

Abstract
Recently, we have shown that the microtubule-associated protein tau is essential for β-amyloid (Aβ)-induced neurotoxicity in hippocampal neurons. However, the mechanisms by which tau mediates Aβ-induced neurite degeneration remain poorly understood. In the present study, we analyzed whether tau cleavage played a role in these events. Our results showed that pre-aggregated Aβ induced the generation of a 17 kDa tau fragment in cultured hippocampal neurons. The generation of this fragment was preceded by the activation of calpain-1. Conversely, inhibitors of this protease, but not of caspases, completely prevented tau proteolysis leading to the generation of the 17 kDa fragment and significantly reduced Aβ-induced neuronal death. Furthermore, the expression of this fragment in cultured hippocampal neurons induced the formation of numerous varicosity-bearing tortuous processes, as well as the complete degeneration of some of those neurite processes. These results suggest that Aβ-induced neurotoxicity may be mediated, at least in part, through the calpain-mediated generation of a toxic 17 kDa tau fragment. Collectively, these results provide insight into a novel mechanism by which tau could mediate Aβ-induced neurotoxicity.
Keywords: Alzheimer, apoptosis, degeneration, hippocampus, neurotoxicity, proteolysis, amyloid
Introduction
Alzheimer's disease (AD), the most common cause of dementia in people 65 years and older, is a neurodegenerative disorder characterized by the presence of senile plaques and neurofibrillary tangles (NFTs) in the affected brain areas. Senile plaques are extracellular deposits of β-amyloid (Aβ), a proteolytic fragment originated in the amyloid precursor protein (Glenner and Wong, 1984; Yankner and Mesulam, 1991; Selkoe, 1994). NFTs, in contrast, are intracellular aggregates of hyperphosphorylated tau that accumulate both in the perikarya and along axonal and dendritic processes (Kosik et al., 1986; Wood et al., 1986; Kondo et al., 1988). These lesions are both considered pathological hallmarks of AD. However, the relationship between them has not been elucidated completely. Evidence of a direct link between these lesions has been obtained recently (Rapoport et al., 2002). This study showed that neurons expressing either mouse or human tau degenerated in the presence of pre-aggregated Aβ, whereas no signs of degeneration were detected in Aβ-treated tau-depleted neurons (Rapoport et al., 2002). Although these results support a key role for tau in Aβ-induced neurodegeneration, the mechanism by which this microtubule-associated protein mediates Aβ neurotoxicity remains poorly understood. A potential link between senile plaques and NFTs involves the Aβ-induced activation of putative tau kinases leading to tau phosphorylation. Indeed, tau hyperphosphorylation has been attributed to the increased activity of kinases, such as cyclin-dependent kinase 5 and glycogen synthase kinase-3β or the mitogen-activated protein kinase, in either young or mature hippocampal neurons treated with pre-aggregated Aβ, respectively (Takashima et al., 1993; Ferreira et al., 1997; Alvarez et al., 1999; Ekinci et al., 1999). More recently, the potential role for the proteolytic tau cleavage in Aβ-induced neuronal degeneration has been investigated. These studies showed that Glu391 truncated tau was present in AD brains but not in age-matched controls (Novak et al., 1993). In addition, the cleavage of tau at Asp421 by caspase-3 has been detected both in neurons treated with pre-aggregated Aβ and in AD brains (Chung et al., 2001; Gamblin et al., 2003).
In the present study, we performed a series of experiments to further elucidate the role of proteolytic tau cleavage in the mechanisms underlying Aβ-induced neurotoxicity. The results described here provided evidence suggesting that pre-aggregated Aβ induced the generation of a 17 kDa tau fragment through the activation of calpain-1 in hippocampal neurons. This proteolytic process was completely blocked by calpain inhibitors [N-acetyl-Leu-Leu-Nle-CHO (ALLN) and carbobenzoxy-valinyl-phenylalaninal (MDL 28,170)] but not by caspase inhibitors (DEVD and VAD). In addition, calpain inhibitors significantly reduced Aβ-induced neuronal death. Conversely, the expression of this tau fragment led to neurite degeneration and cell death in neurons and in non-neuronal cell types. These results suggest that Aβ-induced neurotoxicity may be mediated, at least in part, through the activation of proteases, leading to the generation of the 17 kDa neurotoxic tau fragment in hippocampal neurons.
Materials and Methods
Preparation of hippocampal cultures. Embryonic day 18 rat embryos were used to prepare hippocampal cultures as described previously (Goslin and Banker, 1990). Briefly, hippocampi were dissected and freed of meninges. The cells were dissociated by trypsinization (0.25% for 15 min at 37°C), followed by trituration with a fire-polished Pasteur pipette. The cell suspension was then plated on poly-l-lysine-coated coverslips in MEM with 10% horse serum. After 4 h, the coverslips were transferred to dishes containing an astroglial monolayer and maintained in MEM containing N2 supplements (Bottenstein and Sato, 1979) plus ovalbumin (0.1%) and sodium pyruvate (0.1 mm). For biochemical experiments, hippocampal neurons were plated at high density (500,000 cells/60 mm dish) in MEM with 10% horse serum. After 4 h, the medium was replaced with glia-conditioned MEM containing N2 supplements (Bottenstein and Sato, 1979) plus ovalbumin (0.1%) and sodium pyruvate (0.1 mm).
Aβ aggregation and treatment. Synthetic Aβ(1-40) (Sigma, St. Louis, MO) was dissolved in N2 medium at 0.5 mg/ml and incubated for 4 d at 37°C to pre-aggregate the peptide (Ferreira et al., 1997). Pre-aggregated Aβ was added to the culture medium at a final concentration of 20 μm. For dose-response experiments, hippocampal neurons kept in culture for 21 d were incubated for 24 h with pre-aggregated Aβ at final concentrations ranging from 0.02 to 20 μm. For time course experiments, the neurons were grown in the presence of 20 μm pre-aggregated Aβ for 2, 4, 8, and 24 h.
Protein determination, electrophoresis, and immunoblotting. To prepare heat-stable fractions, cultures were washed twice and scraped in warmed PBS and immediately boiled for 5 min. After centrifugation, the supernatant was diluted 1:1 in Laemmli buffer. To prepare whole-cell extracts, cultures were rinsed twice in warmed PBS, scraped into Laemmli buffer, and homogenized in a boiling water bath for 10 min. The protein concentration was determined by the method of Lowry et al. (1951) as modified by Bensadoun and Weinstein (1976). SDS-polyacrylamide gels were run according to Laemmli (1970). Transfer of protein to Immobilon membrane (Millipore, Bedford, MA) and immunodetection were performed according to Towbin et al. (1979) as modified by Ferreira et al. (1989). The following antibodies were used: anti-α-tubulin (clone DM1A; 1:500,000; Sigma), anti-tau [clone tau-5 (LoPresti et al., 1995); 1:1000], anti-dephosphorylated tau (clone tau-1; 1:100,000; Roche Applied Science, Indianapolis, IN), anti-phosphorylated tau (clone AT8; 1:1000; Biosource International, Foster City, CA), anti-tau truncated at Asp421 (clone tau-C3; 1:1000; Chemicon, Temecula, CA), anti-90 kDa heat shock protein (Hsp90; clone 68; 1:1000; BD Biosciences, San Diego, CA), anti-caspase-3 (1:1000; Cell Signaling Technology, Beverly, MA), anti-cleaved caspase-3 (1:1000; Cell Signaling Technology), anti-calpain-1 (1:5000; Calbiochem, San Diego, CA), and anti-spectrin antibody (1:1000; Chemicon). Secondary antibodies conjugated to horseradish peroxidase (1:1000; Promega, Madison, WI) followed by enhanced chemiluminescence reagents (Amersham Biosciences, Piscataway, NJ) were used for the detection of proteins. Densitometry was performed by using a Bio-Rad (Hercules, CA) 700 flatbed scanner and Molecular Analyst software (Bio-Rad). Films and membranes were scanned at 600 dots per inch by using light transmittance, and pixel volume analysis was performed on the appropriate bands. Densitometric values were normalized using α-tubulin or Hsp90 as internal controls. Scanning of the Western blots demonstrated the curve to be linear in the range used for each antibody.
Caspase-3 activity assay. Caspase-3 activity was measured using the Fluorometric Caspase-3 Activity Assay kit (Calbiochem) according to the manufacturer's instructions. The fluorescence was measured after cleavage of the caspase-3 substrate (DEVD) labeled with a fluorescent molecule, 7-amino-4-trifluoromethyl coumarin (AFC), to AFC by caspase-3. Briefly, hippocampal neurons cultured for 21 d were treated with 20 μm pre-aggregated Aβ for up to 24 h. The neurons were harvested in extraction buffer and incubated on ice for 20 min. After centrifugation at 500 × g for 5 min, the supernatant was incubated with the caspase-3 substrate (DEVD-AFC) for 2 h at 37°C. The fluorescence was assessed using a fluorescent plate reader with a 400 nm excitation and a 505 nm emission. The protein concentration was determined by the method of Lowry et al. (1951) as modified by Bensadoun and Weinstein (1976).
Calpain activity assay. Cleavage of the fluorogenic calpain substrate (Suc-Leu-Tyr-AMC; Calbiochem) to its fluorescent product [amino-methylcoumarin (AMC)] was used to measure calpain activity as described previously (Boland and Campbell, 2003). Briefly, 21-d-in-culture hippocampal neurons treated in the presence or absence of the calpain inhibitor ALLN (50 μm; Santa Cruz Biotechnology, Santa Cruz, CA) or MDL 28,170 (10 μm; Calbiochem) for 1 h were incubated with pre-aggregated Aβ (20 μm) for up to 24 h. These neurons were then harvested in lysis buffer (25 mm HEPES, 5 mm MgCl2, 5 mm DTT, 5 mm EDTA, 2 mm PMSF, and 10 μg/ml pepstatin, pH 7.4), subjected to three freeze-thaw cycles, and centrifuged at 10,000 × g for 10 min at 4°C. The supernatant (90 μl) was incubated with 10 μl of the calpain substrate (500 μm) for 1 h at 30°C. Assay buffer (100 μl; 100 mm HEPES containing 10 mm DTT, pH 7.4) was added, and the fluorescence was assessed measuring excitation at 380 nm and emission at 480 nm using a fluorescence plate reader. The protein concentration was determined by the method of Lowry et al. (1951) as modified by Bensadoun and Weinstein (1976).
Protease inhibitor treatment. To inhibit caspase activation, hippocampal neurons kept in culture for 21 d were pretreated with 50 μm Ac-DEVD-CHO (Calbiochem) or 50 μm Ac-VAD-CHO (Calbiochem) for 1 h as described previously (Canu et al., 1998). Hippocampal neurons were then incubated with 20 μm pre-aggregated Aβ for 4 or 8 h.
To inhibit calpain activation, hippocampal neurons kept in culture for 21 d were pretreated with 50 μm ALLN (Santa Cruz Biotechnology) or 10 μm MDL 28,170 (Calbiochem) for 1 h as described previously (Canu et al., 1998; Boland and Campbell, 2003). Hippocampal neurons were then incubated with pre-aggregated Aβ (20 μm) for 8 or 24 h.
To assess whether calpain inhibitors could prevent Aβ-induced neurodegeneration, 21-d-in-culture hippocampal neurons were pretreated with calpain inhibitor ALLN (50 μm) or MDL 28,170 (10 μm) 1 h before the incubation with pre-aggregated Aβ (20 μm) for 8 or 24 h as described above. The cells were then fixed with 4% paraformaldehyde in PBS containing 0.12 m sucrose for 15 min and rinsed twice in PBS. They were permeabilized in 0.3% Triton X-100 in PBS for 5 min, rinsed twice in PBS, preincubated in 10% BSA in PBS for 1 h at room temperature, and exposed to the neuron-specific tubulin antibody (clone TUJ-1; 1:200) overnight at 4°C. The next day, the hippocampal neurons were rinsed twice in PBS, incubated with Alexa Fluor 568 anti-mouse IgG (Molecular Probes, Eugene, OR) for 1 h at 37°C, rinsed twice in PBS, and mounted on slide glasses. Apoptotic cell death was assessed using the In Situ Cell Death Detection kit, Fluorescein (Roche Applied Science) as described below. Apoptotic cells [terminal deoxynucleotidyl transferase (TdT)-mediated biotinylated UTP nick end labeling (TUNEL+) cells] were counted using a fluorescence microscope (Nikon, Melville, NY) and expressed as a percentage of total cells.
Construction of mutant tau and fragment tau45-230 expression vectors. The plasmid tau-pRC/CMV, which encodes the longest human tau isoform (tau1-441), was obtained from Dr. A. Caceres (Instituto Ferreyra, Cordoba, Argentina). This plasmid was used as a template to generate the mutant tau using the QuikChange Site-Directed Mutagenesis kit (Stratagene, Cedar Creek, TX). One of the known caspase-3 cleavage sites (Asp421) was mutated to glutamic acid, and two of the possible calpain cleavage sites (Leu43 and Val229) were mutated to alanines using the following oligonucleotide primers: 5′-ATCGACATGGTAGAGTCGCCCCAGCTC-3′ and 5′-GAGCTGGGGCGACTCTACCATGTCCAT-3′ (Asp421); 5′-CACGGACGCTGGGCGAAAGAATCTCCCCTG-3′ and 5′-CAGGGGAGATTCTTTCGCGCCAGCGTCCGTG-3′ (Leu43); 5′-AGGTGGCAGTGGCCCGTACTCCACC-3′ and 5′-GGTGGAGTACGGGCCACTGCCACCT-3′ (Val229). Mutations were verified by DNA sequencing.
The 17 kDa tau fragment (tau45-230) was prepared by PCR using full-length tau (tau-pRC/CMV) as a template. The following PCR primers containing the EcoRI (5′ primer) and BamHI (3′ primer) restriction sites that enabled insertion into pEGFP-N1 polylinker were used: 5′-GCCCGAATTCATGAAAGAATCTCCCCTGCAGACC-3′ (5′ primer) and 5′-TTACAGGATCCCGGACCACTGCCACCTTCTT-3′ (3′ primer).
PCR products were gel purified and cloned into pGEM-T Easy vector (Promega). The amplified vector with insert was digested with EcoRI and BamHI, gel purified, and subcloned into the living colors mammalian expression vector [green fluorescent protein (GFP)] and pcDNA3.1(-) (Invitrogen, Grand Island, NY). PCR was performed using the GeneAmp PCR System 2400 (PerkinElmer, Wellesley, MA). PCR conditions were as follows: 94°C for 30 s, 64°C for 30 s, and 72°C for 30 s for 30 cycles, followed by 5 min at 72°C. All amplified inserts were confirmed by DNA sequencing.
Cell culture and DNA transfection. Chinese hamster ovary (CHO) cells were grown in F-12 medium plus 10% fetal bovine serum supplemented with 2 mm glutamine, 100 international units/ml penicillin, and 100 μg/ml streptomycin. CHO cells were transfected with tau-pRC/CMV, D421E-tau-pRC/CMV, L43A-V229A-tau-pRC/CMV, or tau45-230-pcDNA3.1(-) using LipofectAMINE according to the manufacturer's instructions (Invitrogen). Briefly, cells were subcultured to a density of 2 × 105 cells/well in 35 mm dishes. The next day, 2 μg of DNA was incubated with 5 μl of LipofectAMINE at room temperature in the dark for 30 min to form the DNA-lipid complex. Cells were then incubated with DNA-lipid complex for 48 h.
Tau in vitro cleavage assay by caspase-3 and calpain-1. CHO cells were transfected with tau-pRC/CMV or D421E-tau-pRC/CMV. Forty-eight hours later, the cells were washed three times in PBS, scraped in lysis buffer (20 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1 mm dithiothreitol, 5 mm EDTA, 5 mm EGTA, and 1% Triton X-100), and incubated on ice for 1 h. Cell lysates were then centrifuged for 10 min at 16,000 × g, and the supernatants were stored at -20°C until use. The supernatants (10 μl) were then incubated in the presence or absence of recombinant caspase-3 (0.3 μg/μl) in caspase-3 buffer for 60 min at 37°C, as described previously (Brancolini et al., 1997; Estus et al., 1997). The reactions were terminated by adding 1 vol of 2× SDS sample buffer and boiled for 10 min. Samples were subjected to Western blot analysis using tau antibodies.
CHO cells were also transfected with tau-pRC/CMV, L43A-V229A-tau-pRC/CMV, or tau45-230-pcDNA3.1(-) for 48 h. After three washes in PBS, cells were lysed in lysis buffer for 1 h at 0°C. The cell lysates were cleared by centrifugation and stored at -20°C until use. Sixteen microliters of the cell lysates were incubated in the presence or absence of calpain-1 (0.25 U) for 1 h at 30°C. The digestion reaction was stopped by adding 1 vol of 2× SDS sample buffer and boiled for 10 min. Samples were subjected to Western blot analysis using a tau antibody.
Immunocytochemistry and TUNEL assay. Fourteen-day-in-culture hippocampal neurons were transfected with 17Tau-GFP (tau45-230-GFP), Tau-GFP (full-length tau-GFP), or GFP (empty vector) using LipofectAMINE 2000 according to the manufacturer's instructions to assess the potential neurotoxicity of 17Tau-GFP. Briefly, hippocampal neurons were equilibrated in 600 μl of transfection medium [50% feeding medium (N2) and 50% h-medium (MEM plus 10 mm HEPES)] for 1 h and incubated in the presence of DNA complexes (3 μg of DNA with 6 μl of LipofectAMINE 2000 in 50 μl of h-medium kept at room temperature for 15 min) for 1 h at 37°C. The coverslips were returned to the original dishes. After 24 or 48 h, cells were fixed for 15 min in 4% paraformaldehyde in PBS containing 0.12 m sucrose, permeabilized in 0.3% Triton X-100 for 5 min, and stained with an FITC-GFP antibody.
Apoptotic cell death was assessed using the In Situ Cell Death Detection kit, TMR Red (Roche Applied Science). Briefly, cells were fixed for 15 min with 4% paraformaldehyde in PBS containing 0.12 m sucrose and permeabilized in 0.1% Triton X-100 in 0.1% sodium citrate for 2 min, and TMR Red-labeled nucleotide was incorporated at 3′-OH DNA ends using the enzyme TdT. Apoptotic cells (TUNEL+ cells) were counted using a fluorescence microscope (Nikon) and expressed as a percentage of transfected cells.
Results
Pre-aggregated Aβ induced tau cleavage in a dose- and time-dependent manner in mature hippocampal neurons
The mechanisms by which tau mediates Aβ-induced neurotoxicity are not completely elucidated. Recently, it has been shown that the proteolytic cleavage of tau accompanied cell death associated with the deposition of Aβ in immature cortical neurons (Gamblin et al., 2003). However, no information is available regarding tau cleavage or the proteases involved in this cleavage in mature central neurons. To gain insights into such a process, we first studied whether Aβ induced changes in tau content in cultured hippocampal neurons. We have chosen this model system because the hippocampus is one of the most affected brain regions in AD. In addition, when kept in culture for >3 weeks, hippocampal neurons reproduce the functional characteristic and the molecular composition (including the expression of adult, mostly dephosphorylated, tau isoforms) of the ones that develop in vivo (Ferreira et al., 1997). In our first set of experiments, 21-d-in-culture hippocampal neurons were incubated in the presence of pre-aggregated Aβ (20 μm) for 24 h. The cells were then scraped, and heat-stable fractions were prepared. Western blot analysis of tau content in these fractions was performed using a phosphorylation-independent tau antibody [clone tau-5 (LoPresti et al., 1995)]. Pre-aggregated Aβ significantly decreased (∼50%) full-length tau immunoreactivity in Aβ-treated hippocampal neurons compared with untreated controls (Fig. 1A). In addition, a low-molecular weight tau-immunoreactive band (∼17 kDa) was readily detectable in pre-aggregated Aβ-treated hippocampal neurons (Fig. 1A). No comparable band was detected in nontreated controls (Fig. 1A). We next investigated whether the effect of pre-aggregated Aβ on the generation of this 17 kDa tau fragment was dose and/or time dependent. For these experiments, 21-d-in-culture hippocampal neurons were incubated for 24 h with pre-aggregated Aβ at final concentrations ranging from 0.02 to 20 μm. Western blot analysis of heat-stable fractions prepared from these cells revealed the presence of the 17 kDa tau fragment even in cells treated with the lowest dose used in this study. However, the levels of the 17 kDa tau fragment in hippocampal neurons treated with pre-aggregated Aβ at final concentrations below 2 μm were not significantly different from the ones observed in nontreated controls. In contrast, the 17 kDa tau fragment levels were significantly increased in hippocampal neurons treated with 20 μm pre-aggregated Aβ (Fig. 1B,D). In view of these data, we analyzed the time course of the generation of the 17 kDa tau fragment in neurons treated with pre-aggregated Aβ at this final concentration. For these experiments, 21-d-in-culture hippocampal neurons were treated with pre-aggregated Aβ (20 μm) for up to 24 h, and the presence of cleaved tau was assessed by immunoblotting. The levels of the 17 kDa tau fragment were significantly increased as early as 8 h after the addition of pre-aggregated Aβ and continued to increase in a time-dependent manner up to 24 h (Fig. 1C,E). The levels of 17 kDa tau fragments remained elevated even in hippocampal neurons treated with pre-aggregated Aβ for 36 h compared with nontreated controls (data not shown).
Figure 1.

Pre-aggregated Aβ induced tau cleavage in mature hippocampal neurons in a dose- and time-dependent manner. A, Western blot analysis of tau content in heat-stable fractions prepared from 21 d in vitro hippocampal neurons cultured in the absence (C) or in the presence of pre-aggregated Aβ (20 μm) for 24 h using a phosphorylation-independent tau antibody (clone tau-5). Note the decrease in full-length tau and the appearance of a tau-immunoreactive band of ∼17 kDa in Aβ-treated neurons. B, Western blot analysis of tau content in heat-stable fractions prepared from 21 d in vitro hippocampal neurons cultured for 24 h in the presence of pre-aggregated Aβ at final concentrations ranging from 0.02 to 20 μm using a tau antibody (clone tau-5). C, Western blot analysis of heat-stable fractions prepared from 21 d in vitro hippocampal neurons cultured in the presence of pre-aggregated Aβ (20 μm) for up to 24 h using a tau antibody (clone tau-5). Equal amounts of protein were loaded in each lane. The Hsp90 was also used as a loading control. D, E, Graphs showing the levels of the 17 kDa tau fragment in the dose-response (D) and time course (E) experiments performed as described above. Data were obtained using Molecular Analyst software and normalized using full-length tau and Hsp90 as internal controls. Values are expressed as a percentage of untreated controls, considering the values obtained in these neurons as 100%. Each number represents the mean ± SEM from three different experiments. *p < 0.01, different from control.
Pre-aggregated Aβ-induced tau cleavage preceded tau phosphorylation in cultured hippocampal neurons
We next compared the time course of tau cleavage described above with the one of tau phosphorylation in cultured hippocampal neurons. For these experiments, hippocampal neurons were treated with pre-aggregated Aβ for up to 24 h, and heat-stable fractions were prepared. Western blot analysis preformed using a tau antibody that recognizes tau phosphorylated at Ser193 and Thr196 (AT8) showed no increase in full-length tau phosphorylation at 2, 4, or 8 h after the addition of pre-aggregated Aβ. In contrast, a significant increase in AT8 immunoreactivity was observed in hippocampal neurons treated with pre-aggregated Aβ for 24 h compared with nontreated controls (Fig. 2A,B). These results are in agreement with previous results showing the increase in tau phosphorylation under these experimental conditions using antibodies directed to different tau phosphoepitopes (Ferreira et al., 1997). In contrast, the 17 kDa tau fragment was not detected using antibodies directed to phosphorylated tau despite the presence of the fragment as early as 8 h after the addition of pre-aggregated Aβ (Fig. 1C). These results suggested that this tau fragment was dephosphorylated in these residues. To test this hypothesis, we confirmed the presence of this 17 kDa tau fragment in pre-aggregated Aβ-treated hippocampal neurons using a tau antibody directed to the same epitope recognized by AT8, but in its dephosphorylated form [clone tau-1 (Szendrei et al., 1993)]. As expected, an intense tau-1-immunoreactive band was detected 8 h after the addition of pre-aggregated Aβ in mature hippocampal neurons (Fig. 2A,C).
Figure 2.

Pre-aggregated Aβ-induced tau cleavage preceded the increase in tau phosphorylation. A, Western blot analysis of tau content in heat-stable fractions prepared from 21-d-in-culture hippocampal neurons treated with pre-aggregated Aβ (20 μm) for up to 24 h using nonphosphorylation-dependent tau (clone tau-5), phosphorylated tau (clone AT8), and dephosphorylated tau (clone tau-1) antibodies. Note the absence of AT8 immunoreactivity of the 17 kDa tau fragment. Equal amounts of total protein were loaded in each lane. B, C, Graphs showing the time course of tau phosphorylation and tau cleavage in the presence of pre-aggregated Aβ. Densitometric values were normalized using full-length tau (tau-5) and the Hsp90 as internal controls. Values are expressed as a percentage of untreated controls, considering the values obtained in these neurons as 100%. Each number represents the mean ± SEM from three different experiments. *p < 0.05, **p < 0.01, different from control.
Pre-aggregated Aβ activated caspase-3 and calpain-1 in mature hippocampal neurons
Tau contains consensus sequences for cleavage by caspase-3 and calpain, two proteases that seem to play a role in the pathogenesis of AD (Saito et al., 1993; Rohn et al., 2001; Su et al., 2001; Veeranna et al., 2004). However, no information is available regarding the activation of these proteases in our model system. To obtain this information, we first determined the activity of caspase-3 and calpain in 21-d-in-culture hippocampal neurons incubated in the presence of pre-aggregated Aβ for up to 24 h. Immunoblots of whole-cell extracts prepared from these cultures were reacted with antibodies that recognized either uncleaved caspase-3 (total caspase-3) or its cleaved form (active caspase-3). Active caspase-3 levels were significantly higher in pre-aggregated Aβ-treated neurons as early as 4 h after the addition of this peptide compared with untreated controls (Fig. 3A,B). Active caspase-3 levels remained elevated throughout the rest of the period analyzed (Fig. 3A,B).
Figure 3.

Pre-aggregated Aβ induced the activation of caspase-3 and calpain-1 in cultured hippocampal neurons. A, C, Western blot analysis of active caspase-3 (A) and active calpain-1 (C) content in whole-cell extracts prepared from 21 d in vitro hippocampal neurons cultured in the presence of pre-aggregated Aβ (20 μm) for up to 24 h using specific antibodies. Tubulin immunoblots were used as loading controls (B, D, E). The bar graphs show the levels of active caspase-3 (B), active calpain-1 (D), and calpain-specific cleavage products of spectrin (E) in the samples obtained as described above. Note the increase in active calpain-1 levels in samples obtained 8 h after the addition of Aβ and the concomitant decrease in full-length spectrin and increase in the 150 kDa specific spectrin cleaved product (C-E). Densitometric values from active caspase-3, active calpain-1, and cleaved spectrin were normalized using total caspase-3, calpain-1, and full-length spectrin as internal controls. Values are expressed as a percentage of untreated controls, considering the values obtained in these neurons as 100%. Each number represents the mean ± SEM from three different experiments. *p < 0.05, **p < 0.01, different from control.
We then determined whether the treatment with pre-aggregated Aβ also activated calpains in cultured hippocampal neurons. Calpains, a family of calcium-dependent cystein proteases that exist as pro-enzymes in resting cells, are activated by calcium and autolytic processing. Autolysis of the N-terminal region of domain I of calpain leads to the generation of two active proteases of 78 and 76 kDa, respectively. Additional autocatalytic processing of domain IV of this protease leads to the generation of a 58 kDa active form (Zimmerman and Schlaepfer, 1984; Imajoh et al., 1987; Mellgren and Murachi, 1990). To assess calpain activity, we stripped the membranes used to determine the activation of caspase-3 and reprobed them using a calpain antibody that recognizes both calpains. Active calpain (58 kDa) levels were significantly increased 8 h after the addition of pre-aggregated Aβ to mature hippocampal neurons compared with untreated controls (Fig. 3C,D). An increase in active calpain levels was also detected in hippocampal neurons treated with pre-aggregated Aβ for 24 h. However, these levels were lower than the ones observed 16 h earlier (Fig. 3C,D). Calpain activation in Aβ-treated hippocampal neurons was also determined by analyzing spectrin cleavage. Calpain-mediated cleavage of spectrin leads to the generation of a specific 150 kDa cleavage product (Veeranna et al., 2004). Quantitative immunoblot analysis revealed that the generation of this 150 kDa spectrin cleavage product was significantly increased 8 h after the treatment with pre-aggregated Aβ compared with untreated controls (Fig. 3C,E). The time course and extent of calpain activation detected using this method were similar to the ones detected by quantifying the 58 kDa active calpain isoform (Fig. 3C,D).
To confirm the results described above, we analyzed the activation of these proteases in pre-aggregated Aβ-treated neurons using in vitro caspase-3 and calpain assays. The activity of caspase-3 was determined in 21-d-in-culture hippocampal neurons treated with pre-aggregated Aβ for up to 24 h by measuring the fluorescence emitted after the cleavage of substrates to AFC by active caspase-3. As determined by Western blot analysis, caspase-3 activity was significantly increased in hippocampal neurons as early as 4 h after the addition of the peptide (Table 1). In vitro assays also confirmed the activation of calpain in cultured hippocampal neurons treated with pre-aggregated Aβ. For these experiments, hippocampal neurons treated with pre-aggregated Aβ for up to 24 h were harvested in lysis buffer, and the supernatants were incubated with a calpain substrate (Suc-LLVY-AMC) for 1 h at 30°C. The fluorescence emitted after the cleavage of the substrate to AMC by active calpains was measured using a fluorescence plate reader. Calpain activity was significantly higher in cultured hippocampal neurons incubated with pre-aggregated Aβ for 8 h compared with untreated controls (Table 1).
Table 1.
Activation of caspase-3 and calpain by pre-aggregated Aβ in cultured hippocampal neurons
| Pre-aggregated Aβ (20 μm) |
|||||||
|---|---|---|---|---|---|---|---|
| Activity of proteases |
2h |
4h |
8h |
24h |
|||
| Caspase-3 | 122 ± 18 | 154 ± 11** | 127 ± 4* | 113 ± 14 | |||
| Calpain |
81 ± 17 |
83 ± 27 |
163 ± 20*
|
118 ± 25 |
|||
In vitro protease activity assays were performed using cell extracts obtained from 21-d-in-culture hippocampal neurons incubated in the presence of pre-aggregated Aβ from 2 to 24 h. Values are expressed as a percentage of untreated controls, considering the values obtained in these neurons as 100%. The numbers represent the means ± SEM obtained from three experiments. *p<0.05, **p<0.01, different from control.
Protease inhibitors prevented pre-aggregated Aβ-induced tau cleavage in cultured hippocampal neurons
To obtain insights into the role of caspase-3 and calpain in pre-aggregated Aβ-induced tau cleavage leading to the generation of the 17 kDa fragment, we blocked the activation of these proteases using specific inhibitors. For these experiments, hippocampal neurons kept in culture for 21 d were incubated in the presence of Ac-VAD-CHO (an inhibitor of caspase-3 and, to a lesser extent, caspases-1, -4, and -7) or Ac-DEVD-CHO (a specific caspase-3 inhibitor) and ALLN (a calpain inhibitor) 1 h before the addition of pre-aggregated Aβ (20 μm). The cells were scraped up to 24 h later, and whole-cell extracts were analyzed by Western blot using caspase-3, calpain, and tau antibodies. Both VAD and DEVD blocked the activation of caspase-3 induced by pre-aggregated Aβ 4 and 8 h after the addition of the peptide (Fig. 4A,B). In addition, VAD significantly reduced the generation of the 17 kDa tau fragment in the pre-aggregated Aβ-treated cells. On the other hand, DEVD failed to prevent the generation of this tau fragment 8 h after the addition of pre-aggregated Aβ (Fig. 4A,C). In contrast, ALLN completely prevented both the activation of calpain, detected by either the increase in the 58 kDa active calpain-1 or the 150 kDa calpain-cleaved spectrin products, and the generation of this tau fragment in Aβ-treated hippocampal neurons (Fig. 5A,C,E,G). Because ALLN is not only a calpain inhibitor but also a proteasome inhibitor, we repeated these experiments using a more specific calpain inhibitor (MDL 28,170). Mature hippocampal neurons were pretreated with MDL 28,170 for 1 h and incubated with pre-aggregated Aβ for 8 or 24 h. Immunoblots reacted with the calpain, spectrin, and tau antibodies showed that MDL 28,170 completely inhibited both the activation of calpain and the generation of the 17 kDa tau fragment induced by pre-aggregated Aβ (Fig. 5B,D,F,H). Next, we determined whether the incubation of hippocampal neurons with these calpain inhibitors before the addition of pre-aggregated Aβ prevented the activation of this protease as detected by in vitro assays. For these experiments, calpain inhibitor (ALLN, MDL 28,170)-treated hippocampal neurons were incubated with pre-aggregated Aβ for 8 h. The neurons were then scraped in lysis buffer, and the supernatants were incubated with a calpain substrate (Suc-LLVY-AMC) for 1 h at 30°C. The fluorescence emitted after the cleavage of the substrate to AMC by active calpain was measured using a fluorescence plate reader. Calpain activity was significantly lower in calpain inhibitor-treated neurons incubated with pre-aggregated Aβ compared with neurons treated with pre-aggregated Aβ alone (Table 2).
Figure 4.

Caspase inhibitors partially blocked tau cleavage induced by pre-aggregated Aβ. A, Western blot analysis of active caspase-3 and 17 kDa tau fragment content in whole-cell extracts prepared from 21-d-in-culture hippocampal neurons treated with either DEVD (50 μm; specific caspase-3 inhibitor) or VAD (50 μm; general caspase inhibitor) 1 h before the treatment with pre-aggregated Aβ (20 μm) for 4 and 8 h using specific antibodies. B, C, Graphs showing changes in active caspase-3 and 17 kDa tau fragment content in samples obtained as described above. Data were obtained using Molecular Analyst software and normalized using total caspase-3 and tubulin as internal controls. Values are expressed as a percentage of untreated controls, considering the values obtained in these neurons as 100%. Each number represents the mean ± SEM from three different experiments. *p < 0.05, **p < 0.01, different from control.
Figure 5.
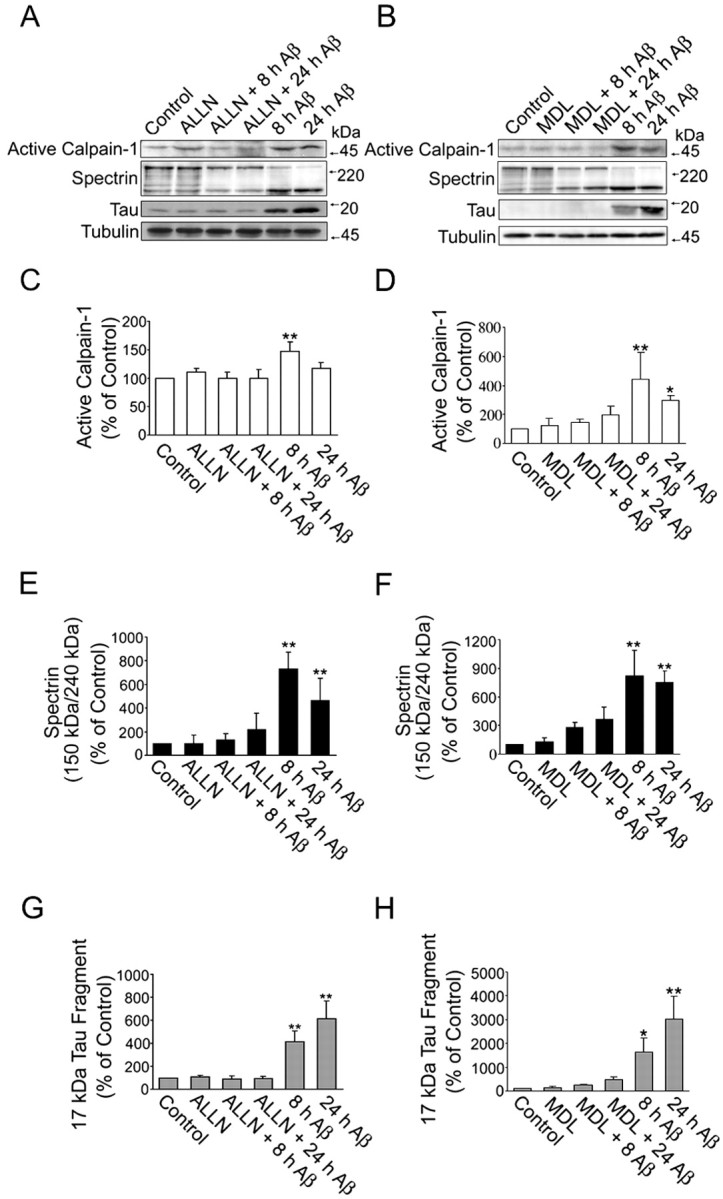
Calpain inhibitors completely blocked the generation of the 17 kDa tau fragment induced by pre-aggregated Aβ in cultured hippocampal neurons. A, B, Western blot analysis of active calpain-1, cleaved spectrin, and 17 kDa tau fragment content in whole-cell extracts prepared from 21-d-in-culture hippocampal neurons treated with either ALLN (A) or MDL 28,170 (B), two calpain inhibitors, 1 h before the treatment with pre-aggregated Aβ (20 μm) for 4 and 8 h using specific antibodies. C-H, Graphs showing changes in the content of active calpain-1 (C, D), calpain-specific cleavage product of spectrin (E, F), and 17 kDa tau fragment (G, H) in hippocampal neurons treated with ALLN (C, E, G) or MDL 28, 170 (D, F, H) before the addition of Aβ. Data were obtained using Molecular Analyst software and normalized using total calpain-1, spectrin, tau, and tubulin as internal controls. Values are expressed as a percentage of untreated controls, considering the values obtained in these neurons as 100%. Each number represents the mean ± SEM from three different experiments. *p < 0.05, **p < 0.01, different from control.
Table 2.
Calpain inhibitors, ALLN and MDL 28170, prevented pre-aggregated Aβ-induced calpain activation in cultured hippocampal neurons
|
|
Treatment |
||||
|---|---|---|---|---|---|
| Protease |
Aβ |
ALLN plus Aβ |
MDL plus Aβ |
||
| Calpain |
236 ± 27*
|
113 ± 17 |
106 ± 19 |
||
In vitro calpain activity assays were performed using cell extracts obtained from 21-d-in-culture hippocampal neurons treated with calpain inhibitors (ALLN or MDL 28,170) 1 h before the incubation with pre-aggregated Aβ (20 μm) for 8 h. Values are expressed as a percentage of untreated controls, considering the values obtained in these neurons as 100%. The numbers represent the means ± SEM obtained from three experiments. *p < 0.01, different from control.
Calpain, but not caspase-3, cleaved tau, generating a 17 kDa fragment in vitro
To obtain more direct information regarding the proteases involved in the generation of the 17 kDa tau fragment in cultured hippocampal neurons treated with pre-aggregated Aβ, we performed a series of in vitro tau cleavage assays. For these experiments, CHO cells were transfected with the full-length tau plasmid (tau-pRC/CMV) and a tau plasmid (D421E-tau-pRC/CMV) with a point mutation (from Asp to Glu) at position 421 to prevent its cleavage by caspase-3. Whole-cell extracts were prepared and treated with recombinant caspase-3 for 1 h at 37°C. As shown in Figure 6A, caspase-3 did cleave full-length tau but not D421E-tau. The cleavage of tau at Asp421 by caspase-3 was confirmed using a tau antibody that recognizes the Asp421 cleavage site (clone tau-C3). However, caspase-3 cleavage failed to generate a 17 kDa tau fragment as the one observed in hippocampal neurons cultured in the presence of pre-aggregated Aβ (Fig. 6A). To determine whether calpain cleavage was able to generate such a tau fragment, we transfected CHO cells with the tau-pRC/CMV or L43A-V229A-tau-pRC/CMV plasmids. From the nine potential calpain cleavage sites, only the cleavage at residues 45 and 230 could generate the 17 kDa tau fragment. Therefore, these residues (Leu43 and Val229) were point-mutated to Ala. CHO cells transfected with the tau-pRC/CMV or L43A-V229A-tau-pRC/CMV plasmid were harvested in lysis buffer and incubated on ice for 1 h. Supernatants following the centrifugation were incubated with calpain-1 for 1 h at 30°C. Incubation of full-length tau with calpain-1 generated a tau-immunoreactive band of ∼17 kDa, identical to that seen in neurons treated with pre-aggregated Aβ for 24 h. In contrast, the digestion of point-mutated tau at 43 and 229 positions by calpain-1 failed to generate such a band (Fig. 6B). To determine whether the 17 kDa tau fragment was further degraded by calpain or whether it was the final cleavage product, the tau45-230-pcDNA3.1(-) plasmid was transfected to CHO cells and the lysates were treated with calpain for 1 h at 30°C. The incubation of lysates with calpain did not generate additional fragments, suggesting that the 17 kDa tau fragment was not further degraded by calpain (Fig. 6C).
Figure 6.

Calpain-1 cleaved tau in vitro, generating a 17 kDa fragment. A, CHO cells were transfected with tau-pRC/CMV or D421E-tau-pRC/CMV using LipofectAMINE. After 48 h of transfection, CHO cells were scraped in lysis buffer. Whole-cell lysates were incubated with buffer control or recombinant caspase-3 at 37°C for 1 h. Reaction mixtures were analyzed by immunoblot with antibodies directed against phosphorylation-independent tau (clone tau-5) or tau truncated at Asp421 by caspase-3 (clone tau-C3) antibodies. Hippocampal neurons treated with pre-aggregated Aβ (20 μm) for 24 h were used as positive controls. Lane 1, Full-length tau with buffer control; lane 2, full-length tau with caspase-3; lane 3, D421E-tau with buffer control; lane 4, D421E-tau with caspase-3; lane 5, hippocampal neurons treated with pre-aggregated Aβ (20 μm) for 24 h. B, Full-length tau or L43A-V229A-tau-pRC/CMV-transfected CHO cells were harvested in lysis buffer. Whole-cell lysates were incubated with either buffer control or calpain-1 at 30°C for 5 or 60 min. There action mixtures were analyzed by Western blot using an antibody directed against tau (clone tau-5). Hippocampal neurons treated with pre-aggregated Aβ (20 μm) for 24 h were used as positive controls. Lane 1, Full-length tau with buffer control; lane 2, full-length tau with calpain-1 for 5 min; lane 3, full-length tau with calpain-1 for 60 min; lane 4, hippocampal neurons treated with pre-aggregated Aβ (20 μm) for 24 h; lane 5, L43A-V229A-tau with buffer control; lane 6, L43A-V229A-tau with calpain-1 for 5 min; lane 7, L43A-V229A-tau with calpain-1 for 60 min. C, Whole-cell lysates of tau45-230-pcDNA3.1(-)-transfected CHO cells were analyzed by immunoblot reacted with an antibody against tau (clone tau-5). Tau45-230, corresponding to the 17 kDa fragment on tau, was incubated with either buffer control (lane 1) or calpain-1 at 30°C for 1 h (lane 2). Hippocampal neurons treated with pre-aggregated Aβ (20 μm) for 24 h were used as positive controls (lane 3).
The 17 kDa Tau fragment had toxic effects both in CHO cells and cultured hippocampal neurons
To assess the potential toxic effects of the 17 kDa tau fragment, we transfected the tau45-230-GFP (17Tau-GFP) plasmid into CHO cells using LipofectAMINE. CHO cells transfected with the GFP (empty vector) were used as controls. CHO cells were fixed 48 h after transfection, and DNA fragmentation was examined using a TUNEL assay. Most of the cells transfected with GFP (empty vector) were TUNEL-. No differences in the number of TUNEL+ cells were detected in GPF-transfected CHO cells compared with nontransfected ones. In contrast, a significantly higher number (>35%) of TUNEL+ cells were detected in 17Tau-GFP-transfected CHO cells compared with GFP (empty vector)-transfected and/or nontransfected cells (Fig. 7).
Figure 7.

The 17 kDa tau fragment induced apoptosis in CHO cells. A-D, Detection of TUNEL+ cells in CHO cells transfected with GFP vector alone (A, B) or 17Tau-GFP (C, D). GFP-transfected (A) and 17Tau-GFP-transfected (C) cells are shown in green, and TUNEL+ cells are shown in red. B and D are phase-contrast pictures of A and C, respectively. Scale bar, 50 μm.
The experiments described above suggested that the 17 kDa tau fragment had toxic effects in CHO cells. To test whether this tau fragment also played a role in neurodegeneration, hippocampal neurons cultured for 14 d were transfected with 17Tau-GFP using LipofectAMINE 2000. Transfections using full-length Tau-GFP or GFP (empty vector) were used as controls. No changes in morphology or cell survival were detected in hippocampal neurons transfected with either full-length Tau-GFP or GFP (empty vector) 24 and 48 h after the transfection (Fig. 8). No morphological changes were detected either in 17 kDa tau-transfected neurons 24 h after the transfection. In contrast, apparent signs of degeneration were detected 48 h after hippocampal neurons were transfected with 17Tau-GFP (Fig. 8). These signs included the formation of tortuous processes, the presence of varicosities along the neurites, and the retraction of neuritic processes.
Figure 8.
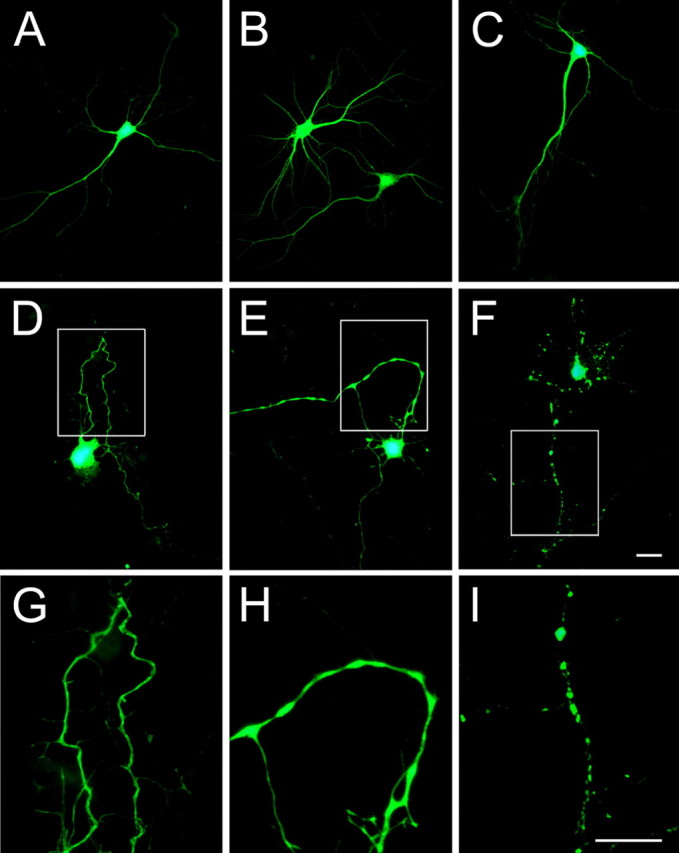
The 17 kDa tau fragment induced neurodegeneration in cultured hippocampal neurons. Fourteen-day-in-culture hippocampal neurons were transfected with GFP (A), full-length Tau-GFP (B), and 17Tau-GFP (C-I). GFP-transfected (A) and full-length Tau-GFP-transfected (B) neurons did not show any sign of degeneration even 48 h after transfection. Normal morphological characteristics were detected also in 17Tau-GFP-transfected cells 24 h after transfection (C). In contrast, hippocampal neurons transfected with 17Tau-GFP for 48 h showed numerous signs of degeneration, including the formation of tortuous processes (D), varicosity along the neurites (E), and the retraction of neurites (F). G-I, High-power magnification of the boxed areas in D-F. Scale bars, 20 μm.
Calpain inhibitors prevented Aβ-induced neurodegeneration in cultured hippocampal neurons
Finally, we analyzed whether calpain inhibitors block Aβ-induced neurotoxicity in cultured hippocampal neurons. For these experiments, 21-d-in-culture hippocampal neurons treated with calpain inhibitors, ALLN (50 μm) or MDL 28,170 (10 μm), were incubated with pre-aggregated Aβ (20 μm). Neuronal morphology was analyzed 24 h later. The signs of neurodegeneration described above were readily observed in hippocampal neurons treated with pre-aggregated Aβ for 24 h (Fig. 9B). In contrast, they were not detectable in untreated controls or in neurons treated with calpain inhibitors before the treatment with pre-aggregated Aβ (Fig. 9A,C,D). We also assessed the effect of calpain inhibitors on neuronal cell death induced by pre-aggregated Aβ. For these experiments, DNA fragmentation was examined using TUNEL assay. Although only a few hippocampal neurons (20 ± 1%) were TUNEL+ in untreated controls, more than one-half of the neurons (60 ± 4%) were TUNEL+ in pre-aggregated Aβ-treated cultures. In contrast, the number of TUNEL+ hippocampal neurons treated with calpain inhibitors, ALLN or MDL 28,170, before pre-aggregated Aβ for 24 h was significantly decreased (30 ± 2 and 27 ± 1%, respectively) compared with neurons treated with Aβ in the absence of calpain inhibitors (Fig. 9E).
Figure 9.
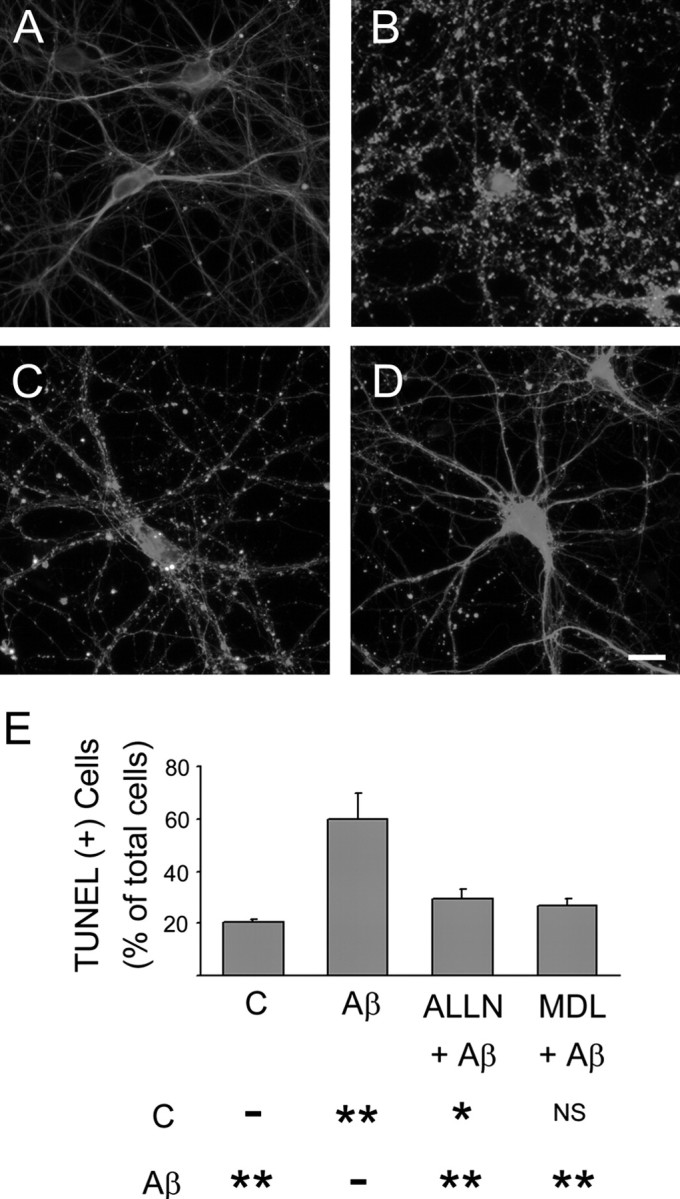
Calpain inhibitors prevented Aβ-induced neurotoxicity in culture hippocampal neurons. Hippocampal neurons cultured for 21 d were treated with calpain inhibitors, ALLN (C) or MDL 28, 170 (D), 1 h before the incubation with pre-aggregated Aβ for 24 h. No signs of neurite degeneration were detected in untreated controls (A). In contrast, severe neurite degeneration was observed in cultures incubated with pre-aggregated Aβ (B). Both calpain inhibitors significantly reduced the appearance of dystrophic neurites induced by Aβ. E, Detection of TUNEL+ cells in hippocampal neurons cultured in the presence or absence of calpain (C) inhibitors and Aβ as described above. Results were expressed as a percentage of the total number of neurons. Each number represents the mean ± SEM from three different experiments. More than 150 neurons were counted for each experimental condition. *p < 0.05, **p < 0.01, different from control. Scale bar, 20 μm. NS, Not statistically different.
Discussion
The results presented here indicated that pre-aggregated Aβ induced tau proteolysis, leading to the generation of a neurotoxic 17 kDa tau fragment in hippocampal neurons. In addition, they suggested that the production of this fragment was the result of the Aβ-induced activation of calpain in these neurons. Furthermore, the inhibition of calpain activation using specific calpain inhibitors prevented Aβ-induced neurodegeneration in cultured hippocampal neurons. Together, these data provided insights into a novel mechanism by which tau could mediate, at least in part, Aβ-induced neurotoxicity in hippocampal neurons.
A consensus exists regarding the toxic effects of pre-aggregated Aβ in neurons that develop in situ and in culture (Flood et al., 1991; Nitta et al., 1994; Busciglio et al., 1995; Copani et al., 1995; Estus et al., 1997). However, the mechanisms by which pre-aggregated Aβ exerts its toxic effects in the CNS remain poorly understood. Oxidative stress, mitochondrial dysfunction, disturbances of calcium homeostasis, and microglial activation have already been identified as mediators of Aβ toxicity (for review, see Canevari et al., 2004). In addition, the role of multiple potential downstream elements of the signaling pathway(s) activated by the deposition of Aβ has been investigated. Recently, we have provided direct evidence indicating that tau is essential for Aβ-mediated neurotoxicity in hippocampal neurons (Rapoport et al., 2002). This result was built on numerous reports suggesting a key role for tau in this neurodegenerative disease. In the past, most of the studies regarding tau and the pathogenesis of AD have been focused on phosphorylation as a mechanism by which this microtubule-associated protein mediates Aβ-induced toxicity. This posttranslational modification has been a tempting explanation for the role of tau in the AD process because of the multiple phosphorylation sites in the tau molecule, the presence of hyperphosphorylated tau as paired helical filaments in the AD brain, and the induction of tau phosphorylation in Aβ-treated neurons (Takashima et al., 1993; Ferreira et al., 1997; Alvarez et al., 1999; Ekinci et al., 1999; Rapoport and Ferreira, 2000). More recently, it has been suggested that the proteolytic cleavage of tau also played a role in AD. Thus, the deposition of pre-aggregated Aβ induced the activation of caspase-3 and the cleavage of tau at residue 421. Truncated tau lacking its C-terminal 20 aa assembled more rapidly into filaments than full-length tau (Gamblin et al., 2003). These results suggested that caspase-cleaved tau might be an important downstream element in the cascade of events triggered by Aβ.
Our results provide evidence of an alternative mechanism by which tau could mediate Aβ-induced neurotoxicity. This mechanism involved the proteolytic cleavage of tau leading to the generation of a neurotoxic 17 kDa tau fragment. The proteolytic cleavage of tau induced by pre-aggregated Aβ could lead to neurite degeneration by reducing the pool of full-length tau available for binding to microtubules. The decrease in tau bound to microtubules could in turn reduce their stability and promote a more rapid depolymerization cycle and therefore the disruption of the microtubule network. In addition, this process could lead to neuronal degeneration by altering the microtubule-based transport of essential materials to the distal ends of the neuronal process. Similar mechanisms have been described as a consequence of a decreased affinity of tau for microtubules resulting from its Aβ-induced hyperphosphorylation (Gong et al., 2000; Yoshida et al., 2004). Although we cannot completely rule out the contribution of these mechanisms to Aβ-induced neuronal degeneration in the presence of cleaved-tau, our results support the view that the 17 kDa tau fragment has direct toxic effects. For example, the transfection of this fragment in CHO cells resulted in the programmed cell death of a significant number of 17 kDa tau-expressing cells. Furthermore, 17 kDa tau-transfected hippocampal neurons showed signs of degeneration such as tortuous processes, the presence of varicosities along the neurites, and retraction of neuritic processes. All of these signs of degeneration were identical to the ones observed in hippocampal neurons treated with pre-aggregated Aβ (Ferreira et al., 1997). The neurotoxic effects of the 17 kDa fragment described in this study are in agreement with previous reports showing the production of other tau fragments with toxic effects under different experimental conditions in cell lines and in central neurons (Canu et al., 1998; Fasulo et al., 2000; Chung et al., 2001).
Our data also suggested that the proteolytic tau cleavage leading to the generation of the 17 kDa fragment might be an early event in the Aβ-induced neurodegeneration. Time-course experiments showed that the generation of the 17 kDa tau fragment preceded tau phosphorylation in Aβ-treated cultured neurons (Busciglio et al., 1995; Ferreira et al., 1997; Rapoport and Ferreira, 2000; Gamblin et al., 2003; present study). Although tau was truncated to the 17 kDa fragment as early as 8 h after the addition of pre-aggregated Aβ, tau phosphorylation was significantly induced 16 h later.
This study provided insights into the protease(s) responsible for the generation of the 17 kDa tau fragment in Aβ-treated hippocampal neurons. Our experiments suggested that calpain might be involved in this tau cleavage. The activation of calpain, easily detectable 8 h after the addition of fibrillar Aβ, was accompanied by a significant increase in the 17 kDa tau fragment levels. Conversely, pretreatment with calpain inhibitors not only completely blocked the generation of the 17 kDa tau fragment in hippocampal neurons but also prevented Aβ-induced neurodegeneration in cultured hippocampal neurons. The role of calpain in the generation of this 17 kDa tau fragment was confirmed by means of in vitro calpain assays. Furthermore, these experiments showed that calpain was able to cleave tau to generate a 17 kDa fragment as a final cleavage product. These data are in agreement with previous reports suggesting that calpain could play a role in AD (Grynspan et al., 1997; Tsuji et al., 1998; Veeranna et al., 2004). Thus, it has been shown that this Ca2+-dependent protease was abnormally activated in AD patients compared with age-matched controls (Saito et al., 1993). In addition, active calpain-2 colocalized with tau filaments in AD, Down syndrome, and frontotemporal dementia brains (Adamec et al., 2002a,b). This close spatial association suggested the involvement of calpain in the proteolysis of this microtubule-associated protein (Adamec et al., 2002a,b). Furthermore, the similarity between tau degradation fragments present in postmortem AD brains and the tau breakdown products generated by calpain in vitro provides additional support for a role of this protease in tau fragmentation in human neurons affected by the AD process (Mercken et al., 1995; Yang and Ksiezak-Reding, 1995).
The mechanisms underlying the activation of calpain in AD are not completely elucidated. However, this increased activity has been considered a result of the massive increase in intracellular free Ca2+ concentration induced by pre-aggregated Aβ (Mattson et al., 1993; Lee et al., 2000; Boland and Campbell, 2003). A similar calpain activation has been obtained by experimentally inducing calcium influx into SH-SY5Y human neuroblastoma cells (Shea et al., 1996).
The data discussed above strongly suggested that calpain might be the main protease responsible for the tau cleavage leading to the generation of the 17 kDa fragment in Aβ-treated hippocampal neurons. However, the experiments performed using caspase-3 inhibitors suggested that this caspase might also play a role in the generation of this fragment. A growing body of evidence suggests that caspases play a pivotal role in apoptosis induced by Aβ (Ivins et al., 1998, 1999; Troy et al., 2000; Allen et al., 2001; Marques et al., 2003). Especially, caspase-3 is considered a key component of Aβ-induced apoptosis in cultured neurons (Harada and Sugimoto, 1999; Marin et al., 2000; Allen et al., 2001). In contrast, the activation time course of caspase-3 in Aβ-treated hippocampal neurons as well as caspase-3 in vitro assays indicated that this caspase might not be directly involved in the generation of this tau fragment. Alternatively, caspases might regulate the production of this tau fragment by inducing the degradation of the endogenous calpain inhibitor calpastatin. This mechanism by which caspases could regulate calpain activity has already been reported in other systems (Wang et al., 1998). If that is the case, caspase inhibitors could reduce the degradation of calpastatin, and this in turn could inhibit the activation of calpain. This synergistic effect of calpain and caspase inhibitors has also been reported in ischemic neuronal death (Rami et al., 2000; Rami, 2003).
Collectively, our results provide insights into a novel mechanism by which tau could mediate Aβ-induced neurotoxicity in hippocampal neurons. They also highlight the importance of the calpain system as a target for therapeutic intervention in this neurodegenerative disease.
Footnotes
This work was supported by National Institutes of Health Grant NS39080 to A.F.
Correspondence should be addressed to Dr. Adriana Ferreira, Institute for Neuroscience, Northwestern University, Searle Building Room 5-474, 320 East Superior Street, Chicago, IL 60611. E-mail: a-ferreira@northwestern.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/255365-11$15.00/0
References
- Adamec E, Mohan P, Vonsattel JP, Nixon RA (2002a) Calpain activation in neurodegenerative diseases: confocal immunofluorescence study with antibodies specifically recognizing the active form of calpain 2. Acta Neuropathol (Berl) 104: 92-104. [DOI] [PubMed] [Google Scholar]
- Adamec E, Murrell JR, Takao M, Hobbs W, Nixon RA, Ghetti B, Vonsattel JP (2002b) P301L tauopathy: confocal immunofluorescence study of perinuclear aggregation of the mutated protein. J Neurol Sci 200: 85-93. [DOI] [PubMed] [Google Scholar]
- Allen JW, Eldadah BA, Huang X, Knoblach SM, Faden AI (2001) Multiple caspases are involved in beta-amyloid-induced neuronal apoptosis. J Neurosci Res 65: 45-53. [DOI] [PubMed] [Google Scholar]
- Alvarez A, Toro R, Caceres A, Maccioni RB (1999) Inhibition of tau phosphorylating protein kinase cdk5 prevents beta-amyloid-induced neuronal death. FEBS Lett 459: 421-426. [DOI] [PubMed] [Google Scholar]
- Bensadoun A, Weinstein D (1976) Assay of proteins in the presence of interfering materials. Anal Biochem 70: 241-250. [DOI] [PubMed] [Google Scholar]
- Boland B, Campbell V (2003) Beta-amyloid (1-40)-induced apoptosis of cultured cortical neurones involves calpain-mediated cleavage of poly-ADP-ribose polymerase. Neurobiol Aging 24: 179-186. [DOI] [PubMed] [Google Scholar]
- Bottenstein JE, Sato GH (1979) Growth of a rat neuroblastoma cell line in serum-free supplemented medium. Proc Natl Acad Sci USA 76: 514-517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brancolini C, Lazarevic D, Rodriguez J, Schneider C (1997) Dismantling cell-cell contacts during apoptosis is coupled to a caspase-dependent proteolytic cleavage of beta-catenin. J Cell Biol 139: 759-771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busciglio J, Lorenzo A, Yeh J, Yankner BA (1995) Beta-amyloid fibrils induce tau phosphorylation and loss of microtubule binding. Neuron 14: 879-888. [DOI] [PubMed] [Google Scholar]
- Canevari L, Abramov AY, Duchen MR (2004) Toxicity of amyloid beta peptide: tales of calcium, mitochondria, and oxidative stress. Neurochem Res 29: 637-650. [DOI] [PubMed] [Google Scholar]
- Canu N, Dus L, Barbato C, Ciotti MT, Brancolini C, Rinaldi AM, Novak M, Cattaneo A, Bradbury A, Calissano P (1998) Tau cleavage and dephosphorylation in cerebellar granule neurons undergoing apoptosis. J Neurosci 18: 7061-7074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung CW, Song YH, Kim IK, Yoon WJ, Ryu BR, Jo DG, Woo HN, Kwon YK, Kim HH, Gwag BJ, Mook-Jung IH, Jung YK (2001) Proapoptotic effects of tau cleavage product generated by caspase-3. Neurobiol Dis 8: 162-172. [DOI] [PubMed] [Google Scholar]
- Copani A, Bruno V, Battaglia G, Leanza G, Pellitteri R, Russo A, Stanzani S, Nicoletti F (1995) Activation of metabotropic glutamate receptors protects cultured neurons against apoptosis induced by beta-amyloid peptide. Mol Pharmacol 47: 890-897. [PubMed] [Google Scholar]
- Ekinci FJ, Malik KU, Shea TB (1999) Activation of the L voltage-sensitive calcium channel by mitogen-activated protein (MAP) kinase following exposure of neuronal cells to beta-amyloid. MAP kinase mediates beta-amyloid-induced neurodegeneration. J Biol Chem 274: 30322-30327. [DOI] [PubMed] [Google Scholar]
- Estus S, Tucker HM, van Rooyen C, Wright S, Brigham EF, Wogulis M, Rydel RE (1997) Aggregated amyloid-beta protein induces cortical neuronal apoptosis and concomitant “apoptotic” pattern of gene induction. J Neurosci 17: 7736-7745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasulo L, Ugolini G, Visintin M, Bradbury A, Brancolini C, Verzillo V, Novak M, Cattaneo A (2000) The neuronal microtubule-associated protein tau is a substrate for caspase-3 and an effector of apoptosis. J Neurochem 75: 624-633. [DOI] [PubMed] [Google Scholar]
- Ferreira A, Busciglio J, Caceres A (1989) Microtubule formation and neurite growth in cerebellar macroneurons which develop in vitro: evidence for the involvement of the microtubule-associated proteins, MAP-1a, HMW-MAP2 and Tau. Brain Res Dev Brain Res 49: 215-228. [DOI] [PubMed] [Google Scholar]
- Ferreira A, Lu Q, Orecchio L, Kosik KS (1997) Selective phosphorylation of adult tau isoforms in mature hippocampal neurons exposed to fibrillar A beta. Mol Cell Neurosci 9: 220-234. [DOI] [PubMed] [Google Scholar]
- Flood JF, Morley JE, Roberts E (1991) Amnestic effects in mice of four synthetic peptides homologous to amyloid beta protein from patients with Alzheimer disease. Proc Natl Acad Sci USA 88: 3363-3366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamblin TC, Chen F, Zambrano A, Abraha A, Lagalwar S, Guillozet AL, Lu M, Fu Y, Garcia-Sierra F, LaPointe N, Miller R, Berry RW, Binder LI, Cryns VL (2003) Caspase cleavage of tau: linking amyloid and neurofibrillary tangles in Alzheimer's disease. Proc Natl Acad Sci USA 100: 10032-10037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glenner GG, Wong CW (1984) Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun 120: 885-890. [DOI] [PubMed] [Google Scholar]
- Gong CX, Lidsky T, Wegiel J, Zuck L, Grundke-Iqbal I, Iqbal K (2000) Phosphorylation of microtubule-associated protein tau is regulated by protein phosphatase 2A in mammalian brain. Implications for neurofibrillary degeneration in Alzheimer's disease. J Biol Chem 275: 5535-5544. [DOI] [PubMed] [Google Scholar]
- Goslin K, Banker G (1990) Rapid changes in the distribution of GAP-43 correlate with the expression of neuronal polarity during normal development and under experimental conditions. J Cell Biol 110: 1319-1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grynspan F, Griffin WR, Cataldo A, Katayama S, Nixon RA (1997) Active site-directed antibodies identify calpain II as an early-appearing and pervasive component of neurofibrillary pathology in Alzheimer's disease. Brain Res 763: 145-158. [DOI] [PubMed] [Google Scholar]
- Harada J, Sugimoto M (1999) Activation of caspase-3 in beta-amyloid-induced apoptosis of cultured rat cortical neurons. Brain Res 842: 311-323. [DOI] [PubMed] [Google Scholar]
- Imajoh S, Kawasaki H, Suzuki K (1987) The COOH-terminal E-F hand structure of calcium-activated neutral protease (CANP) is important for the association of subunits and resulting proteolytic activity. J Biochem (Tokyo) 101: 447-452. [DOI] [PubMed] [Google Scholar]
- Ivins KJ, Bui ET, Cotman CW (1998) Beta-amyloid induces local neurite degeneration in cultured hippocampal neurons: evidence for neuritic apoptosis. Neurobiol Dis 5: 365-378. [DOI] [PubMed] [Google Scholar]
- Ivins KJ, Thornton PL, Rohn TT, Cotman CW (1999) Neuronal apoptosis induced by beta-amyloid is mediated by caspase-8. Neurobiol Dis 6: 440-449. [DOI] [PubMed] [Google Scholar]
- Kondo J, Honda T, Mori H, Hamada Y, Miura R, Ogawara M, Ihara Y (1988) The carboxyl third of tau is tightly bound to paired helical filaments. Neuron 1: 827-834. [DOI] [PubMed] [Google Scholar]
- Kosik KS, Joachim CL, Selkoe DJ (1986) Microtubule-associated protein tau (tau) is a major antigenic component of paired helical filaments in Alzheimer disease. Proc Natl Acad Sci USA 83: 4044-4048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227: 680-685. [DOI] [PubMed] [Google Scholar]
- Lee MS, Kwon YT, Li M, Peng J, Friedlander RM, Tsai LH (2000) Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature 405: 360-364. [DOI] [PubMed] [Google Scholar]
- LoPresti P, Szuchet S, Papasozomenos SC, Zinkowski RP, Binder LI (1995) Functional implications for the microtubule-associated protein tau: localization in oligodendrocytes. Proc Natl Acad Sci USA 92: 10369-10373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurement with the Folin phenol reagent. J Biol Chem 193: 265-275. [PubMed] [Google Scholar]
- Marin N, Romero B, Bosch-Morell F, Llansola M, Felipo V, Roma J, Romero FJ (2000) Beta-amyloid-induced activation of caspase-3 in primary cultures of rat neurons. Mech Ageing Dev 119: 63-67. [DOI] [PubMed] [Google Scholar]
- Marques CA, Keil U, Bonert A, Steiner B, Haass C, Muller WE, Eckert A (2003) Neurotoxic mechanisms caused by the Alzheimer's disease-linked Swedish amyloid precursor protein mutation: oxidative stress, caspases, and the JNK pathway. J Biol Chem 278: 28294-28302. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Tomaselli KJ, Rydel RE (1993) Calcium-destabilizing and neurodegenerative effects of aggregated beta-amyloid peptide are attenuated by basic FGF. Brain Res 621: 35-49. [DOI] [PubMed] [Google Scholar]
- Mellgren RL, Murachi T (1990) Intracellular calcium-dependent proteolysis. Boca Raton, FL: CRC.
- Mercken M, Grynspan F, Nixon RA (1995) Differential sensitivity to proteolysis by brain calpain of adult human tau, fetal human tau and PHF-tau. FEBS Lett 368: 10-14. [DOI] [PubMed] [Google Scholar]
- Nitta A, Itoh A, Hasegawa T, Nabeshima T (1994) Beta-amyloid protein-induced Alzheimer's disease animal model. Neurosci Lett 170: 63-66. [DOI] [PubMed] [Google Scholar]
- Novak M, Kabat J, Wischik CM (1993) Molecular characterization of the minimal protease resistant tau unit of the Alzheimer's disease paired helical filament. EMBO J 12: 365-370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rami A (2003) Ischemic neuronal death in the rat hippocampus: the calpain-calpastatin-caspase hypothesis. Neurobiol Dis 13: 75-88. [DOI] [PubMed] [Google Scholar]
- Rami A, Agarwal R, Botez G, Winckler J (2000) mu-Calpain activation, DNA fragmentation, and synergistic effects of caspase and calpain inhibitors in protecting hippocampal neurons from ischemic damage. Brain Res 866: 299-312. [DOI] [PubMed] [Google Scholar]
- Rapoport M, Ferreira A (2000) PD98059 prevents neurite degeneration induced by fibrillar beta-amyloid in mature hippocampal neurons. J Neurochem 74: 125-133. [DOI] [PubMed] [Google Scholar]
- Rapoport M, Dawson HN, Binder LI, Vitek MP, Ferreira A (2002) Tau is essential to beta-amyloid-induced neurotoxicity. Proc Natl Acad Sci USA 99: 6364-6369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohn TT, Head E, Su JH, Anderson AJ, Bahr BA, Cotman CW, Cribbs DH (2001) Correlation between caspase activation and neurofibrillary tangle formation in Alzheimer's disease. Am J Pathol 158: 189-198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito K, Elce JS, Hamos JE, Nixon RA (1993) Widespread activation of calcium-activated neutral proteinase (calpain) in the brain in Alzheimer disease: a potential molecular basis for neuronal degeneration. Proc Natl Acad Sci USA 90: 2628-2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ (1994) Cell biology of the amyloid beta-protein precursor and the mechanism of Alzheimer's disease. Annu Rev Cell Biol 10: 373-403. [DOI] [PubMed] [Google Scholar]
- Shea TB, Spencer MJ, Beermann ML, Cressman CM, Nixon RA (1996) Calcium influx into human neuroblastoma cells induces ALZ-50 immunoreactivity: involvement of calpain-mediated hydrolysis of protein kinase C. J Neurochem 66: 1539-1549. [DOI] [PubMed] [Google Scholar]
- Su JH, Zhao M, Anderson AJ, Srinivasan A, Cotman CW (2001) Activated caspase-3 expression in Alzheimer's and aged control brain: correlation with Alzheimer pathology. Brain Res 898: 350-357. [DOI] [PubMed] [Google Scholar]
- Szendrei GI, Lee VM, Otvos Jr L (1993) Recognition of the minimal epitope of monoclonal antibody Tau-1 depends upon the presence of a phosphate group but not its location. J Neurosci Res 34: 243-249. [DOI] [PubMed] [Google Scholar]
- Takashima A, Noguchi K, Sato K, Hoshino T, Imahori K (1993) Tau protein kinase I is essential for amyloid beta-protein-induced neurotoxicity. Proc Natl Acad Sci USA 90: 7789-7793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towbin H, Staehelin T, Gordon J (1979) Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA 76: 4350-4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troy CM, Rabacchi SA, Friedman WJ, Frappier TF, Brown K, Shelanski ML (2000) Caspase-2 mediates neuronal cell death induced by β-amyloid. J Neurosci 20: 1386-1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji T, Shimohama S, Kimura J, Shimizu K (1998) m-Calpain (calcium-activated neutral proteinase) in Alzheimer's disease brains. Neurosci Lett 248: 109-112. [DOI] [PubMed] [Google Scholar]
- Veeranna, Kaji T, Boland B, Odrljin T, Mohan P, Basavarajappa BS, Peterhoff C, Cataldo A, Rudnicki A, Amin N, Li BS, Pant HC, Hungund BL, Arancio O, Nixon RA (2004) Calpain mediates calcium-induced activation of the erk1, 2 MAPK pathway and cytoskeletal phosphorylation in neurons: relevance to Alzheimer's disease. Am J Pathol 165: 795-805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang KK, Posmantur R, Nadimpalli R, Nath R, Mohan P, Nixon RA, Talanian RV, Keegan M, Herzog L, Allen H (1998) Caspase-mediated fragmentation of calpain inhibitor protein calpastatin during apoptosis. Arch Biochem Biophys 356: 187-196. [DOI] [PubMed] [Google Scholar]
- Wood JG, Mirra SS, Pollock NJ, Binder LI (1986) Neurofibrillary tangles of Alzheimer disease share antigenic determinants with the axonal microtubule-associated protein tau (tau). Proc Natl Acad Sci USA 83: 4040-4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang LS, Ksiezak-Reding H (1995) Calpain-induced proteolysis of normal human tau and tau associated with paired helical filaments. Eur J Biochem 233: 9-17. [DOI] [PubMed] [Google Scholar]
- Yankner BA, Mesulam MM (1991) Seminars in medicine of the Beth Israel Hospital, Boston. Beta-amyloid and the pathogenesis of Alzheimer's disease. N Engl J Med 325: 1849-1857. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Hastie CJ, McLauchlan H, Cohen P, Goedert M (2004) Phosphorylation of microtubule-associated protein tau by isoforms of c-Jun N-terminal kinase (JNK). J Neurochem 90: 352-358. [DOI] [PubMed] [Google Scholar]
- Zimmerman UP, Schlaepfer WW (1984) Multiple forms of Ca-activated protease from rat brain and muscle. J Biol Chem 259: 3210-3218. [PubMed] [Google Scholar]