concept. Annu. Rev. Phytopathol. 9:275-296. [Google Scholar]
- 18.Gout, L. 2005. Clonage et évolution du gène d'avirulence AvrLm1 chez l'ascomycète phytopathogène Leptosphaeria maculans. Ph.D thesis. Institut National Agronomique Paris-Grignon, Paris, France.
- 19.Hall, R. 1992. Epidemiology of blackleg of oilseed rape. Can. J. Plant Pathol. 14:46-55. [Google Scholar]
- 20.Hammond, K. E., and B. G. Lewis. 1986. The timing and sequence of events leading to stem canker disease in populations of Brassicas napus var. oleifera in the field. Plant Pathol. 35:551-564. [Google Scholar]
- 21.Hammond, K. E., B. G. Lewis, and T. M. Musa. 1985. A systemic pathway in the infection of oilseed rape plants by Leptosphaeria maculans. Plant Pathol. 34:557-565. [Google Scholar]
- 22.Kuswinanti, T., B. Koopmann, and H. H. Hoppe. 1999. Virulence pattern of aggressive isolates of Leptosphaeria maculans on an extended set of Brassica differentials. J. Plant Dis. Prot. 106:12-20. [Google Scholar]
- 23.Li, H., K. Sivasithamparam, and M. J. Barbetti. 2003. Breakdown of a Brassica rapa subsp. sylvestris single dominant blackleg resistance gene in B. napus rapeseed by Leptosphaeria maculans field isolates in Australia. Plant Dis. 87:752-754. [DOI] [PubMed] [Google Scholar]
- 24.Mahuku, G. S., P. H. Goodwin, R. Hall, and T. Hsiang. 1997. Variability in the highly virulent type of Leptosphaeria maculans within and between oilseed rape fields. Can. J. Bot. 75:1485-1492. [Google Scholar]
- 25.McDonald, B. A., and C. Linde. 2002. Pathogen population genetics, evolutionary potential, and durable resistance. Annu. Rev. Phytopathol. 40:349-379. [DOI] [PubMed] [Google Scholar]
- 26.Milgroom, M. G. 1996. Recombination and the multilocus structure of fungal populations. Annu. Rev. Phytopathol. 34:457-477. [DOI] [PubMed] [Google Scholar]
- 27.Nei, M. 1973. Analysis of gene diversity in subdivided populations. Proc. Natl. Acad. Sci. USA 70:3321-3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nei, M. 1972. Genetic distance between populations. Am. Nat. 106:282-292. [Google Scholar]
- 29.Pilet, M. L., R. Delourme, N. Foisset, and R. Renard. 1998. Identification of loci contributing to quantitative field resistance to blackleg disease, causal agent Leptosphaeria maculans (Desm.) Ces. et de Not., in Winter rapeseed (Brassica napus L.). Theor. Appl. Genet. 96:23-30. [Google Scholar]
- 30.Pilet, M. L., G. Duplan, H. Archipiano, P. Barret, C. Baron, R. Horvais, X. Tanguy, M. O. Lucas, M. Renard, and R. Delourme. 2001. Stability of QTL for field resistance to blackleg across two genetic backgrounds in oilseed rape. Crop Sci. 41:197-205. [Google Scholar]
- 31.Pinochet, X., E. Mestries, A. Penaud, R. Delourme, A. M. Chèvre, M. Renard, H. Brun, L. Bousset, M. H. Balesdent, T. Rouxel, and J. N. Aubertot. 2003. Towards a durable management of genetic resistances to Leptosphaeria maculans. Ocl-Ol. Corps. Gras. Li. 10:208-211. [Google Scholar]
- 32.Raymond, M., and F. Rousset. 1995. Genepop (Version-1.2): population genetics software for exact tests and ecumenicism. J. Hered. 86:248-249. [Google Scholar]
- 33.Rice, W. R. 1989. Analyzing tables of statistical tests. Evolution 43:223-225. [DOI] [PubMed] [Google Scholar]
- 34.Rouxel, T., and M. H. Balesdent. 2005. The stem canker (blackleg) fungus, Leptosphaeria maculans, enters the genomic era. Mol. Plant Pathol. 6:225-241. [DOI] [PubMed] [Google Scholar]
- 35.Rouxel, T., A. Penaud, X. Pinochet, H. Brun, L. Gout, R. Delourme, J. Schmit, and M. H. Balesdent. 2003. A 10-year survey of populations of Leptosphaeria maculans in France indicates a rapid adaptation toward the Rlm1 resistance gene of oilseed rape. Eur. J. Plant Pathol. 109:871-881. [Google Scholar]
- 36.Rouxel, T., E. Willner, L. Coudard, and M. H. Balesdent. 2003. Screening and identification of resistance to Leptosphaeria maculans (stem canker) in Brassica napus accessions. Euphytica 133:219-231. [Google Scholar]
- 37.Schneider, S., D. Roessli, and L. Excoffier. 2000. Arlequin, ver 2.000: a software for population genetics data analysis. Genetics and Biometry Laboratory, University of Geneva, Geneva, Switzerland.
- 38.West, J. S., M. H. Balesdent, T. Rouxel, J. P. Narcy, Y. J. Huang, J. Roux, J. M. Steed, B. D. L. Fitt, and J. Schmit. 2002. Colonization of winter oilseed rape tissues by A/Tox+ and B/Tox0 Leptosphaeria maculans (phoma stem canker) in France and England. Plant Pathol. 51:311-321. [Google Scholar]
- 39.West, J. S., P. D. Kharbanda, M. J. Barbetti, and B. D. L. Fitt. 2001. Epidemiology and management of Leptosphaeria maculans (phoma stem canker) on oilseed rape in Australia, Canada, and Europe. Plant Pathol. 50:10-27. [Google Scholar]
- 40.Yeh, F. C., R. C. Yang, T. B. J. Boyle, Z. H. Ye, and J. X. Mao. 1997. POPGENE, the user-friendly shareware for population genetic analysis. Molecular Biology and Biotechnology Centre, University of Alberta, Canada.