

Abstract
Alveolar cell apoptosis is involved in the pathogenesis of emphysema, a prevalent disease primarily caused by cigarette smoking. We report that ceramide, a second messenger lipid, is a critical mediator of alveolar destruction in emphysema. Inhibition of enzymes controlling de novo ceramide synthesis prevented alveolar cell apoptosis, oxidative stress and emphysema caused by blockade of the VEGF receptors in both rats and mice. Emphysema was reproduced with intra-tracheal instillation of ceramide in naïve mice. A feed-forward mechanism of ceramide synthesis due secretory acid sphingomyelinase was supported by the neutralizing effects of ceramide-specific antibody in mice and by sphingomyelinase-deficient fibroblasts. Stimulation of sphingosine-1-phosphate signaling prevented lung apoptosis, implicating that ceramide to sphingosine-1-phosphate balance is required for maintenance of alveolar septal integrity. Finally, increased lung ceramides in patients with smoking-induced emphysema position ceramide upregulation as a critical pathogenetic element and a promising target in this disease lacking effective therapies.
INTRODUCTION
Pulmonary emphysema, which with chronic bronchitis accounts for the chronic obstructive pulmonary diseases, is a highly prevalent lung disease, with no effective treatments. Emphysema involves a destructive and permanent enlargement of distal airspaces and alveolar walls1, ultimately leading to impaired oxygenation. Historically, the pathogenesis of emphysema has been linked to chronic lung inflammation causing protease/antiprotease imbalance2.
It has been recently recognized that alveolar cell apoptosis is a critical step in the cellular disappearance in emphysema3,4,5,6, accounting for the unique nature of alveolar septal destruction in emphysema when compared with other lung diseases also characterized by inflammation and increased matrix proteolysis. The mutual interactions among apoptosis, inflammation, oxidative stress and matrix proteolysis might account for the irrevocable progression of the disease despite smoking cessation4. Because ceramide is a prototypic second messenger molecule which modulates endothelial cell apoptosis7, oxidative stress7, and proteolysis8, we hypothesize that ceramide upregulation engages alveolar cell apoptosis and oxidative stress, amplifying lung destruction and thus causing emphysema.
The signaling mediated by ceramide has been implicated in fundamental eukaryotic cell functions, such as cellular differentiation, stress response, apoptosis, and senescence7,9. Studies have demonstrated a direct role for ceramide in the development of several neurological diseases as well as radiation-induced injury10,11 which share with emphysema a critical role for apoptosis and oxidative stress in its pathogenesis.
The pro-apoptotic effects of ceramide are mediated by a variety of mechanisms e.g. activation of kinase suppressor of ras12, protein phosphatases 1 and 2A13, cathepsin D14, or via direct alteration of plasma15 or mitochondrial16 membrane signaling properties. Enzymatically, ceramide is synthesized primarily via de novo pathway involving serine palmitoyl-CoA transferase and ceramide synthase or from membrane sphingomyelin breakdown via sphingomyelinases (Supplementary fig. 1). The acid sphingomyelinase (ASMase), recently implicated in the development of acute lung edema17, has lysosomal and secretory isoforms, the latter contributing to extracellular increases in ceramide10. A downstream product of ceramide metabolism is sphingosine-1-phosphate (S1-P) which, contrary to ceramide, mediates cell survival and proliferation18. Current evidence indicates that a balance between ceramide and S1-P levels regulates cellular survival and homeostasis19,20.
To investigate the role of ceramide in alveolar wall destruction, we employed the apoptosis- and oxidative stress-dependent emphysema model caused by VEGF receptors (VEGFR) inhibition in rats3 and mice. The advantage of this approach is that both the VEGFR blockade model and human emphysema share the critical pathobiological elements of apoptosis, oxidative stress, decreases in alveolar capillaries, and alveolar destruction2,21. We then translated our findings to the human disease by analyzing the ceramide expression in lungs of patients with cigarette-smoke induced emphysema.
RESULTS
Ceramide upregulation in VEGFR blockade-induced emphysema
SU5416, a specific VEGFR-1 and -2 inhibitor, induces apoptosis- and oxidative stress-dependent alveolar septal destruction and emphysema in rats3,5 and mice 21 days after its administration. VEGFR blockade promptly increased lung ceramide levels, approximately 2-fold at 3d in the mouse lung (Fig. 1a) and 3-fold at 1d in the rat lung (Fig. 1b), compared to vehicle. The enhanced ceramide expression in response to VEGFR inhibition was localized to the alveolar septal cells while expression in bronchial epithelium was similar to that of controls (Fig. 1a). Ceramide increased concurrently with enhanced expression of markers of oxidative stress and apoptosis, such as 8-hydroxy-guanosine and active caspase-3, respectively, which peak at 3d (5 and Supplementary Fig. 2), thus preceding the airspace enlargement (which peaks at 21–30d).
Figure 1. Lung ceramide levels are increased by VEGFR blockade.
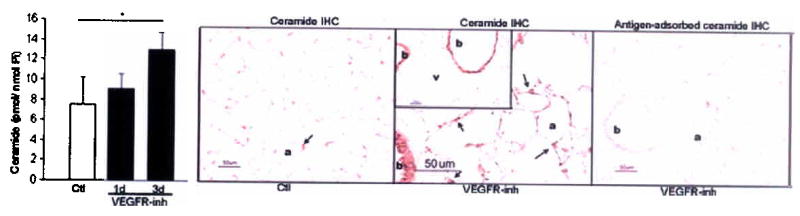
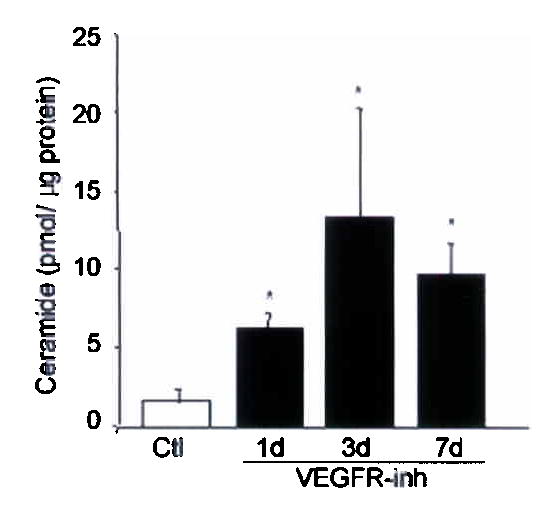




(a) Mouse lung ceramide levels induced by VEGFR inhibitor (VEGFR-inh, SU5416) or vehicle (Ctl); n=4 mice/group; mean +SD. *p=0.01. Mouse lung ceramide expression (brown, arrows) at 3d after treatment (a, alveoli, b, bronchial epithelium, and v large vessels). (b) Rat lung ceramide (n= 3–5 rats/group; mean +SD. *p<0.05). (c–e) Mouse lung enzymatic activities of ceramide synthase (c, n=4/group, +SD, *p=0.02); soluble (secretory) and lysosomal ASMase at 3d (d, n=4/group, +SE, p=0.04); and neutral sphingomyelinase (e, n=4/group; mean +SD). (f) Lung ceramides and ceramide metabolites at 7d after VEGFR-inh (n=4) or Ctl (n=3); mean +SE, *p<0.05.
We then measured the activity of enzymes critical for SU5416-induced lung ceramide synthesis and levels of specific ceramide metabolites at the time of ceramide upregulation. VEGFR blockade activated ceramide synthase rapidly, up to 3-fold at 3d (Fig. 1c). The earliest changes in the sphingomyelinase pathway were noted after 3d, with modest activation of only the secretory (soluble) ASMase, while activities of both the lysosomal ASMase and the neutral sphingomyelinase did not increase above baseline (Fig. 1d, e). VEGFR blockade increased the levels of direct precursors of ceramide in the de novo pathway (Supplementary Fig. 1) dihydrosphingosine (measured as dihydrosphingosine-P) and dihydroceramide at the time of ceramide elevation, while S1-P levels remained unchanged versus controls (Fig. 1f). In aggregate, these results implicate the de novo pathway as the major mechanism of early ceramide synthesis in response to VEGFR blockade.
Inhibition of de novo ceramide synthesis blocks emphysema
We inhibited the early (up to 5d) de novo ceramide production with fumonisin B1 (FB1) and myriocin, two specific inhibitors of ceramide synthase and serine palmitoyl transferase, respectively. FB1 attenuated SU5416-induced alveolar septal destruction in both mice and rats (Fig. 2a). The VEGFR blockade-induced increase in alveolar diameters was inhibited dose-dependently by FB1 pretreatment (Table 1), in mice and most markedly in rats (Fig. 2a, b, Table 1). The inhibitory effect was accounted primarily by a decrease in the number of severely damaged distal airspaces (Supplementary Fig. 3). FB1 expectedly caused significant decreases in lung ceramide induced by the VEGFR blockade by 57% (p=0.04) at 3d and by 32% (p=0.05) at 7d in mouse and rat, respectively (not shown) and markedly decreased both oxidative stress and apoptosis induced by VEGFR blockade (Fig. 2c, d).
Figure 2. Inhibition of de novo ceramide synthesis prevents VEGFR blockade-induced emphysema.
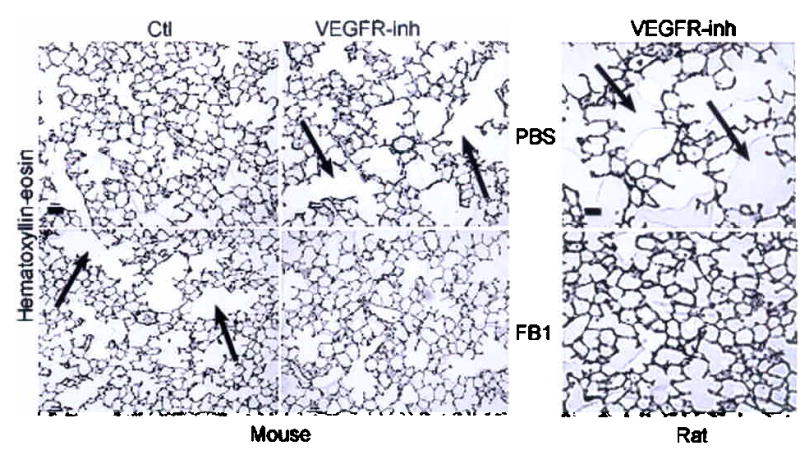




Effect of ceramide synthase inhibition with FB1 on alveolar histology (a) and diameters (b) 21d after VEFGR-inh (arrow: alveolar destruction; bar 50 μm; mean + SD, *p <0.05 versus VEGFR-inh; **p <0.05 versus vehicle. Effect of ceramide synthase inhibition with FB1 on lung oxidative stress (c) detected by 8-hydroxyguanosine (arrows) and on apoptosis (d) detected by active caspase-3 (mean + SE; *p<0.05) at 7d following VEGFR-inh. (e) Effect of S1-P augmentation (S1-P analog FTY720) on mouse lung apoptosis 7d after VEGFR-inh. Mean + SE, *p=0.008 versus Ctl and *p=0.0002 versus VEGFR-inh + FTY720.
Table 1.
Change (%) from baseline in alveolar diameters induced by VEGFR blockade and ceramide synthesis inhibitors.
| Treatment | VEGF R-inh | Vehicle | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Inhibitor (mg/kg) | PBS | FB1 (2.2) | FB1 (1.1) | Myriocin (1.0) | Myriocin (0.3) | PBS | FB1 (2.2) | FB1 (1.1) | Myriocin (1.0) |
| Mouse | 17.8* | 4.3# | 9.3# | −3.3# | −1.8# | 0 | 12.9* | 4.7 | −1.8 |
| Rat | 21.8* | −2.6# | 17.5* | n/a | n/a | 0 | 9.7* | n/a | n/a |
Abbreviations: n-a not assessed.
p <0.05 versus Vehicle + PBS
p <0.05 versus VEGFR-inh + PBS
Surprisingly, control animals treated with high-dose FB1 alone demonstrated increased alveolar size (Fig. 2a and b, Table 1), an effect potentially explained by excessive apoptosis as reported in the kidneys and liver22. Consistent with the hypothesis that FB1 induced cellular apoptosis by inhibiting downstream pro-survival sphingolipids, S1-P supplementation (by concomitant administration of S1-P analogue FTY72023,24) abolished both FB-1-induced apoptosis and airspace enlargement (Supplementary Fig. 3). To address the potential lack of specificity inherent to pharmacological inhibitors such as FB1, we inhibited the upstream enzyme in the de novo pathway, serine palmitoyl transferase with myriocin. Similar to FB1, early ceramide synthesis inhibition with myriocin attenuated SU5416-induced morphometric changes (Table 1) and decreased the number of severely damaged airspaces in mice (Supplementary Fig. 3), supporting our hypothesis that synthesis of abnormally high levels of ceramide cause lung cell apoptosis, oxidative stress and subsequent emphysema in VEGFR-inhibited animals.
We previously reported that caspase-dependent apoptosis was required for airspace enlargement in VEGFR blocker-treated rats3 and that oxidative stress and apoptosis form a mutually interactive feedback loop in this emphysema model5. To clarify the cause-effect relationship between ceramide signaling and alveolar cell apoptosis, we measured ceramide levels in morphometrically-normal rat lung resulting from co-treatment of the VEGFR inhibitor with a caspase inhibitor (z-Asp-CH(2)-DCB), as compared to emphysematous lungs caused by VEGFR inhibition alone. We detected similar elevations in ceramide in both conditions, suggesting that ceramide increases upstream of caspase activation (Supplementary Fig. 3).
Finally, to confirm the causal role of ceramide in emphysema development, we counteracted the biological effects of ceramide by augmenting the S1-P-engaged-signaling at the time of VEGFR blockade-induced lung ceramide. S1-P analogue administration markedly inhibited lung cell apoptosis induced by VEGFR inhibition, implicating a requirement for ceramide/S1-P balance in the maintenance of alveolar cell survival (Fig. 2e).
Augmentation of lung ceramide levels causes acute emphysema
Given that acute increases in ceramide cause apoptosis25, we tested whether directed intra-tracheal (i-t) instillation of ceramide recapitulates the phenotype of the VEGFR blockade, namely alveolar septal cell apoptosis, oxidative stress and emphysema. Because naturally-occurring long- and very-long-chain ceramides have important experimental limitations due to poor cell-permeability, we utilized synthetic short-chain ceramide for exogenous administration. Low-dose (1 mg/kg mixed in perfluorocarbon) or high-dose (10 mg/kg) C12 ceramide was i-t-instilled into mouse lung.
C12 ceramide instillation increased total lung ceramide levels in a dose-dependent manner when normalized by tissue weight (Fig. 3a), or tissue lipid (2-fold increase in the low-dose C12 group (14.5 pmol/nmol lipid Pi) and 4.8-fold increase in the high-dose group versus control (7 pmol/nmol). The increased lung ceramide levels were equivalent to those induced by VEGFR blockade. Impressively, a single lung instillation of C12 caused dose-dependent increases in alveolar diameters at 1d (Fig. 3b), which persisted through 6d (a 7% increase; p =0.01, not shown). Clearly, the alveolar enlargement was ceramide-specific, as dihydroceramide (1mg/kg, i-t), the biologically inactive ceramide precursor, did not alter alveolar diameters (Fig. 3b). Importantly, neither C12 ceramide, nor its negative control dihydroceramide caused alveolar collapse or extensive neutrophilic infiltrate in the time-frame studied.
Figure 3. Ceramide augmentation triggers lung apoptosis and emphysema.







Effect of i-t C12 ceramide (C12Cer) or inactive dihydroceramide (DH-Cer) instillation at 24h on (a) total lung ceramide (mean +SD, *p =0.002); (b) alveolar diameters (mean +SD, *p <0.05); and (c) apoptosis (arrows: active caspase-3; bar 50 μm) quantified as cells per field (mean +SD, #p =0.001; *p <0.05). (d) Mouse lung co-stained for nuclei (DAPI, blue), apoptotic nuclei (TUNEL, green, arrows, arrowheads) and alveolar epithelial II (SPC) or endothelial cells (CD34) in red. C12Cer triggered mostly endothelial, but also epithelial cell apoptosis (arrows). (e) Effect of caspase inhibition (z-Asp-CH(2)-DCB) on C12Cer-triggered lung caspase-3 activation (arrows; bar 25 μm). Effect of C12Cer on lung (f) MMP-12 activation (densitometric units normalized by actin + SD; *p<0.05) detected by immunoblot (inset; HT1080 cells positive control), (g) matrix proteolysis detected by gelatin zymography (lanes 2–4: 24h; 5–7: 72h), and (h) macrophage accumulation (arrows).
C12 ceramide instillation acutely triggered apoptosis, as measured by marked active caspase-3 expression (Fig. 3c), in situ TUNEL, and elevated Bax levels in the lung (Supplementary Fig. 4). Co-staining with cell-specific markers revealed that type II alveolar epithelial cells and mostly endothelial cells underwent apoptosis triggered by ceramide instillation (Fig. 3d). The ceramide-induced airspace enlargement was dependent on caspase activation, as it was prevented by pretreatment with a general caspase inhibitor (Fig. 3e).
Ceramide-induced alveolar cell apoptosis and septal destruction were associated with enhanced oxidative stress (8-hydroxyguanosine, Supplementary Fig. 3). Acute increases in ceramide also activated lung matrix proteolysis, detected by activation of MMP-12 (macrophage metalloelastase) (Fig. 3f), a protease implicated in smoking-induced emphysema26 and by zymography (Fig. 3g). The ceramide-induced MMP-12 activation was accompanied by increase number of resident lung macrophages, the main source of MMP-12 (Fig. 3h).
Positive feedback of ceramide synthesis in emphysema
The functional effects of exogenously-administered short-chain ceramides are explained by either intra-cellular fatty acid chain elongation, or by endogenous ceramide synthesis stimulation.27 The importance of the latter mechanism is in the initiation of a positive feedback of ceramide synthesis, allowing for amplification of apoptosis and lung destruction.
We sought to probe the presence of such mechanism by first measuring ceramide species by liquid chromatography-tandem mass spectroscopy (LC-MS/MS) in the mouse lung at baseline and 24h after C12 ceramide instillation (Fig. 4a). While no changes were detected in levels of unsaturated ceramides, C12 ceramide increased by 20% the endogenous saturated very-long-chain (C24), while decreasing levels of long-chain ceramides (C16, C18). These findings were partly confirmed with cultured lung endothelial cells treated with short-chain ceramide (C8, 10 μM, which causes lung endothelial cell apoptosis, data not shown). C8 ceramide markedly increased the levels of endogenous very-long-chain (C20-C26) and long-chain (C14-C18) ceramides in cultured pulmonary endothelial cells (Fig. 4b).
Figure 4. Self-amplification of lung ceramide synthesis.





Effect of short-chain exogenous ceramide on endogenous ceramide species profile in the (a) lung after C12Cer (1mg/kg, 24h, black) or vehicle (gray) and in (b) bovine pulmonary artery endothelial cells (thin layer chromatography of intra-cellular ceramides detected by DAG kinase assay) after C8Cer (10 μM; n=2). (c) Effect of C12Cer on the ASMase activity in the mouse lung (24h, mean +SD, *p =0.02). Effect of neutralizing ceramide-specific antibody (4mg/kg/d) on (d) lung ceramide species (mean +SE; *p =0.002) and on (e) lung apoptosis detected by active caspase-3 (arrows, bar 50 μm) induced by VEGFR-inh (3d).
The shift towards very-long ceramide synthesis after C12 ceramide instillation was associated with significant activation of lung ASMase activity (1.5- fold, p =0.02) (Fig. 4c), suggesting the sphingomyelin breakdown as a source of amplified endogenous ceramide production.
The sequential activation of the secretory, but not lysosomal ASMase after the activation of ceramide synthase by VEGFR blockade (Fig. 1d) could result from a feed-forward mechanism of ceramide synthesis, thus increasing the pool of paracellular ceramide and amplifying lung injury. To investigate the role of extracellular ceramide released in response to VEGFR blockade, we administered systemically a ceramide-specific antibody during the VEGFR inhibition in mice. The neutralizing ceramide antibody decreased by 30% lung very-long-chain ceramides (Fig. 4d) and attenuated lung apoptosis induced by the VEGFR inhibitor at 3d, as detected by immunohistochemistry (IHC) (Fig. 4e) and caspase-3 activity assay (~20% decrease), but did not inhibit the matrix proteolytic activity induced by SU5416 (not shown). These effects were less potent than those resulting from the inhibition of de novo ceramide synthesis. They however, suggested the presence of an active paracellular ceramide pool, potentially generated from plasma membrane sphingomyelin through a ceramide-triggered positive feedback mechanism.
We investigated in more detail this amplification mechanism utilizing ASMase-deficient human fibroblasts derived from patients with Niemann Pick’s disease. In response to ceramide (C8), but not dihydroceramide, ASMase-deficient fibroblasts failed to increase the amount of endogenous ceramides secreted in the media as compared to wild-type cells (Supplementary Fig. 5). These changes affected the most abundant ceramide species: C16:0,C24:1, and C24:0 (which together constitute >90% of the secreted ceramide), but not C14:0 (~1%). The ASMase-deficient cells exhibited an early resistance to exogenous ceramide-induced apoptosis (4h). These differences persisted albeit as a trend at 8h and 24h (Supplementary Fig. 5). The apoptosis induced by exogenous ceramide in ASMase-deficient cells was de novo synthesis-dependent, as it was blocked by myriocin, whereas wild-type cell apoptosis likely involved myriocin-independent pathways (i.e. ASMase) (Supplementary Fig. 5). These data suggest that ASMase amplifies ceramide synthesis in a paracellular manner and that exogenous ceramide-induced apoptosis requires both de novo and ASMase-mediated endogenous ceramide generation.
Increased lung ceramides in patients with emphysema
To translate our results to human emphysema (primarily caused by cigarette smoking), ceramide levels were measured in lung endothelial cells exposed to cigarette smoke. In bovine endothelial cells, which compared to cultured human endothelial cells are more sensitive to apoptotic stimuli28, cigarette smoke induced rapid, sustained ceramide upregulation, partially inhibited by FB1 (Fig. 5a). Cigarette smoke induced ceramide to a similar extent as did TNF-α (Fig. 5a), a pro-inflammatory cytokine involved in emphysema29 and a classical stimulus of ceramide synthesis in endothelial cells.30
Figure 5. Increased lung ceramide in patients with emphysema.

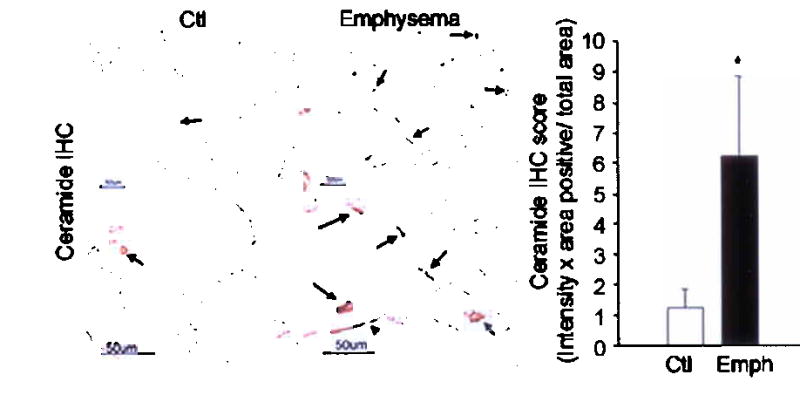



(a) Effect of cigarette smoke (1%) or TNF-α (10 ng/ml; 40 min) ceramide in lung endothelial cells. Ceramide analysis in human emphysema (patient data in Supplementary table 1) by (b) immunostaining (in patients without emphysema (Ctl) ceramide was expressed mostly by alveolar macrophages (arrow), while in emphysema, ceramide was increased in alveolar septal endothelial and epithelial II and I (arrowhead) cells and macrophages; *p =0.02); by (c) DAG kinase assay (means ±SD; *p =0.01); and by (d, e) LC-MS/MS (emphysema, black; non-emphysema non-smokers, light gray; non-emphysema smoker (Smk, gray); *p <0.05.
We next analyzed ceramide expression levels and localization in lung samples from human emphysema (clinical characteristics in Supplementary table 1). Lung ceramide levels were markedly higher in patients with emphysema due to chronic cigarette smoking compared to patients without emphysema, as measured by IHC, DAG kinase (Fig. 5b,c), and HPLC (not shown). In patients with emphysema, the increases in ceramide were almost exclusively localized to alveolar septal cells and alveolar macrophages (Fig. 5b), while the expression of ceramide in large airways and vessels was not different from that of controls (not shown). Further analysis revealed that compared to non-emphysema lungs, human emphysematous lungs exhibit a significant increase in long-chain ceramides (p <0.05) (Fig. 5d, e). Even smoking alone (without a pathological diagnosis of emphysema) changed the lung ceramide expression profile (Fig. 5d), suggesting that not only absolute levels, but also patterns of ceramide species expression may be important in emphysema.
DISCUSSION
Our data indicate that increases in lung ceramide were required and amplified alveolar cell apoptosis, oxidative stress and emphysema caused by VEGFR blockade. Furthermore, we determined that ceramide increases sufficed to cause emphysema and that ceramide functioned proximally to apoptosis. The LC-MS/MS profiles of ceramide species in the lungs from mice, rats, and humans indicated that both an increase in absolute levels and a change in the profiles of ceramide species were important in emphysema development. While the precise functional significance of the latter phenomenon remains unclear31, our findings indicated that an absolute upregulation of ceramide had clear consequence in the disappearance of alveolar septae in emphysema. Endothelial cells are particularly susceptible to ceramide-dependent apoptosis32 and their excessive apoptosis contributes to emphysema pathogenesis3,6,33. As molecular targets of ceramide mediate not only apoptosis, but also matrix proteolysis (via MMPs or cathepsin D14), the effects of ceramide would extend beyond endothelial apoptosis. Similarly, upregulated ceramide may be a novel link between pathological apoptosis and monocyte inflammation (e.g. via CD14 clustering34) in the lung. As seen in our model of ceramide-induced emphysema, ceramide-stimulated macrophages could generate further lung proteolysis and damage. Therefore inflammation, rather than being proximal, might follow excessive apoptosis and, as recently shown in the Nrf-2 knock-out6, oxidative stress which overwhelm cellular maintenance and anti-oxidant defense mechanisms, respectively. Our results firmly position ceramide upstream in the apoptosis cascade and oxidative stress induced by VEGFR-blockade. The hierarchical cause-effect relationship among inflammation, oxidative stress, and apoptosis is critical for the development of susceptibility markers and targets of therapy.
The main mechanism of rapid ceramide upregulation in our model was through de novo pathway. It is conceivable that VEGFR inhibition stimulates de novo ceramide synthesis as part of the stress response35 triggered by lung endothelial cell survival factors deprivation36 or by oxidative stress37,38. We also provide evidence of (delayed) stimulation of the secretory ASMase, directly by the VEGFR inhibitor, or indirectly by the initial ceramide burst. This second wave of ceramide release may contribute to increased ceramide at the exoplasmic plasma membrane, sustaining a feed-forward ceramide production. Utilizing ceramide-neutralizing antibodies, we confirmed the contribution of paracellular ceramide to alveolar cell apoptosis in our model. This ceramide could have originated from dying cells or via secretory ASMase activation. The direct proof of the role of sphingomyelinase vis-à-vis the de novo pathway in the ceramide amplification requires the use of transgenic animals, of which only the ASMase is available.39,40 It should be noted that deletion of the ASMase gene alters cellular and organ structure primarily by excessive accumulation of sphingomyelin. In this mouse model of Niemann-Pick disease, the lung exhibits increased inflammation due to an alveolar macrophage defect in surfactant clearance41, thus exhibiting a complex phenotype with unclear relevance to human emphysema. We expanded and confirmed the in vivo data utilizing human ASMase-deficient fibroblasts from patients with Niemann-Pick. Intriguingly, since endothelial cells have the most abundant levels of secretory ASMase10, injured lung capillary endothelial cells may exacerbate cellular damage in emphysema by this amplification mechanism, a potential explanation for the sustained lung damage present despite discontinuation of smoking42.
Specific blockers of the de novo pathway inhibited emphysema in our model. Nevertheless, excessive ceramide synthase inhibition with FB1 alone triggered emphysema-like changes in both mice and rats. These effects may reflect a biological variation, or toxicity mediated by depletion of cellular pools of sphingolipids. This apparent limitation of the use of FB1 led us to demonstrate that S1-P supplementation not only prevented FB1-induced apoptosis, but also inhibited VEGFR blockade-induced apoptosis. Our results might be interpreted that a tonic pro-survival effect engaged by the ceramide/S1-P pathway, shown in ovary43, is also required for alveolar septal homeostasis.
Can we translate these findings to human disease? In our study of human emphysema, we consistently measured increased ceramide, with intense alveolar septal localization, recapitulating the findings of the VEGFR blockade model. We performed most ceramide measurements in patients with progressive deterioration in lung function despite quitting smoking at least 1 year prior to specimen collection. These late ceramide increases may be the result of ongoing oxidative stress, VEGF depletion, inflammation, and apoptosis, all consequences of previous smoking, all creating a spiraling amplification of lung destruction.
In conclusion, we established that de novo ceramide synthesis is required for the development of murine lung emphysema and that ceramide expression is markedly increased in lungs of patients with cigarette smoke-induced emphysema. Confirming ceramide as a link between excessive apoptosis and inflammation, and extending our studies in human emphysema may uncover novel markers for genetic predisposition and disease severity and offer novel targets to slow the alveolar destruction in this prevalent lung disease.
METHODS
Animal studies were approved by the Johns Hopkins Animal Care and Use Committee. The rat model of emphysema has been previously described3. SU5416 (SUGEN, Inc.) was administered once, 20 mg/kg, subcutaneously to either male Sprague-Dawley rats (3 month old, 300 g, Harlan), or male C57Bl/6 mice (3 mo old, 25 g, Jackson Laboratory). Experiments were performed in the same shipment lot, n= 4–6 rats or mice/group. FB1 (2.2 mg/kg or 1.1 mg/kg in 1 ml (rat) or 300 μl (mouse) in PBS intraperitoneally (i.p.) was administered for 5d. Myriocin was given similarly to mice at doses of 1mg/kg or 0.3 mg/kg. Neutralizing ceramide antibodies (4 mg/kg) or isotype antibodies were dialyzed and then administered i.p to mice daily for 3d after the administration of the VEGFR inhibitor. The lungs of rats given Z-Asp-CH(2)-DCB were from the experiment previously reported3. The S1-P analogue, FTY720, generously provided by Drs. Joe Garcia and Steven Dudek, or vehicle (0.1% DMSO in saline) was given i.p. (0.1 mg/kg)23 for 5d either concomitant with FB1, or following the injection of SU5416. For i-t instillation, ceramide or dihydroceramide was solubilized in ethanol and delivered at the base of the protruded tongue in 80–100 μl perfluorocarbon (1:5 vol/vol). For negative control, we delivered similarly prepared dihydroceramide 1 mg/kg. Z-Asp-CH(2)-DCB (Alexis) was given (3 mg/kg i.p. or vehicle 0.1% DMSO in saline) 1h prior to ceramide instillation. In this set of experiments, ceramide and its control were given by direct i-t injection (30 μl vehicle 1% ethanol, 1% BSA in saline).
Cell culture experiments
Cells were stimulated with cigarette smoke extract (0.5–1% vol:vol) in the culture media44. Human fibroblasts from control subjects or patients with Niemann-Pick’s disease were a gift from Dr. Diane Griffin. (Supplemental Methods) Human lung tissue consisted of samples collected during lung transplantation or surgical biopsy from patients with a pathologic diagnosis of emphysema. (Supplementary table 1). The specimen collection and storage were approved by the Joint Committee on Clinical Investigation, Johns Hopkins University and the Institutional Research Board from the University of Colorado Health Sciences Center.
Chemicals and Reagents
N-Octanoyl-D-erythro-Sphingosine (C8 ceramide), N-Lauroyl-D-erythro-Sphingosine (C12 ceramide), dihydroceramide, sphingomyelin, and purified brain ceramide were from Avanti Polar Lipids. Perfluorocarbon (LiquiVent, Alliance), FB1 (Cayman), myriocin (Biomol Research). Primary antibodies: bax and MMP-12 (Santa Cruz); active caspase-3 (Cell Signaling Technology and Abcam); 8-hydroxy-2’deoxyguanosine (QED Bioscience); anti-ceramide (MID 15B4, Alexis); Anti-mouse IgG-IgM and CD34 (Zymed); SPC (Chemicon); Mac 3 (Tec-1; BD-Pharmingen). HT-1080 cells extract (Oncogene Research Products). All other reagents were from Sigma-Aldrich , unless otherwise specified.
Ceramide determination
We employed complementary methods to measure and to qualify the species of lung tissue ceramide: diacylglycerol (DAG) kinase assay, HPLC, and mass spectroscopy, which relied on the concomitant use of ceramide standards, followed by normalization per tissue wt, protein, or lipid concentration. Additionally, ceramide was detected by IHC. Tissue or cellular lipids were extracted45 and lipid content was assessed by measurements of total lipid phosphorus (Pi)46. DAG kinase assay47 and HPLC48 are detailed in Supplementary methods. We used a modification of the LC-MS/MS by Sullards et al.49 for detecting sphingosine and dihydrosphingosine, seven major ceramide molecular species and 3 major dihydroceramide species. S1-P was analyzed as diacetate derivative to reduce S1-P carryover50. Detection of ceramide by IHC in paraffin-embedded sections with a ceramide-specific antibody (1:60, 1h for mouse tissue and 1:100, 1h for human and rat tissue) with Dako Animal Research Kit (Dako). We used as controls isotype antibody, as well as adsorbed antibody by pre-incubating the ceramide antibody with a saturating C8 ceramide solution. Quantification of ceramide expression is presented in Supplementary Methods. The acid and neutral sphingomyelinase and ceramide synthase activities were measured as described46 and with the Amplex E Red Sphingomyelinase Assay Kit (Molecular Probes).
Morphometric analysis was performed on coded slides as described 5.
Apoptosis
Active caspase-3 IHC was performed utilizing a Catalyzed Signal Amplification kit (Dako)5. Measurements of active caspase-3 were performed on 10–15 images/slide captured by independent observers blinded to the experiment5. The caspase-3 activity was measured by colorimetric (Clontech Laboratories) and fluorometric (Apo-ONE, Promega) assays. TUNEL was performed using a terminal deoxyribonucleotide transferase apoptosis detection kit (Oncogene). For co-localization studies, the TUNEL was performed in parallel with fluorescent IHC for CD34 and SPC, markers for endothelial and type II epithelial cells, respectively. Nucleosomal ELISA kit was from Calbiochem.
Oxidative stress measurements
Detection of 8-hydoxyguanosine was performed by IHC on paraffin-embedded sections5.
Lung matrix proteolytic activity of non-denatured protein extracts form lung tissues was analyzed by gelatin and casein zymogram gels (Novex). MMP-12 levels were analyzed by western blotting. Lung macrophages were detected by Mac3 IHC.
Statistical analysis
Mean values between two groups were compared using Student’s t-test. Morphometry results were analyzed by Kruskal-Wallis One Way Analysis of Variance on Ranks (SigmaStat, SPSS Inc).
Supplementary Material
Acknowledgments
The authors gratefully acknowledge the expert technical assistance of L. Natarajan, S. I. Ramirez, J. Skirball (cell culture and caspase 3 activity assay), and D. Rau (HPLC). We thank D. Griffin who generously provided the acid sphingomyelinase-deficient human fibroblasts. We thank C. Cool with providing additional normal human lung samples and A. M. K. Choi, and J. G. N. Garcia for critically reading our manuscript.
Funding: National Institutes of Health K08 HL04396-04; American Heart Association/Alpha One Foundation Research Grant; and American Lung Association Research Grant (IP); National Institutes of Health RO1 HL71152 (VN); 1S10 RR16798 (WCH); and RO1HL66554 (RMT).
References
- 1.The definition of emphysema. Report of a National Heart, Lung, and Blood Institute, Division of Lung Diseases workshop. Am Rev Respir Dis. 1985;132:182–5. doi: 10.1164/arrd.1985.132.1.182. [DOI] [PubMed] [Google Scholar]
- 2.Shapiro SD. Vascular atrophy and VEGFR-2 signaling: old theories of pulmonary emphysema meet new data. J Clin Invest. 2000;106:1309–10. doi: 10.1172/JCI11344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kasahara Y, et al. Inhibition of VEGF receptors causes lung cell apoptosis and emphysema. J Clin Invest. 2000;106:1311–9. doi: 10.1172/JCI10259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tuder RM, Petrache I, Elias JA, Voelkel NF, Henson PM. Apoptosis and emphysema: the missing link. Am J Respir Cell Mol Biol. 2003;28:551–4. doi: 10.1165/rcmb.F269. [DOI] [PubMed] [Google Scholar]
- 5.Tuder RM, et al. Oxidative stress and apoptosis interact and cause emphysema due to vascular endothelial growth factor receptor blockade. Am J Respir Cell Mol Biol. 2003;29:88–97. doi: 10.1165/rcmb.2002-0228OC. [DOI] [PubMed] [Google Scholar]
- 6.Rangasamy T, et al. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J Clin Invest. 2004;114:1248–59. doi: 10.1172/JCI21146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hannun YA, Obeid LM. The Ceramide-centric universe of lipid-mediated cell regulation: stress encounters of the lipid kind. J Biol Chem. 2002;277:25847–50. doi: 10.1074/jbc.R200008200. [DOI] [PubMed] [Google Scholar]
- 8.Reunanen N, et al. Enhancement of fibroblast collagenase (matrix metalloproteinase-1) gene expression by ceramide is mediated by extracellular signal-regulated and stress-activated protein kinase pathways. J Biol Chem. 1998;273:5137–45. doi: 10.1074/jbc.273.9.5137. [DOI] [PubMed] [Google Scholar]
- 9.Hannun YA, Luberto C. Ceramide in the eukaryotic stress response. Trends Cell Biol. 2000;10:73–80. doi: 10.1016/s0962-8924(99)01694-3. [DOI] [PubMed] [Google Scholar]
- 10.Kolesnick R, Fuks Z. Radiation and ceramide-induced apoptosis. Oncogene. 2003;22:5897–906. doi: 10.1038/sj.onc.1206702. [DOI] [PubMed] [Google Scholar]
- 11.Luberto C, Kraveka JM, Hannun YA. Ceramide regulation of apoptosis versus differentiation: a walk on a fine line. Lessons from neurobiology. Neurochem Res. 2002;27:609–17. doi: 10.1023/a:1020267831851. [DOI] [PubMed] [Google Scholar]
- 12.Zhang Y, et al. Kinase suppressor of Ras is ceramide-activated protein kinase. Cell. 1997;89:63–72. doi: 10.1016/s0092-8674(00)80183-x. [DOI] [PubMed] [Google Scholar]
- 13.Chalfant CE, et al. De novo ceramide regulates the alternative splicing of caspase 9 and Bcl-x in A549 lung adenocarcinoma cells. Dependence on protein phosphatase-1. J Biol Chem. 2002;277:12587–95. doi: 10.1074/jbc.M112010200. [DOI] [PubMed] [Google Scholar]
- 14.Heinrich M, et al. Ceramide as an activator lipid of cathepsin D. Adv Exp Med Biol. 2000;477:305–15. doi: 10.1007/0-306-46826-3_33. [DOI] [PubMed] [Google Scholar]
- 15.Gulbins E, Grassme H. Ceramide and cell death receptor clustering. Biochim Biophys Acta. 2002;1585:139–45. doi: 10.1016/s1388-1981(02)00334-7. [DOI] [PubMed] [Google Scholar]
- 16.Siskind LJ, Kolesnick RN, Colombini M. Ceramide channels increase the permeability of the mitochondrial outer membrane to small proteins. J Biol Chem. 2002;277:26796–803. doi: 10.1074/jbc.M200754200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goggel R, et al. PAF-mediated pulmonary edema: a new role for acid sphingomyelinase and ceramide. Nat Med. 2004;10:155–60. doi: 10.1038/nm977. [DOI] [PubMed] [Google Scholar]
- 18.Cuvillier O, et al. Suppression of ceramide-mediated programmed cell death by sphingosine-1-phosphate. Nature. 1996;381:800–3. doi: 10.1038/381800a0. [DOI] [PubMed] [Google Scholar]
- 19.Payne SG, Milstien S, Spiegel S. Sphingosine-1-phosphate: dual messenger functions. FEBS Lett. 2002;531:54–7. doi: 10.1016/s0014-5793(02)03480-4. [DOI] [PubMed] [Google Scholar]
- 20.Morita Y, Tilly JL. Sphingolipid regulation of female gonadal cell apoptosis. Ann N Y Acad Sci. 2000;905:209–20. doi: 10.1111/j.1749-6632.2000.tb06551.x. [DOI] [PubMed] [Google Scholar]
- 21.Kasahara Y, et al. Endothelial cell death and decreased expression of vascular endothelial growth factor and vascular endothelial growth factor receptor 2 in emphysema. Am J Respir Crit Care Med. 2001;163:737–44. doi: 10.1164/ajrccm.163.3.2002117. [DOI] [PubMed] [Google Scholar]
- 22.Norred WP, Voss KA, Riley RT, Plattner RD. Fumonisin toxicity and metabolism studies at the USDA. Fumonisin toxicity and metabolism. Adv Exp Med Biol. 1996;392:225–36. doi: 10.1007/978-1-4899-1379-1_20. [DOI] [PubMed] [Google Scholar]
- 23.Peng X, et al. Protective effects of sphingosine 1-phosphate in murine endotoxin-induced inflammatory lung injury. Am J Respir Crit Care Med. 2004;169:1245–51. doi: 10.1164/rccm.200309-1258OC. [DOI] [PubMed] [Google Scholar]
- 24.Mandala S, et al. Alteration of lymphocyte trafficking by sphingosine-1-phosphate receptor agonists. Science. 2002;296:346–9. doi: 10.1126/science.1070238. [DOI] [PubMed] [Google Scholar]
- 25.Ravid T, et al. Ceramide accumulation precedes caspase-3 activation during apoptosis of A549 human lung adenocarcinoma cells. Am J Physiol Lung Cell Mol Physiol. 2003;284:L1082–92. doi: 10.1152/ajplung.00172.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hautamaki RD, Kobayashi DK, Senior RM, Shapiro SD. Requirement for macrophage elastase for cigarette smoke-induced emphysema in mice. Science. 1997;277:2002–4. doi: 10.1126/science.277.5334.2002. [DOI] [PubMed] [Google Scholar]
- 27.Ogretmen B, et al. Biochemical mechanisms of the generation of endogenous long chain ceramide in response to exogenous short chain ceramide in the A549 human lung adenocarcinoma cell line. Role for endogenous ceramide in mediating the action of exogenous ceramide. J Biol Chem. 2002;277:12960–9. doi: 10.1074/jbc.M110699200. [DOI] [PubMed] [Google Scholar]
- 28.Petrache I, et al. Caspase-dependent cleavage of myosin light chain kinase (MLCK) is involved in TNF-alpha-mediated bovine pulmonary endothelial cell apoptosis. Faseb J. 2003;17:407–16. doi: 10.1096/fj.02-0672com. [DOI] [PubMed] [Google Scholar]
- 29.Soler N, et al. Airway inflammation and bronchial microbial patterns in patients with stable chronic obstructive pulmonary disease. Eur Respir J. 1999;14:1015–22. doi: 10.1183/09031936.99.14510159. [DOI] [PubMed] [Google Scholar]
- 30.Slowik MR, De Luca LG, Min W, Pober JS. Ceramide is not a signal for tumor necrosis factor-induced gene expression but does cause programmed cell death in human vascular endothelial cells. Circ Res. 1996;79:736–47. doi: 10.1161/01.res.79.4.736. [DOI] [PubMed] [Google Scholar]
- 31.Kroesen BJ, et al. BcR-induced apoptosis involves differential regulation of C16 and C24-ceramide formation and sphingolipid-dependent activation of the proteasome. J Biol Chem. 2003;278:14723–31. doi: 10.1074/jbc.M210756200. [DOI] [PubMed] [Google Scholar]
- 32.Haimovitz-Friedman A, et al. Lipopolysaccharide induces disseminated endothelial apoptosis requiring ceramide generation. J Exp Med. 1997;186:1831–41. doi: 10.1084/jem.186.11.1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Taraseviciene-Stewart, L. et al. An Animal Model of Autoimmune Emphysema. Am J Respir Crit Care Med (2004). [DOI] [PubMed]
- 34.Pfeiffer A, et al. Lipopolysaccharide and ceramide docking to CD14 provokes ligand-specific receptor clustering in rafts. Eur J Immunol. 2001;31:3153–64. doi: 10.1002/1521-4141(200111)31:11<3153::aid-immu3153>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 35.Kolesnick R. The therapeutic potential of modulating the ceramide/sphingomyelin pathway. J Clin Invest. 2002;110:3–8. doi: 10.1172/JCI16127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chavakis E, Dimmeler S. Regulation of endothelial cell survival and apoptosis during angiogenesis. Arterioscler Thromb Vasc Biol. 2002;22:887–93. doi: 10.1161/01.atv.0000017728.55907.a9. [DOI] [PubMed] [Google Scholar]
- 37.Pautz A, et al. Cross-talk between nitric oxide and superoxide determines ceramide formation and apoptosis in glomerular cells. Kidney Int. 2002;61:790–6. doi: 10.1046/j.1523-1755.2002.00222.x. [DOI] [PubMed] [Google Scholar]
- 38.Lavrentiadou SN, et al. Ceramide-mediated apoptosis in lung epithelial cells is regulated by glutathione. Am J Respir Cell Mol Biol. 2001;25:676–84. doi: 10.1165/ajrcmb.25.6.4321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Horinouchi K, et al. Acid sphingomyelinase deficient mice: a model of types A and B Niemann- Pick disease. Nat Genet. 1995;10:288–93. doi: 10.1038/ng0795-288. [DOI] [PubMed] [Google Scholar]
- 40.Otterbach B, Stoffel W. Acid sphingomyelinase-deficient mice mimic the neurovisceral form of human lysosomal storage disease (Niemann-Pick disease) Cell. 1995;81:1053–61. doi: 10.1016/s0092-8674(05)80010-8. [DOI] [PubMed] [Google Scholar]
- 41.Ikegami M, Dhami R, Schuchman EH. Alveolar lipoproteinosis in an acid sphingomyelinase-deficient mouse model of Niemann-Pick disease. Am J Physiol Lung Cell Mol Physiol. 2003;284:L518–25. doi: 10.1152/ajplung.00258.2002. [DOI] [PubMed] [Google Scholar]
- 42.Fletcher C, Peto R. The natural history of chronic airflow obstruction. Br Med J. 1977;1:1645–8. doi: 10.1136/bmj.1.6077.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tilly JL, Kolesnick RN. Sphingolipids, apoptosis, cancer treatments and the ovary: investigating a crime against female fertility. Biochim Biophys Acta. 2002;1585:135–8. doi: 10.1016/s1388-1981(02)00333-5. [DOI] [PubMed] [Google Scholar]
- 44.Tuder RM, Wood K, Taraseviciene L, Flores SC, Voekel NF. Cigarette smoke extract decreases the expression of vascular endothelial growth factor by cultured cells and triggers apoptosis of pulmonary endothelial cells. Chest. 2000;117:241S–2S. doi: 10.1378/chest.117.5_suppl_1.241s. [DOI] [PubMed] [Google Scholar]
- 45.Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Med Sci. 1959;37:911–7. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 46.Dobrowsky RT, Kolesnick RN. Analysis of sphingomyelin and ceramide levels and the enzymes regulating their metabolism in response to cell stress. Methods Cell Biol. 2001;66:135–65. doi: 10.1016/s0091-679x(01)66007-2. [DOI] [PubMed] [Google Scholar]
- 47.Perry DK, Bielawska A, Hannun YA. Quantitative determination of ceramide using diglyceride kinase. Methods Enzymol. 2000;312:22–31. doi: 10.1016/s0076-6879(00)12897-6. [DOI] [PubMed] [Google Scholar]
- 48.Tepper AD, Van Blitterswijk WJ. Ceramide mass analysis by normal-phase high-performance liquid chromatography. Methods Enzymol. 2000;312:16–22. doi: 10.1016/s0076-6879(00)12896-4. [DOI] [PubMed] [Google Scholar]
- 49.Sullards MC, Merrill AH, et al. Analysis of sphingosine 1-phosphate, ceramides, and other bioactive sphingolipids by high-performance liquid chromatography-tandem mass spectrometry. Sci STKE. 2001;2001:PL1. doi: 10.1126/stke.2001.67.pl1. [DOI] [PubMed] [Google Scholar]
- 50.Berdyshev, E.V., Gorshkova, I.A., Garcia, J.G.N., Natarajan, V., and Hubbard, W.C. Quantitative analysis of sphingoid base-1-phosphates as bisacetylated derivatives by liquid chromatography-tandem mass spectrometry. Anal. Biochem. (in the press). [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.