

Abstract
During the last decade, great progress has been made in the discovery of genes that influence risk for epilepsy. However, these gene discoveries have been in epilepsies with Mendelian modes of inheritance, which comprise only a tiny fraction of all epilepsy. Most people with epilepsy have no affected relatives, suggesting that the great majority of all epilepsies are genetically complex: multiple genes contribute to their etiology, none of which has a major effect on disease risk. Gene discovery in the genetically complex epilepsies is a formidable task. It is unclear which epilepsy phenotypes are most advantageous to study, and chromosomal localization and mutation detection are much more difficult than in Mendelian epilepsies. Association studies are very promising for the identification of complex epilepsy genes, but we are still in the earliest stages of their application in the epilepsies. Future studies should employ very large sample sizes to ensure adequate statistical power, clinical phenotyping methods of the highest quality, designs and analytic techniques that control for population stratification, and state-of-the-art molecular methods. Collaborative studies are essential to achieve these goals.
Keywords: Epilepsy, Seizures, Genetics, Phenotype definition, Linkage, Association, Epidemiology
During the last decade, great progress has been made in the discovery of genes that influence risk for epilepsy. However, almost all of the gene discoveries to date have been in epilepsies with Mendelian modes of inheritance, and these comprise only a tiny fraction of all epilepsy. Most people with epilepsy have no affected relatives. Epilepsies that occur in the absence of a significant family history, or where the family history is inconsistent with Mendelian inheritance, have come to be known as “genetically complex.” This paper will describe the meaning of “complex inheritance” as it applies to epilepsy, and discuss research approaches that can be used to study these genetic influences. Implications for clinical practice will also be discussed briefly.
COMPLEX INHERITANCE: A HISTORICAL PERSPECTIVE
The current concept of complex disease implies that multiple genes and environmental factors contribute to etiology, none of which has a major effect on disease risk when acting by itself (1,2). To understand this concept, a brief review of the history of genetic research is useful. In 1866, Gregor Mendel, the Augustinian monk, published his seminal work on breeding experiments in the garden pea, which demonstrated particulate inheritance of discrete units (“genes”) and rules of segregation. This work revolutionized thinking on inheritance at that time, which postulated a blending of the two parental contributions rather than transmission of discrete units. Mendel’s theories were confirmed by subsequent work, and became widely accepted.
Then in the early 1900s, Francis Galton and Karl Pearson noted that many human traits did not appear to follow Mendelian laws of inheritance. The traits they studied were quantitative, such as height, weight, and IQ, and followed a Gaussian (normal) distribution in the population. In contrast, the traits Mendel studied in pea plants were simple dichotomies such as wrinkled versus smooth seeds, and white versus purple flowers. With quantitative traits, offspring trait values were normally distributed, with means midway between the two parental means, instead of segregating into discrete groups as predicted by Mendel’s laws. For a time, geneticists thought that an alternative theory of inheritance was needed to explain this pattern.
However, in 1918, the statistician Sir Ronald Fisher resolved this apparent inconsistency (3). Fisher showed that Mendel’s laws could explain the inheritance pattern of the traits described by Galton and Pearson, like that of other traits. The observed pattern is consistent with the influence of multiple genes, each of which follows Mendel’s laws but has a small effect on the trait, and acts additively with the others (Fig. 1). Environmental factors also contribute, with additive effects of multiple environmental factors. As the number of contributing genes and environmental factors increases, the population distribution of the trait approaches the observed normal distribution. Because multiple genes contribute to these traits, they are called “polygenic,” and because multiple environmental factors also contribute, they are called “multifactorial.”
FIG. 1.
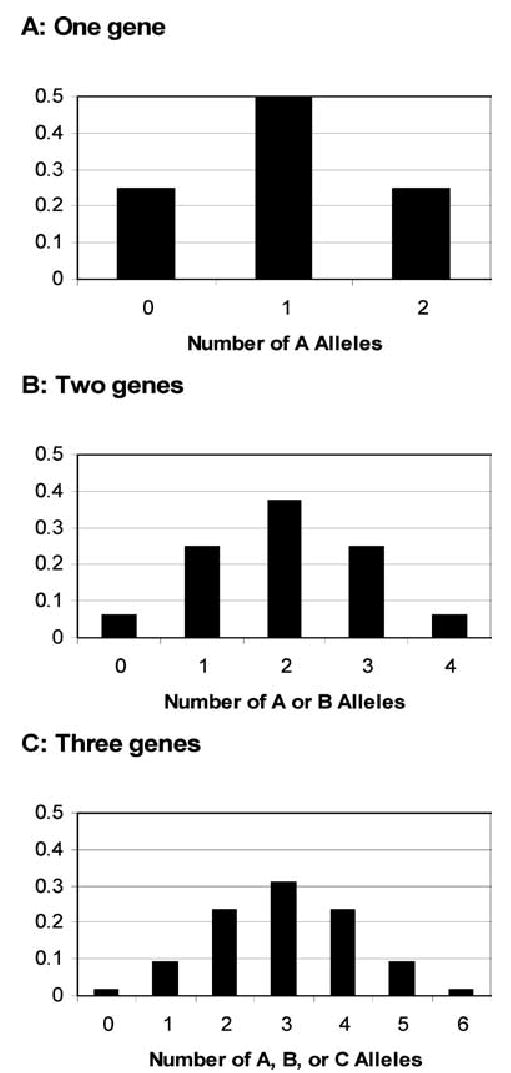
Expected population distribution of a quantitative trait caused by the additive effects of one, two, or three genes, each with two equally frequent alleles (locus A with alleles A and a, locus B with alleles B and b, and locus C with alleles C and c). The phenotype is determined by the number of alleles denoted by “capital letters” an individual inherits. A: the trait is caused by a single gene “A,” and the phenotype is 0 in aa, individuals, 1 in Aa individuals, and 2 in AA individuals. B: the trait is caused by two genes “A” and “B,” and the phenotype is 0 in aabb individuals, 1 in Aabb or aaBb individuals, 2 in AAbb, AaBb, or aaBB individuals, 3 in AABb or AaBB individuals, and 4 in AABB individuals. C: the trait is caused by three genes “A,” “B,” and “C,” and the phenotype ranges from 0 in aabbcc individuals to 6 in AABBCC individuals. As the number of contributing loci increases, the trait distribution in the population more closely approximates a normal distribution.
In 1965, Falconer extended the multifactorial-polygenic model to explain the genetics of diseases like epilepsy, in which people are either unaffected or affected, rather than having measurable quantitative trait values (4). He proposed that in non-Mendelian diseases, the observed patterns of familial risk were consistent with the influence of multiple genes and environmental factors on an unmeasurable, normally distributed quantitative trait called “liability.” People would be affected if their liability exceeded a threshold value.
With epilepsy, this type of liability threshold could be viewed as a “seizure threshold” (Fig. 2). Susceptibility to develop epilepsy (liability), due to the additive effects of many genes and environmental factors, is assumed to be normally distributed in the population. Individuals whose liability exceeds the seizure threshold develop epilepsy. This implies that people who do develop epilepsy have high liability, and to the extent that liability is correlated in families, their relatives will have increased liability, and an increased risk of developing epilepsy, compared with the general population. We could also imagine that the seizure threshold varies among individuals, depending on age, sex, and other factors (5).
FIG. 2.
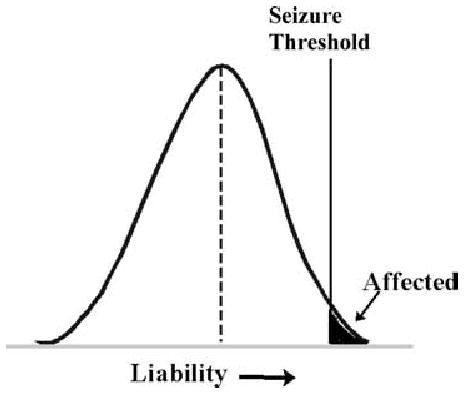
Multifactorial-polygenic model. Liability, an unmeasurable quantitative trait, is normally distributed in the population; and individuals with liability above a threshold value are affected. With epilepsy, the threshold could be conceived of as a seizure threshold. (Modified from Fig. 15 in http://www.uic.edu/classes/bms/bms655/lesson11.html).
The multifactorial-polygenic model provides an excellent framework for thinking about the genetics of complex, non-Mendelian disorders. However, the current concept of complex inheritance has been extended to incorporate additional complexities. Strictly speaking, the multifactorial-polygenic model involves a very large (essentially infinite) number of genes and environmental factors, each of which has a small, additive effect on disease risk. However, some complex diseases probably involve fewer genes, in so-called “oligogenic” models. Also, the assumption that all effects are additive implies no gene–gene or gene–environment interaction, but both types of interactions are likely to be important in many complex diseases. The effects of specific genes on disease risk probably vary depending on genotypes at other loci (gene–gene interaction, or “epistasis”) and on the history of environmental exposures (gene–environment interaction) (6).
EPILEPSY AS A COMPLEX DISEASE
Epilepsy clearly aggregates in families; risk is increased two- to fourfold in the first-degree relatives of people with idiopathic or cryptogenic epilepsy, compared with population incidence (7–9). Yet most people with epilepsy have no affected relatives. In the Epilepsy Family Study of Columbia University (EFSCU) (8–11), we collected family history information on 1,957 people with epilepsy, ascertained from voluntary organizations for epilepsy without regard to their family histories. The proportion of subjects with a positive family history (≥1 first-degree relative with epilepsy) was 15% in those with idiopathic generalized epilepsy (IGE), and 12% in those with cryptogenic localization-related epilepsy (LRE). Moreover, most of those with a family history had just one affected relative (probands with IGE 77%, cryptogenic LRE 79%), and few families appeared consistent with a Mendelian model (12).
In this large group of people with non-Mendelian forms of epilepsy, the genetic influences on risk probably consist mainly of complex disease genes. Two other types of genetic effects may also play a role in some cases. First, some “sporadic” epilepsies (i.e., those occurring in the absence of a family history) may be caused by de novo mutations. This mechanism is important in severe myoclonic epilepsy of infancy (SMEI)—de novo mutations in the gene encoding the alpha 1 subunit of the sodium channel, SCN1A, have been found in 33–100% of patients (13–16). Second, some epilepsies may be caused by somatic mutations occurring in critical brain regions.
Many of the approximately 30,000 genes in the human genome probably influence risk for epilepsy. The contributing genes probably vary widely in the magnitude of their effects on risk, genes with larger effects producing Mendelian patterns of inheritance and genes with smaller effects producing complex patterns. So far, 12 genes influencing risk for idiopathic epilepsy have been identified (Table 1), and all have fallen in the high range of the continuum of genetic effects—the families with mutations contain many affected individuals, in patterns largely consistent with autosomal dominant inheritance. However, there is no clear dividing line between Mendelian and complex disease genes. Although the mutations identified so far raise risk dramatically, penetrance is incomplete, suggesting that other genes or environmental factors influence their effects. Also, the same pathogenic mechanisms may be involved in Mendelian and complex genetic epilepsies. For example, the discovery that most of the Mendelian genes identified so far have encoded voltage-gated or ligand-gated ion channels suggests that variants in ion channel genes are likely to contribute to risk for genetically complex epilepsies.
TABLE 1.
Mendelian idiopathic epilepsy syndromes with genes identified by positional cloning (as of April 1, 2005)
| Epilepsy syndrome | Gene | Chromosomal location | References |
|---|---|---|---|
| Benign familial neonatal seizures | KCNQ2 | 20q13 | (53) |
| KCNQ3 | 8q24 | (54) | |
| Benign familial neonatal–infantile seizures | SCN2Aa | 2q24 | (32) |
| Childhood absence epilepsy with febrile seizures | GABRG2a | 5q31 | (33,34) |
| Autosomal dominant juvenile myoclonic epilepsy | GABRA1 | 5q34 | (21) |
| EFHC1 | 6p12 | (22) | |
| Autosomal dominant idiopathic generalized epilepsy | CLCN2 | 3q26 | (23) |
| Generalized epilepsy with febrile seizures plus | SCN1B | 19q13 | (26) |
| SCN1Ab | 2q24 | (27,28) | |
| SCN2Aa | 2q24 | (29) | |
| GABRG2a | 5q31 | (30,31) | |
| Autosomal dominant nocturnal frontal lobe epilepsy | CHRNA4 | 20q13 | (55) |
| CHRNB2 | 1q21 | (56) | |
| Autosomal dominant partial epilepsy with auditory features | LGI1 | 10q24 | (57,58) |
Mutations identified in more than one epilepsy syndrome.
De novo mutations also identified in severe myoclonic epilepsy of infancy.
Evidence from Mendelian epilepsy syndromes also illustrates complexities in the relations between genotype and phenotype that are likely to be important in non-Mendelian epilepsies. First, genetic heterogeneity is extensive. Mutations in different genes have been found to cause the same syndrome in different families (locus heterogeneity) (Table 1). Moreover, even in epilepsy syndromes with identified genes, many patients have no affected relatives, suggesting that different genetic mechanisms—Mendelian (single gene) and complex—can produce the same syndrome. This makes it impossible to classify syndromes according to genetic mechanisms.
For example, in autosomal dominant partial epilepsy with auditory symptoms (ADPEAF), mutations in the leucine-rich, glioma inactivated 1 gene (LGI1) have been found in approximately 50% of families containing two or more individuals with temporal lobe epilepsy with ictal auditory symptoms (17). However, most patients with these clinical features are sporadic. In two series of sporadic patients screened for mutations in LGI1, none had an inherited mutation (18,19) (although one patient had a de novo mutation (20)). Thus, this syndrome is autosomal dominant in some families, but genetically complex in most. The same principle applies to the IGEs. They are often viewed as genetically complex; yet highly penetrant autosomal dominant genes have been identified in some families (21–23).
A second complication is variable expressivity: mutations in a single gene can produce different epilepsy phenotypes in different individuals. The best example of this is generalized epilepsy with febrile seizures plus (GEFS+), in which the range of seizure disorders within a family with a single mutation in SCN1A can include typical febrile seizures, febrile seizures plus (i.e., febrile seizures persisting beyond age six, or accompanied by afebrile generalized tonic seizures), IGEs, temporal lobe epilepsy, myoclonic-astatic epilepsy, or SMEI (24,25). As with complex inheritance, this variability is likely to result from the modifying effects of other genes or environmental factors.
Figure 3 illustrates the extreme complexity in genotype–phenotype relations in the epilepsies, using the example of locus heterogeneity and variable expressivity in GEFS. Mutations in four different genes have been identified in different families with GEFS+, a syndrome that is, in itself, extremely variable in its phenotypic expression (26–31). Further, mutations in three of these four genes have been identified in other syndromes also. As noted above, de novo mutations in SCN1A have been found in many patients with SMEI (13–16). Many of the de novo mutations have been truncating, whereas the mutations found in GEFS+ families have all been missense. Mutations in SCN2A have been identified in families with benign familial neonatal infantile seizures (32); and mutations in the gene encoding the gamma 2 subunit of the GABA A receptor (GABRG2) have been found in families with childhood absence epilepsy with febrile seizures (33,34). In a GEFS+ family with a GABRG2 mutation, one individual was found to have SMEI (31), confirming the overlap between SMEI and GEFS+ (16).
FIG. 3.
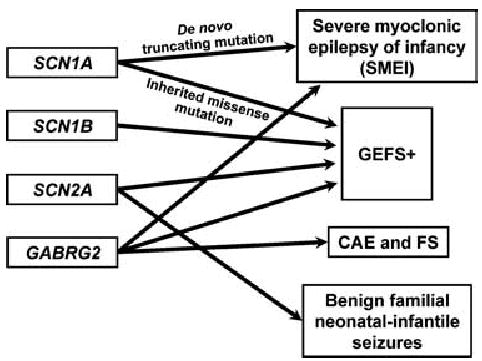
Locus heterogeneity and variable expressivity in GEFS+ and related phenotypes.
RESEARCH ISSUES AND APPROACHES
Two fundamental research questions are especially important in the investigation of genetic contributions to the epilepsies. The first pertains to phenotype definition: what clinical signs and symptoms are produced by specific genetic mechanisms? The second pertains to gene identification: which genes influence risk for human epilepsy, and how do variants in these genes raise seizure risk? The answers to these questions are crucial to the discovery of basic molecular mechanisms leading to epilepsy, and could someday be used to identify people at risk, develop new treatments, or—in the most hopeful scenario of all—develop methods to prevent onset of seizures in susceptible individuals.
Studies of phenotype definition
In understanding the relations between genotype and phenotype in the epilepsies, two alternative models can be envisioned (35,36): the first model postulates that different sets of genes influence risk for different epilepsy syndromes (“distinct genetic influences”), and the second, that the same genes influence risk for different epilepsy syndromes (“shared genetic influences”). Disentangling the shared and distinct genetic influences on different clinically defined subsets of epilepsy is essential for defining which patients should be included in specific types of genetic studies, to maximize genetic homogeneity and thus increase statistical power. Three different approaches have been used to advantage in studying shared and distinct genetic influences. Each of these approaches can also be used to study the possibility of shared genetic susceptibility to epilepsy and other disorders, such as migraine or depression.
In familial aggregation studies, familial risks are examined in a population context. A sample of people with epilepsy (probands) is ascertained and divided into subsets based on syndromes or other clinical features (age at onset, seizure type, etc.). Then the risk of these same subtypes of epilepsy (or other disorders) is examined in the relatives of the probands, and compared with the risk in the population or in the relatives of controls without epilepsy. If the genetic influences on different types of epilepsy are distinct, then among the relatives of probands with a given type, risk will be increased only for the same type as in the proband. On the other hand, if the genetic influences on different types of epilepsy are shared, risk in the relatives will be increased for all types, including those different from that in the proband. In two studies that used this approach, evidence was obtained for shared genetic influences on generalized and localization-related epilepsy. In the relatives of probands with generalized epilepsy, risk for localization-related epilepsy was significantly increased (fourfold), both in population-based data from Rochester, Minnesota (37), and data from the Epilepsy Family Study of Columbia University (38).
However, phenotype research using two other approaches has given different results. In a major twin study, Berkovic et al. (39) found that twins concordant for epilepsy tended to be concordant for syndrome also, suggesting that genetic effects are specific for epilepsy syndrome. Also, Winawer et al. (35,36,40) have used family concordance studies to test hypotheses about shared and distinct genetic influences on different clinically defined subsets of epilepsy. These studies assess the concordance of epilepsy types (syndromes, seizure types, or subsets defined by other clinical features) in families containing multiple affected individuals. The rationale for the analysis is that if some of the genetic influences on different epilepsy types are distinct, families will tend to be concordant—i.e., the proportion of families in which all affected individuals have the same type of epilepsy will exceed that expected by chance. The results of this research have provided evidence for distinct genetic influences on generalized and localization-related epilepsy (36), and, within the IGEs, for distinct genetic influences on myoclonic and absence seizures (40).
Why do the results of familial aggregation studies differ from those of other study designs? The answer to this question is unknown, but one possibility is that shared and distinct genetic effects coexist, and the relative importance of syndrome-specific genes (vs. genes with more general effects) may be greater in families containing multiple affected individuals than in families containing only one affected individual. In epidemiologic studies of familial aggregation, most patients are sporadic, and hence the effects of syndrome-specific genes may be less apparent than in other designs.
Identifying genes that raise risk for epilepsy
Two approaches are being used for gene identification in the epilepsies. The first, positional cloning, has been used to identify all of the genes in Mendelian epilepsy syndromes. It involves three steps: (1) identifying families containing multiple affected individuals, and carefully diagnosing and classifying all affected family members; (2) performing linkage analysis to localize a gene to a small chromosomal region; and (3) sequencing the genes in the linked region to identify a mutation in a specific gene that increases risk. The success of this method depends on the ability to use linkage analysis to localize the gene, and this is much easier with single gene disorders than with complex disorders (2). With complex disorders, statistical power for linkage detection is low, because of the small magnitude of each gene’s effect on disease risk. Also, practical problems complicate the design of linkage studies in complex diseases, such as the scarcity of families containing multiple affected individuals, and uncertainty about the mode of inheritance to be used in the analysis.
In epilepsy as in other disorders, allelic association studies are now being used as an alternative to positional cloning for the detection of complex disease genes (41). These studies are aimed at detecting genetic variants that are more common in people with epilepsy than in unaffected persons from the same population. A significantly increased frequency of a variant in people with epilepsy would suggest either (1) that it directly affects risk for epilepsy, or (2) that it is located very close to a functional variant on the same chromosome, and very often inherited with the functional variant (linkage disequilibrium). The genetic variants examined are usually single nucleotide polymorphisms (SNPs), common DNA sequence variations where one of the four nucleotides is substituted for another, found every 1,000–2,000 nucleotides in the human genome and accounting for about 90% of all DNA polymorphisms. Most allelic association studies have focused on variants in candidate genes with a hypothesized effect on disease risk (such as genes encoding ion channels). Recently, however, investigators have begun to consider seriously the feasibility of genome-wide association studies, and this approach will be used soon (42,43).
Allelic association studies have important advantages for the study of complex diseases. Unlike linkage studies, they do not require families with multiple affected individuals, which are generally so scarce in complex diseases. Also, association studies have greater statistical power than linkage studies for the detection of genes with a small effect on disease risk (2). The difference in statistical power can be very dramatic; for example, for a gene with a twofold effect on risk, one study estimated that 5,382 affected sibling pairs would be needed to detect linkage, whereas only 695 affected individuals and their parents would be needed to detect association (2).
Allelic association studies also have potential limitations. The validity of their basic underlying assumption—that complex diseases result from DNA variations common to a relatively large proportion of cases (the so-called “common disease–common variant hypothesis”) (44)—is still unknown. If many different combinations of risk-raising variants in multiple genes produce similar epilepsy phenotypes, none of the variants may be common enough to be detected through allelic association. Also, the contributions of somatic mutations and de novo mutations to complex epilepsies are unknown.
The potential for population stratification, a special type of confounding, must be considered in the design and interpretation of allelic association studies. It arises when the cases and controls in a study have different genetic ancestries, and the ancestral groups differ in their allelic distributions. As a result, the cases and controls could differ in the frequency of a SNP of interest, for reasons unrelated to the disease. The magnitude of the effect of stratification on current allelic association studies is controversial (45,46), but fortunately, a number of methods have been developed to control for its effects, including family-based association tests (47), genomic control (48), and structured association tests (49).
In the epilepsies, a large number of candidate gene-based allelic association studies has been published, and the pace of publication has increased dramatically in recent years [reviewed in (41)]. Many of the published studies have been plagued by methodologic limitations such as small sample size, lack of control for potential population stratification, and failure to adjust for multiple statistical tests. Few genetic variants have been examined in more than a single study, and among those that have, none has been associated consistently with a form of epilepsy in multiple studies. Thus, although clearly this approach holds great potential, we are still in the earliest stages of its application in the epilepsies.
Another approach, combining linkage and association analysis, has been used in the IGEs with intriguing results. A genome scan of 91 families with genetically complex adolescent onset IGEs provided evidence for a locus common to most IGEs on chromosome 18q21, a locus on chromosome 6p21 for juvenile myoclonic epilepsy, and other loci (on chromosomes 8 and 5) influencing risk for other forms of IGEs (50). The authors suggested that interactions of different combinations of these genes produce the varied phenotypes found in IGE families. In subsequent association studies in the same set of families, they found an association of juvenile myoclonic epilepsy with two SNPs in the promoter region of the BRD2 (RING3) gene on chromosome 6, suggesting that this might be the chromosome 6p21-linked juvenile myoclonic epilepsy gene, although no causative mutations were identified (51). They also found evidence for association of IGEs with a haplotype of SNPs within the malic enzyme 2 gene on chromosome 18, suggesting that this might be the chromosome 18-linked gene predisposing to IGE (52). These findings await confirmation.
RECOMMENDATIONS FOR FUTURE STUDIES
What approaches are likely to have the biggest impact on our understanding of the genetic contributions to the epilepsies? Although Mendelian epilepsy syndromes are rare, identification of the genes that cause them can provide extremely important information about basic epileptogenic mechanisms. Thus, efforts to identify, clinically characterize, and carry out positional cloning efforts in families containing multiple affected individuals should continue. Association studies are very promising for the identification of genes with smaller effects on risk. For these studies collaborative efforts are essential. Sample sizes must be expanded to ensure that statistical power is adequate for the detection of genes of small effects—thousands of people with epilepsy will need to be enrolled. Also, careful consideration must be given to the design of these studies, to ensure that phenotyping methods are of the highest quality (and standardized across collaborating centers), measures are taken to control for population stratification, and state-of-the-art molecular analysis methods are employed.
IMPLICATIONS FOR CLINICAL PRACTICE
In the genetically complex epilepsies, which affect the majority of people with epilepsy, genetic testing is not available; nor is it likely to be available in the near future. This is because multiple genes contribute to susceptibility, none of which has a major effect when acting by itself. The genes that influence risk have not yet been identified, and even when they are, risk prediction based on multigenic models (possibly interacting with environmental factors) will be very difficult. Consequently, in the genetically complex epilepsies, information for genetic counseling must be based on empirical risks from well-designed epidemiologic studies. Patients should be encouraged to participate in genetic studies, so that future generations may benefit from an improved understanding of the genetic contributions to epilepsy.
Acknowledgments
This work was supported by NIH grants R01 NS43472 and R01 NS36319.
References
- 1.Risch NJ. Searching for genetic determinants in the new millennium. Nature. 2000;405:847–56. doi: 10.1038/35015718. [DOI] [PubMed] [Google Scholar]
- 2.Risch N, Merikangas K. The future of genetic studies of complex human diseases. Science. 1996;273:1516–7. doi: 10.1126/science.273.5281.1516. [DOI] [PubMed] [Google Scholar]
- 3.Fisher RA. The correlation between relatives under the supposition of mendelian inheritance. Trans R Soc Edin. 1918;52:399–433. [Google Scholar]
- 4.Falconer DS. The inheritance of liability to certain diseases estimated from the incidence among relatives. Ann Hum Genet. 1965;29:51–76. [Google Scholar]
- 5.Ottman R. Simple test of the Multifactorial-Polygenic Model with sex dependent thresholds. J Chronic Dis. 1987;40:165–70. doi: 10.1016/0021-9681(87)90068-3. [DOI] [PubMed] [Google Scholar]
- 6.Ottman R. Gene-environment interaction: definitions and study designs. Prev Med. 1996;25:764–70. doi: 10.1006/pmed.1996.0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Annegers JF, Hauser WA, Anderson VE, et al. The risks of seizure disorders among relatives of patients with childhood onset epilepsy. Neurology. 1982;32:174–9. doi: 10.1212/wnl.32.2.174. [DOI] [PubMed] [Google Scholar]
- 8.Ottman R, Annegers JF, Risch N, et al. Relations of genetic and environmental factors in the etiology of epilepsy. Ann Neurol. 1996;39:442–9. doi: 10.1002/ana.410390406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ottman R, Lee JH, Risch N, et al. Clinical indicators of genetic susceptibility to epilepsy. Epilepsia. 1996;37:353–61. doi: 10.1111/j.1528-1157.1996.tb00571.x. [DOI] [PubMed] [Google Scholar]
- 10.Ottman R, Susser M. Data collection strategies in genetic epidemiology: the Epilepsy Family Study of Columbia University. J Clin Epidemiol. 1992;45:721–7. doi: 10.1016/0895-4356(92)90049-s. [DOI] [PubMed] [Google Scholar]
- 11.Ottman R, Hauser WA, Susser M. Validity of family history data on seizure disorders. Epilepsia. 1993;34:469–75. doi: 10.1111/j.1528-1157.1993.tb02587.x. [DOI] [PubMed] [Google Scholar]
- 12.Ottman R, Hauser WA, Barker-Cummings C, et al. Segregation analysis of cryptogenic epilepsy and an empirical test of the validity of the results. Am J Hum Genet. 1997;60:667–75. [PMC free article] [PubMed] [Google Scholar]
- 13.Claes L, Del-Favero J, Ceulemans B, et al. De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am J Hum Genet. 2001;68:1327–32. doi: 10.1086/320609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ohmori I, Ouchida M, Ohtsuka Y, et al. Significant correlation of the SCN1A mutations and severe myoclonic epilepsy in infancy. Biochem Biophys Res Commun. 2002;295:17–23. doi: 10.1016/s0006-291x(02)00617-4. [DOI] [PubMed] [Google Scholar]
- 15.Sugawara T, Mazaki-Miyazaki E, Fukushima K, et al. Frequent mutations of SCN1A in severe myoclonic epilepsy in infancy. Neurology. 2002;58:1122–4. doi: 10.1212/wnl.58.7.1122. [DOI] [PubMed] [Google Scholar]
- 16.Wallace RH, Hodgson BL, Grinton BE, et al. Sodium channel alpha1-subunit mutations in severe myoclonic epilepsy of infancy and infantile spasms. Neurology. 2003;61:765–9. doi: 10.1212/01.wnl.0000086379.71183.78. [DOI] [PubMed] [Google Scholar]
- 17.Ottman R, Winawer MR, Kalachikov S, et al. LGI1 mutations in autosomal dominant partial epilepsy with auditory features. Neurology. 2004;62:1120–6. doi: 10.1212/01.wnl.0000120098.39231.6e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bisulli F, Tinuper P, Avoni P, et al. Idiopathic partial epilepsy with auditory features (IPEAF): a clinical and genetic study of 53 sporadic cases. Brain. 2004;127:1343–52. doi: 10.1093/brain/awh151. [DOI] [PubMed] [Google Scholar]
- 19.Flex E, Pizzuti A, Di Bonaventura C, et al. LGI1 gene mutation screening in sporadic partial epilepsy with auditory features. J Neurol. 2005;252:62–6. doi: 10.1007/s00415-005-0599-0. [DOI] [PubMed] [Google Scholar]
- 20.Bisulli F, Tinuper P, Scudellaro E, et al. A de novo LGI1 mutation in sporadic partial epilepsy with auditory features. Ann Neurol. 2004;56:455–6. doi: 10.1002/ana.20218. [DOI] [PubMed] [Google Scholar]
- 21.Cossette P, Liu L, Brisebois K, et al. Mutation of GABRA1 in an autosomal dominant form of juvenile myoclonic epilepsy. Nat Genet. 2002;31:184–9. doi: 10.1038/ng885. [DOI] [PubMed] [Google Scholar]
- 22.Suzuki T, Delgado-Escueta AV, Aguan K, et al. Mutations in EFHC1 cause juvenile myoclonic epilepsy. Nat Genet. 2004;36:842–9. doi: 10.1038/ng1393. [DOI] [PubMed] [Google Scholar]
- 23.Haug K, Warnstedt M, Alekov AK, et al. Mutations in CLCN2 encoding a voltage-gated chloride channel are associated with idiopathic generalized epilepsies. Nat Genet. 2003;33:527–32. doi: 10.1038/ng1121. [DOI] [PubMed] [Google Scholar]
- 24.Singh R, Scheffer IE, Crossland K, et al. Generalized epilepsy with febrile seizures plus: a common childhood-onset genetic epilepsy syndrome. Ann Neurol. 1999;45:75–81. doi: 10.1002/1531-8249(199901)45:1<75::aid-art13>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 25.Mulley JC, Scheffer IE, Petrou S, et al. Channelopathies as a genetic cause of epilepsy. Curr Opin Neurol. 2003;16:171–6. doi: 10.1097/01.wco.0000063767.15877.c7. [DOI] [PubMed] [Google Scholar]
- 26.Wallace RH, Wang DW, Singh R, et al. Febrile seizures and generalized epilepsy associated with a mutation in the Na+-channel beta1 subunit gene SCN1B. Nat Genet. 1998;19:366–70. doi: 10.1038/1252. [DOI] [PubMed] [Google Scholar]
- 27.Escayg A, MacDonald BT, Meisler MH, et al. Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+2. Nat Genet. 2000;24:343–5. doi: 10.1038/74159. [DOI] [PubMed] [Google Scholar]
- 28.Sugawara T, Mazaki-Miyazaki E, Ito M, et al. Nav1.1 mutations cause febrile seizures associated with afebrile partial seizures. Neurology. 2001;57:703–5. doi: 10.1212/wnl.57.4.703. [DOI] [PubMed] [Google Scholar]
- 29.Sugawara T, Tsurubuchi Y, Agarwala KL, et al. A missense mutation of the Na+ channel alpha II subunit gene Na(v)1.2 in a patient with febrile and afebrile seizures causes channel dysfunction. Proc Natl Acad Sci U S A. 2001;98:6384–9. doi: 10.1073/pnas.111065098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baulac S, Huberfeld G, Gourfinkel-An I, et al. First genetic evidence of GABA(A) receptor dysfunction in epilepsy: a mutation in the gamma2-subunit gene. Nat Genet. 2001;28:46–8. doi: 10.1038/ng0501-46. [DOI] [PubMed] [Google Scholar]
- 31.Harkin LA, Bowser DN, Dibbens LM, et al. Truncation of the GABA(A)-receptor gamma2 subunit in a family with generalized epilepsy with febrile seizures plus. Am J Hum Genet. 2002;70:530–6. doi: 10.1086/338710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Heron SE, Crossland KM, Andermann E, et al. Sodium-channel defects in benign familial neonatal-infantile seizures. Lancet. 2002;360:851–2. doi: 10.1016/S0140-6736(02)09968-3. [DOI] [PubMed] [Google Scholar]
- 33.Wallace RH, Marini C, Petrou S, et al. Mutant GABA(A) receptor gamma2-subunit in childhood absence epilepsy and febrile seizures. Nat Genet. 2001;28:49–52. doi: 10.1038/ng0501-49. [DOI] [PubMed] [Google Scholar]
- 34.Kananura C, Haug K, Sander T, et al. A splice-site mutation in GABRG2 associated with childhood absence epilepsy and febrile convulsions. Arch Neurol. 2002;59:1137–41. doi: 10.1001/archneur.59.7.1137. [DOI] [PubMed] [Google Scholar]
- 35.Winawer M, Ottman R, Rabinowitz D. Concordance of disease form in kindreds ascertained through affected individuals. Stat Med. 2002;21:1887–97. doi: 10.1002/sim.988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Winawer MR, Rabinowitz D, Barker-Cummings C, et al. Evidence for distinct genetic influences on generalized and localization-related epilepsy. Epilepsia. 2003;44:1176–82. doi: 10.1046/j.1528-1157.2003.58902.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ottman R, Annegers JF, Hauser WA, et al. Seizure risk in offspring of parents with generalized versus partial epilepsy. Epilepsia. 1989;30:157–61. doi: 10.1111/j.1528-1157.1989.tb05448.x. [DOI] [PubMed] [Google Scholar]
- 38.Ottman R, Lee JH, Hauser WA, et al. Are generalized and localization-related epilepsies genetically distinct? Arch Neurol. 1998;55:339–44. doi: 10.1001/archneur.55.3.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Berkovic SF, Howell RA, Hay DA, et al. Epilepsies in twins: genetics of the major epilepsy syndromes. Ann Neurol. 1998;43:435–45. doi: 10.1002/ana.410430405. [DOI] [PubMed] [Google Scholar]
- 40.Winawer MR, Rabinowitz D, Pedley TA, et al. Genetic influences on myoclonic and absence seizures. Neurology. 2003;61:1576–81. doi: 10.1212/wnl.61.11.1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tan NC, Mulley JC, Berkovic SF. Genetic association studies in epilepsy: “the truth is out there. Epilepsia. 2004;45:1429–42. doi: 10.1111/j.0013-9580.2004.22904.x. [DOI] [PubMed] [Google Scholar]
- 42.Hirschhorn JN, Daly MJ. Genome-wide association studies for common diseases and complex traits. Nat Rev Genet. 2005;6:95–108. doi: 10.1038/nrg1521. [DOI] [PubMed] [Google Scholar]
- 43.Carlson CS, Eberle MA, Kruglyak L, et al. Mapping complex disease loci in whole-genome association studies. Nature. 2004;429:446–52. doi: 10.1038/nature02623. [DOI] [PubMed] [Google Scholar]
- 44.Halushka MK, Fan JB, Bentley K, et al. Patterns of single-nucleotide polymorphisms in candidate genes for blood-pressure homeostasis. Nat Genet. 1999;22:239–47. doi: 10.1038/10297. [DOI] [PubMed] [Google Scholar]
- 45.Thomas DC, Witte JS. Point: population stratification: a problem for case-control studies of candidate-gene associations? Cancer Epidemiol Biomarkers Prev. 2002;11:505–12. [PubMed] [Google Scholar]
- 46.Wacholder S, Rothman N, Caporaso N. Counterpoint: bias from population stratification is not a major threat to the validity of conclusions from epidemiological studies of common polymorphisms and cancer. Cancer Epidemiol Biomarkers Prev. 2002;11:513–20. [PubMed] [Google Scholar]
- 47.Zhao H. Family-based association studies. Stat Methods Med Res. 2000;9:563–87. doi: 10.1177/096228020000900604. [DOI] [PubMed] [Google Scholar]
- 48.Devlin B, Roeder K, Wasserman L. Genomic control, a new approach to genetic-based association studies. Theor Popul Biol. 2001;60:155–66. doi: 10.1006/tpbi.2001.1542. [DOI] [PubMed] [Google Scholar]
- 49.Pritchard JK, Donnelly P. Case-control studies of association in structured or admixed populations. Theor Popul Biol. 2001;60:227–37. doi: 10.1006/tpbi.2001.1543. [DOI] [PubMed] [Google Scholar]
- 50.Durner M, Keddache MA, Tomasini L, et al. Genome scan of idiopathic generalized epilepsy: evidence for major susceptibility gene and modifying genes influencing the seizure type. Ann Neurol. 2001;49:328–35. [PubMed] [Google Scholar]
- 51.Pal DK, Evgrafov OV, Tabares P, et al. BRD2 (RING3) is a probable major susceptibility gene for common juvenile myoclonic epilepsy. Am J Hum Genet. 2003;73:261–70. doi: 10.1086/377006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Greenberg DA, Cayanis E, Strug L, et al. Malic enzyme 2 may underlie susceptibility to adolescent-onset idiopathic generalized epilepsy. Am J Hum Genet. 2005;76:139–46. doi: 10.1086/426735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Singh NA, Charlier C, Stauffer D, et al. A novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns. Nat Genet. 1998;18:25–9. doi: 10.1038/ng0198-25. [DOI] [PubMed] [Google Scholar]
- 54.Charlier C, Singh NA, Ryan SG, et al. A pore mutation in a novel KQT-like potassium channel gene in an idiopathic epilepsy family. Nat Genet. 1998;18:53–5. doi: 10.1038/ng0198-53. [DOI] [PubMed] [Google Scholar]
- 55.Steinlein OK, Mulley JC, Propping P, et al. A missense mutation in the neuronal nicotinic acetylcholine receptor alpha 4 subunit is associated with autosomal dominant nocturnal frontal lobe epilepsy. Nat Genet. 1995;11:201–3. doi: 10.1038/ng1095-201. [DOI] [PubMed] [Google Scholar]
- 56.De Fusco M, Becchetti A, Patrignani A, et al. The nicotinic receptor beta 2 subunit is mutant in nocturnal frontal lobe epilepsy. Nat Genet. 2000;26:275–6. doi: 10.1038/81566. [DOI] [PubMed] [Google Scholar]
- 57.Kalachikov S, Evgrafov O, Ross B, et al. Mutations in LGI1 cause autosomal-dominant partial epilepsy with auditory features. Nat Genet. 2002;30:335–41. doi: 10.1038/ng832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Morante-Redolat JM, Gorostidi-Pagola A, Piquer-Sirerol S, et al. Mutations in the LGI1/Epitempin gene on 10q24 cause autosomal dominant lateral temporal epilepsy. Hum Mol Genet. 2002;11:1119–28. doi: 10.1093/hmg/11.9.1119. [DOI] [PubMed] [Google Scholar]