

Abstract
The adducin family of proteins interacts with the actin cytoskeleton and the plasma membrane in a calcium- and cAMP-dependent manner. Thus, adducins may be involved in changes in cytoskeletal organization resulting from synaptic stimulation. β-Adducin knock-out mice were examined in physiological and behavioral paradigms related to synaptic plasticity to elucidate the role the adducin family plays in processes underlying learning and memory. In situ hybridization for α- and β-adducin demonstrates that these mRNAs are found throughout the brain, with high levels of expression in the hippocampus. Schaffer collateral-CA1 tetanic long-term potentiation decayed rapidly in acute hippocampal slices from β-adducin knock-out mice, although baseline spine morphology and postsynaptic density were normal. Interestingly, the input-output relationship was significantly increased in hippocampal slices from β-adducin knock-out mice. Furthermore, β-adducin knock-out mice were impaired in performance of fear conditioning and the water maze paradigm. The current results indicate that β-adducin may play an important role in the cellular mechanisms underlying activity-dependent synaptic plasticity associated with learning and memory.
Keywords: cytoskeleton, memory, LTP, Morris water maze, fear conditioning, dendritic spines
Introduction
Changes in synaptic strength are thought to underlie learning and memory (Malenka, 2003; Lynch, 2004). Plastic changes have been shown to occur at several levels, including alterations in postsynaptic proteins and receptors (such as composition of receptor types, density, and phosphorylation state), morphological changes in postsynaptic dendrites, and changes in presynaptic function (Yuste and Bonhoeffer, 2001; Contractor and Heinemann, 2002; Sheng and Kim, 2002; Zucker and Regehr, 2002). Modification of spine shape and density occurs with strong stimulation that produces long-term potentiation (LTP) of synaptic transmission in cultured neurons (Engert and Bonhoeffer, 1999; Maletic-Savatic et al., 1999). Identifying proteins involved in cytoskeletal dynamics and understanding mechanisms that alter the stability of actin filaments could provide a clearer insight into the molecular changes associated with plasticity.
Adducins are attractive candidates for involvement in activity-dependent actin change because of their ability to translate inputs from different signaling pathways into structural changes. Adducins cap, bundle, and promote spectrin binding to actin filaments (Gardner and Bennett, 1987; Mische et al., 1987; Kuhlman et al., 1996). Calmodulin binding and phosphorylation by protein kinase A (PKA) and PKC can remove adducin complexes from the barbed ends of actin filaments, allowing them to polymerize or depolymerize (Matsuoka et al., 1996, 1998). In Aplysia californica, ApADD (the Aplysia homolog of mammalian adducin) is phosphorylated by PKC 18 h after induction of long-term facilitation at sensory neuron-motor neuron synapses (Gruenbaum et al., 2003). This supports the idea that adducin mediates cytoskeletal changes associated with synaptic activity, potentially by destabilizing the actin-spectrin meshwork after strong stimulation.
The mammalian adducin family is encoded by three genes (α, β, and γ) with several different splice variants (Suriyapperuma et al., 2000). Functional adducin is composed of heterodimers or heterotetramers of α/β or α/γ subunits (Dong et al., 1995; Hughes and Bennett, 1995). Adducin is found throughout the brain, with high expression of α-adducin in the hippocampus and cerebellum and lower expression throughout the cortex and striatum of rats (Seidel et al., 1995). The β subunit of adducin is specific to brain and erythrocytes (Gilligan et al., 1999).
In the current study, synaptic plasticity was examined in wild-type (WT) mice and knock-out (KO) mice lacking β-adducin at a variety of levels. In morphological studies, in situ hybridization was used to localize adducin mRNA, whereas transmission electron microscopy of the CA1 region of hippocampus was used to evaluate spine morphology and postsynaptic densities (PSDs). Cellular physiology studies examined induction of LTP in acute hippocampal slices from knock-out animals and littermate controls. Finally, mice were tested in fear conditioning and Morris water maze, behavioral tests of learning and memory. Our data suggest that adducin is involved in setting synaptic strength, as well as synaptic plasticity underlying learning and memory. We propose that adducin is an important molecule involved in initiating and establishing structural changes related to synaptic plasticity.
Materials and Methods
Subjects. Male and female β-adducin knock-out mice aged 8-12 months were generated as reported previously (Gilligan et al., 1999). All of the mice were backcrossed onto the C57BL/6J background (The Jackson Laboratory, Bar Harbor, ME) for at least 13 generations. Heterozygous breeding pairs were generated in the laboratory and were the parents for the wild-type and homozygous knock-out mice used in these studies. Sex-matched littermate controls were used for all of the behavioral experiments. Mice were group housed with no more than five animals per cage and kept in a colony room at 22°C on a 12 h light/dark cycle (lights on at 7:00 A.M.). Food and water were available ad libitum. Animals used in behavioral studies were bred at The Jackson Laboratory; all of the other animals used were bred and raised at Yale University. All of the animal experiments were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the Yale Animal Care and Use Committee.
In situ hybridization. Briefly, 14-μm-thick sections were cut on a cryostat at -20°C from fresh frozen mouse brains and thaw mounted onto Superfrost Plus microscope slides (Fisher Scientific, Pittsburgh, PA). Slides were stored at -80°C until being fixed. Sections were fixed for 10 min in 4% paraformaldehyde, for 1 min in PBS/100 mm triethanolamine (TEA), and for 15 min in 100 mm TEA/0.25% acetic anhydride. Slides were then rinsed two times for 2 min each in 2× SSC (2% sodium citrate, 2% sodium chloride), followed by ethanol dehydration (2 min in 30, 75, and 100% ethanol). Slides were allowed to dry before being stored at -80°C until hybridization.
Riboprobes specific to α- (361 bp sequence corresponding to α-adducin mRNA nucleotides 800-1160) and β- (292 bp sequence corresponding to β-adducin mRNA nucleotides 1966-2257) adducin were generated by PCR, followed by cloning into the pSPT 18 vector (Roche Diagnostics, Basel, Switzerland). mRNA was synthesized (SP6/T7 Transcription kit; Roche Diagnostics) using 35S-UTP (PerkinElmer Life Sciences, Wellesley, MA) according to the instructions of the manufacturer, and unincorporated nucleotides were removed by spinning the sample through a mini QuickSpin RNA column (Roche Diagnostics). Slides were prehybridized in 100 μl of buffer (50% formamide, 0.6 m NaCl, 10 mm Tris-HCl, pH 7.4, 1× Denhardt's solution, 10 mm DTT, 250 μg/ml tRNA, 100 μg/ml salmon sperm DNA, and 10% dextran sulfate) for 120 min at 60°C. The probe was then added to the hybridization buffer for an overnight incubation at 60°C. After hybridization, slides were rinsed twice in 2× SSC to remove coverslips, followed by a 30 min incubation in RNase [20 ml of 5 m NaCl, 2 ml of 1 m Tris, pH 8.0, and 400 μl of 0.5 m EDTA, pH 8.0, in 200 ml of Milli-Q purified water (Millipore, Bedford, MA)] to digest unbound riboprobe. Slides were then washed for 10 min in 2× SSC at room temperature, followed by 20 min in 0.2× SSC and 15 min in 0.1× SSC, both at 60°C. Finally, slides were rinsed in 0.1× SSC at room temperature for 1 min, dipped in Milli-Q water, and air dried. Slides were exposed to autoradiographic film (BioMax MR; Eastman Kodak, Rochester, NY) for 7-14 d. Digital images were captured, and density was analyzed using NIH ImageJ software. Average density in brain regions of interest were evaluated from three mice from each genotype and were expressed as arbitrary units. For each in situ hybridization experiment, a set of slides from one knock-out and one wild-type brain from sex-matched siblings was hybridized at the same time, and wild-type and knock-out sections were exposed on the same piece of film. Statistical analysis consisted of an ANOVA comparing regional densitometry between genotypes, with p > 0.05 considered significant.
Electrophysiology. Hippocampal slices were prepared from 3- to 5-week-old knock-out and wild-type littermates. Animals were anesthetized with halothane (Sigma, St. Louis, MO) and killed by decapitation. The brain was quickly removed and submerged in ice-cold artificial CSF (ACSF) solution (in mm: 125 NaCl, 2.5 KCl, 1 MgCl2, 1.25 NaH2PO4, 2 CaCl2, 25 glucose, and 26 NaHCO3). Hippocampi were dissected, and 350 μm transverse slices were cut with a vibratome (VT1000S; Leica, Wetzlar, Germany). Slices were allowed to recover for a minimum of 1 h at room temperature in a submerged chamber containing ACSF bubbled with 95% O2/5% CO2. Slices were transferred to a recording chamber, held submerged between two nylon nets, and constantly perfused at a rate of 2 ml/min with oxygenated ACSF containing 50 mm picrotoxin (Sigma) at room temperature. The recording chamber was mounted on an upright fixed-stage microscope (BX50WI; Olympus Optical, Tokyo, Japan). A cut was made between CA1 and CA3 to prevent the propagation of epileptiform activity. Schaffer collaterals (SCs) were stimulated orthodromically using bipolar tungsten electrodes (Frederick Haer Company, Bowdoinham, ME) with enough current (50 μs pulses) to reliably elicit synaptic responses. Test stimuli were applied at low frequency (0.05 Hz) at a stimulus intensity that elicited a field EPSP (fEPSP) amplitude that was 33% of maximum. All conditioning stimulation was at the same intensity as baseline. Synaptic responses were recorded extracellularly in the stratum radiatum of CA1 (50-100 μm from the stratum pyramidale) with glass microelectrodes (0.5-1 mW) filled with 2 m NaCl. Field potentials were amplified using a DP-301 differential amplifier (Warner Instruments, Hamden, CT), digitized at 10 kHz with pClamp 8.0 software (Axon Instruments, Union City, CA), and analyzed using macros written in IGOR Pro software (WaveMetrics, Lake Oswego, OR). fEPSP magnitude was measured using the initial fEPSP slope (between 10 and 90% points on the rising slope). After establishing a stable baseline, LTP was induced by high-frequency stimulation (two 100 Hz trains for 1 s each, delivered 20 s apart). To generate the input-output curves, slices were prepared as above and stimulated every 20 s with increasing intensity (from 0.0 to 0.2 mA in 0.01 mA increments) using a total of 20 stimuli. Data points were normalized to the mean baseline value and expressed as mean ± SEM. Data were analyzed using repeated-measures ANOVA. In all cases, p < 0.05 was considered statistically significant.
Electron microscopy. All of the reagents were purchased from Electron Microscopy Sciences (Hatfield, PA) unless otherwise noted. Animals were deeply anesthetized with 15% chloral hydrate and perfused intracardially with 0.9% heparinized saline, followed by 4% paraformaldehyde in PBS. Brains were dissected out and postfixed for 1 h in 4% glutaraldehyde and 0.1% paraformaldehyde at 4°C or at room temperature. Sections (700 μm) were cut from fixed brains using a vibratome, saving only those slices that had hippocampus throughout the section. Using a dissection microscope and very fine dissecting scissors, the stratum radiatum of the CA1 region was removed from both hemispheres. CA1 regions were fixed overnight in 4% glutaraldehyde and 0.1% paraformaldehyde at 4°C. The next day, the tissue was washed three times with 0.1 m NaPO4, pH 6, for 10 min at room temperature and then incubated in 1% OsO4 in 0.1% NaPO4, pH6, for 1 h at 4°C. Once again, tissue was washed three times with dH2O and incubated in progressive solutions of EtOH (25, 50, 70, 85, and 95%) at room temperature for 10 min to dehydrate. Subsequently, tissue was incubated with 100% EtOH three times at room temperature for 10 min and then three times in 100% propylene oxide at room temperature for 10 min. Resin [18 ml of EMbed 812 (Electron Microscopy Sciences), 13.4 ml of methyl-5-norbornene-2,3-dicarboxylic anhydride, 5 ml of dodecenyl succinic anhydride, and 0.6 ml of 2,4,6-tri(dimethylaminomethyl)phenol] was diluted both 1:1 and 3:1 (resin/propylene oxide). Tissue was placed in the 1:1 and then 3:1 mixes for 1 h each at room temperature, transferred to 100% resin, and poured into an embedding mold. Molds were baked at 60°C for at least 24 h until fully hardened. Semithin sections were cut, mounted on nickel grids, and counterstained with lead citrate and uranyl acetate. Grids were imaged using a Zeiss (Oberkochen, Germany) electron microscope, and photographs of randomized fields were taken at a magnification of 8000×. Negatives were developed and then scanned into Adobe Photoshop CS (Adobe Systems, Mountain View, CA), in which the values were reversed, and the contrast was adjusted before analysis with NIH ImageJ software. Measurements included asymmetrical synapse PSDs per square micrometer, and spine morphology (length of PSD, width of head at widest point, width of neck at narrowest point, and length of spine from tip of head to base of dendrite) for all of the spines found in which the PSD, head, neck, and parent dendrite were all connected in the field of view. Statistical analysis on synapse density was performed by taking the average density from each animal and comparing across genotype (n = 3 for each genotype, 30 photos per animal) using a Student's t test. Statistical analysis on spine morphology was performed using ANOVA.
Locomotor activity. Mice were placed in 45 × 24 × 20 cm clear plastic cages between two rows of photocells (8 total, 4 cm apart) for 20 min. Locomotor activity was assessed by measuring the number of beam breaks per minute and recorded by an IBM (White Plains, NY) computer using personalized Windows-compatible (Microsoft, Seattle, WA) software. Beam breaks were compiled into four 5 min bins for analysis. Statistics were performed using ANOVA.
Shock reactivity. Mice were placed into the Plexiglas-sided aluminum testing chamber used in fear conditioning experiments and were allowed to habituate for 3 min. Mice were then given a series of 1 s shocks with a 30 s intershock interval, beginning at 0.05 mA and increasing in increments of 0.05 mA, up to 1 mA. Latencies to flinch (any observable response to shock), run, jump, and vocalize were recorded. Mean current thresholds evoking each response were determined for each genotype. Mice were scored by two observers blind to genotype. Data were analyzed using one-way ANOVA.
Visual acuity. Mice were placed in a clear Plexiglas open-field testing chamber (44 × 44 × 20 cm) that was surrounded on all sides with a 1 inch square black and white checkerboard design. One-half of the chamber was suspended over a 44 × 22 × 80 cm open box lined with the same checkerboard design, resulting in the appearance of a visual cliff. Latencies for half-cross (two paws) and full-cross (four paws) of the visual cliff were measured by an observer blind to genotype. Data were analyzed using a Student's t test.
Fear conditioning. Mice were placed into a clear Plexiglas-sided aluminum testing chamber (43 × 17 × 25.5 cm; Med Associates, St. Albans, VT) that was contained in a modified Igloo ice chest (60 × 40 × 33 cm; Igloo Products, Katy, TX) with a clear Plexiglas window (60 × 33 cm) inserted into the front. Mounted on the back wall were a speaker for a computer-generated (8 dB) tone and a fan for ventilation and masking background noise. Before both the training and contextual tests, the chamber was cleaned with 70% ethanol. Isopropanol (70%) was used before the altered context and cued tests. During training, mice were scored for freezing behavior every 10 sec for 2 min before two presentations of a 30 s tone coterminal with a 2 s, 0.5 mA foot shock. There was a 2 min interval between shock pairings, and mice remained in the chamber for 30 s after the last shock, before being returned to their home cage.
Twenty-four hours later, mice were returned to the testing chamber (contextual test) and scored for freezing behavior for 6 min, after which they were returned to their home cage for at least 30 min before altered context and cued testing. For the altered context and cued tests, a clear plastic (29 × 18.5 × 13 cm) cage covered in a filtered lid was used instead of the aluminum/Plexiglas chamber. Additionally, a novel scent of orange extract mixed with water was introduced in a cup beside the cage. Freezing behavior was scored for 3 min (altered context test) before the tone was presented for an additional 3 min (cued test). Freezing behavior was defined as no movement except for respiration for a minimum of 1 s (Caldarone et al., 2000). Statistics were performed using a Student's t test (p < 0.05).
Morris water maze. Animals received four training trials per day between 10:30 A.M. and 2:00 P.M. for 12 d in a circular white plastic tank, 1 m in diameter, filled with water (21-22°C). A clear plastic platform (10 × 10 cm) was submerged 0.5 cm under water and placed in the same location in the tank over the training days. The tank was divided into four quadrants of equal size, with animals starting one training trial in each quadrant on all of the training days. The order of trials was pseudorandomized. Path length, time spent in each quadrant, and latency to find the platform were measured by a PolyTrack video tracking system (San Diego Instruments, San Diego, CA). Animals that did not find the platform within 60 s were placed manually onto the platform. All of the animals were allowed to remain on the platform for 15 s. The intertrial interval was 3.5 min. On days 8 and 13, the platform was removed, and the mice were placed in the middle of the tank and allowed to swim for 60 s (probe trial). Time spent in each quadrant, as well as the number of platform crosses, was recorded. On day 14, the platform was moved to a different quadrant and marked with a flag. Training in the visible task was the same as in the hidden platform procedure. Statistical analysis of swim-time data was performed using two-way ANOVA for repeated measures.
Results
Expression of α-adducin mRNA is unchanged in β-adducin knock-out mice
In situ hybridization was performed on wild-type and knock-out brain sections for both α- and β-adducin mRNA. β-Adducin mRNA is absent in most brain areas in knock-out mice, although some residual hybridization is found in the CA fields and dentate gyrus of the hippocampus (Fig. 1). The region of the β-adducin gene corresponding to the hybridization probe is not eliminated in the construct used to generate the β-adducin knock-out mice and has been shown to form an aberrant mRNA (Gilligan et al., 1999). Thus, the low residual hybridization in hippocampus is not likely to represent functional protein. β-Adducin mRNA is found throughout the forebrain of wild-type mice, and expression levels seem to correlate with cellular density (Fig. 1). For instance, the highest expression of β-adducin mRNA is seen in the stratum pyramidale of all of the hippocampal regions and in the molecular, Purkinje, and granular layers of the cerebellum. Other areas of high cellular density also show high levels of β-adducin mRNA, including the granule layer of the olfactory bulb, piriform cortex, layer 2 of the tenia tecta, bed nucleus of the stria terminalis, and several hypothalamic nuclei, such as the ventromedial preoptic and anteroventral periventricular nuclei. More moderate levels of expression are observed in the cortex and striatum, with a patchier pattern of expression in the striatum compared with relatively even labeling in cortical regions. Very little β-adducin mRNA expression is seen in the thalamus; however, certain nuclei, such as the medial geniculate and reticular and parafascicular nuclei show moderate levels of expression. β-Adducin mRNA expression is higher in cingulate cortex, compared with other cortical regions, and in the ventral striatum (nucleus accumbens) versus the dorsal striatum.
Figure 1.
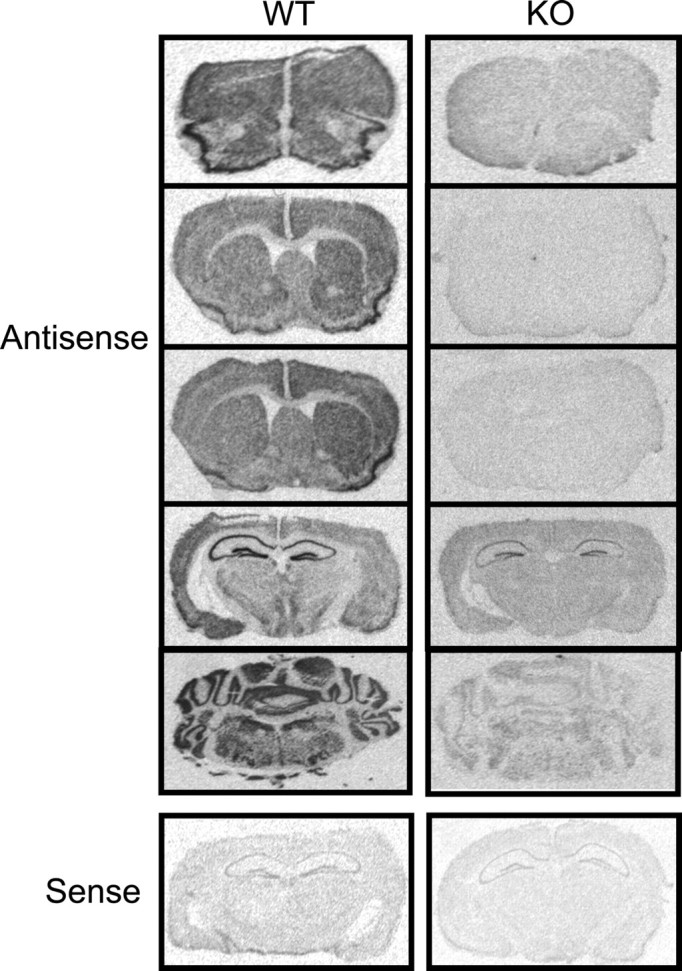
In situ hybridization for β-adducin. β-Adducin is found throughout the brain in wild-type animals, with high expression in the hippocampus, but is mostly absent in the brains of β-adducin knock-out mice. Low levels of probe hybridization seen in knock-out hippocampi indicate the presence of aberrant β-adducin mRNA (Gilligan et al., 1999).
α-Adducin mRNA expression does not differ between knock-out and wild-type mice throughout the orbitofrontal, prelimbic, and cingulate cortices, nucleus accumbens, and ventral tegmental area (Fig. 2, Table 1). Expression levels are also equivalent throughout the hippocampus and amygdala. The expression pattern of α-adducin mRNA is similar to that of β-adducin. α-Adducin mRNA levels are higher in the cingulate cortex than in other cortical regions and in the nucleus accumbens compared with the dorsal striatum. In general, the relative levels and expression pattern of α- and β-adducin mRNA overlap throughout the brain, as might be expected because these subunits combine as heterodimers to form functional adducin molecules. γ-Adducin may be upregulated (Gilligan et al., 1999) and compensate for the loss of β-adducin in knock-out mice, which would account for the similar expression levels of α-adducin seen in knock-out and wild-type mice.
Figure 2.
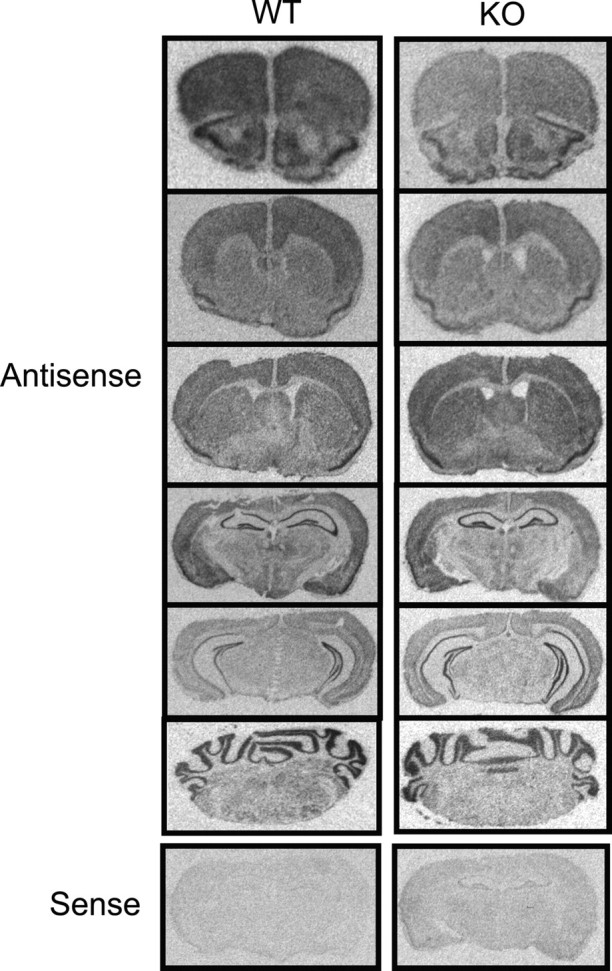
In situ hybridization for α-adducin. α-Adducin mRNA is expressed throughout the brain, with the highest expression found in the hippocampus. Expression levels were not different between wild-type and knock-out animals (p > 0.05 for all of the regions analyzed).
Table 1.
Quantitation of α-adducin in situ hybridization
|
|
Cortex |
NAc |
Amygdala |
Hippocampus |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
PL |
Cin |
Core |
Shell |
BLA |
CeA |
CA1 |
CA3 |
DG |
|||||
| WT | 121 ± 29 | 124 ± 29 | 126 ± 25 | 134 ± 25 | 113 ± 35 | 118 ± 58 | 112 ± 17 | 101 ± 25 | 93 ± 22 | |||||
| KO |
84 ± 6 |
101 ± 16 |
103 ± 13 |
105 ± 12 |
119 ± 21 |
133 ± 24 |
112 ± 17 |
105 ± 24 |
98 ± 19 |
|||||
NAc, Nucleus accumbens; PL, prelimbic; Cin, cingulate; BLA, basolateral amygdala; CeA, central nucleus of the amygdala; DG, dentate gyrus.
CA1 short- and long-term synaptic plasticity is impaired in β-adducin knock-out mice
The localization of β-adducin mRNA in synaptic regions thought to be critically involved in learning and memory suggested that β-adducin may be required for normal hippocampal neurophysiology. We compared extracellular field recordings of the SC-CA1 synapses in acute wild-type hippocampal slices with slices prepared from β-adducin knock-out mice. The fEPSP slope corresponding to a given presynaptic fiber volley did not seem to differ greatly between β-adducin knock-out and slices (Fig. 3A); however, the input-output relationship was significantly increased in slices from β-adducin knock-out mice compared with slices from wild-type mice (Fig. 3B), suggesting that β-adducin is normally involved in regulating synaptic strength in hippocampal neurons.
Figure 3.

β-Adducin knock-out animals do not show tetanic long-term potentiation in the SC-CA1 hippocampal circuit. A, Average presynaptic fiber volley amplitude (in millivolts) plotted against average fEPSP (in millivolts per millisecond) for each slice (WT, n = 17 slices from 7 mice; KO, n = 13 slices from 6 mice). B, Input-output relationships are significantly stronger in hippocampal slices from β-adducin knock-out mice compared with wild-type mice (F(1,39) = 10.113; p < 0.01; WT, n = 16 slices from 3 mice; KO, n = 25 slices from 3 mice). C, Paired-pulse facilitation in slices from β-adducin knock-out mice is similar to that in wild-type slices (F(1,18) = 0.600; p > 0.05; WT, n = 9 slices from 4 mice; KO, n = 10 slices from 5 mice). A slight enhancement of PPF can be seen at the 150 ms time point. Inset, Representative traces from wild-type and β-adducin knock-out slices. D, Long-term potentiation is impaired in β-adducin knock-out slices (WT, n = 17 slices from 7 mice; KO, n = 13 slices from 6 mice). Posttetanic potentiation does not differ between knock-out and wild-type slices (F(1,28) = 0.552; p > 0.05). Within 10 min, posttetanus responses in knock-out slices begin to decline, whereas wild-type responses remain potentiated (F(1,28) = 5.19; p < 0.05). Knock-out slices return to baseline response levels within 1 hr.
To evaluate whether normal presynaptic function requires β-adducin, we examined paired-pulse facilitation (PPF) by measuring fEPSP responses to two stimuli delivered at short interstimulus intervals to the SC inputs. PPF is a transient form of presynaptic plasticity, in which the response to the second stimulus is enhanced because of residual Ca2+ in the presynaptic terminal after the first stimulus. β-Adducin knock-out mice showed slightly enhanced PPF at the 150 ms interstimulus interval (Fig. 3C). A second measure of a short-term form of presynaptic plasticity is posttetanic potentiation (PTP). Stimulation of the SC with high-frequency tetani used to induce LTP results in an elevation of presynaptic Ca2+ and short-term enhancement of transmission caused by mobilization of the reserve pool of synaptic vesicles (Zucker, 1989). Using a tetanic induction protocol, we investigated PTP and the induction of LTP at SC-CA1 synapses in hippocampal slices from β-adducin knock-out mice and littermate controls (Fig. 3D). Immediately after two 1 s trains of 100 Hz stimulation, PTP was observed in slices from β-adducin knock-out mice, but responses decayed to baseline within 90 min. PTP was slightly less robust in slices from wild-type mice compared with knock-out slices, consistent with the data on differences in the input-output relationship, but responses remained potentiated for the duration of the experiment. Together, these experiments indicate that the β-adducin knock-out animals exhibit deficits in both short- and long-term synaptic plasticity.
Dendritic spines on the β-adducin knock-out mice seem normal
Dendritic spines in the stratum radiatum of the CA1 region were analyzed using transmission electron microscopy. The number and morphology of dendritic spines in β-adducin knock-out animals did not differ morphologically from wild-type spines. No differences were seen in PSD length, head width, neck width, or length between genotypes, nor was any difference in PSD observed (Fig. 4, Table 2).
Figure 4.
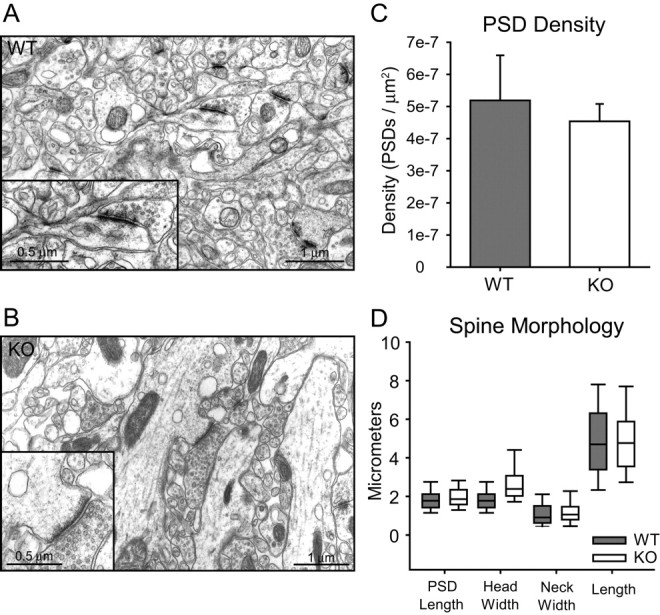
β-Adducin knock-out animals do not show differences in PSD density or spine morphology. Representative transmission electron microscopy images from wild-type (A) and knock-out (B) sections taken at 8000× magnification from CA1 stratum radiatum. Scale bars, 1 μm. C, There is no difference in density of asymmetrical PSDs from wild-type and knock-out animals (p > 0.05). D, PSD length, head width, neck width, and spine length do not differ between wild-type mice and knock-out mice (p > 0.05 for all measurements). Error bars represent SEM.
Table 2.
Electron microscopic analysis of spine characteristics in the CA1 region
|
|
Number of spines analyzed |
Area analyzed (μm2) |
PSDs/μm2 |
Spines/μm2 |
|---|---|---|---|---|
| WT (n = 3) | 188 | 1.70×109 ± 1.17×108 | 5.19×10−7 ± 1.40×10−8 | 3.66×10−8 ± 1.60×10−8 |
| KO (n = 3) |
144 |
1.75×109 ± 2.62×107
|
4.53×10−7 ± 5.42×10−8
|
2.75×10−8 ± 9.31×10−8
|
β-Adducin knock-out mice show generalized learning deficits
β-Adducin knock-out mice and their wild-type littermates showed levels of locomotor activity and habituation similar to a novel environment (Fig. 5A). In contrast, β-adducin knock-out animals were impaired in performance of both the water maze and fear conditioning tests compared with their wild-type littermates. In the fear conditioning paradigm (Fig. 5B), wild-type and knock-out mice had similar freezing responses in the training and altered context tasks; however, the β-adducin knock-out mice froze significantly less than wild-type controls in the context paired previously with foot shock and in response to the shock-paired auditory cue. No difference was observed between knock-out and wild-type mice in the shock reactivity test; therefore, impairment in performance was not attributable to altered pain thresholds in this task (flinch, F(1,14) = 0.572, p = 0.463; run, F(1,14) = 0.033, p = 0.859; jump, F(1,14) = 0.973, p = 0.341; and vocalize, F(1,14) = 0.860, p = 0.369; data not shown). The decreased freezing seen in knock-out mice to both the context and cued tasks suggests that both spatial and general learning are impaired in β-adducin knock-out mice.
Figure 5.

β-Adducin knock-out animals show deficits in the fear conditioning task. A, Baseline (Bsln) locomotor activity is not different between knock-out mice and controls (F(1,35) = 0.439; p > 0.05; WT, n = 14; KO, n = 19). B, Knock-out animals freeze less to both the context (Cx) (t(31) = 4.225; *p < 0.001) and auditory cue (t(31) = 2.920; **p < 0.01) conditions in fear conditioning (WT, n = 14; KO, n = 19). Alt, Altered. Error bars represent SEM.
In accordance with the fear conditioning findings, knock-out and wild-type mice did not differ in distance traveled on the first day of training in the hidden platform task of the Morris water maze; however, on subsequent training days, the distance traveled before finding the platform was significantly longer for knock-out mice than for their wild-type littermates (Fig. 6A). Swim speed did not differ between wild-type and knock-out mice (F(1,35) = 0.261; p = 0.613). Latency to reach the platform was also measured. Knock-out animals consistently lagged their wild-type counterparts by a few seconds throughout the training sessions as a result of a period of brief immobility each knock-out animal displayed when placed into the water, an effect that does not seem to be related to a learning phenotype (Nakamura-Palacios et al., 1996; Diana, 2002). Although β-adducin knock-out mice are not hypertensive (L. L. Peters, personal communication), this effect has also been reported in the spontaneously hypertensive strain of rats, a substrain of Wistar Kyoto, which has polymorphisms in all three of the adducin genes (Nakamura-Palacios et al., 1996; Tripodi et al., 1997; Diana, 2002). The first probe trial was given on day 8 of testing, but both groups performed at chance levels (data not shown). Therefore, training was reinstated for 4 more days before the second probe trial. During the second probe trial, wild-type mice spent 50% of the time in the quadrant containing the platform, whereas knock-out mice spent equivalent amounts of time in each quadrant (wild-type mice, F(1,3) = 75.932, p < 0.001; knock-out mice, F(1,3) = 1.859, p = 0.144) (Fig. 6B). Post hoc analysis of the probe trial data using Tukey's honestly significant difference (HSD) test showed that wild-type mice spent significantly more time in the target and less time in the opposite quadrants (p < 0.001) than in either adjacent quadrant. Knock-out mice did not cross representative platforms in the adjacent or opposite quadrants more often than wild-type mice (Fig. 6D) [wild-type mice, F(1,3) = 21.104, p < 0.001, with the target quadrant differing from all others (Tukey's HSD; p < 0.001); knock-out mice, F(1,3) = 1.743, p = 0.166]. In the visible platform task, knock-out and wild-type mice did not differ in distance traveled on the first day of training, but knock-out mice consistently swam farther to reach the target on subsequent training days (Fig. 6C). This effect cannot be accounted for by a loss of visual acuity in the β-adducin knock-out mice because animals of both genotypes performed similarly in the visual cliff test (t(14) = 0.531; p = 0.603). Although both knock-out and wild-type mice demonstrated steep learning curves for the visible platform task, the knock-out mice swam farther to reach the visible platform (F(1,35) = 15.257; p < 0.001), suggesting, like the fear conditioning data, that β-adducin knock-out animals have procedural learning deficits in addition to impaired spatial learning.
Figure 6.

β-Adducin knock-out animals show impairments in Morris water maze performance. A, Knock-out animals swim farther to find the hidden platform during training than wild-type mice (F(1,35) = 25.324; p < 0.0001; WT, n = 18; KO, n = 19). Data are plotted as path length, because β-adducin knock-out mice were briefly immobile when first placed into the water. Probe trials were given on days 8 and 13. B, Wild-type mice, but not knock-out mice, show preference for the platform-containing quadrant during the probe trial given on day 13 [wild-type mice, F(1,3) = 75.932, p < 0.001, with the target (T) and opposite (O) quadrants differing from both adjacent (Tukey's HSD; *p < 0.001); knock-out mice, F(1,3) = 1.859, p = 0.144; WT, n = 18; KO, n = 19]. Dashed line indicates chance performance. Neither wild-type mice nor knock-out mice showed a preference during the probe trial given on day 8 (data not shown). C, Knock-out mice swim farther to reach the platform on days 2-6 of the visible platform task (F(1,35) = 15.257; p < 0.001; WT, n = 18; KO, n = 19). D, Wild-type animals cross the location of the platform significantly more frequently than knock-out mice during the probe trial [wild-type mice, F(1,3) = 21.104, p < 0.001, with the target quadrant differing from all others (Tukey's HSD; *p < 0.001); knock-out mice, F(1,3) = 1.743, p = 0.166; WT, n = 18; KO, n = 19].
Discussion
The data from this study support the hypothesis that adducins are involved in synaptic plasticity underlying learning and memory. Loss of β-adducin, an isoform expressed highly throughout the brain, results in significant impairments in LTP and performance in behavioral measures of learning.
In situ hybridization studies of α- and β-adducin show that these isoforms are expressed widely throughout the mouse brain, particularly in areas known to undergo synaptic plasticity, such as the hippocampus, cerebellum, amygdala, and cortex. The current findings are consistent with previous studies that detected the expression of α- and β-adducin mRNA and α-adducin protein in the human brain by dot blotting (Gilligan et al., 1999) and immunocytochemistry (Seidel et al., 1995). Although α-adducin mRNA is found in both knock-out and wild-type brains, mRNA encoding β-adducin is primarily absent in the brains of β-adducin knock-out mice, as would be expected; however, some residual hybridization of the β-adducin probe remains in the hippocampus of knock-out animals. The production of an aberrant mRNA that does not result in a protein product has been shown previously (Gilligan et al., 1999), and the restriction of this mRNA to the hippocampus may be a result of higher expression levels in the CA fields. α-Adducin expression in the brain does not differ between knock-out and littermate control mice. This differs from the 30% decrease in α-adducin mRNA observed in red blood cells from β-adducin knock-out mice (Gilligan et al., 1999). An upregulation of γ-adducin expression has been shown previously in red blood cells (Gilligan et al., 1999). Also, α-adducin protein levels seem to be decreased and γ-adducin protein levels seem to be increased in knock-out brain homogenates by Western blot (Gilligan et al., 1999). Compensation by γ-adducin may therefore explain the maintenance of α-adducin mRNA levels in the brains of β-adducin knock-out mice. Alternatively, α-adducin mRNA expression may remain normal in brains of knock-out mice, but the polypeptide may be unstable in the absence of its associated subunit, either β- or γ-adducin.
Long-term changes in synaptic strength are generally accepted to underlie most types of learning and memory, and LTP in the SC-CA1 hippocampal circuit is frequently used as an experimental model of long-lasting synaptic potentiation (Martin and Morris, 2002). Also, dynamic changes in F-actin are necessary for plasticity in this circuit (Kim and Lisman, 1999; Krucker et al., 2000; Okamoto et al., 2004; Zito et al., 2004). Input-output analysis of β-adducin knock-out slices show enhanced basal neurotransmission in the SC-CA1 circuit at almost all of the stimulus intensities that evoke synaptic responses. A similar enhancement in basal neurotransmission in the same region from acute rat slices has been seen after treatment with indolactam V, an activator of all of the PKC isozymes (Hussain and Carpenter, 2003). Our data suggest that this effect may be mediated in part by adducins acting downstream of PKC activation. Enhanced basal transmission may also underlie the impairment in tetanus-induced LTP in β-adducin knock-out slices by occluding additional synaptic potentiation. Although β-adducin knock-out slices show normal posttetanic potentiation, field responses begin to decline 15 min after the tetanus. This is the time point at which postsynaptic changes that may depend on alteration of the actin cytoskeleton, such as AMPA receptor insertion or recruitment of newly synthesized proteins, are thought to occur (Allison et al., 1998; Shi et al., 1999; Hirai, 2000). Therefore, our findings are consistent with the idea that adducins are involved in postsynaptic changes in the actin cytoskeleton that occur as a result of synaptic activation.
PPF is widely regarded to be a measure of presynaptic efficacy (Hess et al., 1987; Zucker and Regehr, 2002). Thus, the small increase in PPF in β-adducin knock-out mice is likely to represent an increase in presynaptic neurotransmitter release. β-Adducin can interact with rabphilin-3a (Miyazaki et al., 1994; Burns et al., 1998), which is involved in synaptic vesicle docking (Geppert et al., 1994). Disruption of endogenous rabphilin-3a function by injection of a C-terminal fragment of rabphilin-3a that binds β-adducin results in altered presynaptic morphology and endocytosis, as well as in an increased number of synaptic vesicles 50-100 nm from the plasma membrane in the squid giant synapse (Burns et al., 1998). Actin in the presynaptic terminal has been proposed to form a network around the readily releasable pool of vesicles (for review, see Doussau and Augustine, 2000), and adducins may be involved in regulating this network and maintaining the readily releasable pool. Alternatively, adducins could affect presynaptic function by regulating the actin network that serves as support for other presynaptic components that regulate vesicle release and reuptake (Mundigl et al., 1998; Colicos et al., 2001; Cremona and De Camilli, 2001).
Based on our physiological findings, we initially hypothesized that disruption of membrane-cytoskeleton interactions in β-adducin knock-out mice could result in a change in spine shape or density. Although many groups have reported changes in spine morphology associated with impairments in LTP after strong stimulation (Engert and Bonhoeffer, 1999; Maletic-Savatic et al., 1999; Chen et al., 2004) and exposure to complex environments (Kolb et al., 2003), dendritic spines in the stratum radiatum of the CA1 region in β-adducin knock-out animals did not differ morphologically from wild-type spines when analyzed using transmission electron microscopy. Therefore, β-adducin may not be necessary for these structural changes, or our measurements may not be sufficiently sensitive to detect subtle differences in spine morphology. Future studies using stimuli known to result in dramatic changes in dendritic spines and the use of more sensitive assays of spine shape and density, such as Golgi staining, will address this issue.
In agreement with the physiological data, β-adducin knock-out animals show impaired performance in behavioral tasks of learning and memory, specifically in fear conditioning and the Morris water maze. The impairment in performance in these tasks may reflect a less-plastic synapse in which incoming information is not adequately encoded. In both the Morris maze and the fear conditioning task, knock-out animals were impaired in both the hippocampal-dependent learning components of the tasks and the cued versions of the tasks. This suggests that plasticity in other brain areas is impaired in β-adducin knock-out mice. α-Adducin and β-adducin, although highly expressed in the hippocampus, are also found in additional brain regions. Specifically, these adducin isoforms are also found throughout the amygdalar complex, which is critical in mediating the response to aversive stimuli such as foot shock or immersion in water (Grace and Rosenkranz, 2002; LeDoux, 2003) and has more recently been implicated in reward-associated learning (Holland and Gallagher, 2004; Kelley, 2004). In addition, impairments in learning performance may result from the loss of adducin in cortical areas because plasticity in these areas is clearly associated with learning as well (Gilbert et al., 2001; Schultz, 2004).
The activity of adducin is regulated by several kinases that are known to be activated as a result of synaptic activity (Matsuoka et al., 1996, 1998; Fukata et al., 1999). Phosphorylation by PKA, PKC, or calmodulin binding causes adducin to dissociate from actin filaments, whereas phosphorylation by Rho kinase increases affinity of adducin for actin filaments. In activated platelets, adducin is phosphorylated by PKC and becomes a better substrate for calpain proteolysis, suggesting that adducin complexes play a role in maintaining the shape of resting platelets but need to be removed from actin filaments during activation (Gilligan et al., 2002). Therefore, adducin could act as an intermediary between synaptic signaling and the resulting cytoskeletal plasticity. Adducin phosphorylation could destabilize the postsynaptic cytoskeleton, allowing changes in the postsynaptic milieu to occur, and could be followed by adducin dephosphorylation, leading to its reassociation with the actin network and thereby to the restabilization of the cytoskeleton afterward. The rapid decay of CA1 LTP in hippocampal slices from β-adducin knock-out animals supports this idea, because these slices show normal potentiation up to the time point at which the cytoskeleton is thought to be involved.
In summary, the current data support the idea that β-adducin is involved in plastic changes at the synapse that occur as a result of synaptic activity. We hypothesize that adducins may be critical players in the regulation of the stability of the postsynaptic actin-spectrin cytoskeleton. The importance of adducin-mediated changes in learning and memory is strongly supported by the poor performance of the β-adducin knock-out animals in several learning tasks. The loss of adducin may result in synapses that are less responsive to stimulation and therefore may impair normal processes underlying learning and memory.
Footnotes
This work was supported by National Institutes of Health Grants DA14331, DA00436 (M.R.P.), MH59800 (R.M.F.), DK55005 (D.M.G.), and HL75714 (L.L.P.) and by an American Psychological Association Diversity Program in Neuroscience grant (N.A.A.). We thank Emily Osterweil, Jennifer Bourne, and David Wells for their assistance with electron microscopy.
Correspondence should be addressed to Marina R. Picciotto, Department of Psychiatry, Yale University School of Medicine, 34 Park Street, 3rd Floor Research, New Haven, CT 06508. E-mail: marina.picciotto@yale.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/252138-08$15.00/0
References
- Allison DW, Gelfand VI, Spector I, Craig AM (1998) Role of actin in anchoring postsynaptic receptors in cultured hippocampal neurons: differential attachment of NMDA versus AMPA receptors. J Neurosci 18: 2423-2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns ME, Sasaki T, Takai Y, Augustine GJ (1998) Rabphilin-3A: a multifunctional regulator of synaptic vesicle traffic. J Gen Physiol 111: 243-255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldarone BJ, Duman CH, Picciotto MR (2000) Fear conditioning and latent inhibition in mice lacking the high affinity subclass of nicotinic acetylcholine receptors in the brain. Neuropharmacology 39: 2779-2784. [DOI] [PubMed] [Google Scholar]
- Chen Y, Bourne J, Pieribone VA, Fitzsimonds RM (2004) The role of actin in the regulation of dendritic spine morphology and bidirectional synaptic plasticity. NeuroReport 15: 829-832. [DOI] [PubMed] [Google Scholar]
- Colicos MA, Collins BE, Sailor MJ, Goda Y (2001) Remodeling of synaptic actin induced by photoconductive stimulation. Cell 107: 605-616. [DOI] [PubMed] [Google Scholar]
- Contractor A, Heinemann SF (2002) Glutamate receptor trafficking in synaptic plasticity. Sci STKE 156: RE14. [DOI] [PubMed] [Google Scholar]
- Cremona O, De Camilli P (2001) Phosphoinositides in membrane traffic at the synapse. J Cell Sci 114: 1041-1052. [DOI] [PubMed] [Google Scholar]
- Diana G (2002) Does hypertension alone lead to cognitive decline in spontaneously hypertensive rats? Behav Brain Res 134: 113-121. [DOI] [PubMed] [Google Scholar]
- Dong L, Chapline C, Mousseau B, Fowler L, Ramsay K, Stevens JL, Jaken S (1995) 35H, a sequence isolated as a protein kinase C binding protein, is a novel member of the adducin family. J Biol Chem 270: 25534-25540. [DOI] [PubMed] [Google Scholar]
- Doussau F, Augustine GJ (2000) The actin cytoskeleton and neurotransmitter release: an overview. Biochimie 82: 353-363. [DOI] [PubMed] [Google Scholar]
- Engert F, Bonhoeffer T (1999) Dendritic spine changes associated with hippocampal long-term synaptic plasticity. Nature 399: 66-70. [DOI] [PubMed] [Google Scholar]
- Fukata Y, Oshiro N, Kinoshita N, Kawano Y, Matsuoka Y, Bennett V, Matsuura Y, Kaibuchi K (1999) Phosphorylation of adducin by Rho-kinase plays a crucial role in cell motility. J Cell Biol 145: 347-361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner K, Bennett V (1987) Modulation of spectrin-actin assembly by erythrocyte adducin. Nature 328: 359-362. [DOI] [PubMed] [Google Scholar]
- Geppert M, Bolshakov VY, Siegelbaum SA, Takei K, De Camilli P, Hammer RE, Sudhof TC (1994) The role of Rab3A in neurotransmitter release. Nature 369: 493-497. [DOI] [PubMed] [Google Scholar]
- Gilbert CD, Sigman M, Crist RE (2001) The neural basis of perceptual learning. Neuron 31: 681-697. [DOI] [PubMed] [Google Scholar]
- Gilligan DM, Lozovatsky L, Gwynn B, Brugnara C, Mohandas N, Peters LL (1999) Targeted disruption of the beta adducin gene (Add2) causes red blood cell spherocytosis in mice. Proc Natl Acad Sci USA 96: 10717-10722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilligan DM, Sarid R, Weese J (2002) Adducin in platelets: activation-induced phosphorylation by PKC and proteolysis by calpain. Blood 99: 2418-2426. [DOI] [PubMed] [Google Scholar]
- Grace AA, Rosenkranz JA (2002) Regulation of conditioned responses of basolateral amygdala neurons. Physiol Behav 77: 489-493. [DOI] [PubMed] [Google Scholar]
- Gruenbaum LM, Gilligan DM, Picciotto MR, Marinesco S, Carew TJ (2003) Identification and characterization of Aplysia adducin, an Aplysia cytoskeletal protein homologous to mammalian adducins: increased phosphorylation at a protein kinase C consensus site during long-term synaptic facilitation. J Neurosci 23: 2675-2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess G, Kuhnt U, Voronin LL (1987) Quantal analysis of paired-pulse facilitation in guinea pig hippocampal slices. Neurosci Lett 77: 187-192. [DOI] [PubMed] [Google Scholar]
- Hirai H (2000) Clustering of delta glutamate receptors is regulated by the actin cytoskeleton in the dendritic spines of cultured rat Purkinje cells. Eur J Neurosci 12: 563-570. [DOI] [PubMed] [Google Scholar]
- Holland PC, Gallagher M (2004) Amygdala-frontal interactions and reward expectancy. Curr Opin Neurobiol 14: 148-155. [DOI] [PubMed] [Google Scholar]
- Hughes CA, Bennett V (1995) Adducin: a physical model with implications for function in assembly of spectrin-actin complexes. J Biol Chem 270: 18990-18996. [DOI] [PubMed] [Google Scholar]
- Hussain RJ, Carpenter DO (2003) The effects of protein kinase C activity on synaptic transmission in two areas of rat hippocampus. Brain Res 990: 28-37. [DOI] [PubMed] [Google Scholar]
- Kelley AE (2004) Ventral striatal control of appetitive motivation: role in ingestive behavior and reward-related learning. Neurosci Biobehav Rev 27: 765-776. [DOI] [PubMed] [Google Scholar]
- Kim CH, Lisman JE (1999) A role of actin filament in synaptic transmission and long-term potentiation. J Neurosci 19: 4314-4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolb B, Gorny G, Soderpalm AH, Robinson TE (2003) Environmental complexity has different effects on the structure of neurons in the prefrontal cortex versus the parietal cortex or nucleus accumbens. Synapse 48: 149-153. [DOI] [PubMed] [Google Scholar]
- Krucker T, Siggins GR, Halpain S (2000) Dynamic actin filaments are required for stable long-term potentiation (LTP) in area CA1 of the hippocampus. Proc Natl Acad Sci USA 97: 6856-6861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlman PA, Hughes CA, Bennett V, Fowler VM (1996) A new function for adducin. Calcium/calmodulin-regulated capping of the barbed ends of actin filaments. J Biol Chem 271: 7986-7991. [DOI] [PubMed] [Google Scholar]
- LeDoux J (2003) The emotional brain, fear, and the amygdala. Cell Mol Neurobiol 23: 727-738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch MA (2004) Long-term potentiation and memory. Physiol Rev 84: 87-136. [DOI] [PubMed] [Google Scholar]
- Malenka RC (2003) Synaptic plasticity and AMPA receptor trafficking. Ann NY Acad Sci 1003: 1-11. [DOI] [PubMed] [Google Scholar]
- Maletic-Savatic M, Malinow R, Svoboda K (1999) Rapid dendritic morphogenesis in CA1 hippocampal dendrites induced by synaptic activity. Science 283: 1923-1927. [DOI] [PubMed] [Google Scholar]
- Martin SJ, Morris RG (2002) New life in an old idea: the synaptic plasticity and memory hypothesis revisited. Hippocampus 12: 609-636. [DOI] [PubMed] [Google Scholar]
- Matsuoka Y, Hughes CA, Bennett V (1996) Adducin regulation. Definition of the calmodulin-binding domain and sites of phosphorylation by protein kinases A and C. J Biol Chem 271: 25157-25166. [DOI] [PubMed] [Google Scholar]
- Matsuoka Y, Li X, Bennett V (1998) Adducin is an in vivo substrate for protein kinase C: phosphorylation in the MARCKS-related domain inhibits activity in promoting spectrin-actin complexes and occurs in many cells, including dendritic spines of neurons. J Cell Biol 142: 485-497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mische SM, Mooseker MS, Morrow JS (1987) Erythrocyte adducin: a calmodulin-regulated actin-bundling protein that stimulates spectrin-actin binding. J Cell Biol 105: 2837-2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki M, Shirataki H, Kohno H, Kaibuchi K, Tsugita A, Takai Y (1994) Identification as beta-adducin of a protein interacting with rabphilin-3A in the presence of Ca2+ and phosphatidylserine. Biochem Biophys Res Commun 205: 460-466. [DOI] [PubMed] [Google Scholar]
- Mundigl O, Ochoa GC, David C, Slepnev VI, Kabanov A, De Camilli P (1998) Amphiphysin I antisense oligonucleotides inhibit neurite out-growth in cultured hippocampal neurons. J Neurosci 18: 93-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura-Palacios EM, Caldas CK, Fiorini A, Chagas KD, Chagas KN, Vasquez EC (1996) Deficits of spatial learning and working memory in spontaneously hypertensive rats. Behav Brain Res 74: 217-227. [DOI] [PubMed] [Google Scholar]
- Okamoto K, Nagai T, Miyawaki A, Hayashi Y (2004) Rapid and persistent modulation of actin dynamics regulates postsynaptic reorganization underlying bidirectional plasticity. Nat Neurosci 7: 1104-1112. [DOI] [PubMed] [Google Scholar]
- Schultz W (2004) Neural coding of basic reward terms of animal learning theory, game theory, microeconomics and behavioural ecology. Curr Opin Neurobiol 14: 139-147. [DOI] [PubMed] [Google Scholar]
- Seidel B, Zuschratter W, Wex H, Garner CC, Gundelfinger ED (1995) Spatial and sub-cellular localization of the membrane cytoskeleton-associated protein alpha-adducin in the rat brain. Brain Res 700: 13-24. [DOI] [PubMed] [Google Scholar]
- Sheng M, Kim MJ (2002) Postsynaptic signaling and plasticity mechanisms. Science 298: 776-780. [DOI] [PubMed] [Google Scholar]
- Shi SH, Hayashi Y, Petralia RS, Zaman SH, Wenthold RJ, Svoboda K, Malinow R (1999) Rapid spine delivery and redistribution of AMPA receptors after synaptic NMDA receptor activation. Science 284: 1811-1816. [DOI] [PubMed] [Google Scholar]
- Suriyapperuma SP, Lozovatsky L, Ciciotte SL, Peters LL, Gilligan DM (2000) The mouse adducin gene family: alternative splicing and chromosomal localization. Mamm Genome 11: 16-23. [DOI] [PubMed] [Google Scholar]
- Tripodi G, Szpirer C, Reina C, Szpirer J, Bianchi G (1997) Polymorphism of gamma-adducin gene in genetic hypertension and mapping of the gene to rat chromosome 1q55. Biochem Biophys Res Commun 237: 685-689. [DOI] [PubMed] [Google Scholar]
- Yuste R, Bonhoeffer T (2001) Morphological changes in dendritic spines associated with long-term synaptic plasticity. Annu Rev Neurosci 24: 1071-1089. [DOI] [PubMed] [Google Scholar]
- Zito K, Knott G, Shepherd GM, Shenolikar S, Svoboda K (2004) Induction of spine growth and synapse formation by regulation of the spine actin cytoskeleton. Neuron 44: 321-334. [DOI] [PubMed] [Google Scholar]
- Zucker RS (1989) Short-term synaptic plasticity. Annu Rev Neurosci 12: 13-31. [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG (2002) Short-term synaptic plasticity. Annu Rev Physiol 64: 355-405. [DOI] [PubMed] [Google Scholar]