

Abstract
The metabolism of vitamin A is a highly regulated process that generates essential mediators involved in the development, cellular differentiation, immunity, and vision of vertebrates. Retinol saturase converts all-trans-retinol to all-trans-13,14-dihydroretinol (Moise, A. R., Kuksa, V., Imanishi, Y., and Palczewski, K. (2004) J. Biol. Chem. 279, 50230–50242). Here we demonstrate that the enzymes involved in oxidation of retinol to retinoic acid and then to oxidized retinoic acid metabolites are also involved in the synthesis and oxidation of all-trans-13,14-dihydroretinoic acid. All-trans-13,14-dihydroretinoic acid can activate retinoic acid receptor/retinoid X receptor heterodimers but not retinoid X receptor homodimers in reporter cell assays. All-trans-13,14-dihydroretinoic acid was detected in vivo in Lrat−/− mice supplemented with retinyl palmitate. Thus, all-trans-13,14-dihydroretinoic acid is a naturally occurring retinoid and a potential ligand for nuclear receptors. This new metabolite can also be an intermediate in a retinol degradation pathway or it can serve as a precursor for the synthesis of bioactive 13,14-dihydroretinoid metabolites.
Metabolites of vitamin A (all-trans-retinol, all-trans-ROL)1 play essential roles in vision, immunity, cellular differentiation, control of gene expression, and development in vertebrates. For example, 11-cis-retinaldehyde (11-cis-RAL) is the chromophore of visual pigments in the photoreceptor cells (1), whereas all-trans-retinoic acid (all-trans-RA) and its 9-cis isomer are important regulators of gene expression via retinoic acid receptors (RAR) and retinoid X receptors (RXR) (2). Other active metabolites of ROL include 14-hydroxy-4,14-retro-ROL (3), anhydroretinol (4), and 13,14-dihydroxy-ROL (5), which regulate growth and cellular survival. The ring-oxidized metabolites 4-oxo-ROL, 4-oxo-RAL, and 4-oxo-RA can activate RAR and RXR receptors and have been implicated in the embryonic development of Xenopus (6–8). Bio-active retinoids continue to be discovered; however, many of the enzymes involved in retinoid metabolism have not been identified.
The oxidation of ROL is both a major metabolic pathway for the synthesis of RAL and RA and a catabolic pathway for the clearance of pharmacological doses of ROL by conversion to polar metabolites that are easier to secrete (9). The enzymes involved in the synthesis and degradation of RA have been extensively described. ROL and RAL can be interconverted by microsomal short-chain dehydrogenase/reductase (SDR) (10, 11) and by class I, III, and IV medium-chain alcohol dehydrogenases (ADH) (12). Irreversible oxidation of RAL to RA is carried out by retinal dehydrogenase (RALDH) types 1–4 (13–18). Cytochrome P450 enzymes CYP26A1, CYP26B1, and CYP26C1 carry out the catabolism of RA to 4-hydroxy-RA, 4-oxo-RA, and 18-hydroxy-RA (19–22).
We recently described a novel enzyme that carries out the saturation of the C13–14 bond of all-trans-ROL to generate all-trans-13,14-dihydro-ROL (all-trans-DROL) (23). The enzyme, ROL saturase (RetSat), is found in many tissues, with the highest levels in the liver, kidney, and intestine. RetSat was shown to convert all-trans-ROL to all-trans-DROL, which was detected in several tissues of unsupplemented animals (23). Shirley et al. (24) have described the conversion of 9-cis-RA to 9-cis-13,14-dihydro-RA (9-cis-DRA) in rats, and others have described 9-cis-4-oxo-13,14-dihydroretinoic acid as a major metabolite in the liver of mice supplemented with ROL palmitate (25). The metabolic pathway responsible for the production of 13,14-dihydroretinoids has not been investigated.
In the current study, we used lecithin:ROL acyltransferase (LRAT)-deficient mice to examine the metabolism of ROL palmitate, all-trans-RA, and all-trans-DROL in vivo, with special attention to the formation of C13–14-saturated retinoids. The pathway was reconstituted in vitro using recombinant enzymes and cells transfected with individual retinoid processing enzymes. Finally, we demonstrated that all-trans-DRA can activate transcription in reporter cell assays through RAR/RXR heterodimers but not RXR homodimers.
MATERIALS AND METHODS
Metabolism of Retinoids in Vivo
All animal experiments employed procedures approved by the University of Washington and conformed to recommendations of the American Veterinary Medical Association Panel on Euthanasia and recommendations of the Association of Research for Vision and Ophthalmology. Animals were maintained on a 12-h light and 12-h dark cycle. All manipulations were done under dim red or infrared light (>560 nm). Most experiments used 6–12-week-old mice. Lrat−/− mice were genotyped as described previously (26). Animals were maintained on a control chow diet up to 1 h prior to oral gavage. The appropriate amount of all-trans-ROL palmitate, all-trans-DROL, or all-trans-RA was dissolved in vegetable oil and administered by oral gavage 3 h prior to analysis.
Analysis of Retinoids
Liver (1 g) from retinoid gavaged or naive mice was homogenized in 2 ml of 137 mm NaCl, 2.7 mm KCl, and 10 mm sodium phosphate (pH 7.4) for 30 s using a Polytron homogenizer. 10 μl of 5 m NaOH was added to 3 ml of the ethanolic extract, and the nonpolar retinoids were extracted using 5 ml of hexane. The extraction was repeated, and the organic phases were combined, dried under vacuum, resuspended in hexane, and examined by normal phase HPLC using a normal phase column (Beckman Ultrasphere Si 5μ, 4.6 × 250 mm). The elution condition was an isocratic solvent system of 10% ethyl acetate in hexane (v/v) for 25 min at a flow rate of 1.4 ml/min at 20 °C with detection at 325 and 290 nm for the detection of nonpolar retinoids and 13,14-dihydroretinoids, respectively. The aqueous phase was acidified with 40 μl of 12 n HCl, and polar retinoids were extracted with 5 ml of hexane. The extraction was repeated, and the organic phases of the polar retinoid extractions were combined, dried, resuspended in solvent composed of 80% CH3CN, 10 mM ammonium acetate, 1% acetic acid, and examined by reverse phase HPLC. Analysis of polar retinoids from tissues was done by reverse phase HPLC using a narrowbore, 120-Å, 5-μm, 2.1 × 250 mm, Denali C18 column (Grace-Vydac, Hesperia, CA). The solvent system was composed of buffer A, 80% methanol, 20% 36 mm ammonium acetate (pH 4.7 adjusted with acetic acid), and buffer B, 100% methanol. The HPLC elution conditions were 0.3 ml/min, 100% buffer A for 40 min, 100% buffer B for 10 min, and 10 min equilibration in buffer A. The elution profiles of RA and DRA were monitored using an online diode array detector set at 350 and 290 nm, respectively. The peaks were identified based on their UV-visible spectra and/or coelution with synthetic or commercially available standards. The measured area of absorbance was converted to picomoles based on a calibration of the HPLC columns using a known amount of all-trans-RA or all-trans-ROL (Sigma) and all-trans-DROL or all-trans-DRA (synthetic standards). The extraction efficiency was monitored by spiking a tissue sample with [3H]RA (PerkinElmer Life Sciences) and monitoring the radioactivity recovered from the HPLC column. In the case of liver samples the extraction efficiency was 95% or better. Mass spectrometry analyses of synthesized retinoids and of natural retinoids purified by HPLC were performed using a Kratos profile HV-3 direct probe mass spectrometer.
Synthesis and Analysis of 13,14-Dihydroretinoids
The synthetic scheme is depicted in supplemental Fig. 7. β-Ionone (I) was first brominated with N-bromosuccinimide in CCl4 followed by substitution of bromine with an acetoxyl group in hexamethylphosphoramide. The acetate ester of ionone was hydrolyzed with K2CO3 in methanol:water, and then the hydroxyl group was protected with tetra-butyldimethylsi-lyl group. The silylated 4-hydroxy-β-ionone (II) was then condensed under Horner-Emmons conditions with triethylphosphonoacetate, and the ester of silyl-protected ethyl 4-hydroxy-β-ionylidene acetate was reduced to alcohol with LiAlH4. The alcohol was acetylated with acetic anhydride in the presence of N,N-dimethylaminopyridine (DMAP); the silyl group was removed by tetrabutylammonium fluoride, and the alcohol was oxidized to a ketone group with MnO2 to give 15-acetoxy-4-oxo-β-ionylidene ethanol (III). Next, ester (III) was hydrolyzed, and the hydroxyl group was brominated with PBr3 in ether. The bromide was reacted with PPh3 to give Wittig salt (IV), which was further condensed with ethyl 4-oxo-3-methylbutyrate under conditions described previously (23) to obtain a mixture of ethyl 13,14-dihydro-4-oxoretinoate isomers (V) with all-trans- as a major compound. The isomers were separated by normal phase HPLC (HP1100, Beckman Ultrasphere Si 5μ, 10 × 250 mm, 5% ethyl acetate:hexane, and detection at 325 nm) and characterized by their UV, mass, and NMR spectra. NMR data were recorded on a Bruker 500-MHz spectrometer using CDCl3 as an internal standard, and their chemical shift values are listed in supplemental Table II. The order of elution was as follows: 9,11-di-cis-, all-trans-, 9-cis, 11-cis-13,14-dihydro-4-oxoretinoate. To obtain free retinoic acid (VI), the ethyl ester was hydrolyzed with NaOH in ethanol:H2O. To obtain 13,14-dihydro-RAL (DRAL), previously prepared ethyl 13,14-dihydroretinoate was reduced with diisobutyl aluminum hydride at − 78 °C. All-trans-4-oxo-DRA has the following UV-visible absorbance spectrum in ethanol, λmax = 328 nm and shoulder at λ = 256 nm, and in hexane, λmax = 314 nm and shoulder at λ = 252 nm.
Cloning and Expression Constructs
Total embryo and liver RNA was obtained from Ambion (Austin, TX) and reverse-transcribed using SuperScript II reverse transcriptase (Invitrogen) and oligo(dT) primers according to manufacturer’s protocol. Embryo cDNA was used to amplify the cDNAs of specific genes using Hotstart Turbo Pfu polymerase (Stratagene, La Jolla, CA) and the following primers: RA-LDH1, forward 5′-CACCGCAATGTCTTCGCCTGCACAAC and reverse 5′-GCTGGCTTCTTTAGGAGTTCTTC; RALDH3 forward CACCTGCG-AACCAGTTATGGCTACC and reverse 5′-GCCTGTTCCTCAGGGGTT-CTT; CYP26B1, forward 5′-CACCAAGCGGCTGCCAACATGC and reverse 5′-GCTGAGACCAGAGTGAGGCTA; and CYP26C1 forward 5′-C-ACCCATTCTCGCCATGATTTCCT and reverse 5′-CCAAGGCTAGAGAAGCAACG. The full-length cDNA of RALDH2 (MGC:76772, IMAGE: 30471325), RALDH4 (MGC:46977, IMAGE:4223059), and CYP26A1 (MGC:13860, IMAGE:4210893) mRNA was obtained from the Mammalian Gene Collection (MGC). These clones were used as templates to amplify the respective cDNAs using Hotstart Turbo Pfu polymerase (Stratagene) and the following primers: RALDH2, forward 5′-CACCATGGCCTCGCTGCAGCTCCTGC and reverse 5′-GGAGTTCTTCTGGGGGATCTTCA; RALDH4, forward 5′-CACCTGTACACAGAGGGCACTTTCC and reverse 5′-GTATTTAATGGTAATGGTTTTTATTTCAGTAAAG; and CYP26A1, forward 5′-CACCATGGGGCTCCCGGCGCTGCT and 5′-GATATCTCCCTGGAAGTGGGTAAAT. The cDNAs for RAL-DH1, -2, -3, and -4 and CYP26A1, -B1, and -C1 were cloned in the pcDNA3.1 Directional TOPO vector under the control of the CMV promoter to express a recombinant protein fused with a C-terminal V5 epitope peptide (GKPIPNPLLGLDST) and a His6 tag (Invitrogen). Both strands of the expression constructs were sequenced to ensure no mutations were present.
Mouse RXR-α was cloned using the primers 5′-GGGCATGAGTTAGTCGCAGA and 5′-AGCTGAGCAGCTGTGTCCA from reverse-transcribed mouse liver cDNA. The RXR-α open reading frame was then subcloned into the pcDNA3.1 Directional TOPO vector (Invitrogen) using the primers 5′-CACCATGGACACCAAACATTTCCT and 5′-AGCTGAGCAGCTGTGTCCA under the control of the CMV promoter. The RXRE from the vector RXR (2) translucent reporter vector (Panomics, Redwood City, CA) was amplified using the primers 5′-CTCAACCCTATCTCGGTCTATTCT and 5′-ATGCCAGCTTCATTATATACCCA and cloned upstream of the minimal promoter and β-galactosidase open reading frame of pBLUE-TOPO (Invitrogen) to create the pRXRE-BLUE expression construct. This construct places five consecutive DR1 elements upstream of β-galactosidase, the expression of which becomes dependent on activation of RXR and formation of RXR homodimers. Both strands of all constructs were sequenced to ensure no mutations were present.
Oxidation of All-trans-ROL and All-trans-DROL Using Liver Alcohol Dehydrogenase
Equine liver ADH (EC 1.1.1.1) was obtained from Sigma and dissolved in 50 mm Tris (pH 8.8) to a concentration of 5 units/ml (8.6 mg/ml). NAD and NADP were mixed together (1:1) at a concentration of 10 mm each. A substrate solution, 2 μl of 2 mm stock of all-trans-ROL or all-trans-DROL in N,N-dimethylformamide, was added to a 1.5-ml Eppendorf tube containing 20 μl of 10% bovine serum albumin, 20 μl of ADH, 2 μl of cofactor mixture, and 50 mm Tris (pH 8.8) to a total volume of 200 μl. The solutions were incubated at 37 °C for 60 min, after which 50 μl of 0.8 m NH2OH solution (pH 7.0) was added, sfollowed by addition of 300 μl of methanol, 15 min at room temperature, and extraction with 300 μl of hexane. The organic phase was dried and analyzed by normal phase HPLC as described in the analysis of non-polar retinoids extracted from tissue samples. As a control for the nonenzymatic reaction, boiled protein (90 °C for 5 min) was used with or without addition of cofactors.
RALDH Oxidation Assay
N-Acetylglucosaminyltransferase I-negative HEK-293S cells, obtained from Dr. G. Khorana (Massachusetts Institute of Technology, Boston) (27) were cultured in Dulbecco’s modified Eagle’s medium, 10% fetal calf serum and maintained at 37 °C, 5% CO2, and 100% humidity. For RALDH enzyme assays, cells were transiently transfected with RALDH1, -2, -3, or -4 expression constructs using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. After 48 h post-transfection, the cells were collected by scraping and were centrifuged. The cell pellet was washed in 137 mm NaCl, 2.7 mm KCl, and 10 mm phosphate (pH 7.4), resuspended in 50 mm Tris (pH 8.0) containing 250 mm sucrose, and homogenized with the aid of a Dounce homogenizer. Cofactors were added to a final concentration of 5 mm NAD, 5 mm NADP, and 1 mm ATP. An aliquot of the cell lysate was boiled for 10 min at 95 °C to provide the control for the nonenzymatic reaction. Substrates in the form of all-trans-RAL or a mixture of isomers of DRAL were added to the cell lysates at a final concentration of 60 μm. The reactions were allowed to proceed for 2 h at 37 °C with shaking and were stopped by the addition of 2 volumes of CH3CN. Samples were treated for 30 min at room temperature with 100 mm NH2OH (final concentration from a freshly made stock of 1 M (pH 7.0)) followed by centrifugation at 12,000 × g for 10 min. The clear supernatant was acidified with 0.1 volume of 0.5 m ammonium acetate (pH 4.0) and examined by reverse phase HPLC system (Zorbax ODS, 5 μm, 4.6 × 250 mm; Agilent, Foster City, CA) with an isocratic mobile phase A of 80% CH3CN, 10 mM ammonium acetate, 1% acetic acid, and a flow rate of 1.6 ml/min held for 15 min. After each run, the column was washed with mixture B (60% tert-butylmethyl ether, 40% methanol) for 10 min at 1.6 ml/min, followed by re-equilibration in phase A. The elution of RA and DRA isomers was monitored at 340 and 290 nm, respectively. The peaks were identified based on their spectra and coelution with standards. The cell lysate was examined for expression of RALDH1–4 by SDS-PAGE and immunoblotting of the V5 epitope-tagged recombinant protein using an anti-V5 epitope monoclonal antibody (Invitrogen).
CYP26A1 Oxidation Assay
N-Acetylglucosaminyltransferase I-negative HEK-293S cells were transiently transfected with cDNAs of CYP26A1, -B1, and -C1 under the control of CMV promoter using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. After 24 h, the transfected cells were split into 12-well plates to ensure an equal number of transfected cells in each assay well. All-trans-RA or all-trans-DRA was added to the cell monolayer at 0.1 mm final concentration in complete media and incubated for 4 h. Media and cells were collected by scraping, and proteins were precipitated with an equal amount of CH3CN by vigorous vortexing followed by centrifugation at 12,000 × g for 10 min. For RA analysis the clear supernatant was acidified with 0.1 volume of 0.5 m ammonium acetate (pH 4.0) and examined by reverse phase HPLC as described for the RALDH assays. The elution of all-trans-RA, all-trans-DRA, and their oxidized metabolites was monitored at 340 and 290 nm. The peaks were identified based on their spectra and coelution with standards. The cell lysate was examined for expression of CYP26A1, -B1, and -C1 by SDS-PAGE and immunoblotting of the V5 epitope-tagged recombinant protein using an anti-V5 epitope monoclonal antibody (Invitrogen).
Conversion of DROL to DRA in RPE
UV-treated RPE microsomes were prepared as described previously (28). Twenty μl of UV-treated RPE microsomes (3 mg/ml) were mixed with 20 μm DROL or ROL substrates, 1% bovine serum albumin, and 50 mm Tris (pH 8.8) and were incubated at 37 °C for 60 min in the presence or absence of NAD NADP cofactor mixture at 50 μm each. In order to stop the reaction, proteins were precipitated by mixing with an equal volume of CH3CN followed by high speed centrifugation. The clear supernatant was acidified with 0.1 volume of 0.5 m ammonium acetate (pH 4.0) and examined by reverse phase HPLC as described for the RALDH assays. A boiled RPE membrane control was used to assay nonenzymatic conversion of DROL. The elution of all-trans-DROL metabolites was monitored at 290 nm.
RARE and RXRE Activation Assay
The RARE reporter cell line F9-RARE-lacZ (SIL15-RA) was a kind gift from Dr. Michael Wagner (State University of New York Downstate Medical Center) and Dr. Peter McCaffery (University of Massachusetts Medical School, E. K. Shriver Center). The RA-responsive F9 cell line was transfected with a reporter construct of an RARE derived from the human retinoic acid receptor-β gene (RARβ) placed upstream of the Escherichia coli lacZ gene (29). Cells were grown in L15-CO2 media containing N-3 supplements and antibiotics. Cells were stimulated for 24 h in the dark at 37 °C and 100% humidity with all-trans-RA or all-trans-DRA dissolved in ethanol at the indicated concentrations, lysed, and assayed for the expression of β-galactosidase using the β-galactosidase enzyme assay system (Promega, Madison WI). For RXRE activation assays N-acetylglucosaminyltransferase I-negative HEK-293S cells were transfected with the pRXRE-BLUE reporter construct with or without the RXRα-expression construct using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. After 24 h, cells were split into 24-well plates to ensure an equal number of trans-fected cells in each assay well. Cells were stimulated with appropriate concentrations of all-trans-RA, 9-cis-RA, or all-trans-DRA. After 48 h, the expression of β-galactosidase was assayed as described above.
RESULTS
Identification of All-trans-DROL and Its Metabolites in the Liver of Lrat−/− Mice Gavaged with All-trans-ROL Palmitate
ROL absorption in mammals is an active process driven by esterification and hydrolysis cycles. Esterification of ROL is carried out mainly by the LRAT enzyme (30). In the absence of LRAT, the equilibrium between ROL and ROL esters is shifted in favor of free ROL. Mice deficient in LRAT expression (Lrat−/−) mice are severely impaired in their ROL uptake and storage capacity (26). Wild type mice, on the other hand, convert most of the ingested ROL to esters, which sequester ROL from circulation and metabolism. Thus, we chose to study the saturation and oxidation of all-trans-ROL to 13,14-dihydroretinoid metabolites in Lrat−/− mice.
Given their similar chemical properties, it is not surprising that all-trans-DROL and all-trans-ROL follow parallel metabolic pathways. Two different groups of Lrat−/− mice were dosed with either 106 units of all-trans-ROL palmitate/kg body weight or 105 units of all-trans-ROL palmitate/kg body weight, and their livers were examined for polar and nonpolar retinoid metabolites at 3 h post-gavage. Reverse phase HPLC analysis of polar hepatic retinoids indicated the presence of all-trans-RA (Fig. 1, A and B, peak 5) and all-trans-DRA (Fig. 1, A and B, peak 4), as well as a cis-DRA isomer (Fig. 1, A and B, peak 2). We also observed another polar DROL metabolite, which eluted earlier than all-trans-DRA, on reverse phase HPLC (Fig. 1, A and B, peak 1) and had the same absorbance spectrum as all-trans-DRA standard (Fig. 1E). This metabolite was not chemically characterized; however, based on its polar character, it could represent a taurine or glucuronide DRA conjugate. The spectra and elution profiles of synthetic all-trans-DRA and all-trans-DRA isolated from liver matched (Fig. 1E). All-trans-DRA was synthesized according to procedures published previously (23) and was characterized by 1H NMR (supplemental Table II).
Fig. 1. Analysis of metabolism of all-trans-ROL palmitate in the liver of Lrat−/− mice.
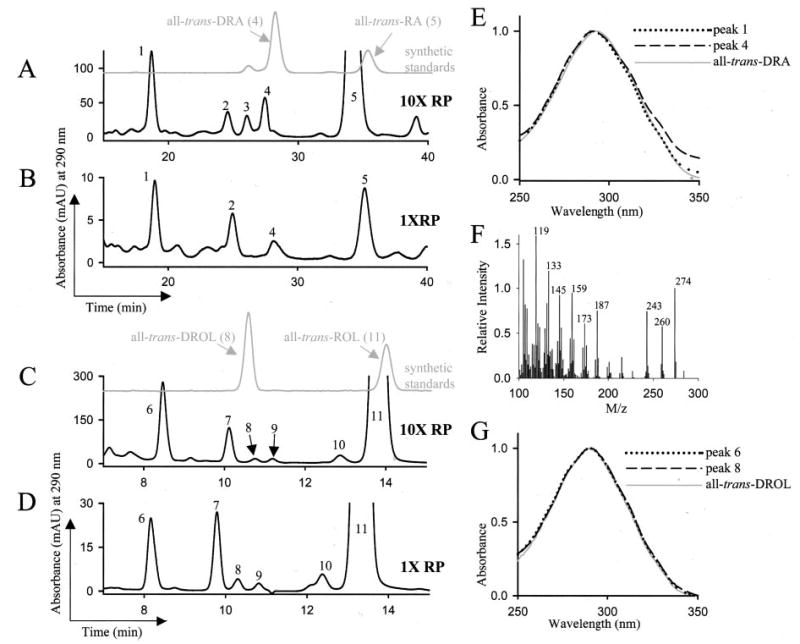
A–D, HPLC analysis of the polar and nonpolar retinoids from the liver of Lrat−/− mice gavaged with all-trans-ROL palmitate. Mice were gavaged with all-trans-ROL palmitate at a high dose of 106 units/kg body weight (marked as 10XRP, n = 3) in A and C or with a lower dose of all-trans-ROL palmitate of 105 units/kg body weight (marked as 1XRP, n = 3) in B and D. Three h after gavage, the polar and nonpolar retinoids from liver were extracted. The retinoids were analyzed by reverse phase HPLC on a narrowbore column system (A and B), and the nonpolar retinoids were analyzed by normal phase HPLC (C and D). Compounds were identified based on comparison with the elution profile and absorbance spectra of authentic standards. E, the spectrum of peak 4 matches that of all-trans-DRA standard, with which it coelutes. The absorbance spectrum of another compound, peak 1, eluting earlier than all-trans-DRA by reverse phase HPLC, also matches that of all-trans-DRA. F, the electron impact mass spectrometry analysis of the compound eluting as peak 6 in C and D indicates it is a possible mixture of compounds with m/z of 274 and 260. G, the compound eluting as peak 6 exhibits a UV-visible absorbance profile identical to the one of biological all-trans-DROL (peak 8) and of synthetic all-trans-DROL. Elution of all-trans-RA was monitored at 350 nm, all-trans-ROL at 325 nm, and all-trans-DROL and all-trans-DRA at 290 nm. Only the absorbance at 290 nm is shown here for simplicity. The extraction efficiency was > 95% and was calculated based on spiking samples with [3H]RA and measuring the radioactivity associated with the RA peak. Based on elution time, absorbance spectra, and comparison with authentic standards, the peaks were identified as the following compounds: peak 2, cis-DRA; peak 3, 13-cis-RA; peak 4, all-trans-DRA; peak 5, all-trans-RA; peak 6, C19-ROL derivative; peak 7, 13-cis-ROL; peak 8, all-trans-DROL; peak 9, 9,13-di-cis-ROL; peak 10, 9-cis-ROL; and peak 11, all-trans-ROL.
We examined the nonpolar hepatic retinoid metabolites by normal phase HPLC. At 3 h post-gavage with ROL palmitate, the livers of the examined mice contained high levels of all-trans-ROL (Fig. 1, C and D, peak 11), whereas all-trans-DROL (Fig. 1, C and D, peak 8) was found at 280–330-fold lower levels (Table I). The absorbance spectra and elution profile of all-trans-DROL matched the synthetic standard prepared according to published procedures (23) and characterized by 1H NMR (Fig. 1, C, D, and G, and supplemental Table II).
Table I. Level of liver retinoids 3 h following gavage with ROL palmitate.
The analysis was carried out as described under “Materials and Methods.”
| Compound identified | 106 IU/kg body weight-dose level of all-trans-ROL palmitate | 105 IU/kg body weight-dose level of all-trans-ROL palmitate |
|---|---|---|
| pmol/g tissue | ||
| All-trans-RA | 9,400 ± 300 | 320 ± 240 |
| All-trans-DRA | 190 ± 17 | 10 ± 2 |
| cis-DRA | 180 ± 53 | 22 ± 4 |
| Fig. 1, A and B, peak 1 | 460 ± 50 | 37 ± 9 |
| All-trans-ROL | 28,000 ± 300 | 7,000 ± 1,200 |
| All-trans-DROL | 100 ± 18 | 21 ± 4 |
| Fig. 1, C and D, peak 6 | 1,800 ± 370 | 200 ± 8 |
Another nonpolar 13,14-dihydroretinoid metabolite (Fig. 1, C and D, peak 6) that was present at higher levels than DROL was identified in the liver of mice gavaged with all-trans-ROL palmitate. The spectra of this compound also matched that of all-trans-DROL (Fig. 1G). The compound does not coelute with cis-DROL isomers and has a different UV-visible absorbance maximum than cis-DROL isomers (not shown). We were able to esterify the compound, whereas NH2OH treatment had no effect on its elution profile (not shown). Thus, we conclude that the functional group of the compound eluting as peak 6 (Fig. 1, C and D) is alcohol. Electron-impact mass spectrometry analysis of the collected fraction corresponding to peak 6 indicates the presence of a compound with an m/z of 274 (Fig. 1F). This suggests that peak 6 could include the chain-shortened C19-ROL derivative (C19H30O, m/z = 274, depicted in Scheme 1).
Scheme 1. Metabolism of all-trans-ROL and all-trans-DROL.
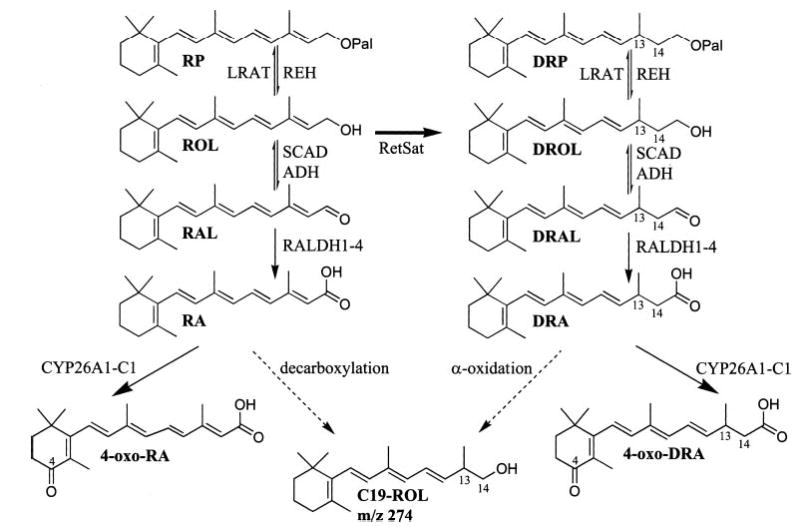
RetSat saturates all-trans-ROL to all-trans-DROL, which was previously shown to be esterified by LRAT. Here we present evidence demonstrating that the oxidative metabolism of DROL closely follows that of ROL. Broad spectrum enzymes such as SDR and ADH carry out the reversible oxidation of all-trans-DROL to all-trans-DRAL. RALDH1, -2, -3, and -4 oxidize all-trans-DRAL to all-trans-DRA. Several members of the cytochrome P450 enzymes CYP26A1, -B1, and -C1 oxidize all-trans-DRA to all-trans-4-oxo-DRA, identified in vivo and in vitro. Other oxidized all-trans-DRA metabolites, which are not depicted, could be all-trans-4-hydroxy-DRA, all-trans-5,6-epoxy-DRA, all-trans-5,8-epoxy-DRA, and all-trans-18-hydroxy-DRA. The short-chain metabolite C19-ROL is shown here with its possible chemical structure. Its synthetic pathway may proceed from either all-trans-RA by decarboxylation and/or from all-trans-DRA via α-oxidation.
Following gavage of Lrat−/− mice with synthetic all-trans-DROL, we observed significant levels of all-trans-DRA and all-trans-4-oxo-DRA. These were identified based on their chromatographic profile, m/z, and absorbance spectra, which matched those of synthetic standards (supplemental Fig. 8A and inset spectra). All-trans-4-oxo-DRA was synthesized according to the scheme depicted in supplemental Fig. 7 and was characterized by 1H NMR (supplemental Table II). The livers of mice gavaged with DROL were also found to contain low levels of C19-ROL (supplemental Fig. 8B, peak 4, and inset spectrum). This is in contrast to the high levels of C19-ROL observed in all-trans-ROL palmitate gavaged mice.
It has been reported that rats can convert exogenously administered 9-cis-RA to 9-cis-DRA and its taurine conjugate (24). We have shown that RetSat does not saturate all-trans-RA (23) or 9-cis-RA.2 This would suggest that another pathway is responsible for saturation of the C13–14 bond of RA to produce DRA. In the current study, we found no evidence of all-trans-DRA or all-trans-4-oxo-DRA formation in the livers of Lrat−/− mice gavaged with all-trans-RA at 3 h post-gavage (supplemental Fig. 9). A compound different from all-trans-DRA (supplemental Fig. 9, marked with *) with a maximum absorbance of 257 nm eluted before the expected elution time of all-trans-DRA. This would suggest that 13,14-dihydroretinoid metabolites can only be derived from all-trans-DROL after saturation of all-trans-ROL by RetSat, emphasizing the key role played by RetSat at this branch of vitamin A metabolism. We also found no evidence of C19-ROL in the livers of Lrat−/− mice gavaged with all-trans-RA at 3 h post-gavage (not shown).
The levels of all-trans-RA, all-trans-DRA, and the compounds eluting as peak 1 in Fig. 1, A and B, and as peak 6 in C and D, are indicated in Table I and reflect the different starting levels of ingested ROL palmitate. The levels of all-trans-DRA are much lower (30–50-fold) than those of all-trans-RA, which could indicate that saturation by RetSat is a limiting step. The low levels of all-trans-DROL in comparison with all-trans-ROL also support this explanation. The levels of all-trans-DROL and all-trans-DRA may also be low because of further processing to shorter chain or to other more oxidized metabolites.
Characterization of the Metabolic Pathway of All-trans-DROL to All-trans-DRA
Given that all-trans-DRA is detected in vivo as a metabolite of all-trans-DROL, we decided to examine its possible mode of synthesis using reconstituted enzyme systems. To oxidize all-trans-DROL to the corresponding aldehyde all-trans-DRAL, we used ADH purified from horse liver (EC 1.1.1.1), which is active toward both primary and secondary alcohols. All-trans-DROL and all-trans-ROL were incubated with purified enzyme and the appropriate cofactors. Following the reaction the samples were treated with NH2OH, extracted into the organic phase, and examined by normal phase HPLC. All-trans-RAL or all-trans-DRAL oximes were identified by comparison with synthetic standards (23). ADH efficiently carried out the conversion of all-trans-ROL to all-trans-RAL and of all-trans-DROL to all-trans-DRAL in the presence of NAD and NADP cofactors (Fig. 2, A and B) and not in their absence (not shown). The boiled enzyme did not exhibit any activity toward either substrate. Next, photoreceptor-specific RDH (prRDH) and RDH12 were tested for ability to catalyze the oxidation of all-trans-DROL to all-trans-DRAL. Both prRDH and RDH12 were active in converting all-trans-ROL to all-trans-RAL but much less so in converting all-trans-DROL to all-trans-DRAL (results not shown).
Fig. 2. Oxidation of all-trans-ROL and all-trans-DROL to the respective aldehyde.
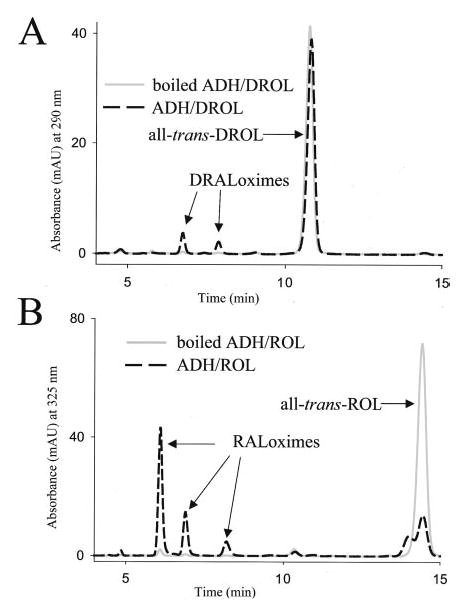
Purified ADH (Sigma) catalyzed the oxidation of all-trans-DROL to all-trans-DRAL (A) and all-trans-ROL to all-trans-RAL (B) in the presence of NAD and NADP. Control reactions using boiled enzyme were negative and show that the conversion is enzymatic. Retinoids were extracted and analyzed by normal phase HPLC. The products of the reaction were syn- and anti-all-trans-DRAL oximes (A) and syn- and anti-all-trans-RAL oximes (B). The experiment was performed in triplicate and repeated.
Conversion of all-trans-DRAL to DRA is mediated by RALDH enzymes. Mouse RALDH1–4 cDNAs were cloned and fused at their C terminus with a tag containing a V5 epitope and His6 stretch. Glycosylation-deficient HEK-293S cells were transiently transfected with the tagged constructs of RALDH1, -2, -3, or -4 under the control of the CMV promoter. These cells allow the reproducible, high level expression of recombinant proteins (27). The cell homogenate of transfected cells was supplemented with NAD, NADP, and ATP cofactors and with all-trans-RAL or all-trans-DRAL substrates. RALDH2 and -3 both efficiently converted all-trans-RAL and all-trans-DRAL into all-trans-RA and all-trans-DRA, respectively (Fig. 3, A and B). The products all-trans-RA and all-trans-DRA were identified based on their elution time, absorbance spectra, and comparison with authentic standards (Fig. 3A, peak 1, and B, peak 6, and inset spectra). Other cis-DRA isomers were also produced as a result of oxidation of cis-DRAL isomers present in the synthetic mixture. The expression level of recombinant protein in transfected cell homogenate was verified by immunoblotting using anti-V5 monoclonal antibody for the presence of V5-tagged RALDH protein. This is shown for RALDH2-V5-His6 in Fig. 3 (top right panel). Based on the intensity of the immunoreactive band, similar expression levels of RALDH1, -2, -3, or -4 were attained in transfected cells (not shown). Homogenates of RALDH1- and RALDH4-transfected cells were less efficient in oxidizing all-trans-RAL or all-trans-DRAL, possibly a consequence of the C-terminal tag affecting some isozymes more than others. Alternatively, some isozymes may be more active than others, as seen for mouse RALDH2 (Km = 0.66 μm for all-trans-RAL) versus mouse RALDH1 (Km = 11.6 μm for all-trans-RAL) (31, 32). Untransfected cells also exhibited significant activity toward both all-trans-RAL and all-trans-DRAL (Fig. 3, gray line chromatogram), suggesting endogenous RALDH activity in HEK-293S cells.
Fig. 3. Oxidation of all-trans-RAL and all-trans-DRAL to all-trans-RA and all-trans-DRA, respectively.
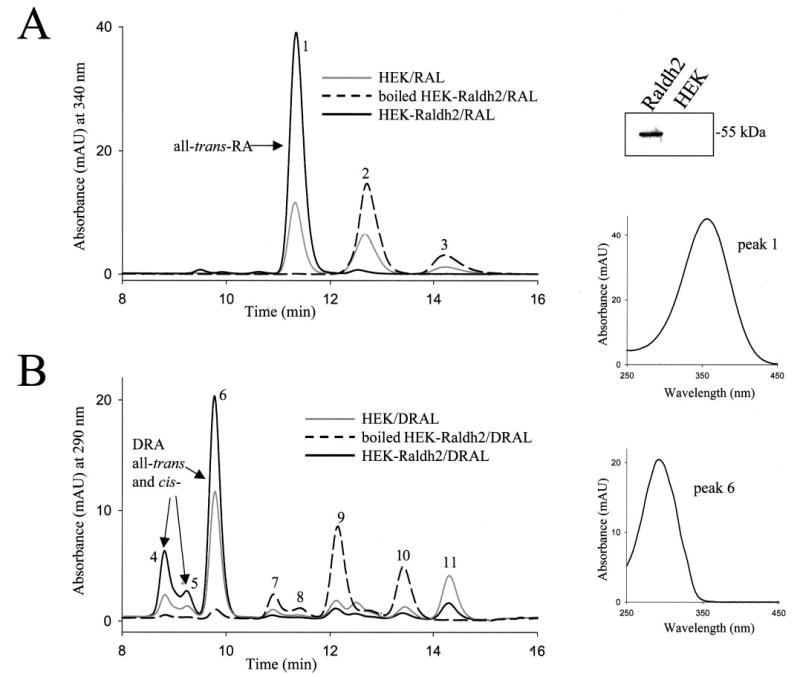
Cells were transiently transfected with vector carrying the cDNA of RALDH2 fused at its C terminus to a V5-His6 tag. The expression of RALDH2-V5-His6-tagged protein was confirmed by immunoblotting with anti-V5 monoclonal antibody and is shown in the top panel on the right in the lane labeled Raldh2. Cell homogenates of transfected HEK-RALDH2 cells (black solid line graph) or untransfected control cells (gray solid line graph) were incubated with all-trans-RAL (A) or all-trans-DRAL (B). Boiled control cells (black dashed line graph) were incubated with substrates under the same conditions. Retinoids were extracted and analyzed by reverse phase HPLC as described under “Methods and Materials.” The products of the reaction were identified based on their absorbance spectra and coelution with available standards. These are as follows: peak 1, all-trans-RA; peaks 2 and 3, syn- and anti-RAL oxime, respectively; peaks 4 and 5, cis-isomers of DRA; peak 6, all-trans-DRA; peaks 7–10, syn- and anti-oximes of several isomers of DRAL; peak 11, all-trans-DROL. The UV-visible absorbance spectra of peak 1 (identified as RA) and peak 6 (identified as DRA) are shown in middle and bottom panels on the right, respectively. The experiment was performed in duplicate and repeated three times. Similar results were obtained with cells transfected with RALDH3 tagged at the C terminus with V5-His6 tag.
Oxidation of All-trans-DRA
The level of RA is tightly controlled by both spatially and temporally regulated synthesis and degradation. RA catabolism is carried out by cytochrome P450 enzymes CYP26A1, -B1, and -C1. It is important to determine whether DRA could also be catabolized in a similar manner. HEK-293S cells were transfected with expression constructs of CYP26A1, -B1, and -C1 fused at their C termini with a V5 epitope and His6 stretch. Transfected and untransfected cells were incubated with all-trans-RA or all-trans-DRA substrate in culture because CYP26A1, -B1, and -C1 activity was adversely affected by homogenization of cells. Oxidized metabolites of all-trans-RA and all-trans-DRA were present in CYP26A1-transfected cells but not in un-transfected cells (Fig. 4, A and B). These metabolites, which could include all-trans-4-oxo-(D)RA, all-trans-4-hydroxy-(D)RA, all-trans-5,8-epoxy-(D)RA, and all-trans-18-hydroxy-(D)RA, were identified as polar compounds eluting shortly after the injection spike (Fig. 4, A and B, peaks 1 and 2 and peaks 7–9, and inset spectra). One of the oxidized all-trans-DRA compounds was identified as all-trans-4-oxo-DRA because it matched the elution profile and absorbance spectrum of a synthetic standard (Fig. 4, lower right, inset panel). The level of tagged enzyme expressed in transfected cells was assayed by SDS-PAGE analysis of transfected cell lysates, followed by immunoblotting using an anti-V5-monoclonal antibody (Fig. 4, top right panel). The level of expression of CYP26A1, -B1, and -C1 in transfected cells was similar, and all three enzymes efficiently carried out the oxidation of all-trans-RA and all-trans-DRA to polar metabolites (not shown).
Fig. 4. Oxidation of all-trans-RA and all-trans-DRA.
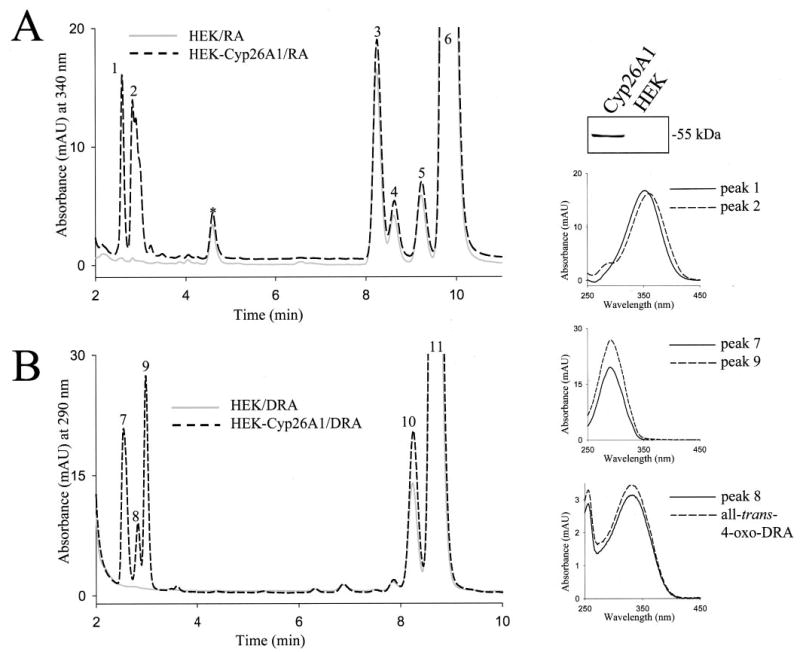
The metabolism of RA and DRA was examined in untransfected cells or cells transfected with CYP26A1. The expression of CYP26A1-V5-His6-tagged protein in transfected cells was examined by SDS-PAGE and immunoblotting with anti-V5 monoclonal antibody and is shown in the top panel on the right in the lane labeled Cyp26A1. Transfected HEK-CYP26A1 cells (black dashed line graph) or untransfected control cells (gray solid line graph) were incubated with RA (A) or DRA (B). Retinoids were extracted and analyzed by reverse phase HPLC as described under “Materials and Methods.” The spectra of oxidized RA metabolites, peaks 1 and 2 (right top inset panel), resemble those of all-trans-4-hydroxy-RA and all-trans-4-oxo-RA, respectively. Peaks 3–5 are cis-isomers of RA; peak 6 is all-trans-RA. Peaks 7 and 9 represent oxidized DRA metabolites and have a λmax = 290 nm shown in the middle inset panel on the right. Peak 8 corresponds to all-trans-4-oxo-DRA based on its absorbance spectra and elution time (absorbance spectra shown in lower inset panel on the right). Peaks 10 and 11 represent cis- and all-trans-DRA, respectively. The experiment was performed in duplicate and repeated three times. Similar results were obtained with cells transfected with CYP26B1 and -C1.
Conversion of All-trans-DROL to All-trans-DRA in RPE
Retinoid metabolism occurs in many embryonic and adult tissues. Thus, it is important to determine whether the entire pathway of synthesis of all-trans-DRA can be reconstituted with tissue extracts. All-trans-DROL (supplemental Fig. 10, peak 2) was efficiently converted to all-trans-DRA (supplemental Fig. 10, peak 1) by microsomes prepared from RPE cells in the presence of dinucleotide cofactors NAD and NADP. All-trans-DRA was identified based on its elution profile and absorbance spectrum in comparison with synthetic all-trans-DRA (supplemental Fig. 10 and inset spectra). RPE microsomes also catalyzed the conversion of all-trans-ROL into all-trans-RA (results not shown), which indicates that adult RPE could be an active all-trans-RA, all-trans-DRA synthesis site. The main ROL oxidizing activity in the RPE is catalyzed by SDR family enzymes. The efficient conversion of all-trans-DROL to all-trans-DRA in the RPE supports the existence of SDR enzymes that can convert all-trans-DROL into all-trans-DRAL. Further studies are required to examine the substrate specificity of the known SDR enzymes from the RPE with respect to all-trans-DROL.
Based on the known all-trans-ROL oxidation pathway and results presented here, we propose that following saturation of all-trans-ROL to all-trans-DROL, all-trans-DROL is oxidized to all-trans-DRA and later to all-trans-4-oxo-DRA and possibly other oxidized metabolites of all-trans-DRA. We showed that the same enzymes involved in the oxidation of ROL to RA are also involved in the oxidation of DROL to DRA as depicted in Scheme 1. All-trans-DROL and other more oxidized metabolites occur naturally and represent a novel and potentially important pathway in the metabolism of vitamin A. This hypothesis is supported by the unequivocal identification of all-trans-DROL and all-trans-DRA in Lrat−/− mice gavaged with all-trans-ROL palmitate.
Characterization of the Transactivation Activity of All-trans-DRA
All-trans-RA binding to RAR and 9-cis-RA binding to RAR or RXR can control the expression of genes containing RA-response element (RARE) sequences within their promoter region. RARE elements are composed of direct repeats (DR) of the canonical sequence PuG(G/T)TCA separated by one to five nucleotides. Activated RAR/RXR heterodimers can associate with RARE composed of DR separated by five nucleotides (DR5), which are found in the promoter region of many genes including the RARβ gene (33).
We studied whether DRA could also control gene expression through RAR activation by using a DR5 RARE-reporter cell line. The F9 teratocarcinoma cell line expresses endogenous RAR and RXR and is exquisitely sensitive to the effects of RA. This cell line has been transfected with lacZ under the control of a minimal promoter and upstream DR5 elements (29). F9-RARE-lacZ cells were treated with different doses of all-trans-RA or all-trans-DRA for 24 h, after which the cells were harvested, and the β-galactosidase activity was evaluated by X-gal staining (Fig. 5, top panels). All-trans-DRA transactivation of DR5-induced β-galactosidase expression was observed at higher concentrations than the equivalent effect produced by RA. All-trans-RA and all-trans-DRA induction activity was quantified by using the soluble substrate o-nitrophenyl β-d-galactopyranoside. The colorless substrate was cleaved by β-galactosidase to yellow colored o-nitrophenol, whose absorbance was measured at 420 nm using a spectrophotometer (Fig. 5). All-trans-DRA induction of DR5 elements is much less efficient than that of all-trans-RA. Induction of DR5 reporter cells with 10−9 M all-trans-RA had a magnitude similar to the one obtained with 10−7 M all-trans-DRA. The response measured in the linear part of the dose-response curve showed that all-trans-DRA is about 100-fold less effective than all-trans-RA in activating DR5-response elements.
Fig. 5. Response of F9-RARE-lacZ reporter cell line to RA and DRA.
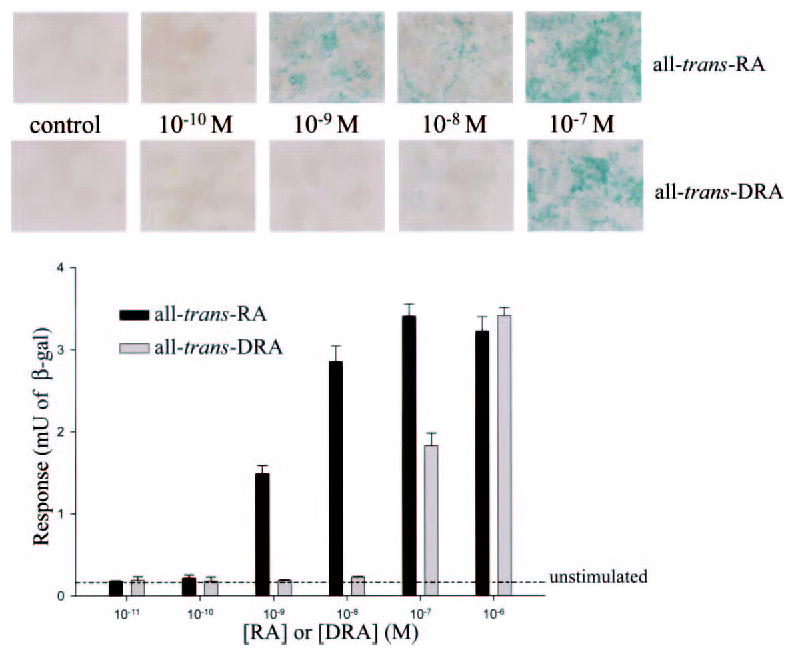
F9-RARE-lacZ cells express endogenous RAR and RXR and were transfected with a construct of lacZ under the control of a minimal promoter and upstream DR5 elements (29). F9-RARE-lacZ cells were treated with different doses of all-trans-RA or all-trans-DRA for 24 h. The RARE-driven lacZ gene produces β-galactosidase, which hydrolyzes X-gal to an insoluble blue product, which was visualized in responder cells by light microscopy (top panels). Alternatively, the response of the cell population was quantified by measuring the β-galactosidase activity using the substrate o-nitrophenyl β-d-galactopyranoside. The colorless substrate was hydrolyzed by β-galactosidase to soluble, yellow-colored o-nitrophenol, whose absorbance was measured at 420 nm using a spectrophotometer (bottom, bar graph). The background β-galactosidase activity in unstimulated cells is indicated by dashed line. The experiment was repeated twice with similar results.
RXR homodimers can be activated by 9-cis-RA (34), phytanic acid (35), docosahexanoic acid (36), and other unsaturated fatty acids (37). RXR homodimers can bind DR1 elements of hexameric motifs separated by a single base pair as found in the CRBP II promoter (38). We studied activation of RXR based on a DR1-reporter cell assay using HEK-293S cells with a construct of lacZ under the control of a minimal promoter and five consecutive upstream DR1 elements, which we termed pRXRE-BLUE. Because HEK-293S cells express little endogenous RXR, there is no induction of DR1 elements by 9-cis-RA in the absence of exogenous RXR (Fig. 6, bottom graph). Thus, we cloned mouse RXRα and expressed it under the control of the CMV promoter in HEK-293S cells, which we cotransfected with pRXRE-BLUE. In our assay 9-cis-RA activates RXR-mediated transcription, whereas all-trans-DRA was a very weak RXR activator (Fig. 6, top graph). Even though all-trans-RA does not bind RXR, we found that addition of all-trans-RA also resulted in robust induction of RXR homodimers in comparison with all-trans-DRA. This result could be a consequence of all-trans-RA isomerization to 9-cis-RA during the overnight incubation.
Fig. 6. Activation of DR1 elements by all-trans-DRA, all-trans-RA, and 9-cis-RA.
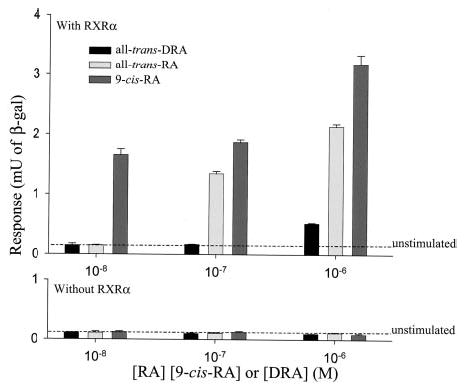
HEK-293S cells were transfected with a construct of lacZ under the control of a minimal promoter and five consecutive upstream DR1 elements. Top, HEK-293S cells were cotransfected with both DR1-reporter construct and mouse RXRα under the control of the CMV promoter. The cells were then treated with the indicated levels of all-trans-RA, 9-cis-RA, or all-trans-DRA for 48 h. The cells were harvested, and β-galactosidase activity was assayed as described under “Materials and Methods.” Bottom, DR1-reporter transfected cells were treated with different doses of all-trans-RA, 9-cis-RA, or all-trans-DRA in the absence of RXR for 48 h. The background β-galactosidase activity in unstimulated cells is indicated by the dashed line in both upper and lower graphs. The cells were harvested, and β-galactosidase activity was assayed as described under “Materials and Methods.” The experiment was repeated twice with similar results.
DISCUSSION
In this study we identify all-trans-DRA and other 13,14-dihydroretinoid metabolites in the tissues of Lrat−/− mice supplemented with ROL palmitate, and we demonstrate that all-trans-DRA can control gene expression in reporter cell assays. All-trans-DRA stimulated expression of a DR5-RARE reporter gene by activating RAR/RXR heterodimers in F9-RARE-lacZ cells. All-trans-DRA did not activate RXR homodimers in HEK-293S cells cotransfected with a DR1-lacZ reporter construct and mouse RXRα. In combination with a previous report on the identification of all-trans-DROL as the product of RetSat (23), this study characterized the enzymatic pathway responsible for the formation of all-trans-DRA from all-trans-ROL. Saturation of the C13–14 bond of all-trans-ROL by RetSat produces all-trans-DROL, which is oxidized to the corresponding retinaldehyde, all-trans-DRAL, by ADH-1 and possibly by SDR family RDHs present in the RPE. All-trans-DRAL is oxidized to all-trans-DRA by RALDH1–4. All-trans-DRA can be oxidized to all-trans-4-oxo-DRA in mice gavaged with all-trans-DROL and in vitro by cytochrome P450 enzymes CYP26A1, -B1, and -C1, suggesting a possible pathway for its degradation (Scheme 1). All the substrates and products of reactions and metabolites isolated from mouse tissues were identified by comparing their UV-visible absorbance spectra and chromatographic profile with authentic synthetic standards characterized by NMR and mass spectrometry. Contrary to a previous report indicating the conversion of 9-cis-RA to 9-cis-DRA (24), we found no evidence of in vivo conversion of all-trans-RA into all-trans-DRA. Thus, all-trans-DRA can only be derived from oxidation of all-trans-DROL, and RetSat is the sole known enzyme responsible for catalyzing the key step in all-trans-DRA formation. These findings indicate that saturation of all-trans-ROL by RetSat is an active and possibly important step in the metabolism of retinoids in vivo.
Synthesis and Degradation of All-trans-DRA
Many of the ADH and SDR families and some RALDHs are expressed in the retina and RPE (17, 39–43). We demonstrate in the current study that a pathway of conversion of all-trans-DROL into all-trans-DRA exists and is efficient in RPE microsomes (supplemental Fig. 10). This implies that all-trans-DRA synthesis can occur in the same tissues where all-trans-RA synthesis occurs and that all-trans-DRA could have a concentration gradient in different tissues. This gradient will be determined by the availability of synthetic and catabolic enzymes as well as the availability of primary substrate, i.e. all-trans-DROL.
RA bioavailability is tightly regulated by the balance between its biosynthesis and catabolism (44). The cytochrome P450-type enzymes, which include ubiquitously expressed CYP26A1, -B1, and -C1 (19, 20, 22, 45), oxidize RA to 4-OH-RA, 4-oxo-RA, 18-OH-RA, and 5,8-epoxy-RA. Thus, CYP26 enzymes are involved in limiting spatial and temporal levels of RA, and in concert with ADH, SDR, and RALDH they guard a desirable level of RA, protecting against fluctuations in the nutritional levels of ROL. As shown here, CYP26A1, -B1, and -C1 enzymes also metabolize all-trans-DRA. This could also contribute to a temporal and spatial gradient of DRA in vivo.
Identification of Chain-shortened ROL Metabolites
In this study we report the identification of an ROL metabolite that contains an alcohol functional group and is saturated at the C13–14 bond and chain-shortened at C-15. Chain-shortened ROL metabolites have been described in early studies that followed the fate of radioactive 14C-labeled RA or all-trans-ROL (46–48). One possible pathway for their synthesis could be through α-oxidation of all-trans-DRA as suggested previously by others (24). The C19-ROL metabolite could be the product of a reduced C19-aldehyde intermediate produced during the α-oxidation of all-trans-DRA (equivalent to the pristanal intermediate of the phytanic acid degradation pathway). Only low amounts of C19-ROL were observed in Lrat−/− mice supplemented with all-trans-DROL compared with the levels obtained in mice gavaged with all-trans-ROL palmitate. This discrepancy might be accounted for by the fact that endogenous all-trans-DROL has access to a different repertoire of enzymes than does all-trans-DROL administered by gavage. The definite pathway of synthesis of the C19-ROL could be established by using knock-out animal models deficient in specific enzymes of this pathway.
Potential Role of 13,14-Dihydroretinoids in Vertebrate Physiology
Based on experiments using mice deficient in specific enzymes involved in retinoid metabolism, it was shown that ADH1 and RALDH1 are involved in a protection mechanism in response to pharmacological doses of ROL. Adh1−/− and Raldh1−/− mice were much more sensitive to ROL-induced toxicity than their wild type counterparts (49, 50). It was proposed that conversion of ROL to RA protects against excess levels of dietary ROL. This idea is counterintuitive considering the well known toxic effects of RA. Here we show that ADH1 and RALDH1 are also involved in DROL oxidation to DRA and that all-trans-DRA is a much weaker activator of RAR- or RXR-mediated transcription compared with all-trans or 9-cis-RA. Thus, it is possible that saturation of the C13–14 bond of all-trans-ROL could be the first step in a degradation pathway, which provides protection against pharmacological doses of all-trans-ROL and circumvents the formation of RA. Our findings show that the combined amounts of hepatic DROL and DROL metabolites amount to less than one-third of the amount of hepatic all-trans-RA at 3 h post-gavage with 106 IU ROL palmitate/kg body weight. This would suggest that saturation by RetSat is a rate-limiting reaction in the metabolic pathway.
Another possibility is that RetSat activity leads to production of novel bioactive 13,14-dihydroretinoids. We identify all-trans-DRA as an activator of RAR/RXR heterodimer-mediated transcription. The tissue concentration and transactivation profile of all-trans-DRA are both lower than those of all-trans-RA. It is possible that all-trans-DRA and other DROL metabolites could have important transactivation activity in certain physiological circumstances. The local concentration of 13,14-dihydroretinoid ligand might reach higher levels as a result of being trapped by receptors or binding proteins. Given that the local concentration and binding affinity are sufficient, all-trans-DRA could be an important endogenous ligand for RAR or possibly for other nuclear receptors. The finding that the same enzymes that were thought to act specifically in the formation of RA are also responsible for the formation of DRA has to be considered in attempts to rescue with RA the phenotype of knockout animal models deficient in these enzymes. In one such example, Raldh2−/− mouse embryos cannot be completely rescued by maternal RA supplementation and die prenatally (51). It is interesting to speculate if other retinoid metabolites, including 13,14-dihydroretinoids, in addition to RA may be necessary for a complete rescue of Raldh2−/− embryos. The identification of the all-trans-DRA metabolic pathway is the first step in this process, and more studies are necessary to establish the physiological role of DRA and other DROL metabolites in controlling gene expression.
In summary, we describe a new metabolic pathway for vitamin A that leads to a new class of endogenous bioactive retinoids. We demonstrate that all-trans-ROL saturation to all-trans-DROL followed by oxidation to all-trans-DRA occurs in vivo. All-trans-DRA can activate transcription of reporter genes by binding RAR but does not bind RXR. The oxidative pathway of all-trans-DROL employs the same enzymes as that of all-trans-ROL. We expect that these previously unknown metabolites will help us better understand the vital functions of retinoids in vertebrate physiology.
Acknowledgments
We are grateful for the F9-RARE-lacZ cells obtained from Dr. Peter McCaffery with permission from Dr. Michael Wagner. We thank Dr. Tadao Maeda for help with oral gavage of animals; Dr. Yoshikazu Imanishi for the Lrat−/− mice and help with light microscopy; Matthew Batten for providing and genotyping Lrat−/− mice; Dr. Michael Gelb for providing facilities for the chemical synthesis; and Dr. Marcin Golczak for insightful comments. We also thank Rebecca Birdsong for proofreading the manuscript. The Department of Ophthalmology, University of Utah, is recipient of a grant from Research to Prevent Blindness, Inc. The University of Utah is the recipient of a Center grant from the Foundation Fighting Blindness.
Footnotes
This work was supported by National Institutes of Health Grants EY08061 and EY08123, a grant from the Macular Vision Research Foundation, and a grant from the E. K. Bishop Foundation.
The on-line version of this article (available at http://www.jbc.org) contains Table II and Figs. 7–10.
The abbreviations used are: ROL, retinol; ROL palmitate, retinyl palmitate; ADH, medium-chain alcohol dehydrogenases; 9-cis-DRA, 9-cis-13,14-dihydroretinoic acid; C19-ROL, (3E,5E,7E)-2,6-dimethyl-8-(2,6,6-trimethylcyclohex-1-enyl)octa-3,5,7-trien-1-ol; DRAL, 13,14-di-hydroretinaldehyde; DROL, 13,14-dihydroretinol; LRAT, lecithin:retinol acyltransferase; RA, retinoic acid; RAL, retinaldehyde; RALDH, RAL dehydrogenase; RAR, retinoic acid receptor; RetSat, all-trans-ROL:all-trans-DROL saturase; RXR, retinoid X receptor; SDR, short-chain dehydrogenase/reductase; RARE, RAR element; RXRE, RXR element; CMV, cytomegalovirus; HPLC, high pressure liquid chromatography; MGC, Mammalian Gene Collection; DR, direct repeats; X-gal, 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside.
A. R. Moise and K. Palczewski, unpublished observations.
References
- 1.Wald G. Science. 1968;162:230–239. doi: 10.1126/science.162.3850.230. [DOI] [PubMed] [Google Scholar]
- 2.Chambon P. FASEB J. 1996;10:940–954. [PubMed] [Google Scholar]
- 3.Buck J, Derguini F, Levi E, Nakanishi K, Hammerling U. Science. 1991;254:1654–1656. doi: 10.1126/science.1749937. [DOI] [PubMed] [Google Scholar]
- 4.Buck J, Grun F, Derguini F, Chen Y, Kimura S, Noy N, Hammerling U. J Exp Med. 1993;178:675–680. doi: 10.1084/jem.178.2.675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Derguini F, Nakanishi K, Hammerling U, Chua R, Eppinger T, Levi E, Buck J. J Biol Chem. 1995;270:18875–18880. doi: 10.1074/jbc.270.32.18875. [DOI] [PubMed] [Google Scholar]
- 6.Achkar CC, Derguini F, Blumberg B, Langston A, Levin AA, Speck J, Evans RM, Bolado J, Jr, Nakanishi K, Buck J, Gudas LJ. Proc Natl Acad Sci U S A. 1996;93:4879–4884. doi: 10.1073/pnas.93.10.4879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blumberg B, Bolado J, Jr, Derguini F, Craig AG, Moreno TA, Chakravarti D, Heyman RA, Buck J, Evans RM. Proc Natl Acad Sci U S A. 1996;93:4873–4878. doi: 10.1073/pnas.93.10.4873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pijnappel WW, Folkers GE, de Jonge WJ, Verdegem PJ, de Laat SW, Lugtenburg J, Hendriks HF, van der Saag PT, Durston AJ. Proc Natl Acad Sci U S A. 1998;95:15424–15429. doi: 10.1073/pnas.95.26.15424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Molotkov A, Deltour L, Foglio MH, Cuenca AE, Duester G. J Biol Chem. 2002;277:13804–13811. doi: 10.1074/jbc.M112039200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duester G. Eur J Biochem. 2000;267:4315–4324. doi: 10.1046/j.1432-1327.2000.01497.x. [DOI] [PubMed] [Google Scholar]
- 11.Haeseleer F, Jang GF, Imanishi Y, Driessen CA, Matsumura M, Nelson PS, Palczewski K. J Biol Chem. 2002;277:45537–45546. doi: 10.1074/jbc.M208882200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duester G, Mic FA, Molotkov A. Chem Biol Interact. 2003;143:201–210. doi: 10.1016/s0009-2797(02)00204-1. [DOI] [PubMed] [Google Scholar]
- 13.Bhat PV, Labrecque J, Boutin JM, Lacroix A, Yoshida A. Gene (Amst) 1995;166:303–306. doi: 10.1016/0378-1119(96)81752-5. [DOI] [PubMed] [Google Scholar]
- 14.Penzes P, Wang X, Sperkova Z, Napoli JL. Gene (Amst) 1997;191:167–172. doi: 10.1016/s0378-1119(97)00054-1. [DOI] [PubMed] [Google Scholar]
- 15.Wang X, Penzes P, Napoli JL. J Biol Chem. 1996;271:16288–16293. doi: 10.1074/jbc.271.27.16288. [DOI] [PubMed] [Google Scholar]
- 16.Zhao D, McCaffery P, Ivins KJ, Neve RL, Hogan P, Chin WW, Drager UC. Eur J Biochem. 1996;240:15–22. doi: 10.1111/j.1432-1033.1996.0015h.x. [DOI] [PubMed] [Google Scholar]
- 17.Mic FA, Molotkov A, Fan X, Cuenca AE, Duester G. Mech Dev. 2000;97:227–230. doi: 10.1016/s0925-4773(00)00434-2. [DOI] [PubMed] [Google Scholar]
- 18.Lin M, Zhang M, Abraham M, Smith SM, Napoli JL. J Biol Chem. 2003;278:9856–9861. doi: 10.1074/jbc.M211417200. [DOI] [PubMed] [Google Scholar]
- 19.Fujii H, Sato T, Kaneko S, Gotoh O, Fujii-Kuriyama Y, Osawa K, Kato S, Hamada H. EMBO J. 1997;16:4163–4173. doi: 10.1093/emboj/16.14.4163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.White JA, Beckett-Jones B, Guo YD, Dilworth FJ, Bonasoro J, Jones G, Petkovich M. J Biol Chem. 1997;272:18538–18541. doi: 10.1074/jbc.272.30.18538. [DOI] [PubMed] [Google Scholar]
- 21.White JA, Ramshaw H, Taimi M, Stangle W, Zhang A, Everingham S, Creighton S, Tam SP, Jones G, Petkovich M. Proc Natl Acad Sci U S A. 2000;97:6403–6408. doi: 10.1073/pnas.120161397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Taimi M, Helvig C, Wisniewski J, Ramshaw H, White J, Amad M, Korczak B, Petkovich M. J Biol Chem. 2004;279:77–85. doi: 10.1074/jbc.M308337200. [DOI] [PubMed] [Google Scholar]
- 23.Moise AR, Kuksa V, Imanishi Y, Palczewski K. J Biol Chem. 2004;279:50230–50242. doi: 10.1074/jbc.M409130200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shirley MA, Bennani YL, Boehm MF, Breau AP, Pathirana C, Ulm EH. Drug Metab Dispos. 1996;24:293–302. [PubMed] [Google Scholar]
- 25.Schmidt CK, Brouwer A, Nau H. Anal Biochem. 2003;315:36–48. doi: 10.1016/s0003-2697(02)00662-0. [DOI] [PubMed] [Google Scholar]
- 26.Batten ML, Imanishi Y, Maeda T, Tu DC, Moise AR, Bronson D, Possin D, Van Gelder RN, Baehr W, Palczewski K. J Biol Chem. 2004;279:10422–10432. doi: 10.1074/jbc.M312410200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reeves PJ, Callewaert N, Contreras R, Khorana HG. Proc Natl Acad Sci U S A. 2002;99:13419–13424. doi: 10.1073/pnas.212519299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stecher H, Gelb MH, Saari JC, Palczewski K. J Biol Chem. 1999;274:8577–8585. doi: 10.1074/jbc.274.13.8577. [DOI] [PubMed] [Google Scholar]
- 29.Wagner M, Han B, Jessell TM. Development (Camb) 1992;116:55–66. doi: 10.1242/dev.116.1.55. [DOI] [PubMed] [Google Scholar]
- 30.Ruiz A, Winston A, Lim YH, Gilbert BA, Rando RR, Bok D. J Biol Chem. 1999;274:3834–3841. doi: 10.1074/jbc.274.6.3834. [DOI] [PubMed] [Google Scholar]
- 31.Gagnon I, Duester G, Bhat PV. Biochem Pharmacol. 2003;65:1685–1690. doi: 10.1016/s0006-2952(03)00150-3. [DOI] [PubMed] [Google Scholar]
- 32.Gagnon I, Duester G, Bhat PV. Biochim Biophys Acta. 2002;1596:156–162. doi: 10.1016/s0167-4838(02)00213-3. [DOI] [PubMed] [Google Scholar]
- 33.Sucov HM, Murakami KK, Evans RM. Proc Natl Acad Sci U S A. 1990;87:5392–5396. doi: 10.1073/pnas.87.14.5392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heyman RA, Mangelsdorf DJ, Dyck JA, Stein RB, Eichele G, Evans RM, Thaller C. Cell. 1992;68:397–406. doi: 10.1016/0092-8674(92)90479-v. [DOI] [PubMed] [Google Scholar]
- 35.Lemotte PK, Keidel S, Apfel CM. Eur J Biochem. 1996;236:328–333. doi: 10.1111/j.1432-1033.1996.00328.x. [DOI] [PubMed] [Google Scholar]
- 36.de Urquiza AM, Liu S, Sjoberg M, Zetterstrom RH, Griffiths W, Sjovall J, Perlmann T. Science. 2000;290:2140–2144. doi: 10.1126/science.290.5499.2140. [DOI] [PubMed] [Google Scholar]
- 37.Goldstein JT, Dobrzyn A, Clagett-Dame M, Pike JW, DeLuca HF. Arch Biochem Biophys. 2003;420:185–193. doi: 10.1016/j.abb.2003.09.034. [DOI] [PubMed] [Google Scholar]
- 38.Mangelsdorf DJ, Umesono K, Kliewer SA, Borgmeyer U, Ong ES, Evans RM. Cell. 1991;66:555–561. doi: 10.1016/0092-8674(81)90018-0. [DOI] [PubMed] [Google Scholar]
- 39.Haeseleer F, Palczewski K. Methods Enzymol. 2000;316:372–383. doi: 10.1016/s0076-6879(00)16736-9. [DOI] [PubMed] [Google Scholar]
- 40.Wagner E, McCaffery P, Drager UC. Dev Biol. 2000;222:460–470. doi: 10.1006/dbio.2000.9719. [DOI] [PubMed] [Google Scholar]
- 41.Mic FA, Molotkov A, Molotkova N, Duester G. Dev Dyn. 2004;231:270–277. doi: 10.1002/dvdy.20128. [DOI] [PubMed] [Google Scholar]
- 42.Fischer AJ, Wallman J, Mertz JR, Stell WK. J Neurocytol. 1999;28:597–609. doi: 10.1023/a:1007071406746. [DOI] [PubMed] [Google Scholar]
- 43.Mey J, McCaffery P, Klemeit M. Res Dev Brain Res. 2001;127:135–148. doi: 10.1016/s0165-3806(01)00127-4. [DOI] [PubMed] [Google Scholar]
- 44.Niederreither K, Abu-Abed S, Schuhbaur B, Petkovich M, Chambon P, Dolle P. Nat Genet. 2002;31:84–88. doi: 10.1038/ng876. [DOI] [PubMed] [Google Scholar]
- 45.MacLean G, Abu-Abed S, Dolle P, Tahayato A, Chambon P, Petkovich M. Mech Dev. 2001;107:195–201. doi: 10.1016/s0925-4773(01)00463-4. [DOI] [PubMed] [Google Scholar]
- 46.Wolf G, Kahn SG, Johnson CB. J Am Chem Soc. 1957;79:1208–1212. [Google Scholar]
- 47.Roberts AB, DeLuca HF. J Lipid Res. 1968;9:501–508. [PubMed] [Google Scholar]
- 48.Yagishita K, Sundaresan PR, Wolf G. Nature. 1964;203:411–412. doi: 10.1038/203410b0. [DOI] [PubMed] [Google Scholar]
- 49.Molotkov A, Duester G. J Biol Chem. 2003;278:36085–36090. doi: 10.1074/jbc.M303709200. [DOI] [PubMed] [Google Scholar]
- 50.Molotkov A, Ghyselinck NB, Chambon P, Duester G. Biochem J. 2004;383:295–302. doi: 10.1042/BJ20040621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Niederreither K, Vermot J, Le Roux I, Schuhbaur B, Chambon P, Dolle P. Development (Camb) 2003;130:2525–2534. doi: 10.1242/dev.00463. [DOI] [PubMed] [Google Scholar]