

Abstract
The social amoebae are exceptional in their ability to alternate between unicellular and multicellular forms. Here we describe the genome of the best-studied member of this group, Dictyostelium discoideum. The gene-dense chromosomes encode ~12,500 predicted proteins, a high proportion of which have long repetitive amino acid tracts. There are many genes for polyketide synthases and ABC transporters, suggesting an extensive secondary metabolism for producing and exporting small molecules. The genome is rich in complex repeats, one class of which is clustered and may serve as centromeres. Partial copies of the extrachromosomal rDNA element are found at the ends of each chromosome, suggesting a novel telomere structure and the use of a common mechanism to maintain both the rDNA and chromosomal termini. A proteome-based phylogeny shows that the amoebozoa diverged from the animal/fungal lineage after the plant/animal split, but Dictyostelium appears to have retained more of the diversity of the ancestral genome than either of these two groups.
The amoebozoa are a richly diverse group of organisms whose genomes remain largely unexplored. The soil-dwelling social amoeba Dictyostelium discoideum has been actively studied for the past fifty years and has contributed greatly to our understanding of cellular motility, signalling and interaction1. For example, studies in Dictyostelium provided the first descriptions of a eukaryotic cell chemo-attractant and a cell-cell adhesion protein2, 3.
Dictyostelium amoebae inhabit forest soil consuming bacteria and yeast, which they track by chemotaxis. Starvation, however, prompts the solitary cells to aggregate and to develop as a true multicellular organism, producing a fruiting body comprised of a cellular, cellulosic stalk supporting a bolus of spores. Thus, Dictyostelium has evolved mechanisms that direct the differentiation of a homogeneous population of cells into distinct cell types, regulate the proportions between tissues and orchestrate the construction of an effective structure for the dispersal of spores4. Many of the genes necessary for these processes in Dictyostelium were also inherited by metazoa and fashioned through evolution for use within many different modes of development.
The amoebozoa are also noteworthy as representing one of the earliest branches from the last common ancestor of all eukaryotes. Each of the surviving branches of the crown group of eukaryotes provides an example of the ways in which the ancestral genome has been sculpted and adapted by lineage-specific gene duplication, divergence and deletion. Comparison between representatives of these branches promises to shed light not only on the nature and content of the ancestral eukaryotic genome, but on the diversity of ways in which its components have been adapted to meet the needs of complex organisms. The genome of Dictyostelium, as the first free-living protozoan to be fully sequenced, should be particularly informative for these analyses.
Mapping, sequencing and assembly
An international initiative to sequence the genome of Dictyostelium discoideum AX4 (references 5, 6) was launched in 1998. The high repeat-content and A+T-richness of the genome (the latter rendering large-insert bacterial clones unstable) posed severe challenges for sequencing and assembly. The response to these challenges was to use a whole-chromosome shotgun (WCS) strategy, partially purifying each chromosome electrophoretically and treating it as a separate project. This approach was supported by novel statistical tools to recover chromosome specificity from the impure WCS libraries, and by highly detailed HAPPY maps that provided a framework for sequence assembly. These approaches have enabled the completion of this difficult genome to a high standard, and are likely to be valuable in tackling the many other genomes which present challenges of composition and complexity.
Genome mapping
To support sequence assembly, we made high-resolution maps of the chromosomes using HAPPY mapping7–9, which relies on analysing the sequence content of single DNA molecules prepared by limiting dilution. A total of 3902 markers selected mostly from the emerging shotgun data were mapped, and maps of all six chromosomes were assembled (see Methods; Table 1;Fig. SI 1; Table SI 1).
Table 1.
Sequence assembly details.
| Chromosome | 1 | 2 | 3 | 4 | 5 | 6 | All chrs |
|---|---|---|---|---|---|---|---|
| Feature | |||||||
| Chromosomal assemblies | |||||||
| Assembly span (bp)1 | 4,919,822 | 8,467,571 | 6,358,352 | 5,430,575 | 5,062,323 | 3,578,828 | 33,817,471 |
| Assembly seq. (bp)2 | 4,911,622 | 8,437,971 | 6,334,852 | 5,397,875 | 5,032,273 | 3,547,128 | 33,661,721 |
| Total contigs | 11 | 40 | 32 | 65 | 107 | 44 | 309 |
| Mean contig size (bp) | 446,511 | 210,949 | 197964 | 83044 | 47031 | 80617 | 108,938 |
| No. of sequence gaps | 4 | 12 | 10 | 34 | 81 | 14 | 155 |
| No. of repeat gaps | 8 | 29 | 23 | 9 | 4 | 11 | 84 |
| No. of clone gaps | 0 | 0 | 0 | 22 | 22 | 20 | 64 |
| Total est. gap size (bp)3 | 8,200 | 29,600 | 23,500 | 32,700 | 30,050 | 31,700 | 155,750 |
| No. HAPPY markers (mean spacing/kb) | 749 (6.6) | 615(12.5)4 | 684 (9.3) | 628 (8.6) | 628 (8.1) | 598 (6.0) | 3902 (8.7) |
| Floating contigs5 | |||||||
| No. of floating contigs | 0 | 22 | 3 | 96 | 0 | 34 | |
| Total size of floating contigs (bp) | 0 | 171,670 | 16,360 | 37309 | 0 | 225,339 | |
| Combined (assemblies plus floating contings) | |||||||
| Total sequence (bp) | 4,911,622 | 8,609,641 | 6,351,212 | 5,416,5296 | 5,050,9286 | 3,547,128 | 33,887,060 |
| Mean coverage (fold) | 9.1 | 6.5 | 6.7 | 9.6 | 9.9 | 10.3 | 8.3 |
Total end-to-end length of the chromosomal assembly, including any gaps.
Sequenced bases covered by chromosomal assembly, not counting gaps.
Sequence, repeat and clone gaps are taken to have average sizes of 50bp, 1000bp and 1000bp, respectively.
Does not include the second copy of the 755kb inverted duplication.
Includes only those contigs which can be assigned to specific chromosomes.
Floating contigs from chromosomes 4 and 5 cannot be distinguished. In calculating total chromosomal sequence, we assume that half of these floatigs are from each of chromosomes 4 and 5.
Genome sequencing and assembly
Two strategies were used to recover chromosome-specific data from impure WCS libraries (see Methods). The first - employed for chromosomes 1, 2 and 3 - used enrichment of the respective libraries as the main statistical indicator of the chromosomal assignment of contigs, and on HAPPY maps to guide assembly. The second - for chromosomes 4, 5 and most of 6 - used mapping data to chromosomally assign sequences initially, with detailed HAPPY maps being used to validate final assemblies. A 1508kb portion of chromosome 6 was sequenced as a pilot project using a combination of approaches (see Methods).
Repetitive tracts complicated assembly. For chromosomes 1, 2 and 3, inspection of polymorphisms, combined with HAPPY maps, allowed unambiguous assembly in many cases. For chromosomes 4, 5 and 6, low-coverage sequencing of AX4-derived YACs alleviated the problems by providing a local dataset within which the troublesome repeat element was present as a single copy. Nevertheless, some repeat tracts proved intractable and remain as gaps. Thirty-four unlinked (‘floating’) contigs of >1kb, totalling 225,339bp, remain unpositioned in the genome, but can be provisionally assigned to specific chromosomes based on their content of reads from the WCS libraries. Most or all of these floating contigs are bounded by repetitive regions. The chromosome 2 sequence in the current assembly supercedes that previously published9, having benefited from further HAPPY mapping and manual sequence finishing. The six chromosomal assemblies span 33,817kb (Table 1), including ~156kb in the form of clone-, sequence- and repeat-gaps. Assuming that the majority of floating contigs lie beyond the termini of the assemblies, the total genome size is estimated at 34,042,810bp. In estimating the completeness of the sequence, we note that of 967 well-characterized D. discoideum genes, 957 (99%) were found initially in the assemblies. Of the remaining ten, seven (cupE, trxA, trxB, trxC, staA, staB, cinB) have close matches, suggesting that their Genbank entries may contain errors or represent alternative alleles. Only three (fcpA, wasA and roco5) had no matches in the initial assemblies, though the first two of these were recovered by searches of unincorporated sequence followed by local reassembly. Of 133,168 ‘qualified’ D. discoideum AX4 ESTs (expressed sequence tags of >200bp and >20% G+C, and not matching mitochondrial sequence; reference 10 and H. Urushihara et al. unpublished), 128,207 (96.3%) are found in the assemblies (the higher proportion of missing sequences amongst the ESTs probably reflects the higher error rate inherent in EST data).
We conclude that the current assembly represents >>95% of chromosomal sequence (less than 1% of which is in floating contigs) and ≥99% of genes, the majority of missing sequence comprising complex or simple repeats. The most stringent test of the medium- to long-range accuracy of the assembly comes from comparison with the HAPPY maps. This is particularly true for chromosomes 4, 5 and 6, where HAPPY markers were used to nucleate contigs but not to guide their assembly or ordering, specifically to allow such a comparison to be made without circularity of argument. As can be seen, good agreement between map and sequence confirms the accuracy of the assembly (Fig. 1).
Figure 1. Chromosomal assemblies compared against HAPPY map data.
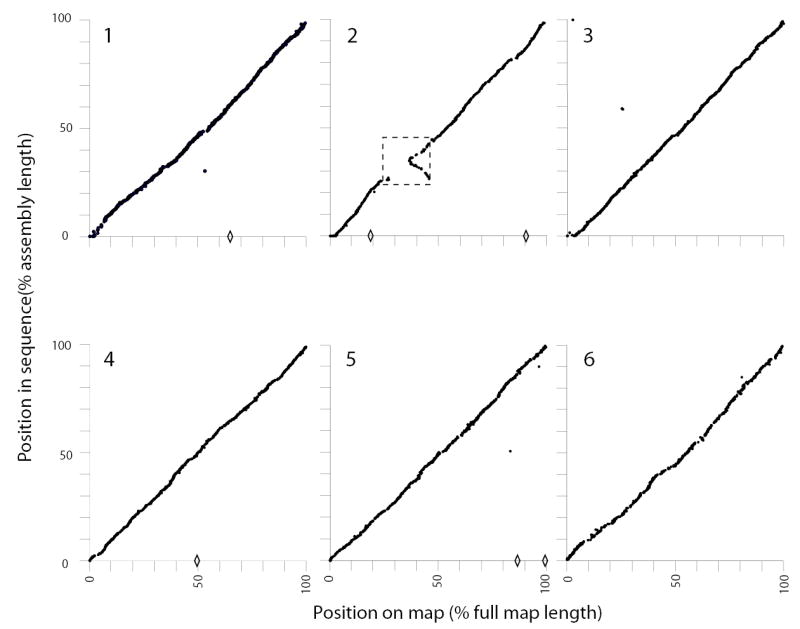
The locations of markers as found in the sequence (vertical axis) are plotted against their location in HAPPY maps (horizontal axis) for chromosomes 1–6. Markers mapped to one chromosome but found in the assembled sequence of another are indicated by diamonds on the horizontal axis. The dashed box indicates a large inverted duplication on Chr2: markers in this region are shown at one of their two possible map locations but are found at two points in the sequence.
The genome - sequence characteristics
The genome is A+T-rich (77.57%) and of broadly uniform composition, apart from the more G+C-rich repeat-dense regions (Fig. 2). On a finer scale, nucleotide composition tracks the distribution of exons (see below). Amongst dinucleotides, CpG is under-represented, not just in absolute terms but relative to its isomer GpC (the former occurring only 62% as often as the latter). This bias normally reflects cytosine methylation at CpG sequences, promoting their mutation to TpG (which isoverrepresented relative to GpT by 38%). Hence, these observations suggest that cytosine methylation may occur in Dictyostelium, contrary to earlier findings11.
Figure 2. (pullout section) The genome of Dictyostelium discoideum.

On each of the six chromosomal assemblies (left) the diameter of the tube represents coding density (proportion of coding bases summed over both strands; centre-weighted sliding window of 100kb; scale on right); coloured bands on the chromosomes represent tRNAs (red), complex repeats (blue), gaps (black) and ribosomal DNA sequences (yellow). G+C content is plotted above each chromosome (centre-weighted sliding window of 100kb; scale on left). The locations of HAPPY markers are indicated by short green ticks immediately below the distance scale. Immediately beneath each chromosome are indicated (short vertical ticks) the locations of genes known to be up-regulated (red), down-regulated (blue) or whose level of expression does not change significantly (grey) in the transition from solitary to aggregative existence (expression data from reference 91); heavy coloured bars below this indicate significant clusters of genes which are preferentially expressed in germinating spores (red), in dedifferentiating cells (green), in prespore cells (blue) or in prestalk cells (yellow). The translucent hourglass on chromosome 2 is centred on a large inverted duplication. The translucent cylinder on chromosome 3 indicates a typical 300kb region which is shown in expanded form in inset panel A (above) to illustrate the clustering of identical tRNA genes (red arrows indicate polarity of tRNA genes); a 50kb section of this region is expanded further in inset panel B, revealing the close association of TRE elements (specific family named above) with tRNAs.
The translucent yellow disc on chromosome 4 indicates the location of the presumed chromosomal master-copy of the rDNA element. In inset panel C (below), the structure of the palindromic extrachromosomal element is shown schematically (i ; magenta bands = rDNA genes, green bands = G+C-rich regions, red end-caps = short repetitive telomere structures; the translucent hoop indicates the central region of asymmetry. (ii) two chromosomal sequence contigs, each carrying an rDNA-like sequence (green or yellow; dotted lines indicate corresponding part of element) flanked by complex repeats (blue). From these contigs, we infer the probable structure (iii) of the genomic master copy (grey=flanking sequence on chromosome 4). This structure suggests a mechanism for regenerating the extrachromosomal copies by transcription of a single strand (iv), hairpin formation and strand extension (v; broken line indicates synthesis of complementary strand), unfolding of the hairpin and synthesis of a fully complementary strand (vi; broken line indicates synthesis of second strand; telomeric caps added post-synthetically).
Simple sequence repeats are abundant and unusual
Simple sequence repeats (SSRs) are more abundant in Dictyostelium than in any other sequenced genome, comprising > 11% of bases (Fig. SI 2). In non-coding sequence, tracts of dinucleotides or longer motifs occur every 392bp on average and comprise 6.4% of the bases. There is a bias towards repeat units of 3–6 bases, whereas dinucleotide tracts predominate in most other genomes. Homopolymer tracts are also abundant, comprising a further 16% of non-coding sequence. The base composition of non-coding SSRs and homopolymer tracts (99.2% A+T) is even more biased than that of their surrounding sequence, suggesting that either selection or the mechanism of repeat expansion favours A+T-rich repeats.
Notably, SSRs are also abundant in protein-coding sequence, occurring on average every 724bp within exons. We consider these coding SSRs in further detail below in the context of proteins.
Transposable elements are clustered
The genome is known to be rich in transposable elements (TEs)9, 12. Completion of the sequence confirms the earlier observation that TEs of the same type are clustered, suggesting their preferential insertion within similar resident elements. However, none of the elements appears to use a specific sequence as a target for insertion: they insert at random within other elements of the same type. Non-LTR (long terminal repeat) retrotransposons are known to insert next to tRNA genes: we find many such instances (Fig. 2), but again no specific sequences were identified as insertion targets.
tRNAs are numerous and paired by specificity
The sequenced genome encodes 390 tRNAs, a number at the upper end of the eukaryotic spectrum (e.g. Plasmodium falciparum=43, Drosophila melanogaster=284, human=496). Allowing for the normal wobble rules in codon-anticodon pairing13, 14 every sense codon can be decoded, apart from the rare alanine codon GCG; we infer that the missing tRNA(s) lie in one or more gaps in the sequence. We also find a possible selenocysteine tRNA in the genome, as well as corresponding selenocysteine insertion targets in two predicted proteins (Supplementary Information; Fig. SI 3).
Dictyostelium, in common only with Acanthamoeba castellanii15, has been shown to lack certain apparently essential tRNAs in its mitochondrial genome16. It therefore seems likely that at least some chromosomally-encoded tRNAs (those for valine, threonine, asparagine and glycine, as well as one arginine and two serine tRNAs) are imported into the mitochondria.
Though the gross distribution of tRNAs is uniform, their organisation on a finer scale is striking: about 20% occur as pairs or triplets with identical anticodons (and usually 100% sequence identity), separated by <20kb and often by <5kb (Fig. 2). There are 41 such groups in the genome; a random distribution would produce few, if any. This pattern is unique amongst sequenced genomes, and suggests a wave of recent duplications. However, tRNA pairs are found in tandem, converging and diverging orientations with comparable frequencies, suggesting no straightforward duplication mechanism; nor is there usually duplication of extensive flanking sequences. Whether the preference of TRE elements for inserting adjacent to tRNAs is related to the large number and unusual distribution of the latter is unclear.
A chromosomal master copy of the extrachromosomal rDNA element
In Dictyostelium, rRNA genes lie on an 88-kb palindromic extrachromosomal element17, present at ~100 copies per nucleus (Fig. 2). Evidence exists also for chromosomal copies: at least the central 3.2 kb of the element is located17 on chromosome 4, whilst chromosome 2 carries both a partial rDNA sequence and a 5S rRNA pseudogene9, 18.
In this study, two unanchored contigs assigned to chromosomes 4/5 were found to contain junctions between rDNA sequences and complex repeats - each of which would confound attempts to extend the sequence and integrate these contigs into the assemblies. We postulate that these contigs represent the junctions between a ‘master copy’ of the rDNA and the remainder of chromosome 4 (Fig. 2). One contig contains sequence matching a region of G+C-rich repeats near the centre of the palindrome, whilst the other matches sequence near the tip of the palindrome arm, adjacent to the one unclosed gap in the rDNA element sequence17. This gap is believed to represent a tandem array of short repeats, probably added post-synthetically to the extrachromosomal elements.
The structure of this master copy suggests a mechanism for generating the extrachromosomal copies by a process of transcription, hairpin formation and second-strand synthesis (Fig. 2). This process would account for the complete absence of sequence variation between the two arms of the palindrome.
Chromosomal mechanics - centromeres, telomeres and rearrangements
Repeat clusters may serve as centromeres
Centromeres mobilize eukaryotic chromosomes during cell division but vary widely in their structure and organisation19, making them difficult to identify. Each Dictyostelium chromosome carries a single cluster of repeats rich in DIRS (Dictyostelium intermediate repeat sequence) elements20, 21 near one end22, and this sole but striking structural consistency suggests that these clusters may serve as centromeres. Although the repetitive nature of the chromosomal termini impeded their assembly, most of the cluster on Chromosome 1 was assembled (Fig. 3) and shows a complex pattern of DIRS and related Skipper elements, each preferentially associated with others of the same type. Frequent insertions and partial deletions have created a mosaic with little long-range order.
Figure 3. DIRS repeat region of chromosome 1.
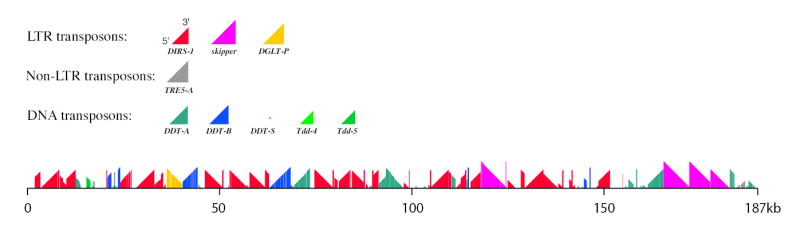
Complete complex repeat units are represented by coloured triangles whose size corresponds to the sequence length of the repeat unit (key, upper); bottom-left and top-right corners of triangle represent 5’ and 3’ ends of repeat, respectively. The arrangement of complete and partial repeat units within the first 187kb of D. discoideum chromosome 1 is shown (lower) by corresponding portions of the triangles; the orientation of the triangles indicates the direction in which each repeat unit lies. Vertical scale (sizes of repeat units) is the same as the horizontal scale (chromosomal distances).
In Dictyostelium cells demonstrating condensed chromosomes characteristic of mitosis, DIRS-element probes hybridise to one end of each chromosome (Fig. SI 4), consistent with the mapping data. DIRS-like elements in other species are more uniformly scattered along the chromosomes23 suggesting that their restricted distribution in Dictyostelium chromosomes is functionally important. Further, the DIRS-containing ends of the chromosomes cluster not only during mitosis, but also during interphase (Fig. SI 4), as has been observed for centromeres in Schizosaccharomyces pombe24.
rDNA sequences appear to act as telomeres
No G+T-rich telomere-like motifs were identified in the sequence. However, earlier findings22 suggested that the chromosomes terminate in the same G+A-rich repeat motif which caps the extrachromosomal rDNA element. We therefore surveyed all shotgun sequence to identify reads containing a junction between complex repetitive elements and rDNA-like sequence. Only 556 such reads were identified, of which 221 could be built into 13 contigs which we refer to as 'C/R (complex-repeat/rDNA) junctions'.
Of the 13 junctions, two represented already-known regions lying internally in the chromosomal assemblies. Of the remaining 11, one had twice the sequence coverage of the others, suggesting that it represents two distinct but identical portions of the genome (a possibility supported by the fact that another two of the junctions differed from each other by only two bases). Hence, we infer that the 11 remaining contigs represent 12 distinct junctions between repetitive elements and rDNA-like sequences - potentially one for every chromosomal end.
Based on their content of sequence-reads from each of the whole-chromosome libraries, we assigned two of the C/R contigs to each of the chromosomes. Chromosomes 4 and 5 cannot be distinguished in this way but three junctions, including the one believed to be present as two copies, are assigned to this chromosome pair. The point in the rDNA palindrome which is represented differs from one junction to the next (Fig. SI 5), but several junctions fall at common parts of the palindrome. This may reflect a preference in the mechanism which forms or maintains the junctions, or may result from an homogenizing recombination between them or with other rDNA sequences. Certainly the low frequency of differences between the rDNA components of the junction fragments and the extrachromosomal rDNA element argues for some process that limits or rectifies mutation. At each junction, we see only the rDNA sequence that immediately adjoins the complex repeat, since further assembly is precluded by the multicopy nature of rDNA. Therefore we cannot tell whether each junctional rDNA sequence extends to the telomere-repeat-carrying tip of the rDNA palindrome sequence, nor whether other sequences lie beyond the rDNA components.
HAPPY mapping of markers derived from six of these C/R junctions confirmed not only the chromosomal assignments which had been made based on the origins of their component sequences, but also their locations at the termini of the mapped regions of the chromosomes. For the other junctions, the absence of unique sequence features precluded such mapping. Taken as a whole, this evidence strongly suggests that rDNA-like elements form part of the telomere structure in D. discoideum and that common mechanisms stabilise both the extrachromosomal rDNA element and the chromosomal termini.
Chromosome 2 duplication - aftermath of a 'breakage-fusion-bridge' event?
Chromosome 2 of D. discoideum AX4 carries a perfect inverted 1.51Mb duplication (Fig. 2; references 9, 25, and this work). This duplication, containing 608 genes, is known25 to be absent from the wild-type isolate NC4 and from one of its direct descendents (AX2), but present in another (AX3); AX4 in turn is derived from AX3. The sequences adjoining the right-hand end of the duplication - a partial copy of a DIRS element (and a partial DDT-A element) and a region identical to part of the rDNA palindrome, both at about 3.74Mb (Fig. 2) – have been implicated in centromeric and telomeric functions, respectively, elsewhere in the genome.
We propose that this duplication arose from a 'breakage-fusion-bridge' cycle as first described in maize26, and since observed in many genomes. The nearby DIRS and rDNA components, in this view, represent abortive attempts to stabilize the halves of the broken chromosome by establishing new telomeres and centromeres, followed by re-fusion of the pieces to create a restored and enlarged chromosome (Fig. SI 6).
Chromosome 2 (the largest of the chromosomes, even discounting the duplication in AX4) may be prone to breakage: in the Bonner isolate of NC4, maintained in vegetative growth for 50 years, chromosome 2 is represented by two smaller fragments27. Comparison with more recent data22 indicates that the break-point in NC4-Bonner lies in the same region as the duplication in AX4, suggesting that NC4-Bonner underwent the early stages of this process, but that the chromosome fragments were stabilized and maintained after the initial breakage. Preliminary results (not shown) from HAPPY mapping also suggest that, whilst wild-type isolates V12M2 and NC4 both lack the duplication seen in AX4, NC4 may carry a duplication of ~300kb near the opposite end of chromosome 2.
The proteome – content and organisation
Prediction of protein-coding genes (see Methods) was performed on the complete set of chromosomes and floating contigs (Table 2). In assessing the completeness and accuracy of the predictions, we find that of the 957 well-characterized D. discoideum genes that are present in the current sequence, 823 (86%) are predicted as transcripts with structures matching the experimentally determined ones. For a further 123 (13%), the predicted transcript differs from the experimentally determined one, about half of these differing only in their 5' boundary; the remaining 11 (1%), though present in the sequence, were not predicted as transcripts. Similarly, of the 128,207 qualified ESTs present in the current sequence, 127,097 (99.1%) fall within predicted transcripts. Combining our estimate of sequence coverage (above) with these estimates of the success of gene prediction, we infer that approximately 98% of all D. discoideum genes are present in the predicted set.
Table 2.
Comparison between the predicted protein-coding gene set of D. discoideum and those of other organisms.
| Species | D.discoideum | P. falciparum | S. cerevisiae | A. thaliana | D. melanogaster | C. elegans | Human |
|---|---|---|---|---|---|---|---|
| Feature | |||||||
| Genome size (Mb) | 34 | 23 | 13 | 125 | 180 | 103 | 2,851 |
| Number of genes | 12,5001 | 5,268 | 5,538 | 25,498 | 13,676 | 19,893 | 22,287 |
| Gene spacing (kbp/gene) | 2.5 | 4.3 | 2.2 | 4.9 | 13.2 | 5.0 | 127.9 |
| Mean gene length (bp) | 1,756 | 2,534 | 1,428 | 2,036 | 1,997 | 2,991 | 27,000 |
| Mean coding size (amino acids) | 518 | 761 | 475 | 437 | 538 | 435 | 509 |
| % genes with introns | 69 | 54 | 5 | 79 | 38 | 5 | 85 |
| Mean intron size (bp) | 146 | 179 | ND | 170 | ND | 270 | 3,365 |
| Mean no. of introns (in spliced genes) | 1.9 | 2.6 | 1.0 | 5.4 | 4.0 | 5.0 | 8.1 |
| Total a.a. encoded (thousands) | 7,021 | 4,009 | 2,471 | 11,143 | 7,358 | 9,038 | 11,333 |
| Codon A+T bias2 | 86 | 83 | 62 | 57 | 50 | 64 | 41 |
| Mean A+T% (exons) | 73 | 76 | 72 | 72 | 45 | 58 | 55 |
| Mean A+T% (introns) | 88 | 87 | 51 | 55 | 38 | 71 | 62 |
| Mean A+T% (intergenic) | 85 | 86 | 51 | 56 | 38 | 72 | 62 |
See text. The estimated number of true transcripts for D. discoideum is given here for comparability with other species; the total predicted gene number of 13,541 is used in calculating the figures below.
Percentage of all codons used which have A/T at their third base.
The level of over-prediction, conversely, is harder to estimate: prediction was performed generously to ensure that most true genes were represented. Of the 13,541 predicted proteins, 47.5% are represented by qualified ESTs, reflecting the inevitable bias in EST sampling. Amongst the shortest predicted proteins, fewer are represented by ESTs (e.g. 21% of those of <60 amino acids), due at least partly to a higher level of overprediction. Based on the simplifying assumption that 50% of all genes <100 amino acids are mis-predictions, we estimate the true number of genes at roughly 12,500. This number is closer to that seen in multicellular organisms than in most unicellular eukaryotes (Table 2). The same relative complexity is seen in the total numbers of amino acids encoded by each genome, which are less biased by shorter (and more dubious) predictions. Introns in Dictyostelium are few and short, and intergenic regions are small, producing a compact genome of which 62% encodes protein.
Genes are distributed approximately uniformly across the genome (Fig. 2). Although we do not see widespread clustering of genes with coordinated expression patterns (see Methods), we do find statistically significant (p<0.01) clusters of genes expressed predominantly at some developmental stages or in specific cell types (Fig. 2).
A+T-richness influences amino-acid composition as well as codon usage
Codon usage in Dictyostelium favours codons of the form NNT or NNA over their NNG or NNC synonyms, the bias being even greater than in the A+T-rich Plasmodium genome. Comparison of tRNA and codon frequencies (Table SI 2) reveals a similar picture to that in human28 and other eukaryotes, suggesting that the same use is made of 'wobble' and of base modifications (for example, of adenine to inosine in some tRNAs) to expand the effective repertoire of tRNAs.
As in Plasmodium29, the extreme A+T-richness is reflected not just in the choice of synonymous codons, but also in the amino acid composition of the proteins. Amino acids encoded solely by codons of the form WWN (W=A or T; N=any base; these are Asn, Lys, Ile, Tyr and Phe), are much commoner in Dictyostelium proteins than in human ones; the reverse is true for those encoded solely by SSN codons (S=C or G; these are Pro, Arg, Ala and Gly).
Geometry reflects phylogeny - tandem duplication in the genome
The predicted gene set of Dictyostelium is rich in relatively recent gene duplications. Of the 13,498 predicted proteins analysed, 3663 fall into 889 families clustered by BLAST-P similarities of e<10−40. Most (538) families contain only two members, but 351 families contain between three and 81 proteins (Table SI 3). Hence, 2774 (20%) of all predicted proteins have arisen by relatively recent duplication, potentially accounting for much of Dictyostelium's excess over typical unicellular eukaryotes.
We tried to infer the mechanisms by which such duplications arise and propagate in the genome. Where members of a family are clustered on one chromosome, the physical distance between family members often (23 of 86 families examined) correlates strongly with their evolutionary divergence (Methods). Where a family is split between different chromosomes, members on the same chromosome are often (23 of 50 families examined) more related to each other than to members on different chromosomes; the reverse is never observed.
These findings suggest that three processes combine to account for most duplications in Dictyostelium: tandem duplication, local inversion, and inter-chromosomal exchange. In this model, gene families expand by tandem duplication of either single genes or blocks containing several consecutive genes, as in an earlier model30; inversions within these expanding clusters may reverse local gene order. An elegant illustration of these two processes is provided by a cluster of Acetyl-coA synthetases on chromosome 2 (Fig. 4). The third process - exchange of segments between chromosomes - may fragment these clusters at any stage. If such an interchromosomal exchange splits a gene family early in its expansion, then each of the two resulting sub-families has a long subsequent period of evolution independently of the other, so similarities will be greatest between genes on the same chromosome. If, conversely, the split occurs later then all family members - whether on the same or on different chromosomes - will tend to resemble each other equally closely. We cannot exclude the possibility of duplication occasionally creating a second copy of a gene, or group of genes, directly on a different chromosome from the first. However, all instances that we have examined can be accounted for without such intermolecular duplication.
Figure 4. Phylogeny of gene family members compared to their physical order.
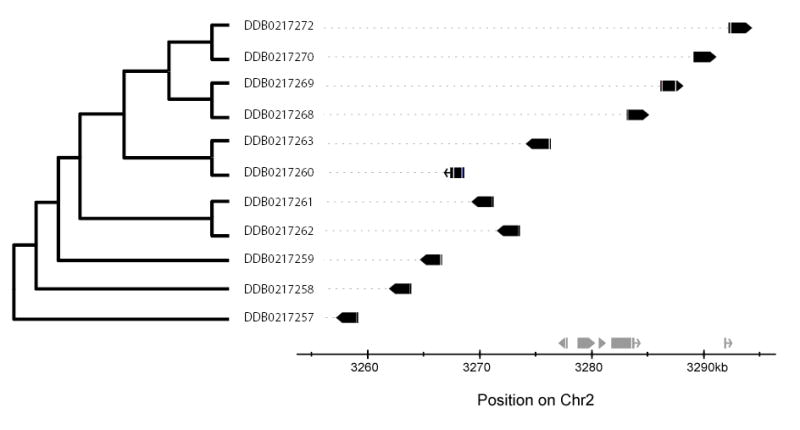
The optimally parsimonious phylogenetic tree of 11 Acetyl-CoA synthase genes , computed using the PHYLIP module 'Protpars' (http://evolution.gs.washington.edu/phylip/doc/protpars.html), is shown to the left; dictyBase ID numbers shown at the end of each branch. The graph (right) indicates the arrangement of these genes on chromosome 2 (solid black boxes; gaps indicate introns, pointed heads indicate direction of transcription; chromosomal distance scale at bottom; other unrelated genes in the same region indicated in grey above the X-axis). The correspondence between phylogeny and physical order implies that the cluster has arisen by a series of segmental tandem duplications and local inversions in parallel with sequence divergence.
Amino acid repeats
Tandem repeats of trinucleotides (and of motifs of 6, 9, 12 etc bases) are unusually abundant in Dictyostelium exons, and naturally correspond to repeated sequences of amino acids. However, at the protein level the situation is even more extreme: there are many further amino-acid repeats which use different synonymous codons, and so do not arise from perfect nucleotide repeats. Amongst the predicted proteins, there are 9582 simple-sequence repeats of amino acids (homopolymers of length ≥10, or ≥5 consecutive repeats of a motif of two or more amino-acids). Of these, the most striking are polyasparagine and polyglutamine tracts of ≥20 residues, present in 2,091 of the predicted proteins. Also abundant are low-complexity regions such as QLQLQQQQQQQLQLQQ: there are 2379 tracts of ≥15 residues composed of only two different amino acids. In total, repeats or simple-sequence tracts of amino acids (even by these conservative definitions) occur in 34% of predicted proteins and encode 3.3% of all amino acids.
It seems likely that these repeats have arisen through nucleotide expansion, but have been selected at the protein level. Evidence for the latter is that any given trinucleotide repeat occurs predominantly in only one of the three reading frames. For example, the repeat ...ACAACAACAACA... is usually translated as polyglutamine ([CAA]n) rather than as polythreonine ([ACA]n) or polyasparagine ([AAC]n). Further evidence comes from the many trinucleotide repeats which have apparently mutated to produce only synonymous codons (e.g. ...GAT,GAC,GAT,GAT,GAC,..., translated as polyaspartate). Moreover, the distribution of repeats and simple-sequence tracts is non-random: most proteins either have no such features (66% of proteins) or have two or more (18% of proteins), suggesting that they are tolerated only in certain types of protein. The polyasparagine- and polyglutamine-containing proteins appear to be over-represented in protein kinases, lipid kinases, transcription factors, RNA helicases and mRNA binding proteins such as spliceosome components (Fig. SI 9). Protein kinases and transcription factors are also over-represented in the polyasparagine- and polyglutamine-containing proteins of S. cerevisiae, so it is possible that these homopolymers serve some functional role in these protein classes. A more detailed analysis of amino acid homopolymers is given in Supplementary Information (tables SI 4–6, Figs. SI 7–10).
Phylogeny, evolution and comparative proteomics
The organisms that diverged from the last common ancestor of all eukaryotes followed different evolutionary paths, but all retained the basic properties of eukaryotic cells. Their genomes have been sculpted by chromosomal deletions and duplications that led to lineage specific gene family expansions, reductions and losses, as well as genes with novel functions32, 33. Our analysis of Dictyostelium’s proteome shows that similar mechanisms have shaped its genome, augmented by horizontal gene transfer from bacterial species.
Phylogeny of eukaryotes based on complete proteomes
Using morphological criteria, early workers were unsure whether to classify Dictyostelids as fungi or protozoa34. Molecular methods indicated that they were amoebozoa and also suggested that Dictyostelium diverged from the line leading to animals at about the same time as plants35, 36. A study of more than 100 proteins suggested that Dictyostelium diverged after the plant/animal split, but before the divergence of the fungi37. The recent finding of a gene fusion encoding three pyrimidine biosynthetic enzymes, shared only by Dictyostelium, fungi and metazoa, indicates that the amoebozoa are a true sister group of the fungi and metazoa38.
To examine the phylogeny of Dictyostelium on a genomic scale, we applied an improved method for predicting orthologous protein clusters to complete eukaryotic proteomes39 (for details, see Supplementary Information). The data were used to construct a phylogenetic tree that confirms the divergence of Dictyostelium along the branch leading to the metazoa soon after the plant/animal split (Fig. 5). Despite the earlier divergence of Dictyostelium, many of its proteins are more similar to human orthologues than are those of S. cerevisiae, probably due to higher rates of evolutionary change along the fungal lineage. Whether the greater similarity between amoebozoa and metazoa proteins translates into a generally higher degree of functional conservation between them compared to the fungi remains to be seen.
Figure 5. Proteome-based eukaryotic phylogeny.

The phylogenetic tree was reconstructed from a database of 5,279 orthologous protein clusters drawn from the proteomes of the 17 eukaryotes shown, and was rooted on 159 protein clusters that had representatives from six archaebacterial proteomes. Tree construction, the database of protein clusters and a model of protein divergence used for maximum likelihood estimation are described in Supplementary Information. The relative lengths of the branches are given Darwins, (1 Darwin= 1/2000 of the divergence between S. cerevisiae and humans). Species that are not specified are Plasmodium falciparum (Malaria Parasite), Chlamydomonas reinhardtii (Green Alga), Oryza sativa (Rice), Zea mays (Maize), Fugu rubripes (Fish), and Anopheles gambiae (Mosquito).
Proteins shared by Dictyostelium and major organism groups
To examine shared functions, we defined eukaryote-specific Superfamily and Pfam protein domains and sorted them according to their presence or absence within 12 completely sequenced genomes to arrive at their distribution amongst the major organismal groups (see Supplementary Information; Fig. SI 11; Tables SI 7–10). The plants, metazoa, fungi and Dictyostelium all share 32% of the eukaryotic Pfam domains (Fig. 6). The protein domains present in Dictyostelium, metazoa and fungi, but absent in plants, are interesting because they probably arose soon after plants diverged and before Dictyostelium diverged from the line leading to animals. The major classes of domains in this group of proteins include those involved in small and large G-protein signalling (e.g., RGS proteins), cell cycle control and other domains involved in signalling (Tables SI 8 and SI 9). It also appears that glycogen storage and utilization arose as a metabolic strategy soon after the plant/animal divergence since glycogen synthetase seems to have appeared in this evolutionary interval.
Figure 6. Distribution of PFAM domains amongst eukaryotes.
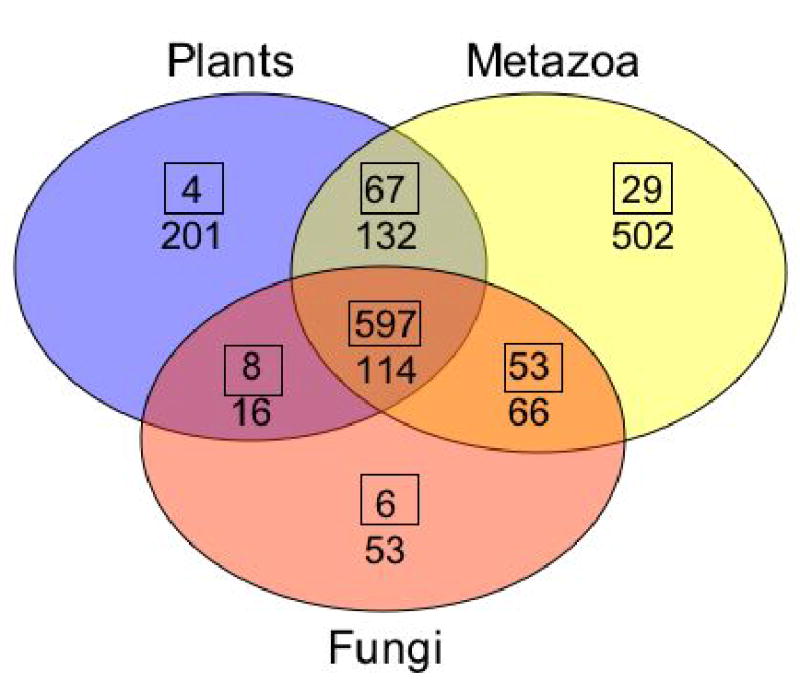
The number of eukaryote-specific Pfam domains present in each group of eukaryotic organisms is shown. The boxed numbers are the domains that are present in Dictyostelium and the other numbers are those domains that are absent from Dictyostelium. The animals are H. sapiens, F. rubripes, C. elegans, D. melanogaster; the fungi are, N. crassa, A. nidulans, S. pombe and S. cerevisiae and the plants are, A. thaliana, O. sativa and C. reinhardtii. A complete listing of the domains can be found in the Supplementary Information.
Particularly striking are the cases where otherwise ubiquitous domains appear completely absent in one group or another. For instance, Dictyostelium appears to have lost the genes that encode collagen domains, the circadian rhythm control protein Timeless and basic helix-loop-helix transcription factors (Table SI 7). Metazoa, on the other hand, appear to have lost receptor histidine kinases that are common in bacteria, plants and fungi, while Dictyostelium has retained and expanded its complement to 14 members40.
Human disease-gene orthologues
An important motivation for sequencing the Dictyostelium genome was to aid the discovery of proteins that would facilitate studies of orthologues in human, with possible implications for human health. Although orthologues of human genes implicated in disease are of course present in many species, Dictyostelium provides a potentially valuable vehicle for studying their functions in a system which is experimentally tractable and intermediate in complexity between the yeasts and the higher multicellular eukaryotes. To assess the usefulness of Dictyostelium for investigating the functions of genes related to human disease we used the protein sequences of 287 confirmed human disease genes as queries and carried out a systematic search for putative orthologues in the Dictyostelium proteome41. At a stringent threshold value of E≤10−40, we identified 64 such proteins. Of these, 33 were similar in length to the human protein and had similarity extending over >70% of the two proteins (Table 3). The number of Dictyostelium orthologues of human disease genes is lower than in D. melanogaster or C. elegans but higher than in S. cerevisiae or S. pombe. Of the 33 putative orthologues of confirmed human disease genes in Dictyostelium, five are absent in both S. cerevisiae or S. pombe (E-value ≤ 10−30), a further four are absent from S. cerevisiae and two are not found in S. pombe.
Table 3.
Dictyostelium genes related to human disease genes
| Disease Category1 | SwissProt2 | dictyBase ID |
|---|---|---|
| Cancer | ||
| Colon Cancer (MSH2) | MSH2_HUMAN | DDB0202539 |
| Colon Cancer (MLH1) | MLH1_HUMAN | DDB0187465 |
| Colon Cancer (MSH3) | MSH3_HUMAN | DDB0204604 |
| Colon Cancer (PMS2) | PMS2_HUMAN | DDB0185791 |
| Xeroderma Pigmentosum (ERCC3) | XPB_HUMAN | DDB0206281 |
| Xeroderma Pigmentosum (XPD) | XPD_HUMAN | DDB0189539 |
| Oncogene (AKT2) | AKT2_HUMAN | DDB0189970 |
| Oncogene (RAS) | RASH_HUMAN | DDB0191937 |
| Cyclin-dependent Kinase 4 (CDK4) | CDK4_HUMAN | DDB0188077 |
| Neurological | ||
| Lowe Oculocerebrorenal (OCRL) | OCRL_HUMAN | DDB0189888 |
| Miller-Dieker Lissencephaly (PAF) | LIS1_HUMAN | DDB0219335 |
| Adrenoleukodystrophy (ABCD1) | ALD_HUMAN (P) | DDB0219834 |
| Angelmann (UBE3A) | UE3A_HUMAN | DDB0188760 |
| Ceroid Lipofuscinosis (CLN2) | TPP1_HUMAN (C, P) | DDB0190668 |
| Tay-Sachs (HEXA) | HEXA_HUMAN (C, P) | DDB0187255 |
| Ceroid Lipofuscinosis (PPT) | PPT1_HUMAN (C) | DDB0186550 |
| Thomsen Myotonia Congenita(CLCN1) | CLC1_HUMAN | DDB0191805 |
| Choroideremia (CHM) | RAE1_HUMAN | DDB0206402 |
| Amyotropic Lateral Sclerosis (SOD1) | SODC_HUMAN | DDB0188850 |
| Parkinson (UCHL1) | UCL1_HUMAN (C, P) | DDB0205083 |
| Cardiovascular | ||
| Hypertrophic Cardiomyopathy | MYH7_HUMAN | DDB0186963 |
| Renal | ||
| Renal tubular acidosis (ATP6B1) | VAB1_HUMAN | DDB0169211 |
| Hyperoxaluria (AGXT) | SPYA_HUMAN (C, P) | DDB0188646 |
| Metabolic/endocrine | ||
| Niemann-Pick Type C (NPC1) | NPC1_HUMAN (P) | DDB0191057 |
| Hyperinsulinism (ABCC8) | ACC8_HUMAN | DDB0187670 |
| McCune-Albright (GNAS1) | GBAS_HUMAN | DDB0185461 |
| Pendred (PDS) | PEND_HUMAN (C) | DDB0202939 |
| Hematological/immune | ||
| G6PD Deficiency (G6PD) | G6PD_HUMAN | DDB0168147 |
| Chronic Granulomatous (CYBB) | C24B_HUMAN (C, P) | DDB0188527 |
| Malformation | ||
| Diastrophic Dysplasia (SLC26A2) | DTD_HUMAN (C) | DDB0202939 |
| Other | ||
| Cystic Fibrosis (ABCC7) | CFTR_HUMAN | DDB0186232 |
| Darier-White (SERCA) | ATA2_HUMAN | DDB0169159 |
| Congenital Chloride Diarrhea (DRA) | DRA_HUMAN (C) | DDB0202939 |
From a list of 287 confirmed human disease protein sequences41. Those listed match a predicted Dictyostelium protein with a BLASTP probability of E≤10−40, are similar in length (+/− 25% in comparison to the Dictyostelium protein) and both proteins align over more than 70% of their respective lengths.
Swiss Prot identifiers for the human proteins. Letters in brackets indicate that the protein has no homologue (BLASTP probability of E≤1.0x10−3) in S. cerevisiae (C) or S. pombe (P)
The best match to the human gene is listed by its dictyBase ID number. Matches with a BLASTP probability of E≤<10−100 are indicated in bold.
Horizontal gene transfer
The acquisition of genes by horizontal transfer from one species to another (HGT) has become increasingly recognized as a mechanism of genome evolution42–44. We identified eighteen potential HGTs by screening Dictyostelium protein domains that are similar to bacteria-specific Pfam domains and that have phyletic relationships consistent with HGT (see Supplementary Information). They encode protein domains that appear to have replaced functions, added new functions or evolved into novel functions (Table 4). The thy1 gene, which encodes an alternative form of thymidylate synthase (ThyX), appears to have replaced the endogenous gene: the conventional thymidylate synthase (ThyA) is not present45. Other HGT domains also have established functions, which are presumably retained and give Dictyostelium the ability to degrade bacterial cell walls (dipeptidase), scavenge iron (siderophore), or resist the toxic effects of tellurite in the soil (terD). Still other HGTs have become embedded within Dictyostelium genes that encode larger proteins. An example of this is the Cna B domain that is found within four large predicted proteins, one of which, colossin A, is predicted to be 1.2 MDa (Fig. SI 12).
Table 4.
Candidate horizontal gene transfers from bacteria
| Function1 | Pfam2 | Number of proteins3 | dictyBase I.D.4 | Length (aa)5 | Region matched6 | E-value7 |
|---|---|---|---|---|---|---|
| Aromatic amino acid lyase | Beta_elim_lyase | 2* | DDB0204031 | 170 | 4–170 | 3.2 x 10−65 |
| Biotin metabolism | BioY | 1 | DDB0184375 | 338 | 145–299 | 5.8 x 10−20 |
| Unknown | Cna_B | 4 | DDB0184530 | 11,103 | multiple8 | 1.1 x 10−10 |
| Peroxidase | Dyp_peroxidase | 1 | DDB0168077 | 306 | 3–303 | 1.4 x 10−82 |
| Insecticide | Endotoxin_N | 2 | DDB0188332 | 628 | 38–210 | 1.2 x 10−32 |
| Isopentenyl transferase | IPT | 1 | DDB0169077 | 283 | 1–63 | 5.1 x 10−12 |
| Siderophore | IucA_IucC | 2 | DDB0219918 | 739 | 183–350 | 2.3 x 10−18 |
| Osmoregulation | OsmC | 2 | DDB0190102 | 156 | 16–156 | 9.8 x 10−22 |
| Dipeptidase/ß-lactamase | Peptidase M15 | 1 | DDB0205124 | 897 | 68–406; 711–879 | 3.4 x 10−16 |
| Dipeptidase/ß-lactamase | Peptidase S13 | 1 | DDB0168572 | 522 | 337–495 | 4.2 x 10−25 |
| Polyphosphate synthesis | PP_kinase | 1 | DDB0192001 | 1053 | 372–1045 | 1.6 x 10−234 |
| Tellurite resistance | TerD | 2 | DDB0169240 | 287 | 152–279 | 2.1 x 10−67 |
| Thymidylate synthesis | Thy1 | 1 | DDB0214905 | 303 | 38–254 | 9.9 x 10−117 |
| Unknown | DUF84 | 1 | DDB0203145 | 179 | 5–175 | 1.6 x 10−20 |
| Unknown | DUF885 | 2 | DDB0205394 | 689 | 318–685 | 1.5 x 10−124 |
| (prespore protein 3B) | DUF1121 | 3* | DDB0169184 | 226 | 1–226 | 8.7 x 10−134 |
| Unknown | DUF1289 | 1 | DDB0204782 | 88 | 29–85 | 3.3 x 10−15 |
| Unknown | DUF1294 | 1 | DDB0186703 | 155 | 2–73 | 8.9 x 10−18 |
Confirmed or proposed function of the prokaryotic ortholog is given. For domains without function information, information on any Dictyostelium protein in the set is given in parentheses.
The Pfam domain designation. (http://www.sanger.ac.uk/Software/Pfam/)
The number of gene models in which the domain appears. Asterisks indicate gene sets where there are pairs of genes that map within 10 kb of each other.
The gene I.D. number for the example given in the rest of the table (release v2.0 at dictyBase.org).
Number of amino acid residues in the predicted Dictyostelium protein containing the domain.
The region of the Dictyostelium protein that matched the prokaryotic domain. The amino acid sequence identity between this region and the most highly related prokaryotic protein was between 21–52 percent.
The E-value for the domain against the Pfam model library used to identify it (see Supplementary Information).
The protein, Colossin A, consists of an array of 91 partial Cna_B domains within 18 larger repeats and the E-value corresponds to one domain.
Dictyostelium ecology
Dictyostelium faces many complex ecological challenges in the soil. Amoebae, fungi and bacteria compete for limited resources in the soil while defending themselves against predation and toxins. For instance, the nematode C. elegans is a competitor for bacterial food and a predator of Dictyostelium amoebae, but also a potential dispersal agent for Dictyostelium spores46. Dictyostelium has expanded its repertoire of several protein classes that are likely to be crucial for such inter-species interactions and for survival and motility in this complex ecosystem.
Polyketide synthases
A small number of natural products have already been identified from Dictyostelium, but the gene content suggests it is a prolific producer of such molecules. Some of them may act as signals during development, such as the dichloro-hexanophenone DIF-1, but others are likely to mediate currently unknown ecological interactions47. Many antibiotics and secondary metabolites destined for export are produced by polyketide synthases, modular proteins of around 3,000 amino acids48. We identified 43 putative polyketide synthases in Dictyostelium (see Supplementary Information). By contrast, S. cerevisiae completely lacks polyketide synthases and Neurospora crassa has only seven. In addition, two of the Dictyostelium proteins have an additional chalcone synthase domain, representing a type of polyketide synthase most typical of higher plants and found to be exclusively shared by Dictyostelium, fungi and plants. In addition to polyketide synthases, the predicted proteome has chlorinating and dechlorinating enzymes as well as O-methyl transferases, which could increase the diversity of natural products made. Thus, Dictyostelium appears to have a large secondary metabolism which warrants further investigation.
ABC transporters
ATP-binding cassette (ABC) transporters are prevalent in the proteomes of soil microorganisms and are thought to provide resistance to xenobiotics through their ability to translocate small molecule substrates across membranes against a substantial concentration gradient49–52. There are 66 ABC transporters encoded by the genome, which can be classified according to the subfamilies defined in humans, ABCA-ABCG, based on domain arrangement and signature sequences53. At least twenty of them are expressed during growth and are probably involved in detoxification and the export of endogenous secondary metabolites.
Cellulose degradation
Curiously, many of the predicted cellulose degrading enzymes in the proteome (see Supplementary Information) that have secretion signals are expressed in growing cells that do not produce cellulose54. The proteome also has one xylanase enzyme that can degrade the xylan polymers that are often found associated with the cellulose of higher plants. Perhaps Dictyostelium uses these enzymes to degrade plant tissue into particles that are then taken up by cells. These enzymes may also aid in the breakdown of cellulose-containing microorganisms upon which Dictyostelium feeds. Alternatively, these enzymes may promote the growth of bacteria that can serve as food, since Dictyostelium’s habitat also contains cellulose-degrading bacteria.
Specializations for cell motility
During both growth and development, Dictyostelium amoebae display motility that is characteristic of human leukocytes55. As a consequence, studies of Dictyostelium have contributed significantly to cytoskeleton research56. Dictyostelium’s survival depends on an ability to efficiently sense, track and consume soil bacteria using sophisticated systems for chemotaxis and phagocytosis. Its multicellular development depends on chemotactic aggregation of individual amoebae and the coordinated movement of thousands of cells during fruiting body morphogenesis. The proteome reveals an astonishing assortment of proteins that are used for robust, dynamic control of the cytoskeleton during these processes. As suggested from the functional parallels to human cells, these proteins are most similar to metazoan proteins in their variety and domain arrangements (Fig. 7; Table SI 11). Surprisingly, although the actin cytoskeleton has been studied for over twenty-five years, 71 putative actin-binding proteins apparently escaped classical methods of discovery. For example, actobindins had not been previously recognized in Dictyostelium. Curiously, the actin depolymerisation factor (ADF) and calponin homology (CH) domain proteins appear to have diversified by domain shuffling, a substantial fraction having domain combinations unique to Dictyostelium (Fig. SI 13; Table SI 12). In addition to 30 actin genes, there are also orthologues of all actin-related protein (ARP) classes present in mammals, as well as three founding members of a new class (Fig. SI 14).
Figure 7. Microfilament system proteins.
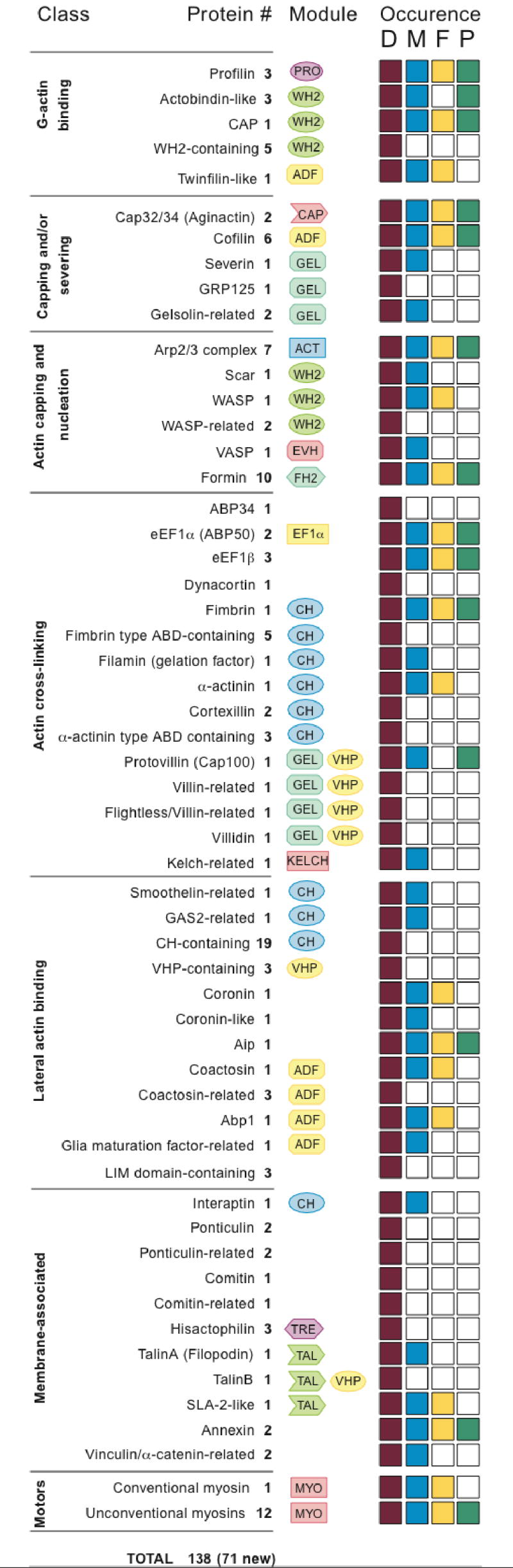
Proteins with probable interactions with the actin cytoskeleton are tabulated by their documented or predicted functions. Coloured boxes indicate the presence of a protein related to the Dictyostelium (D) protein in metazoa (M), fungi (F) or plants (P). Dictyostelium-specific proteins have no recognizable relatives or differ from relatives due to extensions or unusual domain compositions. For details see Supplementary Information. Actin-binding modules: ADF, actin depolymerisation factor/cofilin-like domain; CH, calponin homology domain; EVH, Ena/VASP homology domain 2; FH2, formin homology 2 domain; GEL, gelsolin repeat domain; TRE, trefoil domain; KELCH, Kelch repeat domain; MYO, myosin motor domain; TAL, the I/LWEQ, actin-binding domain of talin and related proteins; VHP, villin head piece; WH2, Wiskott Aldrich syndrome homology region 2.
Cytoskeletal remodelling during chemotaxis and phagocytosis is regulated by a considerable number of upstream signaling components. Of the 15 Rho family GTPases in Dictyostelium, some are clear Rac orthologues and one belongs to the RhoBTB subfamily57. However, the Cdc42 and Rho subfamilies characteristic of metazoa and fungi are absent, as are the Rho subfamily effector proteins. The activities of these GTPases are regulated by two members of the RhoGDI family, by components of ELMO1/DOCK180 complexes and by a surprisingly large number of proteins carrying RhoGEF and RhoGAP domains (>40 of each), most of which show domain compositions not found in other organisms. Remarkably, Dictyostelium appears to be the only lower eukaryote that possesses class I PI 3-kinases, which are at the crossroad of several critical signalling pathways (for details of the regulators and their effectors, see Table SI 13)58. The diverse array of these regulators and the discovery of many additional actin-binding proteins suggests that there are many aspects of cytoskeletal regulation that have yet to be explored.
Multicellularity and development
The evolution of multicellularity was arguably as significant as the origin of the eukaryotic cell in enabling the diversification of life. The common unicellular ancestor of the crown group of organisms must have posessed the basic machinery to regulate nutrient uptake, metabolism, cellular defense and reproduction, and it is likely that these mechanisms were adapted to integrate the functions of cells in multicellular organisms. Dictyostelium achieved multicellularity through a different evolutionary route from the plants and animals, yet the ancestors of these respective groups most likely started with the same endowment of genes and faced the same problem of achieving cell specialization and tissue organisation.
When starved, Dictyostelium develops as a true multicellular organism, organizing distinct tissues within a motile slug and producing a fruiting body comprised of a cellular, cellulosic stalk supporting a bolus of spores4. Thus, Dictyostelium has evolved differentiated cell types and the ability to regulate their proportions and morphogenesis. A broad survey of proteins required for multicellular development shows that Dictyostelium has retained cell adhesion and signalling modules normally associated exclusively with animals, while the structural elements of the fruiting body and terminally differentiated cells clearly derive from the control of cellulose deposition and metabolism now associated with plants. The Dictyostelium genome offers a first glimpse of how multicellularity evolved in the amoebozoan lineage. In the following sections, we consider some of the systems which are particularly relevant to cellular differentiation and integration in a multicellular organism.
Signal Transduction through G-protein coupled receptors
The needs of multicellular development add greatly to those of chemotaxis in demanding dynamically controlled and highly selective signalling systems. G-protein coupled cell surface receptors (GPCRs) form the basis of such systems in many species, allowing the detection of a variety of environmental and intra-organismal signals such as light, Ca2+, odorants, nucleotides and peptides. They are subdivided into six families which, despite their conserved secondary domain structure, do not share significant sequence similarity59. Until recently, in Dictyostelium only the seven CAR/CRL (cAMP receptor/ cAMP receptor-like) family GPCRs had been examined in detail60, 61. Surprisingly, a detailed search uncovered 48 additional putative GPCRs of which 43 can be grouped into the secretin (family 2), metabotropic glutamate/GABA B (family 3) and the frizzled/smoothened (family 5) families of receptors (Fig. 8; see also Supplementary Information). The presence of family 2, 3 and 5 receptors in Dictyostelium was surprising because they had been thought to be animal-specific. Their occurrence in Dictyostelium suggests that they arose before the divergence of the animals and fungi and were later lost in fungi and that the radiation of GPCRs predates the divergence of the animals and fungi. The putative secretin family is particularly interesting because these proteins were thought to be of relatively recent origin, appearing closer to the time of the divergence of animals62. The Dictyostelium protein does not contain the characteristic GPCR proteolytic site, but its transmembrane domains are clearly more closely related to secretin GPCRs than to other families (Fig. 8). Many downstream signalling components that transduce GPCR signals could also be recognized in the proteome, including heterotrimeric G-protein subunits (14 Gα, two Gβ and one Gγ proteins) and seven regulators of G-protein signalling (RGS), most similar to the R4 subfamily of mammalian RGS proteins.
Figure 8. The G-protein coupled receptors.
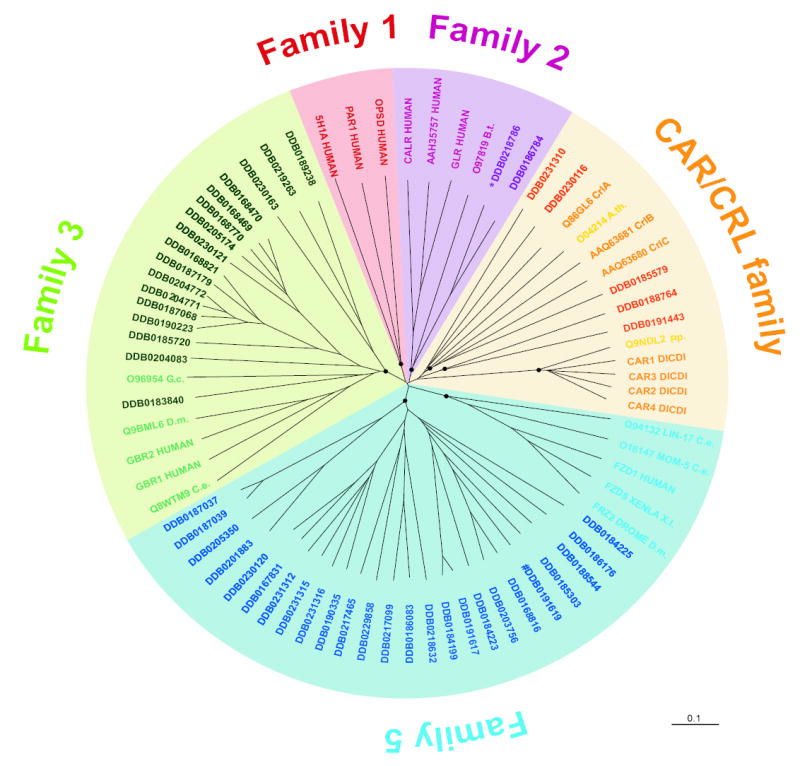
A CLUSTALX alignment of the sequences encompassing the seven transmembrane domains of all Dictyostelium GPCRs, and selected GPCRs from other organisms, was used to create an unrooted dendrogram with the TreeView program. A black circle marks the innermost node of each branch supported by >60% bootstraps. # indicates that this gene model has to be split, and the asterisk indicates a putative pseudogene. dictyBase identifiers (DDB…) were used for the newly discovered Dictyostelium receptors and SwissProt identifiers for all other receptors. CAR/CRL: cAMP receptor/cAMP receptor-like. A.th.: A. thaliana, P.p.: Polysphondylium pallidum, C.e.: C. elegans, D.m.: D. melanogaster; B.t: Bos taurus; X.l.: Xenopus laevis; G.c.: Geodia cydonium.
SH2 domain signalling
In animals, SH2 domains act as regulatory modules of proteins in intracellular signalling cascades, interacting with phosphotyrosine-containing peptides in a sequence-specific manner. Dictyostelium is the only organism, outside the animal kingdom, where SH2 domain-phosphotyrosine signalling has been proven to occur63. What have been lacking in Dictyostelium are the other components of such signalling pathways - equivalents of the metazoan SH2 domain-containing receptors, adaptors and targeting proteins. Three newly predicted proteins are strong candidates for these roles (Fig. SI 15). One of them, CblA, is highly related to the metazoan cbl proto-oncogene product. This is entirely unexpected because it is the first time that a cbl homologue has been observed outside the animal kingdom. The Cbl protein is a “RING finger” ubiquitin-protein ligase that recognizes activated receptor tyrosine kinases and various molecular adaptors64. Remarkably, the Cbl SH2 domain went unrecognised in the protein sequence, but it was revealed when the crystal structure of the protein was determined65. Thus, although SH2 domain proteins are less prevalent in Dictyostelium, there is the potential for the kind of complex interactions that typify metazoan SH2 signalling pathways.
ABC transporter signalling
Dictyostelium, like other organisms, has adapted ABC transporters to control various developmental signalling events. Several ABC transporters, TagA, B and C, are used for peptide-based signalling, akin to that previously observed for mating in S. cerevisiae and antigen presentation in human T cells66–68. The novel domain arrangement of the Tag proteins, a serine protease domain fused to a single transporter domain, suggests that they have been selected for improved efficiency in signal production. Additional ABC transporters are needed for cell fate determination in Dictyostelium, suggesting that this ubiquitous protein family may be used in similar developmental contexts within many different species69.
Kinases and transcription factors
Much cellular signal transduction involves the regulation of protein function through phosphorylation by protein kinases, often leading to the reprogramming of gene transcription in response to extracellular signals. The Dictyostelium proteome contains 295 predicted protein kinases, representing as wide a spectrum of kinase families as that observed in metazoa (Tables SI 14–16; Fig. SI 16). Given the presence of SH2 domain-based signalling it was surprising that no receptor tyrosine kinases could be recognized in the genome. However, Dictyostelium has a number of other receptor kinases such as the histidine kinases and a group of eight novel putative receptor serine/threonine kinases, which are involved in nutrient and starvation sensing70. Most of the ubiquitous families of transcription factors are represented in Dictyostelium, with the notable exception of the otherwise ubiquitous basic helix-loop-helix proteins (Table SI 17; Fig. SI 17). Compared to other eukaryotes, Dictyostelium appears to have fewer transcription factors relative to the total number of genes, suggesting that many transcription factors are yet to be defined, or that the activities of a smaller repertoire of factors are combined and controlled to achieve complex regulation (Table SI 18; Fig. SI 18).
Cell adhesion
Throughout Dictyostelium development, cells must modulate their adhesiveness to the substrate, to the extracellular matrix and to other cells in order to create tissues and carry out morphogenesis. To accomplish this, Dictyostelium uses a surprising number of components that have been normally only associated with animals. For example, disintegrin proteins regulate cell adhesiveness and differentiation in a number of metazoa and at least one Dictyostelium disintegrin, AmpA, is needed throughout development for cell fate specification71. We also identified distant relatives of vinculin and α-catenin – normally associated with adherens junctions - that support the idea that the epithelium-like sheet of cells that surrounds the stalk tube contains such junctions72. Consistent with this, the Dictyostelium genome encodes numerous proteins previously described as components of adherens junctions in metazoa like β-catenin (Aardvark), α-actinin, formins, VASP and myosinVII.
In animals, tandem repeats of immunoglobulin, cadherin, fibronectin III or E-set domains are often present in cell adhesion proteins, although their common protein fold predates the emergence of eukaryotes. EGF/Laminin domains are also found in adhesion proteins but, prior to the analysis of the Dictyostelium genome, no non-metazoan was known to have more than two EGF repeats in a single predicted protein. Dictyostelium has 61 predicted proteins containing repeated E-set or EGF/Laminin domains and many of these contain additional domains that suggest they have roles in cell adhesion or cell recognition, such as mannose-6-phosphate receptor, fibronectin III, or growth factor receptor domains and transmembrane domains (Fig. 9). In support of this idea, four of these proteins, LagC, LagD, AmpA and ComC, have been shown to be required for cell adhesion and signalling during development71, 73–75.
Figure 9. Putative adhesion/signalling proteins.
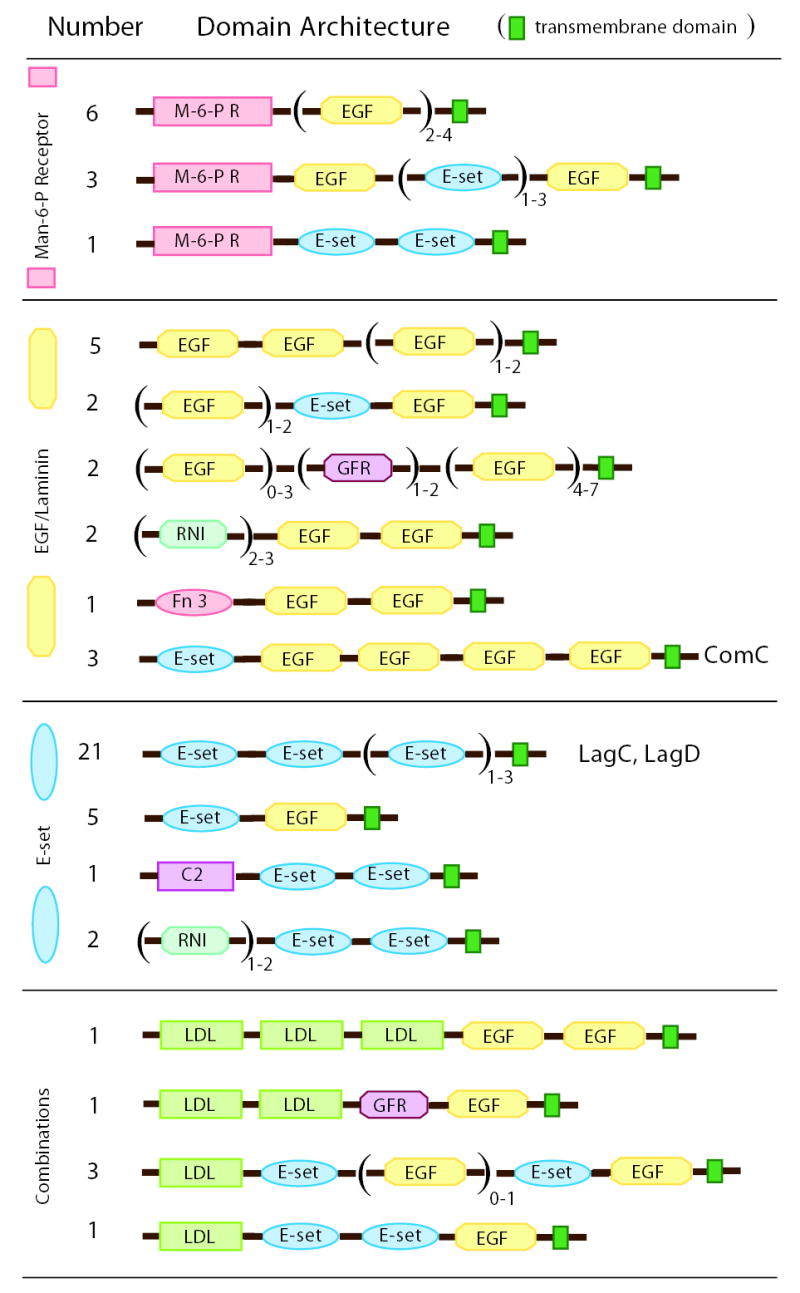
Proteins containing repeated EGF/laminin and/or E-set SCOP Superfamily domains are classified into groups containing mannose-6-phosphate receptor, mainly EGF/laminin, mainly E-set, or combinations of domains. Most of these proteins have predicted transmembrane domains and so are expected to be cell surface proteins. ComC, LagC, and LagD are proteins that have been characterized to have adhesion and/or signalling functions during multicellular development73–75. Other domain abbreviations: M-6-P R, mannose-6-phosphate receptor; GFR, growth factor receptor; RNI, RNI-like; Fn 3, fibronectin type III; C2, Calcium-dependent lipid binding; LDL, L domain-like leucine-rich repeat.
Cellulose-based structures
During development, Dictyostelium cells produce a number of cellulose-based structural elements. Dictyostelium slugs synthesize an extracellular matrix, or sheath, around themselves that is comprised of proteins and cellulose. Several of the smaller sheath proteins bind cellulose and are believed to have a role in slug migration, while the larger, cysteine-rich EcmA protein is essential for full integrity of the sheath and for establishing correct slug shape76, 77. During terminal differentiation, cellulose is deposited in the stalk and in the cell walls of the stalk and spore cells78–80. The first confirmed eukaryotic gene for cellulose synthase was discovered in Dictyostelium and this gene has since been recognized in many plants, N. crassa and the ascidian Ciona intestinalis81. The fungal and urochordate enzymes are more closely related to the Dictyostelium homologue than to plant or bacterial cellulose synthases, indicating that the common ancestor of fungi and animals carried a gene for cellulose synthase that was subsequently lost in most animals. The Dictyostelium genome encodes more than 40 additional proteins that are likely to be involved in cellulose synthesis or degradation and probably are involved in the production and remodelling of cellulose fibres of the slug sheath, stalk tube and cell walls (see Supplementary Information).
The fundamental similarities in cellular cooperation found in Dictyostelium and in the metazoa clearly resulted in a parallel positive selection for structural and regulatory genes required for cell motility, adhesion and signalling. Dictyostelium uses a set of signals and adhesion proteins that are distinct from those employed for similar purposes in metazoa but, like the metazoa, Dictyostelium has maintained a diversity of GPCRs, protein kinases and ABC transporters which enable it to respond to those signals. Dictyostelium has also retained and modified an organizational strategy perfected in plants, basing several structural elements on cellulose. At one level Dictyostelium has achieved multicellularity by employing strategies that are similar to plants and metazoa, but the differences between them suggest convergent evolution, rather than lineal descent from an ancestor with overt or latent multicellular capacities.
Conclusions and prospects
The complete protein repertoire of Dictyostelium provides a new perspective for studying its cellular and developmental biology. At a systems level, Dictyostelium provides a level of complexity that is greater than the yeasts, but much simpler than plants or animals. Thus, high-resolution molecular analyses in this system may reveal control networks that are difficult to study in more complex systems and may presage regulatory strategies used by higher organism82–84. At a practical level, the comparative genomics of Dictyostelium and related pathogens, such as Entamoeba histolytica, should aid in the functional definition of amoebozoa-specific genes that may open new avenues of research aimed at controlling amoebic diseases. Dictyostelium’s adeptness at hunting bacteria also renders it susceptible to infections by intracellular bacterial pathogens85, 86. Dictyostelium and human macrophages display fundamental similarities in their cell biology, which has spurred the use of Dictyostelium as a model host for bacterial pathogenesis. It is also an attractive model in which to study other disease processes: for a number of human disease-related proteins, it provides a test-bed for studying their functions in a model organism which has greater similarity to higher eukaryotes than do the yeasts, yet shares the latter’s experimental tractability.
The high frequency of repeated amino acids tracts in Dictyostelium proteins has long been known anecdotally, but we can now survey their precise nature and number and find them to be more abundant than in any other sequenced genome. Many human diseases result from the expansion of triplet nucleotide repeats, some of which encode polyglutamine tracts that cause cell degeneration87, 88. Learning how Dictyostelium cells tolerate so many proteins with amino acid homopolymers will, we hope, help to elucidate the roles of these motifs in protein function and dysfunction.
Comparative genomic studies in eukaryotes are providing the raw material for global examinations of the evolution of cellular regulation and developmental mechanisms31. Many genes have been lost in one species but retained in others such that each new genome sequence adds to our understanding of the genetic complement of the eukaryotic progenitor. Thus, our understanding of eukaryotes will continue to be refined as more genome sequences become available from representatives of large groups of organisms whose genomes remain largely unexplored, such as the amoebozoa. The surprising molecular diversity of the Dictyostelium proteome, which includes protein assemblages usually associated with fungi, plants or animals, suggests that their last common ancestor had a greater number of genes than had been previously appreciated.
Methods
Details on the availability of reagents can be found in the Supplementary Information. All analyses described here were performed on Version 2.0 of the genome sequence. Updates to the sequence and annotation are available at http://dictybase.org and http://www.genedb.org/genedb/dicty/index.jsp. Further details of analyses not explicitly described below can be found in the Supplementary Information.
HAPPY mapping
A short-range (~100kb), high-resolution (+/−8.54kb) mapping panel was prepared as described9. Briefly, 96 aliquots each containing ±0.52 haploid genome equivalents of sheared AX4 genomic DNA were pre-amplified by PEP (primer extension pre-amplification89). A total of 4913 STS markers (Table SI 1) were typed by 2-phase hemi-nested PCR (multiplexed for up to 1200 markers in the first phase) on aliquots of the diluted PEP products. Maps were assembled from good-quality data essentially as described previously8. A second, longer-range (±150kb) mapping panel was used to confirm some linkages on chromosomes 2 and 5. HAPPY map analysis and PCR primer design for HAPPY mapping was performed using various custom programmes (PHD and ATB unpublished).
Chromosome-purification
Genomic DNA from D. discoideum strain AX4 was prepared and separated by PFGE essentially as described27, 9, except that gels were run in stacked pairs; one member of each pair was stained with ethidium bromide, and bands excised from its unstained counterpart by alignment.
WCS and YAC-subclone libraries
For WCS libraries, gel slices (above) were disrupted by several passages through a 30G syringe needle, digested with beta-agarase (NEB) and phenol-extracted. DNA was concentrated by ethanol precipitation, sonicated, end-blunted using mung bean nuclease and size-fractionated on 0.8% low melting-point agarose gels. Fractions of 1.4-2kb and 2-4kb were excised, DNA extracted as before and ligated into the SmaI site of pUC18 or pUC19. Clone propagation and template preparation followed standard protocols.
For YAC subclone libraries, AX4-derived YACs were identified (and their position and integrity confirmed) by screening the set described by Loomis et al22 using markers from the HAPPY map. Subclones were prepared from PFG-purified YACs essentially as for the WCS libraries; contaminating yeast-derived sequences were filtered out in silico.
Sequencing and assembly
Details of the sequencing and assembly methods can be found in Supplementary Information. Generally, mapped sequence features were used to nucleate sequence contigs assembled from the WCS data, and extended using read-pair information and iterative searches for overlapping sequences, followed by directed gap-closure using a range of approaches.
Fluorescent in situ hybridisation
In situ hybridization was performed as in reference 17
Gene prediction and identification of sequence features
Full details are provided in the Supplementary Information. Briefly, automated gene prediction was performed using a combination of programmes which had been trained on well-characterized D. discoideum genes, and the results integrated with reference to D. discoideum cDNA sequences and homology to genes in other species. Other features in the predicted proteins, and other sequence features, were identified using a variety of software packages.
Analysis of functional gene clustering
Microarray targets54, 90, 91; and N. Van Driessche & G. Shaulsky unpublished) and gene models were mapped onto the genome sequence using BLAST92 and the modified LIS algorithm93. To look for clustering of genes with correlated temporal expression profiles, pairwise correlation coefficients were calculated for genes with known expression profiles on each chromosome91. Blocks of ≥6 consecutive genes were sought for which either (a) all pairwise correlation coefficients were positive and ≥70% were >0.2 (genes with similar developmental trajectories) or (b) each gene had a partner with an absolute correlation coefficient value of >0.6 (tightly co-regulated genes); no statistically significant clusters met these criteria.
To look for clustering of genes associated with specific developmental stages94, 95 or cell types90, 96, the genome was scanned with various sized windows97 for regions with significant (p<0.01) overrepresentation of genes in any one of these groups.
Analysis of duplicated genes
Predicted protein sequences were clustered using TribeMCL98, using a BLAST-P expectation of <10−40 as a cutoff. A χ-squared test invalidated the hypothesis that members of a family are randomly distributed in the genome. Within each family, protein divergences (similarity distances computed using the ‘Protdist’ module of PHYLIP; http://evolution.genetics.washington.edu/phylip.html) and physical intergenic distances between all pairs of family members were tabulated, and the correlation coefficient between the former and latter values was calculated. Analysis was performed on the 86 gene families (representing 155 gene pairs) with at least 10 intrachromosomal distance pairings to provide robust statistical confidence.
Other sequence analysis and graphical representation
Other sequence analyses (nucleotide and dinucleotide composition; identification of simple-sequence repeats in nucleotide and protein sequence; coding density computation; tRNA cluster identification) was performed using a range of custom software (PHD and ATB unpublished). Graphical representation of chromosomes in Fig. 2 was done primarily using Cinema4D-8.5 (Maxon Computer GmbH) after pre-processing using custom software (PHD).
Supplementary Material
Acknowledgments
Sequencing and analysis of chromosomes 1, 2 and 3 was supported by grants from the DFG and by Köln Fortune, and that of chromosomes 4, 5 and 6 in the USA by grants from NICHD/NIH. Work in the UK/EU was supported by a program grant from the MRC to J.W., R.R.K., B.B. and P.H.D. and by the EU. Analyses at DictyBase were supported by a grant from the NIGMS/NIH to R.L.C. The Dictyostelium cDNA project was supported from Research for the Future of JSPS and from Grants-in-Aid for Scientific Research on Priority Areas of MEXT of Japan.
The German team wishes to thank Silke Förste, Nadine Zeisse, Sandra Rothe, Sabine Landmann, Regine Schultz, Christiane Neuhoff and Rolf Müller for expert technical assistance. The USA team is indebted to Steven Kaminsky, Steven Klein and Tyl Hewitt for their scientific foresight in the early stages of this project and for their support of Dictyostelium as a model system, and to Hailey Hosak, Oliver Delgado, Lora Lewis, Keelan Hamilton, Jennifer Hume, Christie Kovar Smith, Dearl Neal, Paul Havlak, K. James Durbin and Paula Burch of the HGSC at Baylor College of Medicine. R.S. thanks Laura Cortez, Emily Joyner and Barry Hill for their assistance during the course of the project. C.B. thanks Michel Veron and Philippe Glaser for their support and fruitful discussions. P.H.D. thanks Helen O’Hare for early involvement in the project, Al Ivens for helpful discussions, the MRC Centre Visual Aids department, Cambridge for advice on graphics. We thank Sharen Bowman and Daniel Lawson for their contribution to the EUDICT region in the initial stages of the project, and David Martin at the Wellcome Trust Biocentre, University of Dundee for running the GOtcha search of our gene models. The Japanese cDNA project deeply thanks Naotake Ogasawara of the Nara Institute of Science and Technology and Ikuo Takeuchi for valuable comments and encouragement, and others who participated in earlier stages of the project.
Footnotes
Correspondence
Correspondence should be sent to P.H.D. (phd@mrc-lmb.cam.ac.uk). Chromosomal sequences are deposited under accession number AAFI00000000.
Competing interests statement
The authors declare that they have no competing financial interests.
References
- 1.Kessin, R.H., Dictyostelium - Evolution, cell biology, and the development of multicellularity. 2001, Cambridge, UK: Cambridge Univ. Press. xiv + 294.
- 2.Konijn TM, et al. The acrasin activity of adenosine-3',5'-cyclic phosphate. Proc Natl Acad Sci USA. 1967;58:1152–1154. doi: 10.1073/pnas.58.3.1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Müller K, Gerisch G. A specific glycoprotein as the target site of adhesion blocking Fab in aggregating Dictyostelium cells. Nature. 1978;274:445–449. doi: 10.1038/274445a0. [DOI] [PubMed] [Google Scholar]
- 4.Raper KB. Pseudoplasmodium formation and organization in Dictyostelium discoideum. J Elisha Mitchell Sci Soc. 1940;56:241–282. [Google Scholar]
- 5.Raper KB. Dictyostelium discoideum, a new species of slime mold from decaying forest leaves. J Agr Res. 1935;50:135–147. [Google Scholar]
- 6.Knecht DA, Cohen SM, Loomis WF, Lodish HF. Developmental regulation of Dictyostelium discoideum actin gene fusions carried on low-copy and high-copy transformation vectors. Mol Cell Biol. 1986;6:3973–3983. doi: 10.1128/mcb.6.11.3973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dear PH, Cook PR. HAPPY mapping - linkage mapping using a physical analog of meiosis. Nucleic Acids Res. 1993;21:13–20. doi: 10.1093/nar/21.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Konfortov BA, Cohen HM, Bankier AT, Dear PH. A high-resolution HAPPY map of Dictyostelium discoideum chromosome 6. Genome Res. 2000;10:1737–1742. doi: 10.1101/gr.141700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Glöckner G, et al. Sequence and analysis of chromosome 2 of Dictyostelium discoideum. Nature. 2002;418:79–85. doi: 10.1038/nature00847. [DOI] [PubMed] [Google Scholar]
- 10.Urushihara H, et al. Analyses of cDNAs from growth and slug stages of Dictyostelium discoideum. Nucleic Acids Res. 2004;32:1647, 1653. doi: 10.1093/nar/gkh262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Smith SS, Ratner DI. Lack of 5-methylcytosine in Dictyostelium discoideum DNA. Biochem J. 1991;277:273–275. doi: 10.1042/bj2770273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Glöckner G, et al. The complex repeats of Dictyostelium discoideum. Genome Res. 2001;11:585–594. doi: 10.1101/gr.162201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Crick FH. Codon-anticodon pairing: the wobble hypothesis. J Mol Biol. 1966;19:548–555. doi: 10.1016/s0022-2836(66)80022-0. [DOI] [PubMed] [Google Scholar]
- 14.Soll, D. & RajBhandary, U. (eds) tRNA: structure, biosynthesis and function. (ASM, Washington DC, 1995).
- 15.Burger G, Plante I, Lonergan KM, Gray MW. The mitochondrial DNA of the amoeboid protozoon, Acanthamoeba castellanii: complete sequence, gene content and genome organization. J Mol, Biol. 1995;3:522–537. doi: 10.1006/jmbi.1994.0043. [DOI] [PubMed] [Google Scholar]
- 16.Ogawa S, et al. The mitochondrial DNA of Dictyostelium discoideum: complete sequence, gene content and genome organization. Molecular and General Genetics. 2000;263:514–519. doi: 10.1007/pl00008685. [DOI] [PubMed] [Google Scholar]
- 17.Sucgang R, et al. Sequence and structure of the extrachromosomal palindrome encoding the ribosomal RNA genes in Dictyostelium. Nucleic Acids Res. 2003;31:2361–2368. doi: 10.1093/nar/gkg348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Szafranski K, Dingermann T, Glöckner G, Winckler T. Template jumping by a LINE reverse transcriptase has created a SINE-like 5S rRNA retropseudogene in Dictyostelium. Mol Genet Genomics. 2004;271:98–102. doi: 10.1007/s00438-003-0961-9. [DOI] [PubMed] [Google Scholar]
- 19.Eichler EE, Sankoff D. Structural dynamics of eukaryotic chromosome evolution. Science. 2003;301:793–797. doi: 10.1126/science.1086132. [DOI] [PubMed] [Google Scholar]
- 20.Cappello J, Cohen SM, Lodish H. F Dictyostelium transposable element DIRS-1 preferentially inserts into DIRS-1 sequences. Mol Cell Biol. 1984;4:2207–2213. doi: 10.1128/mcb.4.10.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cappello J, Handelsman K, Lodish HF. Sequence of Dictyostelium DIRS-1: an apparent retrotransposon with inverted terminal repeats and an internal circle junction sequence. Cell. 1985;43:105–115. doi: 10.1016/0092-8674(85)90016-9. [DOI] [PubMed] [Google Scholar]
- 22.Loomis WF, Welker D, Hughes J, Maghakian D, Kuspa A. Integrated maps of the chromosomes in Dictyostelium discoideum. Genetics. 1995;141:147–157. doi: 10.1093/genetics/141.1.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goodwin TJ, Poulter RT. Multiple LTR-retrotransposon families in the asexual yeast Candida albicans. Genome Res. 2000;10:174–191. doi: 10.1101/gr.10.2.174. [DOI] [PubMed] [Google Scholar]
- 24.Appelgren H, Kniola B, Ekwall K. Distinct centromere domain structures with separate functions demonstrated in live fission yeast cells. J Cell Sci. 2003;116:4035–4042. doi: 10.1242/jcs.00707. [DOI] [PubMed] [Google Scholar]
- 25.Kuspa A, Maghakian D, Bergesch P, Loomis WF. Physical mapping of genes to specific chromosomes in Dictyostelium discoideum. Genomics. 1992;13:49–61. doi: 10.1016/0888-7543(92)90201-3. [DOI] [PubMed] [Google Scholar]
- 26.McClintock B. The production of homozygous deficient tissues with mutant characteristics by means of the aberrant mitotic behaviour of ring-shaped chromosomes. Genetics. 1938;23:315–376. doi: 10.1093/genetics/23.4.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cox EC, Vocke CD, Walter S, Gregg KY, Bain ES. Electrophoretic karyotype for Dictyostelium discoideum. Proc Natl Acad Sci USA. 1990;87:8247–8251. doi: 10.1073/pnas.87.21.8247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–941. [Google Scholar]
- 29.Gardner MJ, Hall N, Fung E, White O, Berriman M, et al. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature. 2002;419:498–511. doi: 10.1038/nature01097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Trusov YA, Dear PH. A molecular clock based on the expansion of gene families. Nucleic Acids Res. 1996;24:995–999. doi: 10.1093/nar/24.6.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rubin GM, et al. Comparative genomics of the eukaryotes. Science. 2000;287:2204–15. doi: 10.1126/science.287.5461.2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kellis M, Birren BW, Lander ES. Proof and evolutionary analysis of ancient genome duplication in the yeast Saccharomyces cerevisiae. Nature. 2004;428:617–24. doi: 10.1038/nature02424. [DOI] [PubMed] [Google Scholar]
- 33.Dujon B, et al. Genome evolution in yeasts. Nature. 2004;430:35–44. doi: 10.1038/nature02579. [DOI] [PubMed] [Google Scholar]
- 34.Raper, K. B. The Dictyostelids. (Princeton Univ. Press, Princeton, NJ, 1984).
- 35.Loomis WF, Smith DW. Consensus phylogeny of Dictyostelium. Experientia. 1995;51:1110–1115. doi: 10.1007/BF01944728. [DOI] [PubMed] [Google Scholar]
- 36.Baldauf SL, Doolittle WF. Origin and evolution of the slime molds (Mycetozoa) Proc Natl Acad Sci USA. 1997;94:12007–12012. doi: 10.1073/pnas.94.22.12007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bapteste E, et al. The analysis of 100 genes supports the grouping of three highly divergent amoebae: Dictyostelium, Entamoeba, and Mastigamoeba. Proc Natl Acad Sci U S A. 2002;99:1414–9. doi: 10.1073/pnas.032662799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nara T, Hshimoto T, Aoki T. Evolutionary implications of the mosaic pyrimidine-biosynthetic pathway in eukaryotes. Gene. 2000;257:209–22. doi: 10.1016/s0378-1119(00)00411-x. [DOI] [PubMed] [Google Scholar]
- 39.Olsen R, Loomis WF. A model of orthologous protein sequence divergence. Journal of Molecular Evolution. 2005 in press. [Google Scholar]
- 40.Thomason P, Kay R. Eukaryotic signal transduction via histidine-aspartate phosphorelay. J Cell Sci. 2000;113:3141–3150. doi: 10.1242/jcs.113.18.3141. [DOI] [PubMed] [Google Scholar]
- 41.Fortini ME, Skupski MP, Boguski MS, Hariharan IK. A survey of human disease gene counterparts in the Drosophila genome. J Cell Biol. 2000;150:F23–30. doi: 10.1083/jcb.150.2.f23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jain R, Rivera MC, Moore JE, Lake JA. Horizontal gene transfer in microbial genome evolution. Theor Popul Biol. 2002;61:489–95. doi: 10.1006/tpbi.2002.1596. [DOI] [PubMed] [Google Scholar]
- 43.Richards TA, Hirt RP, Williams BA, Embley TM. Horizontal gene transfer and the evolution of parasitic protozoa. Protist. 2003;154:17–32. doi: 10.1078/143446103764928468. [DOI] [PubMed] [Google Scholar]
- 44.Iyer LM, Aravind L, Coon SL, Klein DC, Koonin EV. Evolution of cell-cell signaling in animals: did late horizontal gene transfer from bacteria have a role? Trends Genet. 2004;20:292–9. doi: 10.1016/j.tig.2004.05.007. [DOI] [PubMed] [Google Scholar]
- 45.Myllykallio H, et al. An alternative flavin-dependent mechanism for thymidylate synthesis. Science. 2002;297:105–107. doi: 10.1126/science.1072113. [DOI] [PubMed] [Google Scholar]
- 46.Kessin RH, Gundersen GG, Zaydfudim V, Grimson M, Blanton RL. How cellular slime molds evade nematodes. Proc Natl Acad Sci USA. 1996;93:4857–4861. doi: 10.1073/pnas.93.10.4857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Morris HR, Taylor GW, Masento MS, Jermyn KA, Kay RR. Chemical structure of the morphogen differentiation inducing factor from Dictyostelium discoideum. Nature. 1987;328:811–814. doi: 10.1038/328811a0. [DOI] [PubMed] [Google Scholar]
- 48.Cane DE, Walsh CT, Khosla C. Harnessing the biosynthetic code: combinations, permutations, and mutations. Science. 1998;282:63–8. doi: 10.1126/science.282.5386.63. [DOI] [PubMed] [Google Scholar]
- 49.Holland IB, Blight MA. ABC-ATPases, adaptable energy generators fuelling transmembrane movement of a variety of molecules in organisms from bacteria to humans. J Mol Biol. 1999;293:381–99. doi: 10.1006/jmbi.1999.2993. [DOI] [PubMed] [Google Scholar]
- 50.Andrade AC, Van Nistelrooy JG, Peery RB, Skatrud PL, De Waard MA. The role of ABC transporters from Aspergillus nidulans in protection against cytotoxic agents and in antibiotic production. Mol Gen Genet. 2000;263:966–77. doi: 10.1007/pl00008697. [DOI] [PubMed] [Google Scholar]
- 51.Mendez C, Salas JA. The role of ABC transporters in antibiotic-producing organisms: drug secretion and resistance mechanisms. Res Microbiol. 2001;152:341–50. doi: 10.1016/s0923-2508(01)01205-0. [DOI] [PubMed] [Google Scholar]
- 52.Schoonbeek HJ, Raaijmakers JM, De Waard MA. Fungal ABC transporters and microbial interactions in natural environments. Mol Plant Microbe Interact. 2002;15:1165–72. doi: 10.1094/MPMI.2002.15.11.1165. [DOI] [PubMed] [Google Scholar]
- 53.Anjard C, Loomis WF. Evolutionary analyses of ABC transporters of Dictyostelium discoideum. Eukaryot Cell. 2002;1:643–52. doi: 10.1128/EC.1.4.643-652.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Iranfar N, Fuller D, Loomis WF. Genome-wide expression analyses of gene regulation during early development of Dictyostelium discoideum. Eukaryot Cell. 2003;2:664–70. doi: 10.1128/EC.2.4.664-670.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Devreotes PN, Zigmond SH. Chemotaxis in eukaryotic cells: A focus on leukocytes and Dictyostelium. Annu Rev Cell Biol. 1988;4:649–686. doi: 10.1146/annurev.cb.04.110188.003245. [DOI] [PubMed] [Google Scholar]
- 56.Noegel AA, Schleicher M. The actin cytoskleleton of Dictyostelium: a story told by mutants. J Cell Sci. 2000;113:759–766. doi: 10.1242/jcs.113.5.759. [DOI] [PubMed] [Google Scholar]
- 57.Rivero F, Somesh BP. Signal transduction pathways regulated by Rho GTPases in Dictyostelium. J Muscle Res Cell Motil. 2002;23:737–49. doi: 10.1023/a:1024423611223. [DOI] [PubMed] [Google Scholar]
- 58.Merlot S, Firtel RA. Leading the way: directional sensing through phosphatidylinositol 3-kinase and other signalling pathways. J Cell Sci. 2003;116:3471–3478. doi: 10.1242/jcs.00703. [DOI] [PubMed] [Google Scholar]
- 59.Bockaert J, Pin JP. Molecular tinkering of G protein-coupled receptors: an evolutionary success. Embo J. 1999;18:1723–9. doi: 10.1093/emboj/18.7.1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ginsburg GT, et al. The regulation of Dictyostelium development by transmembrane signalling. J Eukaryot Microbiol. 1995;42:200–5. doi: 10.1111/j.1550-7408.1995.tb01565.x. [DOI] [PubMed] [Google Scholar]
- 61.Raisley B, Zhang M, Hereld D, Hadwiger JA. A cAMP receptor-like G protein-coupled receptor with roles in growth regulation and development. Dev Biol. 2004;265:433–45. doi: 10.1016/j.ydbio.2003.09.035. [DOI] [PubMed] [Google Scholar]
- 62.King N, Hittinger CT, Carroll SB. Evolution of key cell signaling and adhesion protein families predates animal origins. Science. 2003;301:361–3. doi: 10.1126/science.1083853. [DOI] [PubMed] [Google Scholar]
- 63.Kawata T, et al. SH2 signaling in a lower eukaryote: A STAT protein that regulates stalk cell differentiation in Dictyostelium. Cell. 1997;89:909–916. doi: 10.1016/s0092-8674(00)80276-7. [DOI] [PubMed] [Google Scholar]
- 64.Thien CB, Langdon WY. Cbl: many adaptations to regulate protein tyrosine kinases. Nat Rev Mol Cell Biol. 2001;2:294–307. doi: 10.1038/35067100. [DOI] [PubMed] [Google Scholar]
- 65.Meng W, et al. Structure of the amino-terminal domain of Cbl complexed to its binding site on ZAP-70 kinase. Nature. 1999;398:84–90. doi: 10.1038/18050. [DOI] [PubMed] [Google Scholar]
- 66.Shaulsky G, Kuspa A, Loomis WF. A multidrug resistance transporter serine protease gene is required for prestalk specialization in Dictyostelium. Genes Devel. 1995;9:1111–1122. doi: 10.1101/gad.9.9.1111. [DOI] [PubMed] [Google Scholar]
- 67.Anjard C, Zeng C, Loomis WF, Nellen W. Signal transduction pathways leading to spore differentiation in Dictyostelium discoideum. Dev Biol. 1998;193:146–155. doi: 10.1006/dbio.1997.8804. [DOI] [PubMed] [Google Scholar]
- 68.Good JR, et al. TagA, a putative serine protease/ABC transporter of Dictyostelium that is required for cell fate determination at the onset of development. Development. 2003;130:2953–65. doi: 10.1242/dev.00523. [DOI] [PubMed] [Google Scholar]
- 69.Good JR, Kuspa A. Evidence that a cell-type-specific efflux pump regulates cell differentiation in Dictyostelium. Dev Biol. 2000;220:53–61. doi: 10.1006/dbio.2000.9611. [DOI] [PubMed] [Google Scholar]
- 70.Chibalina MV, Anjard C, Insall RH. Gdt2 regulates the transition of Dictyostelium cells from growth to differentiation. BMC Dev Biol. 2004;4:8. doi: 10.1186/1471-213X-4-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Blumberg DD, Ho HN, Petty CL, Varney TR, Gandham S. AmpA, a modular protein containing disintegrin and ornatin domains, has multiple effects on cell adhesion and cell fate specification. J Muscle Res Cell Motil. 2002;23:817–28. doi: 10.1023/a:1024440014857. [DOI] [PubMed] [Google Scholar]
- 72.Grimson MJ, et al. Adherens junctions and beta-catenin-mediated cell signalling in a non-metazoan organism. Nature. 2000;408:727–31. doi: 10.1038/35047099. [DOI] [PubMed] [Google Scholar]
- 73.Dynes JL, et al. LagC is required for cell-cell interactions that are essential for cell-type differentiation in Dictyostelium. Genes Devel. 1994;8:948–958. doi: 10.1101/gad.8.8.948. [DOI] [PubMed] [Google Scholar]
- 74.Wang J, et al. The membrane glycoprotein gp150 is encoded by the lagC gene and mediates cell-cell adhesion by heterophilic binding during Dictyostelium development. Dev Biol. 2000;227:734–745. doi: 10.1006/dbio.2000.9881. [DOI] [PubMed] [Google Scholar]
- 75.Kibler K, Svetz J, Nguyen TL, Shaw C, Shaulsky G. A cell-adhesion pathway regulates intercellular communication during Dictyostelium development. Dev Biol. 2003;264:506–521. doi: 10.1016/j.ydbio.2003.08.025. [DOI] [PubMed] [Google Scholar]
- 76.Morrison A, et al. Disruption of the gene encoding the EcmA, extracellular matrix protein of Dictyostelium alters slug morphology. Dev Biol. 1994;163:457–466. doi: 10.1006/dbio.1994.1162. [DOI] [PubMed] [Google Scholar]
- 77.Wang YZ, Slade MB, Gooley AA, Atwell BJ, Williams KL. Cellulose-binding modules from extracellular matrix proteins of Dictyostelium discoideum stalk and sheath. Eur J Biochem. 2001;268:4334–4345. doi: 10.1046/j.1432-1327.2001.02354.x. [DOI] [PubMed] [Google Scholar]
- 78.Freeze H, Loomis WF. Chemical analysis of stalk components of Dictyostelium discoideum. Biochim Biophys Acta. 1978;539:529–537. doi: 10.1016/0304-4165(78)90086-7. [DOI] [PubMed] [Google Scholar]
- 79.Zhang P, McGlynn A, Loomis WF, Blanton RL, West CM. Spore coat formation and timely sporulation depend on cellulose in Dictyostelium. Differentiation. 2001;67:72–79. doi: 10.1046/j.1432-0436.2001.067003072.x. [DOI] [PubMed] [Google Scholar]
- 80.West CM, Zhang P, McGlynn AC, Kaplan L. Outside-in signaling of cellulose synthesis by a spore coat protein in Dictyostelium. Euk Cell. 2002;1:281–292. doi: 10.1128/EC.1.2.281-292.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Blanton RL, Fuller D, Iranfar N, Grimson MJ, Loomis WF. The cellulose synthase gene of Dictyostelium. Proc Natl Acad Sci U S A. 2000;97:2391–6. doi: 10.1073/pnas.040565697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Thomason PA, et al. An intersection of the cAMP/PKA and two-component signal transduction systems in Dictyostelium. EMBO J. 1998;17:2838–2845. doi: 10.1093/emboj/17.10.2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Maeda M, et al. Periodic signaling controlled by an oscillatory circuit that includes protein kinases ERK2 and PKA. Science. 2004;304:875–8. doi: 10.1126/science.1094647. [DOI] [PubMed] [Google Scholar]
- 84.Soler-Lopez M, et al. Structure of an activated Dictyostelium STAT in its DNA-unbound form. Mol Cell. 2004;13:791–804. doi: 10.1016/s1097-2765(04)00130-3. [DOI] [PubMed] [Google Scholar]
- 85.Solomon JM, Rupper A, Cardelli JA, Isberg RR. Intracellular growth of Legionella pneumophila in Dictyostelium discoideum, a system for genetic analysis of host-pathogen interactions. Infect Immun. 2000;68:2939–47. doi: 10.1128/iai.68.5.2939-2947.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Skriwan C, et al. Various bacterial pathogens and symbionts infect the amoeba Dictyostelium discoideum. Int J Med Microbiol. 2002;291:615–624. doi: 10.1078/1438-4221-00177. [DOI] [PubMed] [Google Scholar]
- 87.Zoghbi HY, Orr HT. Glutamine repeats and neurodegeneration. Ann Rev Neurosci. 2000;23:217–47. doi: 10.1146/annurev.neuro.23.1.217. [DOI] [PubMed] [Google Scholar]
- 88.Brown LY, Brown SA. Alanine tracts: the expanding story of human illness and trinucleotide repeats. Trends Genet. 2004;20:51–8. doi: 10.1016/j.tig.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 89.Zhang L, Cui X, Schmitt K, Hubert R, Navidi W, Arnheim N. Whole genome amplification from a single cell: Implications for genetic analysis. Proc Natl Acad Sci USA. 1992;89:5487–5851. doi: 10.1073/pnas.89.13.5847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Iranfar N, et al. Expression patterns of cell-type-specific genes in Dictyostelium. Mol Biol Cell. 2001;12:2590–2600. doi: 10.1091/mbc.12.9.2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Van Driessche N, et al. A transcriptional profile of multicellular development in Dictyostelium discoideum. Development. 2002;129:1543–1552. doi: 10.1242/dev.129.7.1543. [DOI] [PubMed] [Google Scholar]
- 92.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 93.Zhang H. Alignment of BLAST high-scoring segment pairs based on the longest increasing subsequence algorithm. Bioinformatics. 2003;19:1391–1396. doi: 10.1093/bioinformatics/btg168. [DOI] [PubMed] [Google Scholar]
- 94.Katoh M, et al. An orderly retreat: dedifferentiation is a regulated process. Proc Natl Acad Sci USA. 2004;101:7005–7010. doi: 10.1073/pnas.0306983101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Xu Q, et al. Transcriptional transitions during Dictyostelium spore germination. Euk Cell. 2004;3:1101–1110. doi: 10.1128/EC.3.5.1101-1110.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Maeda M, et al. Changing patterns of gene expression in Dictyostelium prestalk cell subtypes recognized by in situ hybridization with genes from microarray analyses. Euk Cell. 2003;2:627–37. doi: 10.1128/EC.2.3.627-637.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cohen BA, Mitra RD, Hughes JD, Church GM. A computational analysis of whole-genome expression data reveals chromosomal domains of gene expression. Nature Genet. 2000;26:183–186. doi: 10.1038/79896. [DOI] [PubMed] [Google Scholar]
- 98.Enright AJ, Van Dongen S, Ouzounis CA. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002;30:1575–1584. doi: 10.1093/nar/30.7.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.