

Abstract
rRNA accessibility is a major sensitivity issue limiting the design of working probes for fluorescence in situ hybridization (FISH). Previous studies empirically highlighted the accessibility of target sites on rRNA maps by grouping probes into six classes according to their brightness levels. In this study, a recently proposed mechanistic model of FISH, based on the thermodynamics of secondary nucleic acid interactions, was used to evaluate the accessibility of the 16S rRNA of Escherichia coli to fluorescein-labeled oligonucleotides when thermodynamic and kinetic barriers were eliminated. To cover the entire 16S rRNA, 109 probes were designed with an average thermodynamic affinity (ΔGooverall) of −13.5 kcal/mol. Fluorescence intensity was measured by flow cytometry, and a brightness threshold between classes 3 and 4 was used as the requirement for proof of accessibility. While 46% of the probes were above this threshold with conventional 3-h hybridizations, extending the incubation period to 96 h dramatically increased the fraction of bright probes to 86%. Insufficient thermodynamic affinity and/or fluorophore quenching was demonstrated to cause the low fluorescence intensity of the remaining 14% of the probes. In the end, it was proven that every nucleotide in the 16S rRNA of E. coli could be targeted with a bright probe and, therefore, that there were no truly inaccessible target regions in the 16S rRNA. Based on our findings and mechanistic modeling, a rational design strategy involving ΔGooverall, hybridization kinetics, and fluorophore quenching is recommended for the development of bright probes.
Since its introduction as a cultivation-independent molecular technique in 1989 (14), fluorescence in situ hybridization (FISH) has been widely accepted as a powerful tool for the detection of individual cells in various environments (2, 3, 30, 44). In FISH, either the small-subunit (SSU) or large-subunit rRNA is typically selected as the phylogenetic marker (49) and probed in situ with fluorophore-labeled DNA oligonucleotides. FISH experiments are successful only if the fluorescent signal received from target cells is sufficient for discrimination of these cells from the background and nontarget cells. Because a significant proportion of newly designed DNA probes fail to give satisfactory signal intensity (3, 7, 17), sensitivity is considered one of the major challenges in FISH (44).
When the ribosome content and permeability of cell walls are not limiting, the lack of signal intensity can be attributed to quenching of the fluorophore or to the inefficiency of hybridization. While studies addressing the significance of the former factor in FISH have just started (5), the latter factor has been investigated extensively (7, 16-19, 50). The ability of probes to reach their target sites within the three-dimensional structure of the ribosome has been regarded as the major factor for hybridization efficiency (3). Accordingly, the differential accessibilities of target sites on the SSU (17) and large-subunit (19) rRNA of Escherichia coli were systematically studied by Fuchs et al., and the accessibility of target sites on the SSU rRNA of three other organisms was studied by Behrens et al. (7). Although the accessibility maps from these studies have reportedly been used for selection of potential target sites (8, 12, 21), their predictive power regarding the hybridization efficiency of newly designed probes is limited, as probe brightness can vary remarkably with small shifts at the targeted region (17) or when different organisms are targeted (7).
More recently, we proposed a mechanistic model of FISH to provide a mathematical basis for the development of efficiently hybridizing probes (50). The key parameter was the thermodynamic affinity of a probe, defined as the overall free energy change (ΔGooverall) of a comprehensive probe-target reaction mechanism involving not only the intermolecular DNA-rRNA interactions of the hybrid but also the intramolecular DNA-DNA and rRNA-rRNA interactions within the probe and target structure, respectively. Using this model, it was found that ΔGooverall was a powerful predictor of fluorescence intensity and that probe brightness could be improved by increasing the probe affinity even at seemingly inaccessible target sites (50). Moreover, our data suggested that kinetic limitations imposed by structural restraints in the ribosome, which could actually be defeated by extending the incubation period or using formamide, may be the reason for the dim signals obtained with some probes (50). These results led us to hypothesize that all parts of the 16S rRNA molecule could be made accessible to FISH probes provided that thermodynamic and kinetic barriers were overcome. In this study, we aimed to check the validity of this hypothesis with a large set of fluorescein-labeled probes targeting the entire length of 16S rRNA of E. coli. The analysis took into account the thermodynamic affinity of probes, potential kinetic limitations, and fluorophore quenching.
MATERIALS AND METHODS
DNA oligonucleotide probes.
Probes used in this study are listed together with their specifications in Tables S1 and S2 in the supplemental material and in Table 1. The design of new probes was based on the 16S rRNA sequence of E. coli K-12 (GenBank accession no. AE000460). Oligonucleotides were synthesized and monolabeled at the 5′ end by the University of Wisconsin Biotechnology Center. All probes were labeled with fluorescein phosphoramidite, and for six probes there were additional versions with Cy3-CE phosphoramidite.
TABLE 1.
Oligonucleotide probes used to demonstrate the effect of probe elongation and moving the location of the fluorescein label
| Probe namea | Sequence (5′-3′)d | Free energy change (kcal/mol)
|
Brightness (CBU)b
|
Quadrante
|
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ΔGo1 | ΔGo2 | ΔGo12 | ΔGo3 | ΔGooverall | 3 hc | 96 hc | Original | Elongated | ||
| E86-109 | TCCGCCACTCGTCAGCAAAGAAGC | −27.1 | 0.2 | −26.8 | −13.0 | −13.8 | 63 ± 1 | 70 ± 4 | Q1 | Q3 |
| E251-271 | GTGAGCCGTTACCCCACCTAC | −27.4 | 1.2 | −27.3 | −9.8 | −17.5 | NDf | 24 ± 2 | Q4 | Q4 |
| E312-332 | CCGTGTCTCAGTTCCAGTGTG | −25.1 | 1.9 | −25.1 | −5.8 | −19.3 | 87 ± 1 | 80 ± 6 | Q4 | Q3 |
| E325-346 | CGTAGGAGTCTGGACCGTGTCT | −25.7 | 0.4 | −25.4 | −11.5 | −13.9 | 48 ± 2 | 63 ± 4 | Q1 | Q3 |
| E483-506 | CCGGTGCTTCTTCTGCGGGTAACG | −29.0 | −0.2 | −28.5 | −8.5 | −20.0 | 50 ± 1 | 81 ± 5 | Q4 | Q3 |
| E595-618 | GCCCGGGGATTTCACATCTGACTT | −28.4 | 0.5 | −28.1 | −16.1 | −12.1 | 5 ± 4 | 63 ± 3 | Q1 | Q3 |
| E615-634 | GCAGTTCCCAGGTTGAGCCC | −27.6 | 0.2 | −27.3 | −12.4 | −14.9 | 9 ± 3 | 67 ± 4 | Q1 | Q3 |
| E631-655 | TCAAGCTTGCCAGTATCAGATGCAG | −26.0 | 0.5 | −25.8 | −10.5 | −15.3 | 8 ± 2 | 62 ± 3 | Q1 | Q3 |
| E731-750 | GTCTTCGTCCAGGGGGCCGC | −29.0 | 0.0 | −28.6 | −8.4 | −20.1 | ND | 28 ± 2 | Q4 | Q4 |
| E839-859 | CCGGAAGCCACGCCTCAAGGG | −26.6 | 0.2 | −26.3 | −11.8 | −14.5 | 6 ± 4 | 56 ± 3 | Q1 | Q3 |
| E958-980 | GGTTCTTCGCGTTGCATCGAATT | −24.3 | 0.0 | −23.8 | −6.2 | −17.6 | 53 ± 1 | 56 ± 2 | Q4 | Q3 |
| E1149-1172 | GTTTATCACTGGCAGTCTCCTTTG | −25.9 | 0.5 | −25.7 | −8.7 | −17.0 | ND | 28 ± 2 | Q4 | Q4 |
| E1379-1400 | GGTGTGTACAAGGCCCGGGAAC | −25.8 | 0.8 | −25.6 | −4.4 | −21.2 | 59 ± 2 | 58 ± 2 | Q4 | Q3 |
| E1425-1449 | GGTTAAGCTACCTACTTCTTTTGCA | −26.9 | 0.2 | −26.5 | −9.6 | −16.9 | 16 ± 2 | 52 ± 2 | Q4 | Q3 |
| E1449-1469 | GTGGTAAGCGCCCTCCCGAAG | −25.4 | 0.6 | −25.2 | −6.2 | −18.9 | 16 ± 3 | 28 ± 2 | Q4 | Q3 |
| E255-278 | CGCCTAGGTGAGCCGTTACCCCAC | −32.9 | 0.4 | −32.6 | −12.4 | −20.2 | 121 ± 4 | 97 ± 1 | Q4 | Q3 |
| E734-755 | CGTCAGTCTTCGTCCAGGGGGC | −29.1 | 0.4 | −28.8 | −7.7 | −21.1 | 6 ± 6 | 63 ± 1 | Q4 | Q3 |
| E1153-1178 | CCTCCAGTTTATCACTGGCAGTCTCC | −32.0 | −2.8 | −29.3 | −10.1 | −19.2 | 8 ± 4 | 86 ± 1 | Q4 | Q3 |
“E” in the probe name indicates E. coli, and the numbers indicate the position of the target site on the 16S rRNA (5′ to 3′).
Brightness values were converted to the scale of Fuchs et al. (17). The values are means ± standard deviations of the means.
Hybridization period.
Underlining indicates bases added to elongate the original versions shown in Table S2 in the supplemental material.
Positions of the original and elongated probes according to Fig. 4.
ND, not determined.
Whole-cell hybridization.
Aerobically grown cells from a pure culture of E. coli K-12 were harvested during the mid-exponential phase (optical density at 600 nm, 0.3 to 0.4). Fixation, hybridization, and washing steps were carried out as described previously (50). In brief, cells were fixed with 3% paraformaldehyde, incubated at 46°C in hybridization buffer (0.9 M NaCl, 0.1% sodium dodecyl sulfate, 20 mM Tris [pH 7.2], 0.25 μM probe), and washed at 46°C for 20 min in probe-free hybridization buffer. Subsequently, samples were transferred to 1 ml of cold (4°C) phosphate-buffered saline (0.138 M NaCl and 0.0027 M KCl [pH 7.4]; Sigma Aldrich Co., St. Louis, MO) with 0.01% Tween 20 (Fisher Scientific, Fair Lawn, NJ) and analyzed by flow cytometry on the same day. Cells could be stored in phosphate-buffered saline (130 mM NaCl and 10 mM Na2HPO4 [pH 7.2]) at 4°C for up to 2 weeks after fixation with no significant change in probe brightness.
Flow cytometry and data acquisition.
The fluorescence intensities of hybridized samples were measured using a FACSCalibur flow cytometer (Becton Dickinson, San Jose, CA). A 488-nm argon ion laser was used for all excitations. Forward angle light scatter, right angle light scatter, and green (FL1) and yellow (FL2) fluorescence detectors were used at logarithmic amplification. FL1 and FL2 were detected with 530- ± 15-nm and 585- ± 21-nm band-pass filters, which captured the signals from fluorescein and Cy3, respectively. FACSFLow (Becton Dickinson) was used as the sheath fluid. The performance of the flow cytometer was monitored on a daily basis with CaliBRITE beads (Becton Dickinson) using the FACSComp software (Becton Dickinson). Data acquisition was managed with the CellQuest Pro software (Becton Dickinson). A total of 10,000 events falling into a bacterial gate loosely defined on the forward angle light scatter-right angle light scatter plot for E. coli were collected for each measurement.
Calculation of probe brightness.
Due to variable levels of clumping in samples and overlapping of positive and background events for dim probes, it was necessary to evaluate brightness with methods that were more elaborate than simple statistics (35). To this end, the overall population of positives was deconvoluted by curve fitting into two normal distributions (32, 37, 47), one representing the subpopulation of clumped hybridized cells and the other representing the subpopulation of nonclumped hybridized cells. The best-fit mean for the latter subpopulation defined the unbiased fluorescence intensity of the given probe, which was designated Brightness+ and expressed in brightness units (BU). When the distribution of positives overlapped background events (i.e., cellular and noncellular debris), a third normal distribution was added to represent the background events. Curve fitting was generally in good agreement with the data, with R2 of >0.8 for >99% of the measurements (the measurements not satisfying this criterion were rejected). See the supplemental material for a detailed explanation of probe brightness calculations.
Negative controls were routinely prepared using the complement to the EUB probe (nonEUB; 5′-ACTCCTACGGAGGCAGC-3′), which is not supposed to bind to rRNA (45). The overall negative population could be represented by a single normal distribution whose mean was used to calculate the brightness of nonEUB in BU (Brightness−). Typical histograms of negative events for 3-h and 96-h hybridizations are shown in Fig. S2 in the supplemental material.
Three to five independent tests were performed per probe. The net brightness of a probe in BU (Brightnessnet) was calculated by subtracting the average Brightness− value for the relevant hybridization period from the average Brightness+ value. The fourth and fifth independent tests were done only if the standard error for Brightnessnet was more than 7% of its mean. After the fourth and fifth tests, independent Brightness+ measurements that differed by more than 3 standard deviations from the average of the rest of the measurements were discarded as outliers (<6% of the data).
Hybridization scheme and calculation of standard Gibbs free energy changes.
In situ hybridization of DNA probes with 16S rRNA was modeled according to the reaction scheme reported previously (50), which involved probe-rRNA duplex formation (reaction 1), probe folding and unfolding (reaction 2), and target folding and unfolding (reaction 3). The corresponding free energy changes (i.e., ΔGoi with i = 1, 2, or 3 for the respective reactions) were also estimated as described in the previous study (50). In brief, ΔGo1 was calculated based on the thermodynamics of base stacking in DNA-RNA hybridizations (33, 40), and ΔGo2 and ΔGo3 were predicted using mfold (28, 51). From four different methods proposed for estimation of ΔGo3 (50), the approach that required no prior knowledge of the secondary structure of the 16S rRNA was adopted. Thus, one of the four domains (11, 48) or two adjacent domains of 16S rRNA which fully encompassed the target site of the probe were used as the input for in silico folding of the target via mfold. Once the three ΔGo values had been determined, the overall binding affinity of a probe (ΔGooverall) for FISH conditions was calculated using equation 1, where R is the ideal gas constant (1.99 × 10−3 kcal/mol K and T is the hybridization temperature (319.15 K).
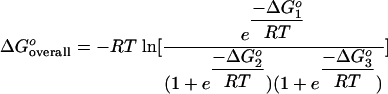 |
(1) |
Thermodynamics-based design of DNA probes.
A set of 109 probes was developed to cover the entire length of the 16S rRNA (see Table S2 in the supplemental material). Significant changes in thermodynamic affinity due solely to variations in the probe sequence were avoided by restraining the design with the ΔGo1 and ΔGo2 values. These free energy changes were used to calculate the combined parameter ΔGo12 (using equation 1 with a ΔGo3 of ≫0.0 kcal/mol), which was restricted to be in the narrow range between −20.5 kcal/mol and −22.5 kcal/mol (see Table S2 in the supplemental material). In addition, to minimize possible effects of probe length and structure, the length of most probes (>90%) was constrained to be between 17 to 21 nucleotides, and most of them (>90%) were designed to be unstructured (i.e., ΔGo2 ≥ 0.0 kcal/mol). Since it was not possible to target every residue on the 16S rRNA with these constraints, the length and structure criteria were relaxed for some probes. In the final set, the probe length was between 14 and 25 nucleotides and the ΔGo2 was ≥−1.0 kcal/mol (see Table S2 in the supplemental material). Other probes designed in this study were elongated forms of a subset of these 109 probes (Table 1).
RESULTS
We designed the set of 109 probes mentioned above so that their affinities to hypothetical perfectly accessible target sites were virtually the same (i.e., 20.5 kcal/mol > ΔGo12 > −22.5 kcal/mol), but we allowed the overall affinity to vary according to the stability of the secondary rRNA interactions (i.e., ΔGo3). The evaluation of accessibility was based on the fluorescence intensity of hybridized probes, as reported on a scale adopted from Fuchs et al. (17). To convert our probe brightness values expressed in BU (see Materials and Methods) to this scale, we tested 23 probes from the study of Fuchs et al. and derived a linear relationship (see supplemental material for details). Thus, BU values multiplied by a conversion factor of 0.24, expressed in what we called converted brightness units (CBU), were used to categorize the probes according to the following six brightness classes defined by Fuchs et al.: class 1, ≥81 CBU; class 2, 61 to 80 CBU; class 3, 41 to 60 CBU; class 4, 21 to 40 CBU; class 5, 6 to 20 CBU; and class 6, 0 to 5 CBU. As an arbitrary and conservative definition of accessibility, we assumed that unequivocal proof of accessibility was obtained if we could hybridize a target site with a probe corresponding to classes 1 to 3. For comparison, this cutoff represented less than one-half of the probes used in the pioneering work of Fuchs et al. (17).
Three-hour hybridizations.
The 3-h hybridization period was selected to represent conventional hybridization times since it has been used in many FISH studies with flow cytometry (32, 36, 45, 46), including those in which the SSU rRNA accessibility maps were developed (7, 17). The brightness levels obtained with 3-h hybridizations are shown on the secondary structure of the 16S rRNA in Fig. 1 (for a complete list of brightness values, see Table S2 in the supplemental material). This figure parallels the accessibility map provided by Fuchs et al. for fluorescein-labeled probes (17), except that the probe design strategies are different. Consistent with the study of Fuchs et al., the upper 3′ major domain (helices 35 through 45) and helices 22 and 23 seemed inaccessible, as they had clusters of probes in the three dimmest classes (classes 4 to 6), while the 5′ domain allowed efficient hybridization at most locations. A detailed analysis of other locations revealed discrepancies between the two studies (e.g., helix 24 is not totally accessible in the map of Fuchs et al., while it is in Fig. 1), which were anticipated not only because of differences in probe affinities but also because fluorescence may be potentially affected by different levels of quenching of fluorescein in the DNA-rRNA hybrids (13, 27, 31, 41). In total, 59 of 109 target sites (ca. 54%) were targeted by probes in classes 4 to 6 and hence were considered, as a first approximation, to require kinetic and/or thermodynamic enhancement of hybridization efficiency to be made accessible.
FIG. 1.

Representation of the brightness values for 109 probes after 3 h of hybridization on the secondary structure of E. coli 16S rRNA. The scale used is equivalent to that of Fuchs et al. (17). The arrows indicate the bordering nucleotides between adjacent domains (i.e., positions 566, 912, and 1396).
Effects of hybridization period.
We have previously shown that probe access to some target sites may be kinetically limited due to the higher-order structure of the ribosome (50). Increasing the hybridization period can improve brightness in such cases, and the extent of brightness is expected to vary depending on the target region. Here, using seven probes that targeted different regions on the 16S rRNA, we monitored the variation in signal intensity with increasing hybridization time (Fig. 2). In addition to six probes arbitrarily selected from Table S2 in the supplemental material, our probe set included E146 (5′-CCGTTTCCAGTAGTTATCCC-3′) from our previous work (50) to provide a connection between the two studies and the nonEUB probe as a control of nonspecific binding.
FIG. 2.

Effect of hybridization time on probe brightness for selected probes.
The trends shown in Fig. 2 revealed that virtually none of the probes reached the highest brightness level by the end of 3 h. While overnight (24-h) hybridization was sufficient for three of the bright probes (E16-36, E146, and E962-980) to reach equilibrium, the dimmest probes (E617-634, E734-750, and E1153-1172) did not attain a clear steady state even after 100 h. Also noteworthy is that the relative brightness of probes changed over time. While after 1.5 h of hybridization the order of fluorescence intensity for the four brightest probes was E1235-1255 < E16-36 < E146 < E962-980, it changed to E16-36 < E962-980 < E146 < E1235-1255 after 100 h of hybridization.
Ninety-six-hour hybridizations.
Given the effect of incubation period demonstrated in Fig. 2, our data set for 3-h hybridizations clearly underestimated the actual accessibility of the target sites analyzed. Thus, we decided to use a 96-h incubation period to represent near-equilibrium conditions for most target sites. The brightness results from 96-h hybridizations are shown in Fig. 3. Compared with Fig. 1, Fig. 3 clearly demonstrates that probe brightness improved in most regions of the 16S rRNA when the length of the hybridization period was increased from 3 to 96 h. This was especially remarkable for the upper 3′ major domain (helices 35 to 45), where all probes except E1068-1089 belonged to the three dimmest classes (classes 4 to 6) with 3-h hybridizations but belonged to the upper three classes (classes 1 to 3) (except for E1153-1172) with 96-h hybridizations. In contrast, helix 22 maintained a relatively low hybridization efficiency. Although class 6 probes were completely eliminated with 96-h hybridizations, 16 relatively dim probes (classes 4 and 5) were still present (15% of the total), suggesting that some target sites could be considered practically inaccessible; therefore, in further analyses we focused on these sites.
FIG. 3.

Representation of the brightness values for 109 probes after 96 h of hybridization on the secondary structure of E. coli 16S rRNA. The scale used is equivalent to that of Fuchs et al. (17). The arrows indicate the bordering nucleotides between adjacent domains (i.e., positions 566, 912, and 1396).
Thermodynamic analysis of relatively inaccessible sites.
Since kinetic limitations due to the target structure are likely overcome with a 96-h hybridization period (Fig. 2), seemingly inaccessible sites in Fig. 3 may indicate local thermodynamic barriers. Therefore, it is useful to use the free energy of binding (ΔGooverall) as a guide for further analysis of the accessibility at these sites. The relationship between ΔGooverall and brightness is shown for the complete set of 109 probes in Fig. 4a. A theoretical curve, based on the fraction of hybridized target molecules and calculated from equilibrium chemistry (50), is also shown in the same plot. To facilitate the analysis, we divided the ΔGooverall-brightness plot into quadrants with one horizontal line and one vertical line. The horizontal line was positioned based on the brightness of probe E1302-1320 (40.2 ± 0.8 CBU) (see Fig. S1-c and S1-f in the supplemental material for flow cytometry histogram representations of the brightness of this probe), on the border of classes 3 and 4. Data points below this line correspond to our definition of dim probes. The vertical line was placed on a ΔGooverall value of −10.2 kcal/mol, which is only −0.5 kcal/mol to the left of the theoretical melting point of probe-rRNA hybrids and forms a natural boundary to the right of an intense cluster of high-brightness data points. The first quadrant (Q1) included six probes with low affinities and dim signals. Q2 contained 10 probes that were brighter than expected based on ΔGooverall. Q3 contained the majority of the probes (83 of 109 probes), all with high affinity and satisfactory fluorescence intensity. Finally, Q4 comprised nine probes with high affinities but dim signals.
FIG. 4.

(a) Relationship between affinity and brightness (96 h) for the original set of 109 probes. (b) Effect of increasing affinity on dim probes. (c) Effect of 5′ end elongation on presumably quenched probes. The dashed line indicates theoretical hybridization according to the previously described mechanistic model (50) for a probe concentration of 250 nM, when the lower and upper limits of brightness were taken as 0 and 100 CBU, respectively. Horizontal and vertical lines divide the plots into quadrants (Q1 through Q4). The numbers indicate probes shown in Fig. 5. Open circles, default data points; solid circles, probes not enhanced in brightness after initial elongation; solid squares, unsuccessful extensions; open squares, successful extensions. The error bars indicate standard deviations of the means.
To validate our hypothesis that all regions of the 16S rRNA can be made accessible to single oligonucleotides, it was necessary to determine whether the relatively dim probes under the horizontal line in Fig. 4a could be modified to obtain brightness values above this threshold. Using the elongation strategy described previously (50), we designed extended versions of the dim probes (Table 1 shows the complete list of extended probes). Figure 4b shows that all low-affinity probes from Q1 shifted to Q3 when they were modified to acquire higher ΔGooverall. In Q4, however, increasing the already high free energies of binding did not increase the brightness of three of nine probes. Thus, all but three dim probes in Q1 and Q4 moved into Q3 when they were extended (Fig. 4b), which left only 3 of the 109 preselected target sites (E. coli positions 255 to 271, 734 to 750, and 1153 to 1172) not proven to be accessible.
Moving the position of fluorescein.
Quenching of fluorescein in its microenvironment is a possible explanation for the low brightness observed for probes in Q4 and for the lack of improvement on three of them when they were elongated. To remove quenching as a potential factor causing dim signals, the probes for which elongation did not improve hybridization efficiency were extended by addition of at least five nucleotides to the 5′ end so that the fluorophores of the new versions (i.e., E255-278, E734-755, and E1153-1178 [Table 1]) would be positioned distant from the presumed quenching envelope. This was different from the initial elongation (i.e., E251-271, E731-750, and E1149-1172 [Table 1]), which did not change the position of the fluorophore. As shown in Fig. 4c, this strategy worked for development of bright probes targeting the sites that previously seemed to be inaccessible and allowed us to complete the demonstration that all target sites could be hybridized with bright probes.
Changing the fluorophore.
Since 5′ extensions in the probe sequences changed both the position of fluorescein and probe affinity, whether quenching played a role in dim signals was not clear from the analysis described above. For more direct assessment of quenching, the three extended probes that did not yield gains in probe brightness (i.e., E251-271, E731-750, and E1149-1172) were resynthesized and labeled with Cy3, a fluorophore that is not quenched by nucleotides (27, 41). Hybridizations performed in parallel (Fig. 5) revealed that the three relevant target sites were as accessible to Cy3-labeled probes as the target site of the commonly used bright bacterial probe EUB338, contrary to the implications of the results obtained with fluorescein-labeled probes. To rule out a general sensitivity effect of switching to Cy3, we also tested two low-affinity low-brightness probes (from Q1 in Fig. 4; E635-655 and E597-616) as controls. As expected, there was no significant improvement in brightness (Fig. 5), which is consistent with the mechanistic idea that the low affinity of these probes should limit hybridization efficiency.
FIG. 5.

Fluorescence intensities of probes with Cy3 labeling (striped columns) and fluorescein labeling (gray columns) as normalized by using the brightness of EUB338. The bars indicate the averages from two independent experiments, and the error bars indicate standard deviations. The numbers match the probes indicated in Fig. 4.
DISCUSSION
Based on probe-conferred brightness, all of the 109 target sites selected in this study were shown to allow efficient hybridization, verifying our hypothesis that all regions of the 16S rRNA of E. coli can be made accessible. More specifically, our results prove that all nucleotides on this molecule are potentially accessible to single oligonucleotides, as the probe set covered the full length of the molecule. This result can have a significant impact on the design of new FISH protocols, as it suggests that one should not a priori eliminate rRNA target regions even if they have previously been reported to be seemingly inaccessible. Rather, regardless of the location of the target region, one should be able to develop working probes.
Our strategy for making all target sites accessible was based on our previous study (50), which demonstrated with a limited number of target regions that for efficient hybridization, probes should be designed with reasonable affinity and kinetic limitations due to the higher-order structure of the ribosome should be overcome. In the beginning, we reasoned that if these two factors were optimized, inefficient hybridization would correspond to truly inaccessible regions, although we did not know if such regions existed. Accordingly, the results shown in Fig. 3 had both theoretical driving forces in effect for most probes, since equilibrium was virtually established (Fig. 2) and ΔGooverall values were generally greater than the theoretical melting point, ca. −10 kcal/mol (Fig. 4a; see Table S2 in the supplemental material). Although the data points in Fig. 4a were not in good agreement with the theoretical curve (see below), there was a significant correlation between affinity and brightness (r = −0.30; P < 0.01), with most probes following the expected high hybridization efficiency with high probe affinity (Q3) and low hybridization efficiency with low probe affinity (Q1). This allowed us to improve the brightness of dim probes by increasing the ΔGooverall (Fig. 4b and c).
While 96-h hybridizations proved the accessibility of all target sites, 3-h hybridizations (Fig. 1) were particularly useful for analyzing the impact of hybridization time. The average brightness in our 3-h data set (ca. 41 CBU) was about 18% greater than that in the corresponding data set of Fuchs et al. (ca. 35 CBU), which is consistent with the fact that the average affinity of our probes (ca. −13.5 kcal/mol) was about 2 kcal/mol higher (based on a subset of 176 probes from Fuchs et al. evenly distributed over the 16S rRNA [7, 50]). However, Fig. 6 shows that the class distribution of our 3-h brightness values was comparable to the class distribution in the data set of Fuchs et al. (17). Considering the errors in analysis due to conversions and differences in the details of the protocol used (i.e., the pH of the flow cytometry buffer, cell storage, etc.), the improvements compared with the accessibility map of Fuchs et al. might be thought to be insignificant when the hybridization period is same. Indeed, a much more noticeable shift of the probe population to the brighter side was achieved by increasing the length of the hybridization period to 96 h (Fig. 6). However, although our analysis indicates the importance of kinetic limitations, it is not independent from thermodynamic considerations, as the use of low-affinity probes would likely result in low signal intensities regardless of the hybridization time.
FIG. 6.

Frequency distributions of probes for the six brightness classes. The data sets included are the data sets from experiments with 3 h (gray bars) and 96 h (striped bars) of hybridization and the data set of Fuchs et al. (dotted bars) (17).
Moreover, the ratio of probe brightness for 3-h hybridization to probe brightness for 96-h hybridization (referred to below as the 3-h/96-h ratio) was useful in highlighting the potential local impact of increasing the hybridization period, as shown in Fig. 7 for only the probes that yielded satisfactory brightness signals (i.e., classes 1 to 3) with 96-h hybridizations. By definition, the lower the 3-h/96-h ratio, the larger the signal improvement due to prolonged hybridization at a target site. Another important property of this parameter is that it should not vary with fluorophore quenching, the effect of which is likely eliminated when the ratio of brightness values is used. Therefore, Fig. 7 serves as a putative kinetic map of 16S rRNA.
FIG. 7.

Putative kinetic accessibility map of 16S rRNA of E. coli based on the ratio of brightness with 3-h hybridizations to brightness with 96-h hybridizations. Light areas represent high 3-h/96-h ratios, while dark areas represent low 3-h/96-h ratios. Only the data from probes yielding brightness values of >40 CBU for 96-h hybridizations were used. Residues targeted by multiple probes were assigned the average 3-h/96-h ratio for all the probes. The arrows indicate the bordering nucleotides between adjacent domains (i.e., positions 566, 912, and 1396).
Mechanistic aspects of accessibility.
Accessibility of target sites is thought to be a function of secondary and tertiary rRNA-rRNA and protein-rRNA interactions (3, 6, 17, 50). Possible thermodynamic and kinetic roles of these interactions have been discussed previously (50). This study provided useful data for a more elaborate discussion of potential mechanisms. For instance, unfolding of the secondary rRNA structure is theoretically the rate-limiting step of probe-rRNA hybridization (43, 50). We are now able to support this view with a significant correlation between the 3-h/96-h ratios and ΔGo3 (r = 0.43, P < 0.01). Thus, one should expect less complete hybridization with conventional incubation periods for more stable secondary rRNA-rRNA interactions at the target site (i.e., more negative ΔGo3). On the other hand, the thermodynamic role of secondary structure is evident from the significant correlation between ΔGo3 and 96-h brightness values for the set of 109 original probes (r = 0.33, P < 0.01).
Our analysis also revealed evidence of the role of proteins in FISH. Interestingly, the 3′ major domain, which seems to be kinetically the most restrained domain (Fig. 7), is the most intensive domain among the four domains with regard to protein-rRNA interactions, while the 5′ domain, most of which is accessible with 3-h hybridizations, is largely free of proteins (9, 48). Furthermore, using the structural accessibility methods described previously (50) and the same atomic model of the SSU rRNA of E. coli (42), we found a statistically significant (P < 0.05) but weak (r = −0.21) correlation between protein blockage at target sites and the 3-h/96-h ratios. This implies that the presence of proteins may impose kinetic limitations, but not necessarily via direct blockage of target sites. It is tempting to speculate based on preliminary evidence and theory (43, 50) that the relatively stable secondary rRNA-rRNA interactions are primarily responsible for the kinetic limitations of hybridization, while proteins may play a secondary role by keeping the tertiary structure packed (48) and thereby restraining the rearrangements in the secondary structure during the unfolding of the target region. Certainly, FISH is also open to complications beyond these simple kinetic concepts. For instance, irreversible denaturation and/or removal of proteins by sodium dodecyl sulfate in hybridization buffer (6) might be a cause of signal improvement with prolonged incubation. In any case, we expect that our findings and mechanistic ideas will evoke systematic analyses necessary for the elucidation of complex mechanisms behind the kinetics of FISH.
Besides their potential kinetic effects, tertiary rRNA-rRNA and protein-rRNA interactions might also impose thermodynamic limitations. While tertiary rRNA-rRNA structures are known to be relatively unstable (43), the stability of protein-rRNA interactions is not clear. Whether these unpredictable thermodynamic barriers could be defeated by high-affinity probes was crucial in this study. It seemed in the end that these interactions were either not sustainable throughout the incubation or not significantly stable compared to ΔGooverall values achievable by reasonable elongations in the probe sequence (50), so that all target sites could be made accessible.
Fluorophore quenching.
Elimination of potential mechanistic limitations led us to investigate quenching as a potential sensitivity problem for at least three probes (Fig. 4c and 5). Quenching of fluorophores due to the electron donation properties of nucleobases is well documented (13, 27, 31, 34, 41). In particular, fluorescein is known to be severely quenched by nearby guanine nucleotides or G-C base pairs (13, 27, 31). Interestingly, all three probes that seemed to experience quenching had the fluorescein attached to a guanine at the 5′ end (Table 1). Moreover, covariance analyses (analyses of variance) with high-affinity probes (i.e., the probes in Q3 and Q4 in Fig. 4) confirmed that a guanine at the 5′ end of a probe sequence was associated with a dim signal and further suggested that a guanine located in the nonhybridized section of the rRNA and two nucleotides away from the 5′ end of the probe would be the next most influential nucleotide (data not shown). Altogether, our independent findings imply that fluorophore quenching was one of the major sensitivity problems that had to be overcome if the accessibility hypothesis in this study were to be proved. This is in contrast with the results of Behrens et al. (5), who, based on the only relevant systematic analysis so far, concluded that quenching of fluorescein may not be significant for FISH protocols. While we were able to provide the first strong evidence of significant quenching in FISH, whether this indicates a general quenching phenomenon requires further experimental analyses outside the scope of this study.
Predictive power of the mechanistic model.
Our initial probe design intentionally clustered most probes around a ΔGooverall of −13.0 kcal/mol (roughly −10 to −16 kcal/mol), where the theoretical equilibrium curve reaches its plateau (Fig. 4a). However, large variations in ΔGo3 resulted in a broader range of ΔGooverall values (−6.6 to −19.4 kcal/mol), which made it possible to assess the predictive power of the hybridization model. An obvious potential cause of unaccounted variability in Fig. 4a is fluorophore quenching. Among the other factors discussed above, unpredictable thermodynamic stability of protein-rRNA and tertiary rRNA-rRNA interactions may contribute to the deviations from the theoretical trend. Finally, the relatively large uncertainties in ΔGo3 estimates (50) may explain part of the data scattering. For example, the affinities of probes in Q2 are clearly underestimated, likely due to overestimation of ΔGo3 using mfold. As these data points are near the transition region of the theoretical curve, small errors in prediction can cause large deviations. Nonetheless, since most probes in Q3 and Q4 are above the brightness threshold and since analyses of variance suggested that most of the variability observed here is potentially explicable in terms of quenching, we think that ΔGooverall is a strong predictor of hybridization efficiency. An affinity above the theoretical threshold of −13.0 kcal/mol for maximum hybridization efficiency (valid for typical probe concentrations in FISH) should be targeted for rational design of FISH probes to maximize the possibility of satisfactory sensitivity. It is important to note that this level of affinity is not new to FISH, as shown in Table 2 with a set of commonly used probes that have different levels of taxonomic specificity.
TABLE 2.
Thermodynamic affinities of selected probes
| Probea | General target | Target molecule/ positionb | Sequence | Selected target organismc | Degeneracyd | ΔGooverall (kcal/mol) | Reference |
|---|---|---|---|---|---|---|---|
| EUB338 | Bacteria | 16S/338-355 | GCTGCCTCCCGTAGGAGT | E. coli | −13.8 | 4 | |
| ALF1B | Alphaproteobacteria | 16S/19-35 | CGTTCGYTCTGAGCCAG | Rhodobacter capsulatus | Y = T | −14.8 | 26 |
| BET42a | Betaproteobacteria | 23S/1027-1043 | GCCTTCCCACTTCGTTT | Burkholderia mallei | −14.8 | 26 | |
| GAM42a | Gammaproteobacteria | 23S/1027-1043 | GCCTTCCCACATCGTTT | E. coli | −15.3 | 26 | |
| RHC439 | Rhodocyclus spp. | 16S/439-456 | CNATTTCTTCCCCGCCGA | Rhodocylus tenuis | N = A | −13.2 | 22 |
| Nso190 | Ammonia-oxidizing betaproteobacteria | 16S/189-207 | CGATCCCCTGCTTTTCTCC | Nitrosomonas europaea | −15.6 | 29 | |
| PAO462 | Candidatus “Accumulibacter phosphatis” | 16S/462-485 | CCGTCATCTACWCAGGGTATTAAC | Candidatus “Accumulibacter phosphatis” | W = T | −15.6 | 12 |
| Goam192 | Gordona amarae | 16S/189-207 | CACCCACCCCCATGCAGG | Gordona amarae | −15.5 | 15 | |
| CF319a | Cytophaga-Flavobacterium group | 16S/319-336 | TGGTCCGTGTCTCAGTAC | Flavobacterium aquatile | −14.5 | 25 | |
| ARC915 | Archaea | 16S/915-934 | GTGCTCCCCCGCCAATTCCT | Methanococcus jannaschii | −25.7 | 39 | |
| EURY498 | Euryarchaeota | 16S/498-511 | CTTGCCCRGCCCTT | Methanococcus jannaschii | R = A | −10.8 | 10 |
| CREN499 | Crenarchaeota | 16S/499-516 | CCAGRCTTGCCCCCCGCT | Desulfurococcus mobilis | R = A | −15.3 | 10 |
General probe name according to probeBase (24).
Position based on E. coli numbering.
Selected arbitrarily from organisms with perfect complementarity to the probe.
Nucleotides at the target site of the selected organism corresponding to the degenerate nucleotide in the probe.
Specificity concerns.
Since using probes with very high ΔGooverall values might enhance the affinity of the probe also to nontarget organisms with few mismatches, increasing ΔGooverall values far beyond the −13.0-kcal/mol threshold is not recommended (50). Specificity at an affinity slightly greater than −13.0 kcal/mol can be expected to be achieved by moderate levels of stringency when formamide is used, because the probes are close to the theoretical dissociation point (Fig. 4). As shown in Table 2, the recommended design value should not compromise specificity, while it can eliminate unnecessary experimentation with potential probes that, although highly specific, are unlikely to bind to the target site due to poor affinity.
On the other hand, the use of long periods of hybridization may also cause specificity problems. First, increased nonspecific binding may increase background fluorescence. In this study, negative controls with the nonEUB probe, used in most hybridization experiments, ruled this out as a significant contributor to fluorescence intensity. As shown in Fig. 2, nonEUB brightness exhibited a negligible increase with extended hybridization periods compared to the gains in the signal intensity of regular probes. Figure S2 in supplemental material demonstrates that the average nonEUB brightness did not change significantly from 3 h- to 96 h-hybridizations. The level of signal for 96 h corresponded to less than 1.1 CBU in the brightness scale used, which is insignificant compared to the signals obtained with efficient probes (40.2 CBU was the lowest brightness accepted as proof of accessibility). Consequently, strong fluorescent signals obtained in this study can unequivocally be attributed to specific and efficient hybridization between the probes and their target sites, and therefore, they can be correlated with target site accessibility. Nonetheless, nonspecific binding may cause sensitivity problems in complex systems like activated sludge, and hence, prolonged hybridization with such samples should be handled with care. Another concern with long hybridization periods is that mismatch discrimination may be hampered, although there are currently no data or theory to suggest this. Thus, we recommend that the incubation time be extended only to a point where a satisfactory signal is obtained from target cells. It is also important to note that formamide, which is generally required for adjusting stringency (26, 39), may reduce the time needed for efficient hybridization by denaturing the potentially rate-limiting higher-order structure of the ribosome (20, 50).
Concluding remarks.
Given the significant departure here from the prevailing idea that some regions in the 16S rRNA molecule are practically inaccessible to single DNA oligonucleotides (7, 17-19), we recommend the following steps for rational design of FISH probes to achieve high fluorescence intensity. (i) Locate a target segment on rRNA that is specific to the organisms of interest (2, 3, 39, 44). (ii) Determine the length of the probe so that ΔGooverall has a value of ca. −13 kcal/mol. (iii) Starting with conventional periods (i.e., 2 to 5 h [1, 45]), extend the hybridization time until the brightness reaches satisfactory levels. When rapid protocols are preferred and multiple specific target sites are available, kinetically accessible sites can be selected during step i according to previously published accessibility maps (7, 17, 19), which are already linked to other available design tools (23), or to Fig. 7 in this study. (iv) If step iii does not work with reasonable hybridization times, return to step ii, increase the magnitude of ΔGooverall, and repeat the subsequent steps. (v) If steps ii to iv do not work, quenching might be the cause, so consider switching to a new dye or changing its position (e.g., from the 5′ end to the 3′ end). (vi) If steps ii to iv require prolonged hybridizations or the design of high-affinity probes, consider using nonlabeled competitor oligonucleotides (26) to maximize probe specificity.
Here, steps i to v of this strategy were proven to work when the 16S rRNA of E. coli was used as the model phylogenetic marker. From a mechanistic point of view, our conclusions could be extended to other rRNA molecules and to other organisms (50), although experimental demonstration of such extension is still needed. The application of the proposed design strategy to other organisms should eventually provide evidence that supports its universality or underscore additional fundamental concepts that need to be incorporated into the mechanistic model of FISH that we have developed.
Supplementary Material
Acknowledgments
This research was supported by National Science Foundation grant BES-0302618.
Footnotes
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1.Amann, R. 1996. In situ identification of microorganisms by whole cell hybridization with rRNA-targeted nucleic acid probes, p. 1-15. In A. D. L. Akkermans, D. J. van Elsas, and F. J. de Bruijn (ed.), Molecular microbial ecology manual. Kluwer Academic Publishers, Dordrecht, The Netherlands.
- 2.Amann, R., B. M. Fuchs, and S. Behrens. 2001. The identification of microorganisms by fluorescence in situ hybridisation. Curr. Opin. Biotechnol. 12:231-236. [DOI] [PubMed] [Google Scholar]
- 3.Amann, R., W. Ludwig, and K.-H. Schleifer. 1995. Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol. Rev. 59:143-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Amann, R. I., B. J. Binder, R. J. Olson, S. W. Chisholm, R. Devereux, and D. A. Stahl. 1990. Combination of 16S rRNA-targeted oligonucleotide probes with flow cytometry for analyzing mixed microbial populations. Appl. Environ. Microbiol. 56:1919-1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Behrens, S., B. M. Fuchs, and R. Amann. 2004. The effect of nucleobase-specific fluorescence quenching on in situ hybridization with rRNA-targeted oligonucleotide probes. Syst. Appl. Microbiol. 27:565-572. [DOI] [PubMed] [Google Scholar]
- 6.Behrens, S., B. M. Fuchs, L. Mueller, and R. Amann. 2003. Is the in situ accessibility of the 16S rRNA of Escherichia coli for Cy3-labeled oligonucleotide probes predicted by a three-dimensional structure model of the 30S ribosomal subunit? Appl. Environ. Microbiol. 69:4935-4941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Behrens, S., C. Rühland, J. Inácio, H. Huber, A. Fonseca, I. Spencer-Martins, B. Fuchs, and R. Amann. 2003. In situ accessibility of small-subunit rRNA of members of the domains Bacteria, Archaea, and Eucarya to Cy3-labeled oligonucleotide probes. Appl. Environ. Microbiol. 69:1748-1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brett, J. B., M. A. Lutz, S. C. Dawson, P. L. Bond, and J. F. Banfield. 2004. Metabolically active eukaryotic communities in extremely acidic mine drainage. Appl. Environ. Microbiol. 70:6264-6271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brodersen, D. E., W. M. Clemons, A. P. Carter, B. T. Wimberly, and V. Ramakrishnan. 2002. Crystal structure of the 30S ribosomal subunit from Thermus thermophilus: structure of the proteins and their interactions with 16S RNA. J. Mol. Biol. 316:725-768. [DOI] [PubMed] [Google Scholar]
- 10.Burggraf, S., T. Mayer, R. Amann, S. Schadhauser, C. R. Woese, and K. O. Stetter. 1994. Identifying members of the domain Archaea with rRNA-targeted oligonucleotide probes. Appl. Environ. Microbiol. 60:3112-3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cannone, J. J., S. Subramanian, M. N. Schnare, J. R. Collett, L. M. D'Souza, Y. Du, B. Feng, N. Lin, L. V. Madabusi, K. M. Muller, N. Pande, Z. Shang, N. Yu, and R. R. Gutell. 2002. The Comparative RNA Web (CRW) Site: an online database of comparative sequence and structure information for ribosomal, intron, and other RNAs. BMC Bioinformatics 3. [Online.] http://www.biomedcentral.com/1471-2105/3/15. [DOI] [PMC free article] [PubMed]
- 12.Crocetti, G., P. Hugenholtz, P. Bond, A. Schuler, J. Keller, D. Jenkins, and L. Blackall. 2000. Identification of polyphosphate-accumulating organisms and design of 16S rRNA-directed probes for their detection and quantitation. Appl. Environ. Microbiol. 66:1175-1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crockett, A. O., and C. T. Wittwer. 2001. Fluorescein-labeled oligonucleotides for real-time PCR: using the inherent quenching of deoxyguanosine nucleotides. Anal. Biochem. 290:89-97. [DOI] [PubMed] [Google Scholar]
- 14.DeLong, E., G. Wickham, and N. Pace. 1989. Phylogenetic stains: ribosomal RNA-based probes for the identification of single cells. Science 243:1360-1363. [DOI] [PubMed] [Google Scholar]
- 15.de los Reyes, F.-L., W. Ritter, and L. Raskin. 1997. Group-specific small-subunit rRNA hybridization probes to characterize filamentous foaming in activated sludge systems. Appl. Environ. Microbiol. 63:1107-1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frischer, M. E., P. J. Floriani, and S. A. Nierzwicki-Bauer. 1996. Differential sensitivity of 16S rRNA targeted oligonucleotide probes used for fluorescence in situ hybridization is a result of ribosomal higher order structure. Can. J. Microbiol. 42:1061-1071. [DOI] [PubMed] [Google Scholar]
- 17.Fuchs, B., G. Wallner, W. Beisker, I. Schwippl, W. Ludwig, and R. Amann. 1998. Flow cytometric analysis of the in situ accessibility of Escherichia coli 16S rRNA for fluorescently labeled oligonucleotide probes. Appl. Environ. Microbiol. 64:4973-4982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fuchs, B.-M., F.-O. Glockner, J. Wulf, and R. Amann. 2000. Unlabeled helper oligonucleotides increase the in situ accessibility to 16S rRNA of fluorescently labeled oligonucleotide probes. Appl. Environ. Microbiol. 66:3603-3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fuchs, B. M., K. Syutsubo, W. Ludwig, and R. Amann. 2001. In situ accessibility of Escherichia coli 23S rRNA to fluorescently labeled oligonucleotide probes. Appl. Environ. Microbiol. 67:961-968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gamper, H. B., G. D. Cimino, and J. E. Hearst. 1987. Solution hybridization of crosslinkable DNA oligonucleotides to bacteriophage M13 DNA. J. Mol. Biol. 197:349-362. [DOI] [PubMed] [Google Scholar]
- 21.Haruta, S., M. Kondo, K. Nakamura, H. Aiba, S. Ueno, M. Ishii, and Y. Igarashi. 2002. Microbial community changes during organic solid waste treatment analyzed by double gradient-denaturing gradient gel electrophoresis and fluorescence in situ hybridization. Appl. Microbiol. Biotechnol. 60:224-231. [DOI] [PubMed] [Google Scholar]
- 22.Hesselmann, R. P. X., C. Werlen, D. Hahn, J. van der Meer, and A. J. B. Zehnder. 1999. Enrichment, phylogenetic analysis and detection of a bacterium that performs enhanced biological phosphate removal in activated sludge. Syst. Appl. Microbiol. 22:454-465. [DOI] [PubMed] [Google Scholar]
- 23.Kumar, Y., R. Westram, S. Behrens, B. Fuchs, F. O. Glockner, R. Amann, H. Meier, and W. Ludwig. 2005. Graphical representation of ribosomal RNA probe accessibility data using ARB software package. BMC Bioinformatics 6. [Online.] http://www.biomedcentral.com/1471-2105/6/61. [DOI] [PMC free article] [PubMed]
- 24.Loy, A., M. Horn, and M. Wagner. 2003. probeBase—an online resource for rRNA-targeted oligonucleotide probes. Nucleic Acids Res. 31:514-516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Manz, W., R. Amann, W. Ludwig, M. Vancanneyt, and K.-H. Schleifer. 1996. Application of a suite of 16S rRNA-specific oligonucleotide probes designed to investigate bacteria of the phylum Cytophaga-Flavobacter-Bacteroides in the natural environment. Microbiology 142:1097-1106. [DOI] [PubMed] [Google Scholar]
- 26.Manz, W., R. Amann, W. Ludwig, M. Wagner, and K.-H. Schleifer. 1992. Phylogenetic oligodeoxynucleotide probes for the major subclasses of proteobacteria: problems and solutions. Syst. Appl. Microbiol. 15:593-600. [Google Scholar]
- 27.Marras, S. A. E., F. R. Kramer, and S. Tyagi. 2002. Efficiencies of fluorescence resonance energy transfer and contact-mediated quenching in oligonucleotide probes. Nucleic Acids Res. 30:e122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mathews, D., J. Sabina, M. Zuker, and D. Turner. 1999. Expanded sequence dependence of thermodynamic parameters improves prediction of RNA secondary structure. J. Mol. Biol. 288:911-940. [DOI] [PubMed] [Google Scholar]
- 29.Mobarry, B., M. Wagner, V. Urbain, B. Rittmann, and D. Stahl. 1996. Phylogenetic probes for analyzing abundance and spatial organization of nitrifying bacteria. Appl. Environ. Microbiol. 62:2156-2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moter, A., and U. B. Gobel. 2000. Fluorescence in situ hybridization (FISH) for direct visualization of microorganisms. J. Microbiol. Methods 41:85-112. [DOI] [PubMed] [Google Scholar]
- 31.Nazarenko, I., R. Pires, B. Lowe, M. Obaidy, and A. Rashtchian. 2002. Effect of primary and secondary structure of oligodeoxyribonucleotides on the fluorescent properties of conjugated dyes. Nucleic Acids Res. 30:2089-2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Perez Feito, R., J. Peccia, and D. R. Noguera. Comparison between direct microscopy and flow cytometry for quantifiable RNA-based population analysis of activated sludge. Water Environ. Res., in press. [DOI] [PubMed]
- 33.SantaLucia, J., Jr. 1998. A unified view of polymer, dumbbell, and oligonucleotide DNA nearest-neighbor thermodynamics. Proc. Natl. Acad. Sci. USA 95:1460-1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seidel, C. A. M., A. Schulz, and M. M. H. Sauer. 1996. Nucleobase-specific quenching of fluorescent dyes. 1. Nucleobase one-electron redox potentials and their correlation with static and dynamic quenching efficiencies. J. Phys. Chem. 100:5541-5553. [Google Scholar]
- 35.Shapiro, H. M. 2003. Practical flow cytometry, 4th ed. John Wiley & Sons, Hoboken, N.J.
- 36.Simon, N., N. LeBot, D. Marie, F. Partensky, and D. Vaulot. 1995. Fluorescent in situ hybridization with rRNA-targeted oligonucleotide probes to identify small phytoplankton by flow cytometry. Appl. Environ. Microbiol. 61:2506-2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sladek, T., and J. Jacobberger. 1993. Flow cytometric titration of retrovial expression vectors: comparison of methods for analysis of immunofluorescence histograms derived from cells expressing low antigen levels. Cytometry 43:461-470. [DOI] [PubMed] [Google Scholar]
- 38.Sohail, M., S. Akhtar, and E. M. Southern. 1999. The folding of large RNAs studied by hybridization to arrays of complementary oligonucleotides. RNA 5:646-655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stahl, D., and R. Amann. 1991. Development and application of nucleic acid probes, p. 205-248. In Nucleic acid techniques in bacterial systematics. John Wiley & Sons Ltd., West Sussex, United Kingdom.
- 40.Sugimoto, N., S. Nakano, M. Katoh, A. Matsumura, H. Nakamuta, T. Ohmichi, M. Yoneyama, and M. Sasaki. 1995. Thermodynamic parameters to predict stability of RNA/DNA hybrid duplexes. Biochemistry 34:11211-11216. [DOI] [PubMed] [Google Scholar]
- 41.Torimura, M., S. Kurata, K. Yamada, T. Yokomaku, Y. Kamagata, T. Kanagawa, and R. Kurane. 2001. Fluorescence-quenching phenomenon by photoinduced electron transfer between a fluorescent dye and a nucleotide base. Anal. Sci. 17:155-160. [DOI] [PubMed] [Google Scholar]
- 42.Tung, C., S. Joseph, and K. Y. Sanbonmatsu. 2002. All-atom homology model of the Escherichia coli 30S ribosomal subunit. Nat. Struct. Biol. 9:750-755. [DOI] [PubMed] [Google Scholar]
- 43.Turner, D. 2000. Conformational changes, p. 259-334. In V. A. Bloomfield, D. M. Crothers, and I. J. Tinoco (ed.), Nucleic acids: structures, properties, and functions. University Science Books, Sausalito, Calif.
- 44.Wagner, M., M. Horn, and H. Daims. 2003. Fluorescent in situ hybridisation for the identification and characterization of prokaryotes. Curr. Opin. Microbiol. 6:302-309. [DOI] [PubMed] [Google Scholar]
- 45.Wallner, G., R. Amann, and W. Beisker. 1993. Optimizing fluorescent in situ hybridization with rRNA-targeted oligonucleotide probes for flow cytometric identification of microorganisms. Cytometry 14:136-143. [DOI] [PubMed] [Google Scholar]
- 46.Wallner, G., R. Erhart, and R. Amann. 1995. Flow cytometric analysis of activated sludge with rRNA-targeted probes. Appl. Environ. Microbiol. 61:1859-1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Watson, J. V. 1991. Introduction to flow cytometry. Cambridge University Press, New York, N.Y.
- 48.Wimberly, B.-T., D.-E. Brodersen, V.-M.-J. Clemons, R. Morgan-Warren, C. von Rhein, T. Hartsch, and V. Ramakrishnan. 2000. Structure of the 30S ribosomal subunit. Nature 407:327-339. [DOI] [PubMed] [Google Scholar]
- 49.Woese, C. R. 1987. Bacterial evolution. Microbiol. Rev. 51:221-271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yilmaz, L., and D. R. Noguera. 2004. Mechanistic approach to the problem of hybridization efficiency in fluorescent in situ hybridization. Appl. Environ. Microbiol. 70:7126-7139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zuker, M. 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 31:3406-3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.