

Abstract
Lecithin:retinol acyltransferase (LRAT) is believed to be the predominant if not the sole enzyme in the body responsible for the physiologic esterification of retinol. We have studied Lrat-deficient (Lrat−/−) mice to gain a better understanding of how these mice take up and store dietary retinoids and to determine whether other enzymes may be responsible for retinol esterification in the body. Although the Lrat−/− mice possess only trace amounts of retinyl esters in liver, lung, and kidney, they possess elevated (by 2–3-fold) concentrations of retinyl esters in adipose tissue compared with wild type mice. These adipose retinyl ester depots are mobilized in times of dietary retinoid insufficiency. We further observed an up-regulation (3–4-fold) in the level of cytosolic retinol-binding protein type III (CRBPIII) in adipose tissue of Lrat−/− mice. Examination by electron microscopy reveals a striking total absence of large lipid-containing droplets that normally store hepatic retinoid within the hepatic stellate cells of Lrat−/− mice. Despite the absence of significant retinyl ester stores and stellate cell lipid droplets, the livers of Lrat−/− mice upon histologic analysis appear normal and show no histological signs of liver fibrosis. Lrat−/− mice absorb dietary retinol primarily as free retinol in chylomicrons; however, retinyl esters are also present within the chylomicron fraction obtained from Lrat−/− mice. The fatty acyl composition of these chylomicron retinyl esters suggests that they are synthesized via an acyl-CoA-dependent process suggesting the existence of a physiologically significant acyl-CoA:retinol acyltransferase.
Retinoids3 have important roles in mediating or facilitating many essential physiologic functions within the body (1). In vision, 11-cis-retinal serves as the chromophore for the visual pigments present in the rod and cone photoreceptor cells (2, 3). Retinoids are also needed to maintain cell proliferation and normal differentiation, normal immune response, normal reproduction, and normal fetal development (4). It has been suggested in the literature that over 500 different genes may be transcriptionally responsive to retinoids (5). The transcriptional regulatory activities of retinoids are thought to result from the actions of all-trans- and 9-cis-retinoic acid (6–8). These actions of retinoic acid are mediated through six distinct ligand-dependent transcription factors as follows: three retinoic acid receptors (RARα,4 RARβ, and RARγ) and three retinoid X receptors (RXRα, RXRβ, and RXRγ) (6–8). Ultimately, all retinoid must be acquired from the diet either as preformed retinoid (primarily as dietary retinol or retinyl ester) or as proretinoid carotenoid (primarily as β-carotene) (9, 10). The different dietary retinoid forms are processed within the enterocyte and packaged along with other dietary lipids as retinyl esters in nascent chylomicrons (9, 10). Approximately 66–75% of dietary retinoid is taken up and stored as retinyl ester in the liver (9, 10), primarily in the nonparenchymal hepatic stellate cells (also called Ito cells, lipocytes, and fat-storing cells) (11). These hepatic stores can be called upon and mobilized into the circulation as retinol bound to plasma retinol-binding protein (RBP) (12, 13). Tissues acquire retinol from the circulating retinol-RBP complex or postprandially from chylomicrons (9, 10, 12–14) and are able to oxidize enzymatically this retinoid to retinal and retinoic acid (14–16). Most tissues also possess some capacity to esterify and thus store retinol prior to its use for the synthesis of retinal or retinoic acid (9, 10, 14).
It is generally agreed upon in the literature that retinol is esterified primarily through the actions of the enzyme lecithin:retinol acyltransferase (LRAT) (17–20). LRAT is broadly expressed in tissues and at relatively high levels in the intestine, liver, and eye where it has been proposed to catalyze the transesterification of retinol with an acyl group present in the A1 position of membrane lecithin (17–23). Recently, the gene encoding LRAT was knocked out in mice (20). It was clear from the initial studies of Lrat-deficient (Lrat−/−) mice that LRAT is the preponderant and possibly sole enzyme responsible for retinyl ester formation in the eye and liver (20). Lrat−/− mice when maintained on a control diet develop normally and show no obvious phenotypic differences when compared with wild type mice. However, at an early age these mutant mice do show a phenotype of severely attenuated rod and cone visual functions. Moreover, only trace levels of all-trans-retinyl esters were detected in the liver, eye, and blood of Lrat−/− mice. The literature suggests the existence of an additional enzymatic activity within cells and tissues that is able to esterify retinol in an acyl-CoA-dependent manner. This enzymatic activity has been termed acyl-CoA:retinol acyl-transferase (ARAT) (24). The physiological significance of this ARAT activity has not been established.
We are interested in studying the importance of retinyl esters and LRAT in the uptake of dietary retinoid and its storage in the liver and other tissues and in understanding how retinyl ester accumulation influences normal retinoid-dependent functions in the body. For instance, the loss of hepatic stellate cell retinyl ester stores occurs with stellate cell activation and the development of hepatic fibrosis (25, 26). Similarly, dietary retinoid is normally absorbed almost exclusively as retinyl ester, but humans afflicted with abetalipoproteinemia must absorb retinoids through another route because they are unable to package retinyl esters in chylomicrons (27). The mechanism of retinoid uptake in abetalipoproteinemia patients has yet to be defined. Below we report further characterizations of the Lrat−/− mice that are relevant for understanding the role of LRAT and retinyl esters in maintaining normal retinoid-dependent function and preventing disease.
MATERIALS AND METHODS
Diet and Animal Husbandry
Animals were maintained on a standard rodent chow diet. For all of our studies, male and female wild type and Lrat−/− mice at 3–4 months of age were employed. The genotypes of these mice were determined by a PCR protocol and tail clip DNA according to a protocol described previously (20). To investigate the effects of a retinoid-deficient diet on wild type and Lrat−/− mice, we employed a purified, nutritionally complete control retinoid-deficient diet (Purified Test Diet 5755; W. F. Fisher and Son, Inc.) containing < 0.22 IU of retinol/g but otherwise nutritionally complete diet. Prior to administration of this diet to Lrat−/− and wild type mice at 3 months of age, these mice were maintained from weaning on a standard nutritionally complete chow diet (W. F. Fisher and Sons, Inc.). For all of our studies, both diet and water were available to the animals on an ad libitum basis. Mice were maintained on a 12-h dark-light cycle, with the period of darkness between 7:00 a.m. and 7:00 p.m. All mice used in our studies were sacrificed in the morning between 9:30 and 11:30 a.m. The animal experimentation described in this study was conducted in accordance with the National Research Council (50) and was approved by the Institutional Committee on Animal Care, Columbia University.
HPLC Analysis of Retinoids
Tissue and serum retinol and retinyl ester levels were determined by reverse phase HPLC procedures that have been described previously (28). Briefly, serum and tissues (liver, lung, kidney, testis, perigonadal (epididymal or ovarian) adipose tissue, and heart) were flash-frozen in liquid N2 after their dissection from the experimental mice. For this analysis, tissues were first homogenized in 10 volumes of PBS (10 mm sodium phosphate, pH 7.2, 150 mm sodium chloride) using a Polytron homogenizer (Brinkmann Instruments) set at half-maximal speed for 10 s. Homogenates (or 200-μl aliquots of the homogenates in the case of liver) were then treated with an equal volume of absolute ethanol containing known amounts of retinyl acetate as an internal standard, and the retinoids present in the homogenates were extracted into hexane. The extracted retinoids were separated on a 4.6 × 250-mm Ultrasphere C18 column (Beckman, Fullerton, CA) preceded by a C18 guard column (Supelco, Bellefonte, PA), using 70% acetonitrile, 15% methanol, 15% methylene chloride as the running solvent flowing at 1.8 ml/min. Retinol and retinyl esters (retinyl palmitate, oleate, linoleate, and stearate) were detected at 325 nm and identified by comparing the retention times and spectral data of experimental compounds with those of authentic standards. Concentrations of retinol and retinyl esters in the tissues were quantitated by comparing integrated peak areas for those of each retinoid against those of known amounts of purified standards. Loss during extraction was accounted for by adjusting for the recovery of the internal standard added immediately after homogenization of the tissues.
Tissue retinoic acid determinations were carried out using procedures we have described earlier (29). All extraction and analytical procedures were carried out under dim yellow light in brown glass tubes to protect the retinoids from exposure to light. Plasma samples were diluted with equal volumes of PBS prior to extraction. Tissues were homogenized in PBS (2 ml of PBS/g of tissue) using three 15-s pulses of a Brinkmann Polytron PT 300 homogenizer (Brinkmann Instruments), at setting 5 on the homogenizer. An internal standard consisting of a known amount of all-trans-7-(1,1,3,3,-tetramethyl-5-indanyl)-3-methyl-octa-2,4,6-trienoic acid (provided by Hoffmann-LaRoche) was added in 0.1 ml of ethanol to each plasma or tissue sample in order to monitor the recovery of retinoic acid during the extraction and HPLC procedures. This extraction procedure is gentle and does not cause retinoyl-β-glucuronide hydrolysis. Plasma and tissue homogenates were extracted twice with chloroform/methanol (2:1), and the chloroform extracts were combined and concentrated, under a gentle stream of N2, to a final volume of less than 1 ml. The retinoid-containing chloroform extract was then applied to 100 or 500 mg of aminopropyl solid phase extraction columns (Baxter Laboratories Inc., Chicago) that had been equilibrated previously with hexane. Under these chromatographic conditions, most lipids are retained by the column. The neutral lipids were eluted first from the column with 5 ml of chloroform/isopropyl alcohol (2:1). After the neutral lipids were eluted, the retinoic acid was eluted from the aminopropyl column with 5 ml of 2% acetic acid in diethyl ether containing 0.01% butylated hydroxytoluene as an antioxidant. The acetic acid/diethyl ether eluates were collected, evaporated to dryness under a gentle stream of N2, and redissolved in HPLC mobile phase (hexane/acetonitrile/acetic acid, 99.5:0.4:0.1) for injection onto the HPLC. All-trans-retinoic acid levels were determined by normal phase HPLC employing two silica columns linked in tandem. The silica columns consisted of a 3.9 × 150-mm 5-μm Resolve (Waters) followed by a 4.6 × 150-mm 3-μm Supelcosil LC-Si (Supelco Inc., Bellefonte, PA). The first column was preceded by a Waters Silica Guard-PAK guard column. For chromatography, we employed an isocratic system using the mobile phase described above flowing at 1.8 ml/min. The mobile phase was made fresh daily and filtered and degassed immediately prior to use. Retinoic acid mass was detected at 350 nm using a Waters 996 photodiode array detector. All-trans-retinoic acid levels were quantitated from the integrated area under its peak using a standard curve, constructed with authentic standards of all-trans-retinoic acid of known mass. For standards, authentic all-trans- and 9-cis-retinoic acid were obtained as a gift (Hoffmann-La Roche), and authentic 13-cis-retinoic acid was obtained from Sigma. Low limits of detection for all-trans-, 13-cis-, and 9-cis-retinoic acid in our HPLC assay were estimated to be less than 1.5 pmol/g tissue.
Histology and Electron Microscopy
Tissues (liver and adipose tissue) from 3-month-old wild type and Lrat−/− mice were fixed overnight in 10% buffered formalin. The samples were processed and paraffin-embedded in a core facility at the Department of Pathology, Columbia University Medical Center. Sections were applied to glass slides and stained with hematoxylin and eosin and Masson trichrome. For immunohistochemical analysis, 5-μm sections were applied to Fisherbrand Superfrost/Plus Slides. Monoclonal antibodies against α-smooth muscle actin (α-SMA) (dilution 1:400; Dako, Carpinteria, CA), desmin (dilution 1:100; Dako), and glial fibrillary acidic protein (GFAP) (dilution 1:100; Novocastra, Newcastle upon Tyne, UK) were employed. Prior to staining, heat-induced antigen retrieval was performed in 10 mm sodium citrate, pH 6.0. Staining was performed on a Dako autostainer using Dako Envision Plus with diaminobenzidine as chromagen. Hematoxylin was used as a counterstain.
Western Blot Analysis of CRBPI and CRBPIII
Polyclonal antibodies were raised in rabbits against the C terminus of both mouse CRBPI and mouse CRBPIII as described previously (30). The resulting anti-CRBPI and anti-CRBPIII were immunoaffinity-purified, respectively, on CRBPI-Sepharose and CRBPIII-Sepharose to yield anti-CRBPI and anti-CRBPIII preparations that did not cross-react (30). Cytosolic extracts of liver and perigonadal adipose tissue samples were prepared from age- and sex-matched wild type and Lrat−/− mice for Western blot analysis according to procedures we have described previously (30). Monoclonal antibody directed against β-actin was used as a control to confirm protein load (Sigma). Immunoreactive proteins were visualized using a secondary antibody conjugated to horseradish peroxidase followed by chemiluminescence (Pierce).
Radioimmunoassay (RIA) for RBP
Analyses of tissue and blood RBP levels were carried out by RIA. For these analyses, we employed antiserum against rat RBP and the standard procedures we reported earlier (31).
In Vitro DGAT1 Assay
HEK293 cells were transfected with the mammalian expression vector pCR3.1 harboring a cDNA encoding human DGAT1 (a gift from Drs. Henry Ginsberg and Steven Sturley of Columbia University) using a calcium phosphate transfection protocol (32). Membrane fractions containing DGAT1 were prepared from the postmitochondrial fraction of HEK293 cells. Briefly, cells in monolayer were washed twice with cold PBS and then scraped into ice-cold homogenization buffer (20 mm HEPES, pH 7.4, 1 mm CaCl2, 1 mM MgCl2, 1 mM dithiothreitol, and a mixture of protease inhibitor mixture (Sigma) containing 4-(2-aminoethyl)benzenesulfonyl fluoride, aprotinin, leupeptin, bestatin, pepstatin A, and E-64. Cells were allowed to swell on ice for 10 min before homogenization employing 10 strokes of a Dounce homogenizer. A 0.25 volume of 30% sucrose was added to the sample immediately following homogenization. The homogenization mixture was then centrifuged at 1,500 × g for 10 min at 4 °C. The supernatant was centrifuged again at 150,000 × g for 1 h at 4 °C. The membrane pellet was homogenized and resuspended in a buffer containing 20 mm HEPES, pH 7.4, 0.25 m sucrose, and the protease inhibitor mixture. Protein concentrations were determined using the DC protein assay kit (Bio-Rad) according to the supplier’s instructions.
DGAT1 activity was measured by modifying a method described previously (33). Briefly, 15 μg of membrane protein was added to a 200-μl reaction mixture containing 100 mm Tris-Cl pH 7.5, 250 mm sucrose, 10 mm MgCl2, 0.8 mM EDTA, 1 mg/ml fatty acid free bovine serum albumin, 25 μm palmitoyl-CoA, and 16 μm all-trans-retinol added in a small volume of ethanol. The reaction mixture was preincubated at 37 °C for 10 min. To assess the effects of CRBPI on CoA-dependent retinol esterification, incubations were carried out similarly except that retinol was added to the reaction mixture to a final concentration of 16 μm purified holo-His-tagged mouse CRBPI. Microsomal protein was then added to the reaction mixture and allowed to incubate further for 10 min at 37 °C. The enzymatic reaction was stopped by addition of 200 μl of ice-cold ethanol. Samples were extracted into hexane, and the retinyl palmitate content was analyzed by reverse phase HPLC as described above for tissue retinoid analysis.
Expression and Purification of Recombinant Mouse CRBPI and CRBPIII
To obtain purified CRBPI and CRBPIII protein, cDNAs encoding each of the proteins were expressed in Escherichia coli using the PetVector expression system (Novagen, Madison, WI). CRBPI and CRBPIII expression vector induction, expression, and purification of recombinant proteins containing the 3′ His tags were performed as described earlier (30). Briefly, E. coli expressing a specific recombinant protein was extracted into B-FER Bacterial Protein Extraction Reagent (Pierce), and this extract was sonicated on ice until it was no longer viscous. The extract was clarified by centrifugation at 12,000 × g and applied to a column (1 cm diameter, 7.5 ml volume) packed with 1 ml of His-Bind resin (Novagen, Madison, WI) according to the manufacturer’s instruction. The column was washed with 3 volumes of sterile deionized water and 5 volumes of charge buffer (50 mm NiSO4), followed by 3 volumes of binding buffer (0.5 m NaCl, 20 mm Tris-HCl, 5 mm imidazole, pH 7.9). The column was loaded with the supernatant, washed with 10 volumes of binding buffer, and washed again with 6 volumes of wash buffer (0.5 m NaCl, 20 mm Tris-HCl, 60 mm imidazole, pH 7.9). The recombinant His-tagged CRBPs were eluted from the resin with 6 volumes of elution buffer (1 m imidazole, 0.5 m NaCl, 20 mm Tris-HCl, pH 7.9). The purity of each protein was determined by 12% SDS-PAGE prior to its use in our experiments.
Isolation of Chylomicrons
Three-month-old female and male wild type and Lrat−/− mice received an intraperitoneal injection of the lipase inhibitor, P-407 (1 g/kg body weight) ~12 h before the experiment began (34, 35). At this dose the mice become very hyperlipidemic for a time period exceeding that of our experiments (~4–5 days) (34, 35). The morning after P-407 administration, the mice received an oral bolus of all-trans-[3H]retinol (2 × 106 cpm/6 μg) in 50 μl of peanut oil. Plasma samples were obtained from blood that had been collected in a tube containing EDTA upon centrifugation at 14,000 μg. Samples from individual male and female Lrat−/− and wild type mice were pooled (4 mice/pool) according to gender and genotype. These pools (containing ~1.5 ml of plasma) were then overlaid with ~10 ml of PBS in an ultracentrifuge tube and centrifuged in an SW 40 rotor at 20 °C for 60 min at 39,000 rpm in a Beckman L8-80M ultracentrifuge. The chylomicrons floating at the top of the PBS layer were aspirated and used for further analysis. To assess the total [ 3H]retinoid in the chylomicrons, 10 μl of each chylomicron sample was transferred to a scintillation vial and dissolved in 10 ml of Hydroflor liquid scintillation counting solution. To assess individual retinoids present in the chylomicrons, the chylomicrons were extracted and analyzed by reverse phase HPLC as described above for tissue homogenates. Individual fractions were collected at 0.5-min intervals throughout the entire HPLC run, and [ 3H]retinoid for each fraction was obtained as described above. The 3H counts/min present in the fractions was measured in a Beckman LS 1800 liquid scintillation counter.
RESULTS
LRAT Is Not the Sole Enzyme That Catalyzes Retinyl Ester Formation in Vivo
In their initial description of Lrat−/− mice, Batten et al. (20) reported that only trace amounts of retinyl esters were detected in liver, lungs, eyes, and serum from the mutant mice. This finding clearly establishes the importance of LRAT in catalyzing retinyl ester formation in the liver, lung, and eye. To assess the importance of LRAT for catalyzing retinol esterification and storage in other tissues, we investigated retinyl ester levels in Lrat−/− mice for other tissues that are known to contain retinyl esters in wild type mice. TABLE ONE shows a list of tissue values for male and female mice at 3 months of age. Consistent with the report of Batten et al. (20), we could only detect occasional trace amounts of retinyl esters in the livers of the Lrat−/− mice. Only trace amounts of retinyl esters also were detected in the lungs, testes, or kidneys of Lrat−/− mice, in sharp contrast to wild type mice. One tissue from Lrat−/− mice where retinyl esters were present was adipose tissue. TABLE ONE shows the levels of the retinol and retinyl esters present in the adipose tissue from Lrat−/− and wild type male and female mice at 3 months of age. Both adipose retinol and retinyl ester stores were markedly elevated in the Lrat−/− compared with wild type mice, possibly in compensation for the lack of retinyl ester stores elsewhere. Levels of both retinol and retinyl esters in adipose tissue were elevated by ~3-fold for male mice. For female Lrat−/− mice, adipose levels of both retinol and retinyl esters were also elevated, albeit to a lesser degree (by ~2-fold) than was observed for male Lrat−/− mice. Most interestingly, we also detected retinyl esters in milk obtained from lactating Lrat−/− dams, although the absolute amount of retinyl present in Lrat−/− milk appeared to be less than for wild type dams (data not shown).
TABLE ONE. Levels of retinol and retinyl esters in tissues from 3-month-old male and female Lrat−/− and wild type mice.
All values are given as means ± 1 S.D. The number of individual mice (n) used for each measure is given in the parentheses following each total retinol value. ND signifies no detectable retinyl esters were present in the HPLC profiles, whereas the value 0.0 signifies that retinyl esters were detectable in the HPLC profiles but at levels below 0.05 nmol/g tissue.
| Male (−/−) | Male (+/+) | Female (−/−) | Female (+/+) | |
|---|---|---|---|---|
| nmol/g tissue wet weight | ||||
| Liver | ||||
| Retinol | 1.6 ± 0.3 | 81.5 ± 46.7 | 2.2 ± 0.8 | 49.3 ± 14.2 |
| Retinyl ester | 0.0 ± 0.0 | 595.0 ± 253.8 | 0.1 ± 0.1 | 1030.6 ± 323.4 |
| Total retinol | 1.6 ± 0.3 (10) | 676.5 ± 261.1 (8) | 2.3 ± 0.9 (4) | 1079.9 ± 313.4 (5) |
| Lung | ||||
| Retinol | 1.3 ± 0.4 | 11.4 ± 0.9 | 0.9 ± 0.5 | 16.6 ± 3.2 |
| Retinyl ester | 0.0 ± 0.0 | 725.0 ± 252.5 | 0.3 ± 0.8 | 867.0 ± 274.5 |
| Total retinol | 1.3 ± 0.4 (5) | 739.0 ± 253.9 (4) | 1.2 ± 1.3 (5) | 883.6 ± 277.7 (4) |
| Testis | ||||
| Retinol | 0.9 ± 0.4 | 0.5 ± 0.4 | ||
| Retinyl ester | N.D. | 1.0 ± 1.9 | ||
| Total retinol | 0.9 ± 0.4 (4) | 1.5 ± 2.0 (4) | ||
| Kidney | ||||
| Retinol | 2.2 ± 0.5 | 1.2 ± 0.1 | 1.4 ± 0.2 | 1.2 ± 0.2 |
| Retinyl ester | 0.1 ± 0.1 | 1.4 ± 1.5 | 0.1 ± 0.0 | 0.0 ± 0.0 |
| Total retinol | 2.3 ± 0.6 (4) | 2.6 ± 1.6 (4) | 1.4 ± 0.2 (5) | 1.2 ± 0.2 (4) |
| Heart | ||||
| Retinol | 1.5 ± 0.2 | 0.7 ± 0.2 | 1.1 ± 0.3 | 1.1 ± 0.1 |
| Retinyl ester | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.0 ± 0.0 |
| Total retinol | 1.5 ± 0.2 (4) | 0.7 ± 0.2 (4) | 1.1 ± 0.3 (5) | 1.1 ± 0.1 (4) |
| Adipose | ||||
| Retinol | 8.2 ± 1.7 | 2.9 ± 0.7 | 3.6 ± 2.2 | 1.9 ± 0.4 |
| Retinyl ester | 17.1 ± 3.5 | 5.2 ± 2.2 | 9.9 ± 2.8 | 4.2 ± 3.4 |
| Total retinol | 25.3 ± 4.2 (8) | 8.2 ± 2.3 (6) | 13.5 ± 2.2 (5) | 6.1 ± 3.7 (5) |
| Seruma | ||||
| Retinol (μm) | 0.7 ± 0.3 (5) | 0.8 ± 0.1 (9) | 0.4 ± 0.2 (5) | 0.5 ± 0.2 (4) |
The serum retinol concentrations are given in terms of μm.
Histological examination of adipose tissue from 3-month-old Lrat−/− and wild type mice indicated that the size of adipocytes present in the tissue were not different for the two strains. Moreover, the wet weights of the dissected adipose depots obtained from age- and sex-matched mice from the two strains were not different (data not shown). To confirm further the notion that the adipose depots of Lrat−/− and wild type mice were not markedly different, we examined by quantitative real time PCR the expression levels of several genes known to be important for maintaining adipocyte differentiation and function. These genes included the transcription factors PPARα, PPARγ, and SREBP and the triglyceride-synthesizing enzymes diacylglycerol acyltransferase 1 (DGAT1) and diacylglycerol acyltransferase 2 (DGAT2). For total RNA isolated from adipose tissue from five male Lrat−/− mice and five male age-matched wild type mice, we observed no statistically significant differences in PPARα, PPARγ, SREBP, DGAT1, or DGAT2 mRNA levels (data not shown). The elevation in total retinol levels observed in the adipose depots of Lrat−/− mice appeared to be a specific retinoid-related response directly arising from the absence of LRAT.
There were, however, other striking retinoid-specific differences in adipose tissue from Lrat−/− mice as compared with wild type mice. Western blot analysis of tissue cytosol fractions indicated that the levels of CRBPIII were elevated in Lrat−/− mice. Fig. 1 shows that the levels of CRBPIII protein are increased 3–4-fold in adipose tissue from Lrat−/− mice as compared with adipose tissue from wild type controls. No change was observed in the levels of CRBPI protein in either adipose tissue or liver of Lrat−/− mice. Because CRBPIII has been postulated to facilitate retinol uptake by cells (30), this elevation may account, at least in part, for the elevation of both retinol and retinyl ester concentrations observed in Lrat−/− mice.
FIGURE 1. CRBPIII but not CRBPI levels are elevated in adipose tissue from 3-month-old Lrat−/− mice.
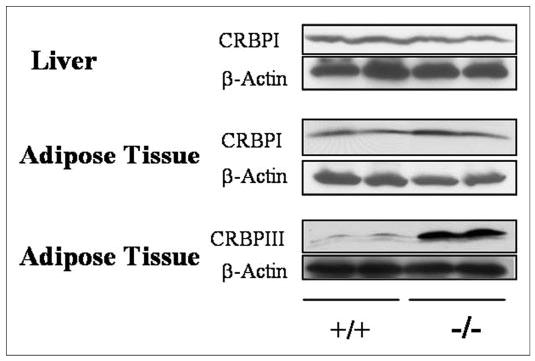
Western blots for CRBPI in the liver and epididymal adipose tissue and for CRBPIII in epididymal adipose tissue for two male wild type (+/+) and two male Lrat−/− (−/−) mice are shown.
In order to establish whether adipose retinol and retinyl esters are in fact real stores that can be mobilized in times of dietary retinoid insufficiency, we placed 3-month-old male and female Lrat−/− and wild type mice on a purified totally retinoid-deficient diet that was otherwise nutritionally complete for 1 month. Adipose, serum, and liver were collected and analyzed for retinoid content at the end of this period. We observed a sharp reduction in the levels of free retinol and retinyl esters in the adipose tissue of the Lrat−/− mice (Fig. 2, panel A). This observation indicates that these retinoids in the adipose tissue of Lrat−/− mice are indeed stores that are mobilized in times of dietary insufficiency. Most interestingly, the wild type controls also showed a large reduction in the level of retinyl esters in adipose tissue when maintained under the same dietary regime. These data suggest that adipose tissue may be an important primary store of retinyl esters that is readily mobilized in times of insufficient dietary retinoid intake. The levels of retinol present in the livers of Lrat−/− mice were also markedly reduced for mice maintained on the retinoid-deficient diet. This observation was not surprising considering the relatively low levels of retinol that are present in the livers of Lrat−/− mice receiving the control diet. A similar large drop in the level of total retinol was not observed for livers of wild type mice (Fig. 2, panel B).
FIGURE 2. Effects of a totally retinoid-deficient diet on adipose tissue and liver levels of total retinol (retinol + retinyl ester) for wild type (+/+) and Lrat−/− (−/−) mice.
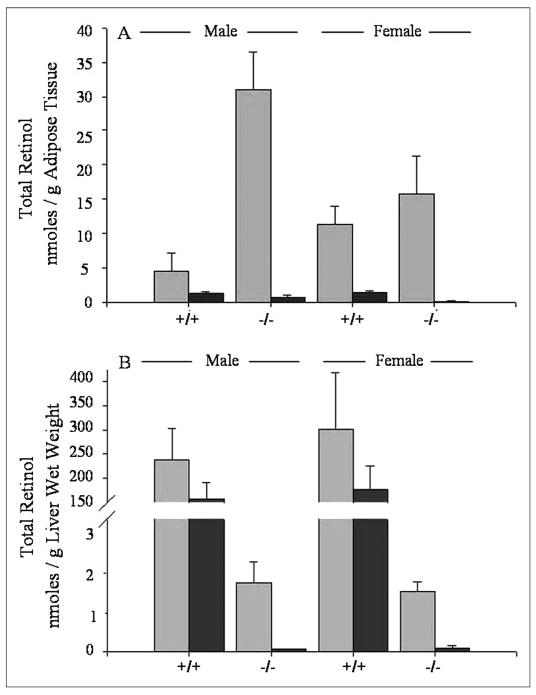
Panel A shows the levels of total retinol (retinol + retinyl esters) in perigonadal adipose tissue obtained from male and female wild type and Lrat−/− mice. The groups receiving the control diet are indicated by light gray bars and the groups receiving the retinoid-deficient diet are indicated by dark gray bars. Panel B shows the levels of total retinol (retinol + retinyl esters) in the livers of male and female wild type and Lrat−/− mice receiving a control diet (light gray bars) or a retinoid-deficient diet (dark gray bars). All mice were maintained on a chow diet through the first 3 months of age, and at this point they were then randomly assigned to groups and maintained for 4 additional weeks on either the same chow diet or a totally retinoid-deficient purified diet.
Tissue Retinoic Acid Levels Are Not Different for Wild Type and Lrat−/− Mice
Because retinyl ester levels in tissues of Lrat−/− mice are markedly different from those observed in age- and sex-matched wild type mice, we investigated whether retinoic acid levels might also be different in liver, adipose tissue, and testes of 3-month-old male Lrat−/− mice compared with male wild type mice (TABLE TWO). No differences in liver, adipose tissue, or testis levels of retinoic acid were observed for Lrat−/− mice. Only all-trans-retinoic acid could be detected in these tissues. If 13-cis- or 9-cis-retinoic acid were present in the tissues, the levels of these retinoic acid isomers were below the detection limit of our HPLC assay (< 1.5 pmol/g tissue).
TABLE TWO. Levels of retinoic acid in tissues from different strains of 3-month-old male mice.
Values are given as means ± 1 S.D. The number of individual mice (n) used for each measure is given in the parentheses following each value.
| All-trans-retinoic acid | |
|---|---|
| pmol/g tissue wet weight | |
| Liver | |
| Wild type | 7.9 ± 1.6 (10) |
| Lrat−/− | 12.1 ± 6.3 (5) |
| Adipose tissue | |
| Wild type | 34.7 ± 11.2 (10) |
| Lrat−/− | 34.2 ± 10.1 (5) |
| Rpb−/− | 29.2 ± 8.0 (5) |
| Testis | |
| Wild type | 28.5 ± 4.4 (5) |
| Lrat−/− | 28.9 ± 4.0 (4) |
Intestinal Absorption of Retinol Is Impaired in the Absence of LRAT
LRAT has long been thought to be critical to the intestinal absorption of retinol, converting it to retinyl esters for incorporation into the nascent chylomicrons (9, 10). Nevertheless, Lrat−/− mice are able to take up retinoid from the diet. To understand intestinal retinoid absorption better, we investigated the mechanisms by which the Lrat−/− mice were absorbing retinol. We first asked whether retinyl esters are present in the chylomicrons of Lrat−/− following a challenge with a gavage dose of retinol provided in peanut oil. Because we wanted to optimize our chances of detecting chylomicron retinyl esters, we employed the total lipase inhibitor P-407 to prevent hydrolysis and clearance of nascent chylomicrons generated in these gavage studies (34, 35). The evening before the administration of the retinol-containing gavage, mice were given a dose of P-407 (1 g/kg body weight) that is known to block chylomicron clearance for a period of up to several days (30, 31). The following morning, Lrat−/− and wild type mice were given a gavage dose containing ~2 × 106 cpm all-trans-[3H]retinol and 6 μg of unlabeled all-trans-retinol in a peanut oil vehicle. We estimate that this is the amount of retinol that is consumed in 1 day by mice receiving a standard chow diet. Three hours after administration of the gavage, mice were sacrificed, and serum was collected for chylomicron isolation. When the retinoid content of the chylomicrons was analyzed by HPLC, we detected retinyl esters in the chylomicrons of both the Lrat−/− and wild type mice (Fig. 3). However, unlike the wild type mice, the retinyl ester composition of the chylomicrons from Lrat−/− mice reflected the fatty acyl composition of the peanut oil vehicle. The most abundant fatty acyl moieties present in peanut oil are linoleate, oleate, and palmitate. As can be seen in Fig. 3, the most abundant retinyl esters present in chylomicrons from Lrat−/− are retinyl linoleate, retinyl oleate, and retinyl palmitate, corresponding to the acyl composition of peanut oil. As expected for wild type mice expressing LRAT, the most abundant chylomicron retinyl ester was retinyl palmitate, with much lower concentrations of retinyl linoleate and retinyl oleate also present. The close correlation between the fatty acyl composition of the peanut oil vehicle and the retinyl ester composition observed for chylomicrons from Lrat−/− mice suggests that retinyl ester formation in the intestine of these mice involves an acyl-CoA-dependent enzyme.
FIGURE 3. Reverse phase HPLC profiles showing the distribution of retinol and retinyl esters and 3H counts/min present in chylomicrons obtained from wild type (panel A) and Lrat−/− (panel B) mice following administration of a gavage dose of retinol (6 μg containing 2 × 106 3H counts/min) in peanut oil.
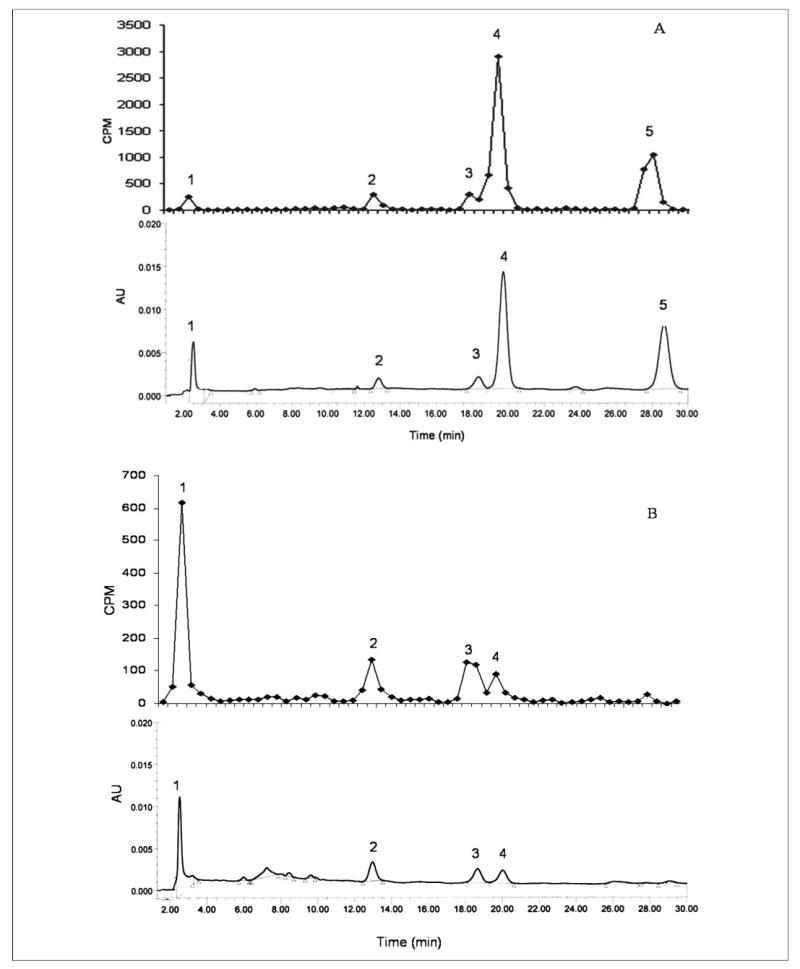
Panels A and B, the upper profiles show the distribution of [3H]retinoids and the lower profiles the UV absorbance of the retinoids. Note that for panels A and B the lower profiles are scaled to reflect at full scale the same absorbance units (AU). The extracted retinoids were separated on a 5-μm 4.6 × 250-mm Ultrasphere C18 column preceded by a C18 guard column, using 70% acetonitrile, 15% ethanol, 15% methylenechloride as the running solvent flowing at 1.8 ml/min. The numbers above the HPLC peaks indicate the following: 1, retinol; 2, retinyl linoleate; 3, retinyl oleate; 4, retinyl palmitate; and 5, retinyl stearate.
The efficiency of intestinal retinoid uptake by the Lrat−/− mice was less than that observed for wild type mice. Under our experimental conditions, the percentages of the retinol present in the gavage dose that were detected in sera obtained from individual male and female Lrat−/− mice were 5.2 ± 1.4 and 3.0 ± 0.5%, respectively. For male and female wild type mice, the percentages of the retinol dose present in sera were 8.8 ± 2.3 and 5.8 ± 2.2%. Thus, it appears that the absence of intestinal LRAT results in less efficient intestinal uptake of dietary retinoid.
We also detected relatively greater levels of unesterified retinol in the chylomicrons obtained from Lrat−/− mice. In the wild type mice, the majority of the chylomicron retinoid 3H counts/min and mass associated with the fractions corresponding to retinyl ester peaks on the HPLC profile (Fig. 3, panel A). Only ~10% of both the retinoid 3H counts/min and mass were present as free retinol in chylomicrons obtained from the wild type mice. However, for Lrat−/− mice, the majority of the 3H counts/min and retinoid mass (~60%) appeared in the fraction corresponding to free retinol (Fig. 3, panel B, peak 1). We take these data to indicate that Lrat−/− mice package greater quantities of free retinol into chylomicrons upon its absorption from the diet.
DGAT1 Can Catalyze Acyl-CoA-dependent Esterification of Reti-nol
DGAT1 is expressed in both the intestine and adipose tissue, and we wondered whether this enzyme might account for the synthesis of retinyl esters that are formed/present in these tissues. By using recombinant human DGAT1 expressed in HEK293 cells, we investigated the retinol-esterifying capabilities of DGAT1 in an in vitro enzyme assay. These assays were performed both in the presence and absence of purified recombinant holo-CRBPI and holo-CRBPIII. Our studies show that DGAT1 is capable of catalyzing the acyl-CoA-dependent esterification of free retinol in vitro (Fig. 4). DGAT1 will not utilize retinol bound to CRBPI as a substrate for retinol esterification. However, DGAT1 will use retinol bound to CRBPIII, albeit less well than free retinol. When we provided free retinol or holo-CRBPIII, each at a final concentration of 16 μm, we observed for the DGAT1-expressing HEK293 microsomes specific activities that were respectively, 72.9 ± 3.8 ng of retinyl ester formed per mg of protein/min (n = 3) and 26.3 ± 0.4 ng of retinyl ester formed per mg of protein/min (n = 3). When the concentration of free retinol or holo-CRBPIII was increased in the assay to 30 μm, the respective specific activities of the HEK293 microsomes were 117.5 ± 14.6 ng of retinyl ester formed per mg of protein/min (n = 3) and 76.9 ± 8.4 ng of retinyl ester formed per mg of protein/min (n = 3).
FIGURE 4. CRBPIII- but not CRBPI-bound retinol enables human DGAT1 catalysis of acyl-CoA-dependent esterification of retinol.
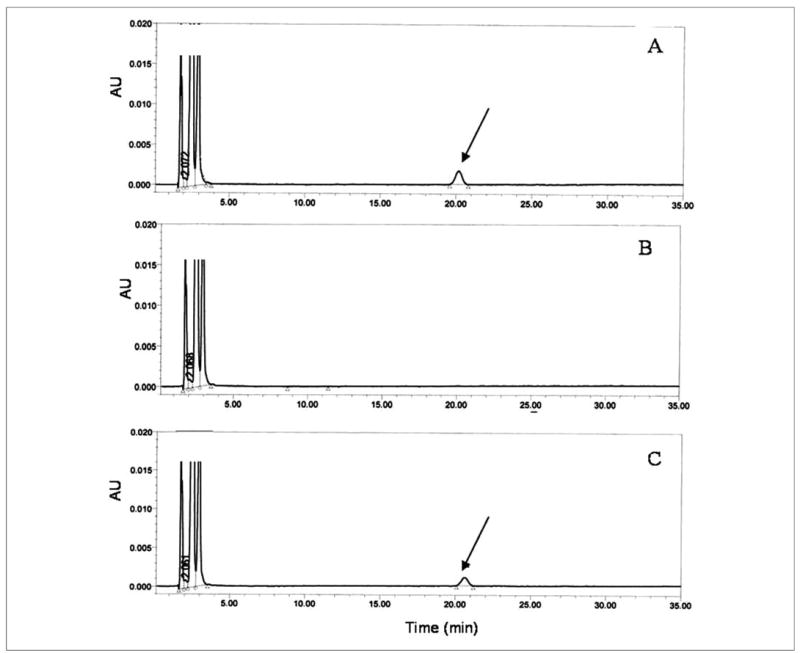
Microsomes isolated from HEK293 cells transfected with a cDNA encoding human DGAT1 were incubated either in the presence of 16 μm free all-trans-retinol (panel A) or 16 μm all-trans-retinol bound to CRBPI (panel B) or 16 μm all-trans-retinol bound to CRBPIII (panel C) for 10 min. The HPLC peak identified with the arrow is that of all-trans-retinyl palmitate formed upon DGAT1 action.
In preliminary studies, we also explored the possibility that human DGAT2 is able to catalyze retinyl ester formation. In keeping with findings reported by Yen et al. (33), we were unable to detect significant retinyl ester formation when recombinant human DGAT2 was incubated in the presence of free retinol.
LRAT Is Required for Retinoid Storage and Lipid Droplet Formation in Hepatic Stellate Cells
Lrat−/− mice contain only trace amounts of retinyl esters in their livers (20) (TABLE ONE). The predominant cellular site of hepatic retinyl ester storage is the hepatic stellate cell, located perisinusoidally in the space of Disse (11). Stellate cells are characterized by their large lipid droplets, in which the majority of hepatic retinyl esters is stored (11). Immunohistochemical staining for desmin, a marker used to identify hepatic stellate cells, indicates that the relative number, distribution, and light microscopic morphologic characteristics of stellate cells within the livers of Lrat−/− mice are similar to those of wild type mice. The staining pattern for desmin is shown in Fig. 5, panel A. We also undertook transmission electron microscopy studies of livers of wild type and Lrat−/− mice at 3 months of age. The wild type livers show the characteristic lipid droplets in hepatic stellate cells (Fig. 5, panel B, left). However, there is a striking absence of these large lipid droplets in stellate cells of Lrat−/− mice (Fig. 5, panel B, right). The lipid droplets present in the hepatocytes of Lrat−/− mice do not appear morphologically different from those of wild type mice (data not shown).
FIGURE 5. The hepatic stellate cells of Lrat−/− mice lack lipid droplets that are a morphologic hallmark of these cells.
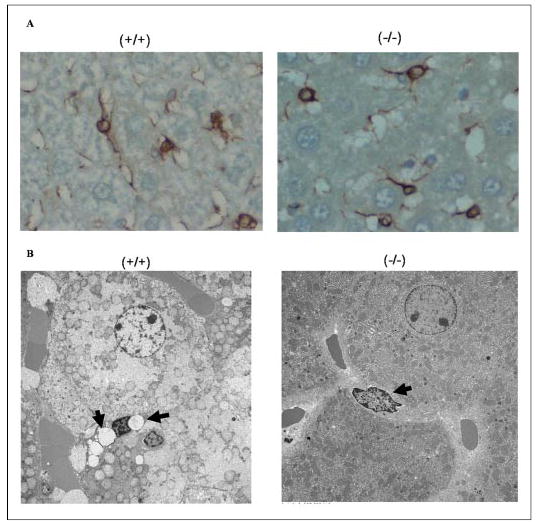
Panel A shows the immunohistochemical staining pattern for desmin, a marker for hepatic stellate cells, in liver sections from 3-month-old male wild type (+/+) and Lrat−/− (−/−) mice. Panel B shows electron micrographs of liver sections prepared from 3-month-old male wild type and Lrat−/− mice. The electron micrographs show the presence of characteristic retinyl ester-containing lipid droplets in hepatic stellate cells of wild type mice (−/−) and their absence in livers from Lrat−/− mice (−/−). The arrows indicate the presence (+/+) or absence (−/−) of lipid droplets in hepatic stellate cells. The large adjoining cells are hepatocytes.
Because the absence/loss of stellate cell lipid droplets is associated with the development of liver fibrosis, we next asked whether there was any indication of liver fibrosis in 3-month-old Lrat−/− mice. To assess this possibility, we stained liver sections for α-SMA and GFAP, markers used to assess hepatic stellate cell activation and Masson trichrome stain to assess collagen deposition and for identifying hepatic fibrosis. This analysis indicated that at this age, the livers of the Lrat−/− mice are histochemically identical to wild type controls (data not shown). Thus, 3-month-old Lrat−/− mice exhibit no histological signs of hepatic fibrosis.
Adult Lrat−/− Mice Are More Susceptible to Developing Retinoid Deficiency than Wild Type Mice
As described above, we carried out an experiment in which both 3-month-old Lrat−/− and wild type mice were maintained on a totally retinoid-deficient diet for 1 month. Prior to initiation of the mice onto this diet, they had received a control retinoid-sufficient chow diet from the time of weaning. After 1 month on the retinoid-deficient diet, we sacrificed the mice and performed RIAs to quantitate the levels of RBP in serum and liver. The Lrat−/− mice placed on the retinoid-deficient diet at 3 months of age for 1 month showed a drop in their serum retinol and RBP levels and a dramatic 10-fold increase in hepatic RBP levels (Fig. 6). These striking changes in retinol and RBP levels were not observed for wild type mice. A decline in serum retinol and RBP levels and a 3–10-fold elevation in hepatic RBP levels are seen in animals experiencing retinoid deficiency (12, 13). We take these observations to indicate that the Lrat−/− mice, unlike wild type mice, are beginning to experience retinoid deficiency after only 1 month of maintenance on a totally retinoid-deficient diet.
FIGURE 6. RBP levels are elevated in the livers of Lrat−/− mice but not wild type mice fed a totally retinoid-deficient diet for 1 month.
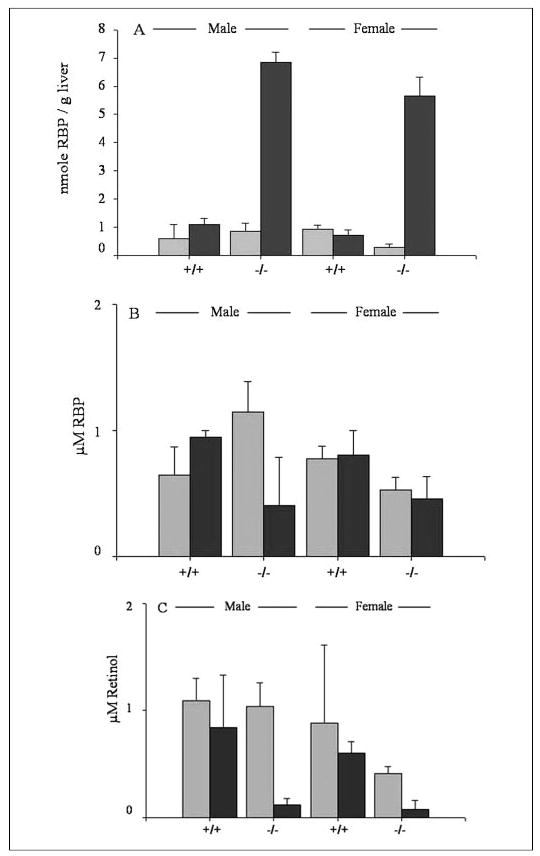
Panel A shows RBP concentrations in livers from male and female wild type (+/+) and Lrat−/− (−/−) mice receiving either a nutritionally complete control diet (light gray bars) or a totally retinoid-deficient diet (dark gray bars). Panel B shows RBP concentrations in serum from male and female wild type (+/+) and Lrat−/− (−/−) mice receiving either a nutritionally complete control diet (light gray bars) or a totally retinoid-deficient diet (dark gray bars). Panel C shows serum retinol concentrations for male and female wild type (+/+) and Lrat−/− (−/−) mice receiving either a nutritionally complete control diet (light gray bars) or a totally retinoid-deficient diet (dark gray bars).
DISCUSSION
Our present studies of Lrat−/− mice offer four major new insights regarding the physiologic actions of LRAT within the body and/or the unique physiology of Lrat−/− mice. First, LRAT is not the sole physiologically significant enzyme that catalyzes retinyl ester formation within the body. The intestine, adipose tissue, mammary tissue, and possibly other tissues possess metabolic machinery that allows for the synthesis and accumulation of retinyl esters. Second, retinyl ester levels are elevated in the perigonadal fat pads of Lrat−/− mice. Elevation of adipose retinyl ester content is accompanied by a 3–4-fold elevation in the level of CRBPIII in adipose tissue. This effect appears to be retinoid- and/or LRAT-specific because we were unable to detect morphological or other biochemical differences between the fat pads of Lrat−/− and wild type mice. Third, the absence of LRAT renders hepatic stellate cells devoid of their characteristic intracellular lipid droplets. Fourth, primarily because of the absence of hepatic retinoid stores in stellate cells, the Lrat−/− mice are susceptible for developing retinoid deficiency. Unlike wild type mice that require many months to display biochemical symptoms of retinoid deficiency (36, 37), the adult Lrat−/− mice show evidence of developing retinoid insufficiency after only 1 month of intake of a retinoid-insufficient diet.
Lrat−/− Mice Absorb Dietary Retinoid as Both Retinol and Retinyl Ester
Even in the complete absence of LRAT, the mutant mice are able to absorb dietary retinoid in chylomicrons. Under our experimental conditions, absorption of physiological doses of retinol in chylomicrons by the Lrat−/− mice is only 50–60% as efficient as retinoid absorption in age- and sex-matched wild type mice. Moreover, ~60% of the retinoid present in chylomicrons obtained from the Lrat−/− mice is in the form of free retinol (see Fig. 3). The remainder is as retinyl esters, primarily retinyl linoleate, retinyl oleate, and retinyl palmitate, reflecting the acyl group composition of the fat load administered to the mice. This retinoid composition is unlike wild type mice in which less than 10% of the retinoid present in chylomicrons is as free retinol and where the retinyl ester acyl group composition is independent of that of the fat load used in administering the retinol. It is clear however from these data that the intestine possesses an enzymatic activity that is able to catalyze retinyl ester formation even when LRAT is absent. However, retinyl ester formation must be suboptimal because a relatively high percentage of the retinoid in chylomicrons is present as free retinol. There is precedence in the literature for absorption of dietary retinoid as retinol. This precedence comes from the observation that abetalipoproteinemia patients who are afflicted with a genetic disease that renders them unable to make triglyceride-rich chylomicrons do absorb some dietary retinoid (27). It has been postulated that these patients absorb retinol on apoRBP present in the portal circulation. We also wondered whether some retinol also might be absorbed by apoRBP in the Lrat−/− mice and are presently exploring this possibility. To undertake these studies, we have generated Rbp−/− Lrat−/− double knock-out mice. These double knock-out mice are viable and appear to be phenotypically normal, but we have not yet challenged them with dietary retinoid manipulations.
An ARAT Activity May Be Physiologically Significant for Catalyzing Retinyl Ester Formation in Lrat−/− Mice
Although LRAT is undoubtedly the most physiologically significant enzyme involved in the synthesis of retinyl esters, it is not the sole enzyme. An enzyme(s) present in the intestine and adipose and mammary tissues can also catalyze retinol esterification. The literature has long suggested, based on in vitro studies, that an ARAT activity exists in tissues; however, the molecular identification and characterization of ARAT has remained elusive (24, 38–41). Ross and co-workers (24, 38, 39) suggested that, aside from mammary tissue, the ARAT activity of tissues only becomes physiologically relevant when excessive and possibly toxic concentrations of retinol are present in the tissues. Moreover, these investigators have demonstrated that retinol bound to CRBPI and/or CRBPII is not a substrate for tissue ARAT activity (17, 18, 38–41). This observation implies that tissue ARAT activity only becomes active when the retinol binding capacity of CRBPI or CRBPII has been exceeded. Yen et al. (33) convincingly demonstrated that mouse DGAT1 possesses ARAT activity through in vitro studies when free retinol is provided as a substrate and when retinol is provided to COS-7 cells overexpressing mouse DGAT1. Our data confirm the work of Yen et al. (33) and extend it by demonstrating that retinol bound to CRBPI is not a substrate for human DGAT1 and that retinol bound to CRBPIII is a relatively poor substrate for the enzyme. The finding that chylomicrons from Lrat−/− mice contain retinyl esters whose acyl group composition mirrors the acyl moiety composition of the peanut oil used to administer the retinol-containing gavage suggests strongly that the retinyl esters are synthesized though the actions of an acyl-CoA-utilizing enzyme. Considering that DGAT1 is expressed in the intestine as well as adipose tissue and mammary tissue (42), the two other tissues in Lrat−/− mice that are able to synthesize retinyl esters, it seems likely to us that DGAT1 acts in vivo as a physiologically significant ARAT.
Lrat Disruption Results in an Elevation of CRBPIII Levels in Adipose Tissue
Adipose tissue retinyl ester levels and levels of CRBPIII are elevated by severalfold in Lrat−/− mice compared with age-matched wild type mice. Most interestingly, the level of CRBPI is not elevated in liver or adipose tissue of Lrat−/− mice. The substantial increase in the levels of CRBPIII protein in adipose tissue suggests the possibility that this is a mechanism for increasing the uptake of retinol into adipose tissue that results in the observed increased retinoid levels. Our data, however, do not provide us with a biochemical basis for understanding why the levels of CRBPIII are elevated in adipose tissue. We recently reported (30) that CRBPI levels are elevated in adipose tissue but not liver of CRBPIII-deficient mice and conversely that CRBPIII levels are elevated in muscle and adipose tissue of CRBPI-deficient mice (CRBPIII is not normally expressed in liver and does not become expressed there in the CRBPI knock-outs). Although we did not study CRBPIII expression in muscle of Lrat−/− mice, our present data clearly suggest a specific linkage between LRAT expression or actions and CRBPIII expression in adipose tissue, but they do not provide insight into the interesting regulatory mechanisms that underlie these changes in expression. Moreover, they provide clear evidence that the regulation of retinyl ester storage and metabolism is different in adipose tissue compared with the liver.
Lrat−/− Mice Are More Susceptible to Developing Retinoid Deficiency than Wild Type Mice
When Lrat−/− and wild type mice were placed on a retinoid-deficient diet for 1 month, adipose tissue retinol and retinyl esters declined. For wild type mice, adipose retinoid levels declined before a drop in hepatic retinyl ester stores was observed. These observations indicate that adipose tissue retinoids are indeed available as stores that can be accessed in times of dietary retinoid insufficiency. Previous research has demonstrated that adipocytes express and secrete RBP (43, 44). Therefore, it seems that retinol is being mobilized from the adipose tissue of Lrat−/− and wild type mice bound to RBP, as from the liver. These data indicate that adipose tissue is a metabolically active and physiological relevant storage site of retinyl esters and that these adipose tissue stores may in fact be the first tissue retinoid stores mobilized in times of dietary scarcity.
Our studies also provide promising evidence that the Lrat−/− mice will become a useful mouse model to study retinoid deficiency. Because Lrat−/− mice have only adipose tissue retinyl ester stores and completely lack the normally large liver and lung stores of wild type mice, these mice should in principle be more susceptible to developing retinoid deficiency more readily than most other mouse strains. It is difficult to make wild type mice retinoid-deficient, and often investigators resort to depleting dams of retinoid and then maintaining their progeny throughout life on a retinoid-deficient diet (36, 37). These studies often take many months to undertake and complete. After 1 month on a retinoid-deficient diet, the adipose retinoid stores of Lrat−/− mice became depleted and serum levels of retinol and RBP declined, and hepatic RBP levels were elevated by ~10-fold. Hepatic RBP becomes elevated when retinol is unavailable because pre-RBP cannot be released from the endoplasmic reticulum into the secretory pathway (45). Consequently, serum levels of RBP decline and hepatic levels increase by 3–10-fold. These changes in RBP levels are routinely measured as a biochemical marker for the development of retinoid deficiency (12). Thus, it appears that the adult Lrat−/− mice maintained on a retinoid-deficient diet for 1 month were becoming retinoid-deficient. This observation suggests that the Lrat−/− mouse will be a convenient model for studying retinoid-dependent functions, especially in the adult animal.
Lrat−/− Mice Lack the Large Lipid Droplets That Are Characteristic of Hepatic Stellate Cells
The hepatic stellate cells are the primary cellular site of hepatic retinoid storage (9–11). Hepatic stellate cells are characterized by large lipid droplets that in healthy well nourished animals account for ~70% of the retinoid stored in the liver. On a per mass basis, the stellate cell lipid droplets consist of ~30–40% retinyl ester with the remainder of the mass contributed by triglyceride, cholesterol, cholesteryl ester, phospholipids, and free fatty acids (46, 47). Our electron micrographs (Fig. 5, panel B) indicate a striking absence of these large lipid droplets in the stellate cells of Lrat−/− mice. The absence of lipid droplets is surprising because the lipid droplets consist mainly of other lipids whose levels should not be affected by the absence of LRAT and retinyl ester formation. These data suggest that LRAT and/or retinyl esters are in some manner critically needed to allow for the genesis of lipid droplets within stellate cells.
The loss of stellate cell retinyl esters and lipid droplets is one of the first events that occurs following an insult to the liver that gives rise to liver disease and fibrosis (25, 26, 48, 49). It is well established that as hepatic stellate cells become activated in response to liver injury, the retinyl ester containing lipid droplets are quickly lost from the cells. However, it is not established whether the loss of stellate cell retinyl esters and lipid droplets is causal to the development of hepatic fibrosis or simply a result of the process of stellate cell activation (47, 48). Because the hepatic stellate cells of Lrat−/− mice have no lipid droplets, we wondered whether this absence predisposed the mice to the development of hepatic fibrosis. To investigate this possibility, we stained liver sections from Lrat−/− and wild type mice with desmin, a marker used to identify stellate cells, α-SMA and GFAP, markers of hepatic stellate cell activation, and Masson trichrome stain, a marker used to identify fibrosis development. If the Lrat−/− livers were undergoing stellate cell activation and fibrosis, we would have expected to see a marked increase in α-SMA and trichrome staining, but we did not. These data indicate that at 3 months of age the livers of Lrat−/− mice are exhibiting no signs of hepatic fibrosis and suggest that lipid droplet loss from stellate cells is not causal for fibrosis development. We are presently continuing this line of investigation by exploring whether Lrat−/− mice are more susceptible to the development of experimentally induced liver disease.
Acknowledgments
The assistance of Dr. Li-Shin Huang (Department of Medicine, Columbia University) in carrying out quantitative real time PCR analysis of PPARα, PPARγ, SREBP, DGAT1, and DGAT2 mRNA levels in adipose tissue is gratefully acknowledged. The gift of the lipase inhibitor P-407 from Dr. T. P. Johnston (Division of Pharmaceutical Sciences, University of Missouri School of Pharmacy, Kansas City) is gratefully acknowledged.
Footnotes
This work was supported by National Institutes of Health Grants R01 DK061310, R01 DK068437, R01 DK067512, R01 EY08123, and R01 EY09339, United States Department of Defense Grant BC031116, and a Foundation Fighting Blindness grant.
In the text when the cis- or trans-isomeric configuration of a retinoid is not specifically designated, we are referring collectively to all of the different geometric configurations (cis-species + trans-species) for the retinoid.
The abbreviations used are: RAR, retinoic acid receptor; ARAT, acyl-CoA:retinol acyltransferase; α-SMA, α-smooth muscle actin; CRBPI, cellular retinol-binding protein, type I; CRBPIII, cellular retinol-binding protein, type III; DGAT1, diacylglycerol acyltransferase 1; DGAT2, diacylglycerol acyltransferase 2; GFAP, glial fibrillary acidic protein; HPLC, high performance liquid chromatography; LRAT, lecithin:retinol acyltransferase; Lrat−/− mice, Lrat-deficient mice; RIA, radioimmunoassay; RXR, retinoid X receptor; RBP, retinol-binding protein; Rbp−/− mice, Rbp-deficient mice; SREBP, sterol response element-binding protein.
References
- 1.Moore, T. (1957) Vitamin A, Elsevier Publishing Co., Amsterdam
- 2.Wald G. Nature. 1968;219:800–807. doi: 10.1038/219800a0. [DOI] [PubMed] [Google Scholar]
- 3.McBee JK, Palczweski K, Baehr W, Pepperberg DR. Prog Retin Eye Res. 2001;20:469–529. doi: 10.1016/s1350-9462(01)00002-7. [DOI] [PubMed] [Google Scholar]
- 4.Gudas, L. J., Sporn, M. B., and Roberts, A. B. (1994) in The Retinoids, Biology, Chemistry and Medicine (Sporn, M. B., Roberts, A. B., and Goodman, D. S., eds) 2nd Ed., pp. 443–520, Raven Press, Ltd., New York
- 5.Balmer JE, Blomhoff R. J Lipid Res. 2002;43:1773–1808. doi: 10.1194/jlr.r100015-jlr200. [DOI] [PubMed] [Google Scholar]
- 6.Mangelsdorf, D. J., Umesono, K., and Evans, R. M. (1994) in The Retinoids, Biology, Chemistry and Medicine (Sporn, M. B., Roberts, A. B., and Goodman, D. S., eds) 2nd Ed., pp. 319–350, Raven Press, Ltd., New York
- 7.Chambon P. Cell Biol. 1994;5:115–125. doi: 10.1006/scel.1994.1015. [DOI] [PubMed] [Google Scholar]
- 8.Chambon P. FASEB J. 1996;10:940–954. [PubMed] [Google Scholar]
- 9.Blaner, W. S., and Olson, J. A. (1994) in The Retinoids, Biology, Chemistry and Medicine (Sporn, M. B., Roberts, A. B., and Goodman, D. S., eds) 2nd Ed., pp. 229–256, Raven Press, Ltd., New York
- 10.Vogel S, Gamble M, Blaner W. Handb Exp Pharmacol. 1999;139:31–96. [Google Scholar]
- 11.Geerts, A., Bleser, P. D., Hautekeete, M. L., Niki, T., and Wisse, E. (1994) in The Liver: Biology and Pathobiology (Arias, I. M., Boyer, J. L., Fausto, N., Jakoby, W. B., Schachter, D., and Shafritz, D. A., eds) 3rd Ed., pp. 819–837, Raven Press, Ltd., New York
- 12.Goodman, D. S. (1984) in The Retinoids (Sporn, M. B., Roberts, A. B., and Goodman, D. S., eds) Vol. 2, pp. 41–88, Academic Press, Orlando, FL
- 13.Soprano, D. R., and Blaner, W. S. (1994) in The Retinoids, Biology, Chemistry and Medicine (Sporn, M. B., Roberts, A. B., and Goodman, D. S., eds) 2nd Ed., pp. 257–282, Raven Press, Ltd., New York
- 14.O’Byrne, S. M., and Blaner, W. S. (2005) in Carotenoids and Retinoids, Molecular Aspects and Health Issues (Packer, L., Obermüller-Jevic, Kraemer, K., and Sies, H., eds) pp. 1–22, AOCS Press, Champaign, IL
- 15.Duester G. Eur J Biochem. 2000;267:4315–4324. doi: 10.1046/j.1432-1327.2000.01497.x. [DOI] [PubMed] [Google Scholar]
- 16.Napoli JL. Prog Nucleic Acids Res Mol Biol. 1999;63:139–188. doi: 10.1016/s0079-6603(08)60722-9. [DOI] [PubMed] [Google Scholar]
- 17.Herr FM, Ong DE. Biochemistry. 1992;31:6748–6755. doi: 10.1021/bi00144a014. [DOI] [PubMed] [Google Scholar]
- 18.Yost RW, Harrison EH, Ross AC. J Biol Chem. 1988;263:18693–18701. [PubMed] [Google Scholar]
- 19.Saari JC, Bredberg DL. J Biol Chem. 1989;264:8636–8640. [PubMed] [Google Scholar]
- 20.Batten ML, Imanishi Y, Maeda T, Tu DC, Moise AR, Bronson D, Possin D, Van Gelder RN, Baehr W, Palczewski K. J Biol Chem. 2004;279:10422–10432. doi: 10.1074/jbc.M312410200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kurlandsky SB, Duell EA, Kang S, Voorhees JJ, Fisher GJ. J Biol Chem. 1996;271:15346–15352. doi: 10.1074/jbc.271.26.15346. [DOI] [PubMed] [Google Scholar]
- 22.Ruiz A, Winston A, Lim YH, Gilbert BA, Rando RR, Bok D. J Biol Chem. 1999;274:3834–3841. doi: 10.1074/jbc.274.6.3834. [DOI] [PubMed] [Google Scholar]
- 23.Guo Z, Ruiz A, Rando RR, Bok D, Gudas LJ. Carcinogenesis. 2000;21:1925–1933. doi: 10.1093/carcin/21.11.1925. [DOI] [PubMed] [Google Scholar]
- 24.Ross AC. J Biol Chem. 1982;257:2453–2459. [PubMed] [Google Scholar]
- 25.Friedman SL. J Biol Chem. 2000;275:2247–2250. doi: 10.1074/jbc.275.4.2247. [DOI] [PubMed] [Google Scholar]
- 26.Geerts A. Semin Liver Dis. 2001;21:311–335. doi: 10.1055/s-2001-17550. [DOI] [PubMed] [Google Scholar]
- 27.Berriot-Varoqueaux N, Aggerbeck LP, Samson-Bouma ME, Wetterau JR. Annu Rev Nutr. 2000;20:663–697. doi: 10.1146/annurev.nutr.20.1.663. [DOI] [PubMed] [Google Scholar]
- 28.Blaner WS, Obunike JC, Kurlandsky SB, Al-Haideri M, Piantedosi R, Deck-elbaum RM, Goldberg IJ. J Biol Chem. 1994;269:16559–16565. [PubMed] [Google Scholar]
- 29.Kurlandsky SB, Gamble MV, Ramakrishnan R, Blaner WS. J Biol Chem. 1995;270:17850–17857. doi: 10.1074/jbc.270.30.17850. [DOI] [PubMed] [Google Scholar]
- 30.Piantedosi R, Ghyselinck N, Blaner WS, Vogel S. J Biol Chem. 2005;280:24286–24292. doi: 10.1074/jbc.M503906200. [DOI] [PubMed] [Google Scholar]
- 31.Blaner WS. Methods Enzymol. 1990;189:270–281. doi: 10.1016/0076-6879(90)89298-v. [DOI] [PubMed] [Google Scholar]
- 32.Liang JJ, Oelkers P, Guo C, Chu PC, Dixon JL, Ginsberg HN, Sturley SL. J Biol Chem. 2004;279:44938–44944. doi: 10.1074/jbc.M408507200. [DOI] [PubMed] [Google Scholar]
- 33.Yen CLE, Monetti M, Burri BJ, Farese RV. J Lipid Res. 2005;46:1502–1511. doi: 10.1194/jlr.M500036-JLR200. [DOI] [PubMed] [Google Scholar]
- 34.Wasan KM, Subramanian R, Kwong M, Goldberg IJ, Wright T, Johnston TP. J Pharm Pharm Sci. 2003;6:189–197. [PubMed] [Google Scholar]
- 35.Johnston TP. J Cardiovasc Pharmacol. 2004;43:595–606. doi: 10.1097/00005344-200404000-00016. [DOI] [PubMed] [Google Scholar]
- 36.Dickman ED, Thaller C, Smith SM. Development (Camb) 1997;124:3111–3121. doi: 10.1242/dev.124.16.3111. [DOI] [PubMed] [Google Scholar]
- 37.Molotkov A, Deltour L, Foglio MH, Cuenca AE, Duester G. J Biol Chem. 2002;277:13804–13811. doi: 10.1074/jbc.M112039200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Randolph RK, Winkler KE, Ross AC. Arch Biochem Biophys. 1991;288:500–508. doi: 10.1016/0003-9861(91)90227-a. [DOI] [PubMed] [Google Scholar]
- 39.Ross AC. J Lipid Res. 1982;23:133–144. [PubMed] [Google Scholar]
- 40.Ong DE, MacDonald PN, Gubitosi AM. J Biol Chem. 1988;263:5789–5796. [PubMed] [Google Scholar]
- 41.Muller H, Norum KR. Br J Nutr. 1986;55:37–41. doi: 10.1079/bjn19860007. [DOI] [PubMed] [Google Scholar]
- 42.Cases S, Smith SJ, Zheng YW, Myers HM, Lear SR, Sande E, Novck S, Collins C, Wesch CB, Lusis AJ, Erickson SK, Farese RV. Proc Natl Acad Sci U S A. 1998;95:13018–13023. doi: 10.1073/pnas.95.22.13018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tsutsumi C, Okuno M, Tannous L, Piantedosi R, Allen M, Goodman DS, Blaner WS. J Biol Chem. 1992;267:1805–1810. [PubMed] [Google Scholar]
- 44.Zovich DC, Orologa A, Okuno M, Kong LW, Talmage DA, Piantedosi R, Goodman DS, Blaner WS. J Biol Chem. 1992;267:13884–13889. [PubMed] [Google Scholar]
- 45.Suhara A, Kato M, Kanai M. J Lipid Res. 1990;31:1669–1681. [PubMed] [Google Scholar]
- 46.Yamada M, Blaner WS, Soprano DR, Dixon JL, Kjeldbye HM, Good-man DS. Hepatology. 1987;7:1224–1229. doi: 10.1002/hep.1840070609. [DOI] [PubMed] [Google Scholar]
- 47.Moriwaki H, Blaner WS, Piantedosi R, Goodman DS. J Lipid Res. 1988;29:1523–1534. [PubMed] [Google Scholar]
- 48.Friedman SL. N Engl J Med. 1993;328:1828–1835. doi: 10.1056/NEJM199306243282508. [DOI] [PubMed] [Google Scholar]
- 49.Li D, Friedman SL. J Gastroenterol Hepatol. 1999;14:618–633. doi: 10.1046/j.1440-1746.1999.01928.x. [DOI] [PubMed] [Google Scholar]
- 50.National Research Council (1996) Guide for the Care and Use of Laboratory Animals, 7th Ed., National Academy Press, Washington, D. C.