

Abstract
N-Isopropylammelide isopropylaminohydrolase, AtzC, the third enzyme in the atrazine degradation pathway in Pseudomonas sp. strain ADP, catalyzes the stoichiometric hydrolysis of N-isopropylammelide to cyanuric acid and isopropylamine. The atzC gene was cloned downstream of the tac promoter and expressed in Escherichia coli, where the expressed enzyme comprised 36% of the soluble protein. AtzC was purified to homogeneity by ammonium sulfate precipitation and phenyl column chromatography. It has a subunit size of 44,938 kDa and a holoenzyme molecular weight of 174,000. The Km and kcat values for AtzC with N-isopropylammelide were 406 μM and 13.3 s−1, respectively. AtzC hydrolyzed other N-substituted amino dihydroxy-s-triazines, and those with linear N-alkyl groups had higher kcat values than those with branched alkyl groups. Native AtzC contained 0.50 eq of Zn per subunit. The activity of metal-depleted AtzC was restored with Zn(II), Fe(II), Mn(II), Co(II), and Ni(II) salts. Cobalt-substituted AtzC had a visible absorbance band at 540 nm (Δɛ = 84 M−1 cm−1) and exhibited an axial electron paramagnetic resonance (EPR) signal with the following effective values: g(x) = 5.18, g(y) = 3.93, and g(z) = 2.24. Incubating cobalt-AtzC with the competitive inhibitor 5-azacytosine altered the effective EPR signal values to g(x) = 5.11, g(y) = 4.02, and g(z) = 2.25 and increased the microwave power at half saturation at 10 K from 31 to 103 mW. Under the growth conditions examined, our data suggest that AtzC has a catalytically essential, five-coordinate Zn(II) metal center in the active site and specifically catalyzes the hydrolysis of intermediates generated during the metabolism of s-triazine herbicides.
The environmental fate of agricultural chemicals is dependent upon their metabolism by microorganisms. s-Triazine herbicides figure prominently in chemical weed control. Atrazine (2-chloro-4-ethylamino-6-isopropylamino-1,3,5-s-triazine) is one of the most widely used s-triazine herbicides for the control of broadleaf weeds in corn (50). Bacteria that metabolize and use s-triazine herbicides as their sole source of nitrogen for growth have only recently been isolated and characterized (9, 27, 34, 46, 48, 49, 52). Metabolism of the s-triazine herbicide atrazine has been extensively studied by using Pseudomonas sp. strain ADP (27) (Fig. 1). Metabolism is initiated via a dechlorination reaction catalyzed by atrazine chlorohydrolase (AtzA) (16, 17). In the second reaction of the degradation pathway, AtzB catalyzes the hydrolysis of hydroxyatrazine to yield N-isopropylammelide (8). The third metabolic step utilizes N-isopropylammelide isopropylaminohydrolase (AtzC) to hydrolytically remove N-isopropylamine and generate cyanuric acid (38). Nearly identical genes encoding these enzymes have been found in a wide variety of gram-negative and gram-positive bacteria (9, 15, 34, 36, 49) and have been localized to a recently sequenced catabolic plasmid, pADP-1, in Pseudomonas sp. strain ADP (30). While atrazine metabolism is relatively rare among microorganisms, cyanuric acid can be metabolized by many soil bacteria (12, 13, 18, 22). Thus, the advent of bacterial atrazine catabolism is thought to have required the relatively recent evolution of three new enzymes: AtzA, AtzB, and AtzC (41).
FIG. 1.

Atrazine catabolic pathway in Pseudomonas sp. strain ADP.
Interestingly, AtzA, AtzB, and AtzC have all been identified as members of the amidohydrolase protein superfamily (38). Amidohydrolases are distributed throughout the three domains of living organisms: Eubacteria, Archaea, and Eucarya. Members of the superfamily catalyze the hydrolysis of amides or the C—N bond of amines (19). Moreover, superfamily members are responsible for key steps in different metabolic pathways, such as purine-pyrimidine metabolism and the degradation of histidine and cytosine (19), and include cytosine deaminase, urease, adenosine deaminase, phosphotriesterase, and ammelide aminohydrolase. These and other amidohydrolase superfamily members have been found to contain mononuclear or binuclear metal centers (19). This has shifted attention to looking for putative metal-coordinating amino acids during sequence analysis of newly identified members of the amidohydrolase superfamily.
AtzC shows only modest sequence identity, 27%, to the cytosine deaminase CodA (38). However, sequence analyses of the 35 amino acids that are proposed to reside near or at the four residues which are ligands to a putative metal center show that AtzC and cytosine deaminase have 61% overall sequence identity and 85% sequence similarity. CodA is active with bound Fe(II), Mn(II), or Co(II), and removal of these metals causes loss of activity (33).
In this study, we describe the purification and characterization of AtzC. The enzyme was found to cleave the side chain carbon-nitrogen bonds in a variety of s-triazine substrates. Since none of the tested pyrimidines was a substrate for AtzC, this is consistent with the idea that this enzyme has evolved specifically to catabolize s-triazine pesticides. The purified enzyme contained 0.50 zinc atoms per subunit as isolated. Removal of zinc with chelators resulted in a proportional loss of enzyme activity, and metal-depleted AtzC could be reactivated with cobalt. Sequence alignments and spectral analyses suggested that this enzyme contains a five-coordinate metal center.
MATERIALS AND METHODS
Chemicals.
2-Isopropylamino-4,6-dihydroxy-1,3,5-triazine (N-isopropylammelide), 2-isopropylamino-4-ethylamino-6-hydroxy-1,3,5-triazine (hydroxyatrazine), and 2-amino-4,6-dihydroxy-1,3,5-triazine (ammelide) (Table 1) were graciously provided by Syngenta Crop Protection (Greensboro, N.C.). Commercially available 2-amino-4,6-dihydroxypyrimidine and 2-amino-5-nitropyrimidine were purchased from Aldrich Chemical Co. (Milwaukee, Wis.), and 2-amino-4-hydroxy-1,3,5-triazine (5-azacytosine) and 2-oxy-4-amino pyrimidine (cytosine) were purchased from Acros Organics-Fisher Scientific (Pittsburgh, Pa.). All other compounds were synthesized specifically for this study and were shown to be >99% pure by gas chromatography-mass spectrometry (GC-MS) by using a Hewlett-Packard (San Fernando, Calif.) model 6890/5973 instrument. Ammelide and ammelide derivatives (N-methylammelide, N-ethylammelide, N-hydroxyethylammelide, N-cyclopropylammelide, N-t-butylammelide, and N,N-dimethylammelide) (Table 1) were prepared as follows. First, 2,4-dichloro-6-amino-1,3,5-triazine and 2,4-dichloro-6-(N-substituted amino)-1,3,5-triazines were prepared from cyanuric chloride as described by Thurston et al. (47) and then the amino-dichloro compounds were hydrolyzed to yield the parent ammelides as described by Smolin and Rapoport (45). Ammeline derivatives (N-ethylammeline, N-hydroxyethylammeline, and N-isopropylammeline) (Table 2) were prepared as follows. Cyanuric chloride, purchased from Aldrich Chemical Co., was used to prepare (N-substituted amino)-amino-chloro-s-triazines as described by Thurston et al. (47). These chloro compounds were then hydrolyzed to the ammelines in 20% (vol/vol) acetic acid, catalyzed with potassium bromide, and buffered with sodium acetate.
TABLE 1.
AtzC kinetic parameters for several substrates

TABLE 2.
AtzC kinetic parameters for several inhibitors

LS, no kinetic analysis was done due to the low solubility of the compound in the reaction buffer.
Chemical analytical methods.
The purity and structures of the hydrolyzed ammelide and ammeline products were determined by derivatization and analysis via GC-MS. Approximately 1 mg of each compound was treated with a mixture of 25 μl of dimethyl sulfoxide and 25 μl of 1,1,1,3,3,3-hexamethyldisilazane (Aldrich Chemical Co.) and incubated under nitrogen on a rotary shaker for 15 to 20 min. Heat was applied, as needed, in order to dissolve the compound. Vials were flushed with N2 to remove effluxing ammonia and then treated with 25 μl of N,O-bis(trimethylsilyl)acetamide (Aldrich Chemical Co.) for an additional 15 to 20 min on a rotary shaker. After dilution with 1 ml of dichloromethane, samples were analyzed using a Hewlett-Packard GC-MS equipped with an HP-5 5% phenylmethylsilicone capillary column (30 m long, 250-μm inside diameter, 0.25-μm film thickness).
AtzC overexpression.
The atzC gene from Pseudomonas sp. strain ADP was amplified, without its native promoter, by PCR with primers 391f 5′-GAATTCAAGGAGGCTCGAAGTTAC-3′ and 1700r 5′-AAGTGATTCGTGCAGACCAAGCTT-3′. The primers contained EcoRI and HindIII restriction enzyme cloning sites in their 5′ and 3′ regions, respectively. The gene was cloned downstream of the tac promoter in pKK223-3 (Amersham Pharmacia Biotech, Piscataway, N.J.) and transformed into Escherichia coli JM109, and its sequence was verified. E. coli JM109 (pKK223-3::atzC) was grown in Luria-Bertani medium (40) containing 50 μg of ampicillin per ml at 30°C with shaking at 150 rpm. When the culture reached an optical density at 600 nm of 0.5, 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) was added, and then the induced cells were grown overnight under the same conditions.
Enzyme assay.
Enzymatic activity was routinely measured by monitoring absorbance at 240 nm by using a model DU 64 spectrophotometer (Beckman Coulter, Inc., Fullerton, Calif.) with 0.75 mM N-isopropylammelide (ɛ240 = 2.2 × 10−3 μM−1 cm−1). The products, cyanuric acid and isopropylamine, had no absorbance at 240 nm. Reactions were conducted in 25 mM HEPES buffer, pH 7.6, at 25°C. One activity unit was defined as the hydrolysis of 1 μmol of N-isopropylammelide per min.
Protein purification.
E. coli (pKK223-3::atzC) cultures were centrifuged at 10,000 × g for 10 min at 4°C and washed three times in 0.85% NaCl, and cell pellets (5 g [wet weight]) were resuspended in 40 ml of 25 mM 3-(N-morpholine)propanesulfonate (MOPS) buffer, pH 6.9. Cells were broken by four sonication cycles (3 min at 70% intensity and a 50% duty cycle), with intermittent cooling on ice for 5 min by using a Heat Systems-Ultrasonics model W-225R sonicator and a microtip probe. Crude cell extracts were obtained by centrifugation at 18,000 × g for 90 min at 4°C. Extracts were cooled to 4°C, and solid (NH4)2SO4 was added to 60 to 75% saturation with stirring for 1 h at 4°C. Suspensions were centrifuged at 12,000 × g for 15 min, and the precipitated material was collected and resuspended in 25 mM MOPS buffer, pH 6.9. The resuspended material was loaded onto a 1.75- by 30-cm column containing 50 ml of ToyoPearl (TosoHaas, Montgomeryville, Pa.) phenyl 650 M resin. The column was washed with 2.6 M (NH4)2SO4 in 25 mM MOPS buffer, pH 6.9, and protein was eluted by using a 250-ml linear descending gradient of 2.0 to 0.5 M (NH4)2SO4 in 25 mM MOPS buffer, pH 6.9, at a flow rate of 0.5 ml min−1. Protein eluting from the column was monitored at 280 nm with a Beckman DU 64 spectrophotometer and assayed for N-isopropylammelide hydrolase activity. The protein was analyzed for purity by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (Bio-Rad Laboratories, Hercules, Calif.). The relative amount of AtzC in crude cell extract and the percentage purity were estimated from SDS-PAGE gels by using the densitometry UN-Scan-It program (Silk Scientific Inc., Orem, Utah).
Gel filtration chromatography.
The holoenzyme molecular weight was estimated by gel filtration chromatography on a Superose 12 high-resolution column (Pharmacia, Uppsala, Sweden) by using a fast performance liquid chromatography system (Pharmacia). The column was equilibrated with 25 mM MOPS buffer, pH 6.9, containing 0.15 M KCl at a flow rate of 0.1 ml min−1. The column was calibrated with the following proteins and compounds of known molecular weight: thyroglobulin, 670,000; gamma globulin, 158,000; chicken ovalbumin, 44,000; horse myoglobin, 17,000; and vitamin B-12, 1,350.
Amino acid analysis and protein concentration determination.
The N-terminal amino acid sequence of purified AtzC was determined with a model 241 protein sequencer (Hewlett-Packard Company, Palo Alto, Calif.) at the University of Minnesota Microchemical Facility. Protein concentration for quantitative studies was determined by Scientific Research Consortium, Inc. (St. Paul, Minn.) with a model 7300 amino acid analyzer (Beckman Coulter, Inc.) and was similar to those determined by using the Bio-Rad protein reagent. The latter method was used for routine determination of protein concentration. Bovine serum albumin was used as the standard.
Metal analysis.
Purified AtzC (2.0 mg) was dissolved in 4 ml of 10% HCl in 25 mM MOPS buffer, pH 6.9. The sample was incubated overnight at 90°C, and the metal content was determined by inductively coupled plasma emission spectroscopy (ICP) at the University of Minnesota Soils Analytical Laboratory (St. Paul, Minn.).
Effect of chelators on AtzC activity.
Purified AtzC was incubated for 30 min with various concentrations of chelators (from 0.01 to 5 mM), and the activity of the enzyme was measured as described above. The chelators and other potential metal ligands tested were 1,10-phenanthroline, 8-hydroxyquinoline-5-sulfonic acid, EDTA, sodium azide, NaF, and KCN. To determine if chelation was time or pH dependent, the enzyme was incubated with 1 mM 1,10-phenanthroline in 25 mM HEPES buffer at pH 6.0 or 7.6 for various periods of time at 4°C. The activity of the metal-depleted enzyme was measured as described above.
Metal addition to metal-depleted enzyme.
Metal-depleted AtzC was prepared by incubating purified enzyme with 1 mM 1,10-phenanthroline in 25 mM MOPS buffer, pH 6.9, for 60 min at 25°C. Protein was separated from the free and metal-bound 1,10-phenanthroline by passage through a Sephadex G-25 column (1.5 by 30 cm) equilibrated with 25 mM MOPS buffer, pH 6.9. The metal-depleted enzyme eluting from the column was concentrated by using a Centricon-10 filtration unit (Amicon, Beverly, Mass.). Metal-depleted enzyme was incubated for 30 min at 25°C in 25 mM HEPES buffer, pH 7.6, containing 0.1 mM ZnSO4, CuCl2, FeSO4, MnSO4, MgSO4, CoCl2, or NiCl2 or in mixtures containing either 0.1 mM NiCl2 or CoCl2 with 1 mM MgSO4 or NaHCO3. Activity was measured as described above. To maintain Fe(II), reaction mixtures were supplemented with 0.5 mM cysteine and 1.5 mM dithiothreitol as reductants. The influence of Zn(II) on metal-depleted enzyme was measured more precisely by using a range of Zn(II) concentrations. Metal-depleted enzyme at 20 μM was incubated with 1.0 to 100 μM ZnSO4, and the activity was measured as described above. Activity was calculated as a percentage of the initial enzyme activity.
Electron paramagnetic resonance (EPR) and visible spectroscopy of Co(II)-reconstituted enzyme.
Approximately 475 μM metal-depleted AtzC was incubated for 30 min at 25°C with 420 μM CoCl2 (0.9 eq), with or without the addition of 1 mM 5-azacytosine, a competitive inhibitor. Visible spectra (300 to 800 nm) of the samples were acquired by using a Beckman DU 64 spectrophotometer. The calculated extinction coefficient for Co(II)-reconstituted enzyme was based on 1 mol of bound cobalt. Controls were metal-depleted enzyme, 1,10-phenanthroline-Co(II) complex, buffer alone, buffer plus CoCl2, and CoCl2 plus 5-azacytosine.
The same samples were used for X-band EPR spectroscopy by employing a Bruker (Billerica, Mass.) model E500 spectrometer equipped with an Oxford Instruments ESR-10 liquid helium cryostat. The EPR samples (175 μl each) were placed in 4-mm-outside-diameter quartz EPR tubes and frozen by slow immersion in a liquid nitrogen bath. EPR spectra were corrected for the contribution of the quartz tubes by subtracting the spectra of them when filled with deionized water. The Co(II) spin concentration was determined by double integration in comparison with a CoCl2 standard in buffer by using spectra obtained under identical conditions with nonsaturating power. EPR spectra were manipulated with the WinEPR program (version 2.11; Bruker) and simulated following a protocol for high-spin S = 3/2 Co(II) spectra (3, 4). To establish geff values, simulations were performed with the WinEPR Simfonia program (version 1.25; Bruker), treating the S = 3/2 high-spin Co(II) species as an effective S = 1/2 system. The Rhombo program, kindly provided by W. R. Hagen (Delft University of Technology), was then used to determine the closest theoretically allowed geff values from an isotropic g tensor and a rhombicity (E/D), providing a theoretical basis for the simulation. The equivalence between the simulated and theoretical geff values indicated the quality of the simulation. Simulated geff(x,y,z) values are presented in this paper and are followed by those from theory in parentheses.
Power saturation results were plotted as P versus normalized log(I/P0.5) (10), where I is the signal intensity and P is microwave power, and iteratively fitted to the equation
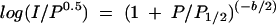 |
by using Origin version 5.0 (OriginLab Corporation, Northampton, Mass.). P1/2 is the microwave power at half saturation. The variable b is the inhomogeneity parameter (37), with a value from 1 to 3 (39).
Substrate specificity analysis.
Solutions of various s-triazine and pyrimidine compounds were prepared in distilled water. The compounds were incubated with purified AtzC in 25 mM HEPES buffer, pH 7.6, for 30 min, and changes in the UV spectra were recorded. In cases where changes in the spectra were detected, a wavelength with maximal absorbance difference between the spectra of the substrate and product was chosen for further kinetic study. The extinction coefficient of each substrate was monitored and found to be as follows: N-methylammelide, ɛ240 = 8.0 × 10−4 μM−1 cm−1; N-ethylammelide, ɛ240 = 2.0 × 10−3 μM−1 cm−1; N-hydroxyethylammelide, ɛ240 = 1.7 × 10−3 μM−1 cm−1; N-isopropylammelide, ɛ240 = 2.2 × 10−3 μM−1 cm−1; N-cyclopropylammelide, ɛ245 = 1.7 × 10−3 μM−1 cm−1; N-t-butylammelide, ɛ243 = 1.7 × 10−3 μM−1 cm−1; N,N-dimethylammelide, ɛ243 = 2.0 × 10−3 μM−1 cm−1; and ammelide, ɛ240 = 1.1 × 10−3 μM−1 cm−1. The reaction was confirmed in each case by high-pressure liquid chromatography (HPLC) detection of the product, cyanuric acid. The kinetic parameters were calculated from the initial hydrolysis rates at different concentrations (Table 1). Analyses of control samples without enzyme were conducted in parallel. Compounds that were not hydrolyzed were examined as inhibitors of AtzC. Inhibitors were tested by standard steady-state analysis. Kinetic parameters of AtzC were calculated by using the Hanes-Woolf equation: [S]/V0 = [S]/Vmax + Km/Vmax (43). Linear regression of the plot [S]/V0 versus [S] was used to determine the Vmax and Km parameters. The kcat value was calculated by dividing Vmax by the moles of AtzC subunits present. For steady-state kinetic determinations, concentrations used generally ranged from 32 μM to 1.5 mM.
RESULTS
Purification and subunit structure of AtzC.
The IPTG-induced expression of atzC, under the control of the strong tac promoter, resulted in the production of AtzC representing 36% of the soluble protein in E. coli. The enzyme was purified 2.8-fold to an estimated purity of 94% by ammonium sulfate precipitation and phenyl column chromatography (Table 3). SDS-PAGE revealed that the purified enzyme had a subunit molecular weight of approximately 44,000, which was consistent with the calculated molecular weight of 44,938 based on the gene sequence. N-terminal sequence analysis of purified AtzC showed that the first 12 amino acids (SKDFDLIIRNAY) were identical to those predicted by the translation of the atzC gene, except for the absence of an N-terminal methionine. The molecular weight of the native protein was estimated by gel filtration chromatography on a Superose 12 high-resolution column to be 174,000, which suggests that the native enzyme is a homotetramer.
TABLE 3.
Purification of AtzC
| Fraction | Total protein (mg) | Total activity (U)a | Sp act (U mg−1) | Yield (%) | n-Fold purification |
|---|---|---|---|---|---|
| Crude | 412 | 2,380 | 5.80 | 100 | |
| Ammonium sulfate | 86.9 | 1,020 | 11.7 | 42.6 | 2.0 |
| Phenyl column | 27.9 | 452 | 16.2 | 19.0 | 2.8 |
1 U of enzyme activity is defined as the conversion of 1 μmol of N-isopropylammelide to cyanuric acid and isopropylamine in 1 min.
Substrate specificity and kinetic parameters.
Enzyme reactions were carried out in 25 mM HEPES buffer, pH 7.6. Under these conditions, the Km and kcat values of AtzC with N-isopropylammelide were 406 μM and 13.3 s−1, respectively, based on the molecular weight of a single protein subunit (Table 1).
AtzC also catalyzed the deamination of other s-triazine compounds (Table 1). The reaction was confirmed in each case by UV spectroscopy and HPLC detection of the product, cyanuric acid. Although N-isopropylammelide is the substrate for AtzC in the Pseudomonas sp. strain ADP atrazine degradation pathway (38), the enzyme was much more efficient in the deamination of N-ethylammelide. The kcat/Km values were 21-fold higher for N-ethylammelide than those for N-isopropylammelide, at 679 and 32.9 mM−1 s−1, respectively. Moreover, when the amino group in position 6 was not replaced (as in ammelide) or was replaced with a relatively bulky alkyl group, like N-t-butylammmelide, the kcat/Km values were low, at 1.25 and 0.23 mM−1 s−1, respectively. When N-isopropylammelide was compared to a structurally related compound with a cyclopropyl-replaced amino group (N-cyclopropylammelide), it was found that they had similar Km values but that the latter gave a higher kcat.
The s-triazine compounds with an amino group on position 2 and hydroxyl group on position 4 were not substrates but, rather, inhibited AtzC activity when assayed in an admixture with N-isopropylammelide. 2-Amino-4-hydroxy-1,3,5-s-triazine (5-azacytosine), N-hydroxyethylammeline, and ammeline were found to be competitive inhibitors of AtzC, with Ki values of 15, 129, and 687 μM, respectively (Table 2). N-Ethylammeline and N-isopropylammeline also inhibited the enzyme, but kinetic data were not obtained due to the low water solubility of these inhibitors. When the s-triazine compound had amino groups and no hydroxyl groups (e.g., 2,4,6-triamino-s-triazine), no inhibition or turnover was observed (data not shown). Pyrimidines were neither substrates nor inhibitors for AtzC under the experimental conditions used in this study. Pyrimidines that were tested and showed no effect on the enzyme were cytosine, 4-amino-6-hydroxy-2-mercaptopyrimidine, 4,6-dihydroxy-2-mercaptopyrimidine, 2-methylthio-4,6-dihydroxypyrimidine, 4,6-dihydroxy-5-nitropyrimidine, uracil, barbituric acid, and 2,4,5-trihydroxypyrimidine. Creatinine was also found to be neither a substrate nor an inhibitor for AtzC.
Homology of AtzC to proteins from the amidohydrolase superfamily.
The AtzC amino acid sequence was compared with those of other proteins in GenBank using BLASTP. The enzyme showed less than 30% identity and 46% similarity with any protein in the database other than those involved in s-triazine catabolism. The greatest identities were observed with ammelide aminohydrolase (TrzC) from Enterobacter cloacae (28% identity, 44% similarity), creatinine deaminase (CRDI) from Mesorhizobium loti (28% identity, 46% similarity) and cytosine deaminase (CodA) from E. coli (27% identity, 45% similarity). However, pairwise alignments of these proteins and other members of the amidohydrolase superfamily showed much higher sequence identity in the region known from the crystal structure of cytosine deaminase (CodA) to be involved in active-site metal coordination (Fig. 2). In this region, four histidines and one aspartic acid were conserved residues known to be involved as metal ligands and with the activation of a water molecule in the hydrolytic active site of some members of the amidohydrolase superfamily (19, 20, 51).
FIG. 2.

Alignment of amino acid sequences of AtzC and other enzymes from the amidohydrolase superfamily: ammelide aminohydrolase (TrzC), cytosine deaminase (CodA), creatinine deaminase (CRDI), adenosine deaminase (Ada), and phosphotriesterase (Opd). The alignment was done in the regions known to include the amino acid metal ligands in CodA from E. coli. Conserved residues are numbered, and the four proposed direct metal-coordinating amino acids in AtzC are indicated with surrounding boxes.
Evidence that AtzC is a metalloenzyme.
ICP tested for the presence of Ca, Mg, Cd, Mn, Fe, Zn, Cu, Ni, and Co. Analysis of AtzC revealed that the enzyme contained 0.50 Zn atoms (varied from 0.48 to 0.50 atoms with different enzyme preparations) and 0.09 Cu atoms per 44,938-kDa subunit. The ratio of the metal-to-protein subunit for every other metal was below 0.06.
The effect of chelators and potential metal ligands on AtzC activity was tested to determine if the metal was important in catalysis. Potential metal ligands such as sodium azide, NaF, and KCN showed no effect on AtzC activity after 30 min of incubation at concentrations up to 1 mM. EDTA slightly inhibited the enzyme, with a less than 10% loss of activity after 30 min of exposure to 5 mM EDTA (Fig. 3). However, 5 mM concentrations of 1,10-phenanthroline 8-hydroxyquinoline-5-sulfonic acid had significant inhibitory effects on enzyme activity (Fig. 3). These compounds diminished AtzC activity by 65% compared to that for the control enzyme in buffer alone. Less than 5% activity was lost when the enzyme was incubated under the same conditions in the absence of metal chelators. The metal content of the 1,10-phenanthroline-treated enzyme, measured by ICP, was 0.25 Zn atoms per subunit. The inhibitory effect of 1,10-phenanthroline on AtzC purified from different batches varied between 60 and 80%. Extending the incubation time of the enzyme with the chelator and reducing the buffer pH did not result in further activity loss (data not shown).
FIG. 3.

Influence of chelators on AtzC activity after 30 min of incubation. ▪, 1,10-phenanthroline; •, 8-hydroxyquinoline-5-sulfonic acid; ▴, EDTA. Error bars represent the standard error of the mean.
Reconstitution of metal-depleted enzyme.
Recovery of AtzC activity was obtained by incubating metal-depleted AtzC with metal solutions at final concentrations of 0.1 mM, with the exception of Mg(II), which was used at a final concentration of 1.0 mM (Fig. 4). Incubation with Cu(II) led to complete loss of activity, suggesting that the trace copper found associated with native AtzC is not needed for its catalytic activity. Incubation with 0.1 mM Zn(II) depressed activity by 10% (Fig. 4). However, during titration of AtzC with Zn(II), lower concentrations (0.5 to 10 μM) were found to be stimulatory, restoring activity to up to 70% of the activity of the native enzyme (data not shown). Incubation with 0.1 mM Fe(II) reconstituted activity to 63% of that of the native enzyme, while Mn(II) increased activity to 80% of the initial activity. Co(II) and Ni(II) restored activity to nearly the same level as that of the native enzyme. A mixture of either Co(II) or Ni(II) metal with 1 mM Mg(II) increased activity to more than 100% of that of the native enzyme. Addition of bicarbonate in admixture with Co(II) or Ni(II) under conditions known to aid reconstitution of the binuclear metal centers of certain amidohydrolases had no further stimulatory effect on the enzyme activity.
FIG. 4.

The effect of 0.1 mM metal addition on activity with N-isopropylammelide by metal-depleted AtzC. MgSO4 concentration was 1 mM. Error bars represent the standard error of the mean.
Electronic spectroscopy of Co(II)-reconstituted enzyme.
Electronic spectra (300 to 800 nm) of metal-depleted (Fig. 5) and native (data not shown) AtzC were essentially featureless. To probe the metal-binding site, the spectroscopically silent Zn(II) was replaced with Co(II), which has well-studied spectroscopic signatures. Adding 0.9 eq of Co(II) per subunit of metal-depleted AtzC yielded enzyme absorbance with a peak centered at 540 nm. Spectra were taken over time until no further changes were observed. The difference spectrum of reconstituted enzyme minus metal-depleted enzyme yielded a roughly symmetrical single peak with a Δɛ540 of 84 M−1 cm−1 (Fig. 5B). CoCl2 alone in buffer had an absorbance maximum at 509 nm and a ɛ540 of 3.0 M−1 cm−1. Moreover, the addition of 5-azacytosine to the Co(II)-reconstituted enzyme did not change its spectrum.
FIG. 5.

(A) UV-Vis spectra of Co(II)-reconstituted AtzC. · · ·, metal-depleted enzyme; —, Co(II)-reconstituted enzyme. (B) Difference of the Co(II)-reconstituted AtzC spectrum minus the spectrum of the metal-depleted enzyme.
EPR spectroscopy of Co(II)-reconstituted enzyme
EPR spectroscopy was performed on Co(II)-reconstituted enzyme in the absence or presence of the inhibitor 5-azacytosine. Both samples exhibited an axial-type signal centered around g = 5 (Fig. 6A and C). Neither metal-depleted AtzC nor buffer alone had a significant EPR signal. Co(II) spin concentrations were found to be within ±20% of the expected concentrations. Further, EPR spectra of buffer plus Co(II) with or without the inhibitor 5-azacytosine were appreciably different from those observed with the reconstituted enzyme (data not shown). The Co(II)-reconstituted enzyme without inhibitor had geff values of 5.18 (5.17), 3.93 (3.94), and 2.24 (2.24). These values correspond to an Ms of | ± 1/2〉 ground-state transition with a greal of 2.292 and an E/D of 0.091. Addition of inhibitor to the Co(II)-reconstituted enzyme shifted the geff values to 5.11 (5.11), 4.02 (4.02), and 2.25 (2.25). These values correspond to an Ms of | ± 1/2〉 ground-state transition with a greal of 2.295 and an E/D of 0.081. The change suggests that the competitive inhibitor alters the environment of the metal-binding site. It was considered that the addition of inhibitor changed the affinity of the metal-binding site for Co(II), which could then equilibrate with Co(II) in solution. However, the line-shape changes seen could not be accounted for by simply altering the ratio of unbound-to-bound Co(II). The differences between experimental and calculated spectra (data not shown) indicated that approximately 10% of the Co(II) added remained unbound in solution. Washing samples with buffer removed 10 to 20% of the Co(II) that had been free in solution, although the procedure also removed a considerable fraction of the bound Co(II) as well.
FIG. 6.

EPR spectra of Co-reconstituted AtzC. (A) Co(II)-reconstituted AtzC (475 μM). (B) Simulation of A with geff values of 5.18, 3.93, and 2.24. (C) Co(II)-reconstituted AtzC (475 μM) with the addition of 1 mM 5-azacytosine. (D) Simulation of C with geff values of 5.11, 4.02, and 2.25. Extraneous Cu(II) signals for both panels A and C are evident in the g = 2 region. Buffer for the samples was 25 mM MOPS (pH 6.9). EPR parameters: temperature, 10.0 K; microwave power, 781 μW; modulation amplitude, 10 G; microwave frequency, 9.604 GHz.
Power saturation studies were conducted to provide additional evidence regarding whether Co(II) was bound near the active site and was thus affected by the binding of inhibitor. At 2 K, P1/2 values for Co(II)-reconstituted AtzC with and without inhibitor were essentially the same at 240 and 300 μW, respectively. For 10 K, the P1/2 values were 103 mW for inhibited and 31 mW for uninhibited Co(II)-reconstituted AtzC.
DISCUSSION
Here, we describe the purification and characterization of N-isopropylammelide isopropylaminohydrolase. The enzyme was known to hydrolyze N-isopropylammelide (38), an intermediate generated during metabolism of the s-triazine herbicide atrazine (Fig. 1). The product, cyanuric acid, can be metabolized by many different soil bacteria (12, 13, 18, 22). Other commercial s-triazine herbicides vary in the N-alkyl side chain substituents. Thus, their metabolism to carbon dioxide and ammonia is largely predicated on the ability of AtzC to remove the N-alkyl substituent and generate cyanuric acid. The herbicides simazine, terbuthylazine, and cyromazine contain N-ethyl, N-tertiary butyl, and N-cyclopropyl substituents, respectively. AtzC hydrolyzed all of these and several other N-alkyl substituents. In short, all eight ammelides tested were substrates. In contrast, all the ammelines tested, as well as 5-azacytosine, proved to be inhibitors. For the three out of five compounds that had water solubility sufficient to make accurate kinetic measurements, the inhibition was found to be competitive (Table 2). The identification of a competitive inhibitor provided a tool for probing the substrate-binding site in the spectroscopic studies discussed below.
AtzC was not reactive with compounds that were not metabolic intermediates of herbicides. Hydroxydiaminotriazines and structurally analogous pyrimidines were not substrates. These data are consistent with the idea that AtzC specifically functions in the catabolism of s-triazine herbicides. Supporting this conclusion is the genetic linkage of atzC with other herbicide metabolism genes on plasmid pADP-1 in Pseudomonas sp. strain ADP (30). Additionally, nearly identical atzC genes have been identified in different atrazine-metabolizing bacteria found on four continents (15).
AtzC had a <30% amino acid identity with any protein in the databases other than those involved in s-triazine herbicide metabolism. However, AtzC has five key residues of the N-terminal dihydroorotase signature pattern (Prosite entry PS00482), D-X-H-X-H (38), and contains other putative metal ligands found in members of the amidohydrolase superfamily. This superfamily contains well-studied metalloenzymes such as adenosine deaminase (11), hydantoinase (24), and cytosine deaminase (33). Additionally, there are other amidohydrolase superfamily members involved in the bacterial catabolism of pesticides and industrial chemicals: s-triazine chlorohydrolase, TrzA (48); melamine deaminase, TriA (42); and phosphotriesterase (35). Members of the superfamily catalyze biochemically distinct reactions, including ring deamination, amide bond hydrolysis, phosphotriester hydrolysis, and dechlorination reactions (41).
Pairwise alignment of AtzC with a few members of the superfamily (Fig. 2) suggested that amino acid residues His 60, His 62, His 217, His 249, and Asp 303 in AtzC might be involved in coordinating a transition metal and activating a molecule of water necessary for the hydrolytic mechanism, as in the enzymes cytosine deaminase (20) and adenosine deaminase (51). ICP analysis revealed that the enzyme contains zinc as the predominant metal with a Zn subunit ratio of 0.50. This partial occupancy per subunit may be due to a partial loss of bound zinc metal during the purification process.
Since Zn(II) is silent in EPR and visible absorption spectroscopies, we decided to replace the metal in native AtzC with one amenable to spectroscopic study yet that retained enzyme activity. To this end, metal-depleted enzyme was reconstituted with different divalent metals and the enzyme activity was measured (Fig. 4). Ni(II) and Co(II) have been found to be the most efficient replacements for Zn(II) in other proteins (21). The high activities reported with Ni(II) and Co(II) are probably due to their strong Lewis acid characteristics (21), which make a metal-bound hydroxide more reactive in catalysis. Ni(II) and Co(II) both restored the activity level of AtzC at or above that for the wild-type (Fig. 4), consistent with a metal-bound hydroxide acting in the catalytic cycle of AtzC.
Co(II) is similar in chemistry to Zn(II) (7) and has well-studied spectroscopic signatures (6, 25). Co(II) is a 3d7 ion and so is expected to be colored (7). When substituting for Zn(II) in metalloproteins, Co(II) gives rise to many electronic transitions in the near infrared-visible region (28). Spectroscopic studies were done with Co(II)-reconstituted AtzC to learn more about the location and coordination of the metal site.
Co(II) can be either low (S = 1/2) or high (S = 3/2) spin (7) and has almost always been found to be high spin (28) when substituting for metals in metalloproteins. Since Co(II) has a noninteger spin and is therefore paramagnetic, EPR was a natural choice for studying the metal environment. The EPR spectrum of Co(II)-reconstituted AtzC was unique and could be simulated closely. The competitive inhibitor with the lowest Ki value, 5-azacytosine (Table 2), was chosen to probe the active site. The shifts in EPR g values of the Co(II)-reconstituted enzyme upon addition of 5-azacytosine (Fig. 6), although not large, are of a magnitude similar to those that have been reported for Co(II)-substituted carboxypeptidase A (29). In AtzC, the rhombicity of the line-shape decreased from an E/D of 0.091 to an E/D of 0.081 upon inhibitor addition, indicating that the metal ligands became slightly more symmetrical. Also, the greater than threefold change in P1/2 values at 10 K upon inhibitor addition is comparable to what has been observed previously in Co(II)-substituted carboxypeptidase A and liver alcohol dehydrogenase (23, 26). The observed effects of the inhibitor on the EPR spectra are consistent with Co(II) being present either in the active site or at a distance from the active site where it is influenced by gross protein conformational changes upon inhibitor binding. The former option, that the metal is in the active site, is the most likely for the following reasons. First, sequence alignments with phosphotriesterase, adenosine deaminase, and cytosine deaminase proteins, which are also in the amidohydrolase superfamily, reveal conservation of the metal ligands. X-ray studies of these enzymes showed that their active-site metals activate a molecule of water, which attacks the substrate (5, 20, 51). Second, as in previous reports on the reconstitution of active-site zinc in metalloenzymes (21), Co(II) and Ni(II) recoup high activity upon reconstituting metal-depleted AtzC.
Comparison of the visible spectra of Co(II)-reconstituted AtzC and the metal-depleted AtzC (Fig. 5) showed a broad and roughly symmetrical single peak between 450 and 600 nm due to Co(II) binding, having a maximum at 540 nm with a Δɛ540 of 84 M−1 cm−1. The magnitude of the extinction coefficient has been shown empirically to correlate with the coordination number in Co(II) enzymes: in a four-coordinate site, the extinction coefficient is more than 300 M−1 cm−1; a five-coordinate site is between 50 and 300 M−1 cm−1; and a six-coordinate site is below 50 M−1 cm−1 (2, 6). The magnitude of the extinction coefficient obtained in this study suggests that the Co(II) in AtzC is five-coordinate. This finding is supported by the sequence alignment of AtzC with cytosine deaminase and adenosine deaminase, for which X-ray structures are now available (20, 51). Sequence alignments (Fig. 2) have identified 4 amino acids in AtzC that are homologous to the catalytic metal ligands in cytosine deaminase and adenosine deaminase: H60, H62, H217, and D303. In these enzymes, a molecule of water is the fifth ligand to the catalytic metal. As in cytosine deaminase and adenosine deaminase, H249 (AtzC numbering) could hydrogen bond with this molecule of water, positioning it to act as a general base.
Additional evidence supporting the metal ligand scheme proposed here is the absence of a ligand-to-metal charge transfer band around 310 to 350 nm in the cobalt-substituted enzyme (data not shown). A ligand-to-metal charge transfer band is expected in this region when cysteine is a ligand for Co(II) (1, 14, 32). Additionally, since almost all structural zinc sites have at least one cysteine ligand (31), the absence of a cysteine ligand reinforces the finding that the zinc has a catalytic, not a structural, role. Therefore, it is suggested that the metal ligand coordination in AtzC is analogous to those of cytosine deaminase and adenosine deaminase.
The present data are most consistent with a mononuclear metal center in AtzC; members of the amidohydrolase superfamily contain either mononuclear or binuclear metal centers. For the latter, bicarbonate is known to be required for effective reconstitution of the metal center (44). Bicarbonate had no effect on the reconstitution of metal-depleted AtzC. During reconstitution, 0.9 eq of CoCl2 was added to the metal-depleted AtzC, which still contained 0.25 eq of Zn(II) per enzyme subunit, to give maximal activity. A stoichiometry of 1 Zn(II) or Co(II) per subunit predicts that 0.15 eq of Co(II) per subunit, or 15%, would remain unbound. Consistent with this, simulations indicated that approximately 10% of the EPR signal comes from [Co(H2O)6]2+, Co(II) not complexed with protein. Additionally, washing samples with metal-free buffer showed that 10 to 20% of the total Co(II) was not complexed to the protein. The 5% differences between prediction and observation are probably within the margin of error for our measurements.
In summary, AtzC is specifically reactive with intermediates generated during the metabolism of s-triazine pesticides. AtzC is a homotetramer with Zn(II) in the active site. Further studies with methods such as X-ray crystallography and EXAFS (extended X-ray absorbance fine structure) need to be done in order to address the coordination issue more directly, to solidify whether the metal center is mononuclear or binuclear, and to investigate the catalytic mechanism of AtzC.
Acknowledgments
This work was partially supported by a postdoctoral fellowship from the United States-Israel Binational Agricultural Research and Development (BARD) fund (grant number FI-295-99) (to N.S.) and grants from the U.S. Department of Energy's Natural and Accelerated Bioremediation Program (to J.P.O.), Syngenta Crop Protection (to L.P.W. and M.J.S.), and the University of Minnesota Agricultural Experiment Station (to N.S. and M.J.S.).
We also thank John Lipscomb for making the EPR spectrometer available to us and Matt Wolfe for technical assistance with the EPR.
REFERENCES
- 1.Alexander, R. S., L. L. Kiefer, C. A. Fierke, and D. W. Christianson. 1993. Engineering the zinc binding site of human carbonic anhydrase II: structure of the His-94 → Cys apoenzyme in a new crystalline form. Biochemistry 32:1510-1518. [DOI] [PubMed] [Google Scholar]
- 2.Banci, L., A. Bencini, C. Benelli, D. Gatteschi, and C. Zanchini. 1982. Spectral-structural correlations in high-spin cobalt(II) complexes. Struct. Bonding 52:37-86. [Google Scholar]
- 3.Bennett, B., and R. C. Holz. 1997. EPR studies on the mono- and dicobalt(II)-substituted forms of the aminopeptidase from Aeromonas proteolytica. Insight into the catalytic mechanism of dinuclear hydrolases. J. Am. Chem. Soc. 119:1923-1933. [Google Scholar]
- 4.Bennett, B., and R. C. Holz. 1997. Spectroscopically distinct cobalt(II) sites in heterodimetallic forms of the aminopeptidase from Aeromonas proteolytica: characterization of substrate binding. Biochemistry 36:9837-9846. [DOI] [PubMed] [Google Scholar]
- 5.Benning, M. M., J. M. Kuo, F. M. Raushel, and H. M. Holden. 1995. Three-dimensional structure of the binuclear metal center of phosphotriesterase. Biochemistry 34:7973-7978. [DOI] [PubMed] [Google Scholar]
- 6.Bertini, I., and C. Luchinat. 1984. High spin cobalt(II) as a probe for the investigation of metalloproteins. Adv. Inorg. Biochem. 6:71-111. [PubMed]
- 7.Bertini, I., C. Luchinat, and M. S. Viezzoli. 1986. Metal substitution as a tool for the investigation of zinc proteins, p. 27-47. In H. Garry and I. Bertini (ed.), Zinc enzymes. Birkhäuser Verlag, Boston, Mass.
- 8.Boundy-Mills, K. L., M. L. de Souza, R. T. Mandelbaum, L. P. Wackett, and M. J. Sadowsky. 1997. The atzB gene of Pseudomonas sp. strain ADP encodes the second enzyme of a novel atrazine degradation pathway. Appl. Environ. Microbiol. 63:916-923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bouquard, C., J. Ouazzani, J. C. Prome, Y. Michel-Briand, and P. Plesiat. 1997. Dechlorination of atrazine by a Rhizobium sp. isolate. Appl. Environ. Microbiol. 63:862-866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brudvig, G. W. 1995. Electron paramagnetic resonance spectroscopy. Methods Enzymol. 246:536-554. [DOI] [PubMed] [Google Scholar]
- 11.Chang, Z., P. Nygaard, A. C. Chinault, and R. E. Kellems. 1991. Deduced amino acid sequence of Escherichia coli adenosine deaminase reveals evolutionarily conserved amino acid residues: implications for catalytic function. Biochemistry 30:2273-2280. [DOI] [PubMed] [Google Scholar]
- 12.Cook, A. M., P. Bellstein, H. Grossenbacher, and R. Hutter. 1985. Ring cleavage and degradative pathway of cyanuric acid in bacteria. Biochem. J. 231:25-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cook, A. M. 1987. Biodegradation of s-triazine xenobiotics. FEMS Microbiol. Rev. 46:93-116. [Google Scholar]
- 14.Crowder, M. W., Z. Wang, S. L. Franklin, E. P. Zovinka, and S. J. Benkovic. 1996. Characterization of the metal-binding sites of the β-lactamase from Bacteroides fragilis. Biochemistry 35:12126-12132. [DOI] [PubMed] [Google Scholar]
- 15.de Souza, M. L., J. Seffernick, B. Martinez, M. J. Sadowsky, and L. P. Wackett. 1998. The atrazine catabolism genes atzABC are widespread and highly conserved. J. Bacteriol. 180:1951-1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Souza, M. L., L. P. Wackett, K. L. Boundy-Mills, R. T. Mandelbaum, and M. J. Sadowsky. 1995. Cloning, characterization, and expression of a gene region from Pseudomonas sp. strain ADP involved in the dechlorination of atrazine. Appl. Environ. Microbiol. 61:3373-3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de Souza, M. L., M. J. Sadowsky, and L. P. Wackett. 1996. Atrazine chlorohydrolase from Pseudomonas sp. strain ADP: gene sequence, enzyme purification, and protein characterization. J. Bacteriol. 178:4894-4900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Erickson, L. E., and K. H. Lee. 1989. Degradation of atrazine and related s-triazines. Crit. Rev. Environ. Control 19:1-14. [Google Scholar]
- 19.Holm, L., and C. Sander. 1997. An evolutionary treasure: unification of a broad set of amidohydrolases related to urease. Proteins 28:72-82. [PubMed] [Google Scholar]
- 20.Ireton, G. C., G. McDermott, M. E. Black, and B. L. Stoddard. 2002. The structure of Escherichia coli cytosine deaminase. J. Mol. Biol. 315:687-697. [DOI] [PubMed] [Google Scholar]
- 21.Jackman, J. E., C. H. R. Raetz, and C. A. Fierke. 1999. UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine deacetylase of Escherichia coli is a zinc metalloenzyme. Biochemistry 38:1902-1911. [DOI] [PubMed] [Google Scholar]
- 22.Jutzi, K., A. M. Cook, and R. Hutter. 1982. The degradative pathway of the s-triazine melamine. The steps to ring cleavage. Biochem. J. 208:679-684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuo, L. C., and M. W. Makinen. 1985. Ground term splitting of high-spin Co+2 as a probe coordination structure. 2. The ligand environment of the active site metal ion of carboxypeptidase A in ester hydrolysis. J. Am. Chem. Soc. 107:5225-5261. [Google Scholar]
- 24.LaPointe, G., S. Viau, D. Leblanc, N. Roberts, and A. Morin. 1994. Cloning, sequencing, and expression in Escherichia coli of the d-hydantoinase gene from Pseudomonas putida and distribution of homologous genes in other microorganisms. Appl. Environ. Microbiol. 60:888-895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Makinen, M. W., L. C. Kuo, M. B. Yim, G. B. Wells, J. M. Fukuyama, and J. E. Kim. 1985. Ground term splitting of high-spin Co+2 ion as a probe of coordination structure. 1. Dependence of the splitting on coordination geometry. J. Am. Chem. Soc. 107:5245-5255. [Google Scholar]
- 26.Makinen, M. W., and M. B. Yim. 1981. Coordination environment of the active-site metal ion of liver alcohol dehydrogenase. Proc. Natl. Acad. Sci. USA 78:6221-6225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mandelbaum, R. T., D. L. Allan, and L. P. Wackett. 1995. Isolation and characterization of a Pseudomonas sp. that mineralizes the s-triazine herbicide atrazine. Appl. Environ. Microbiol. 61:1451-1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maret, W., and B. L. Vallee. 1993. Cobalt as probe and label of proteins. Methods Enzymol. 226:52-71. [DOI] [PubMed] [Google Scholar]
- 29.Martinelli, R. A., G. R. Hanson, J. S. Thompson, B. Holmquist, J. R. Pilbrow, D. S. Auld, and B. L. Vallee. 1989. Characterization of the inhibitor complexes of cobalt carboxypeptidase A by electron paramagnetic resonance spectroscopy. Biochemistry 28:2251-2258. [DOI] [PubMed] [Google Scholar]
- 30.Martinez, B., J. Tomkins, L. P. Wackett, R. Wing, and M. J. Sadowsky. 2001. Complete nucleotide sequence and organization of the atrazine catabolic plasmid pADP-1 from Pseudomonas sp. strain ADP. J. Bacteriol. 183:5684-5697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McCall, K. A., C. Huang, and C. A. Fierke. 2000. Function and mechanism of zinc metalloenzymes. J. Nutr. 130:1437S-1446S. [DOI] [PubMed]
- 32.Orellano, E. G., J. E. Girardini, J. A. Cricco, E. A. Ceccarelli, and A. J. Vila. 1998. Spectroscopic characterization of a binuclear metal site in Bacillus cereus β-lactamase II. Biochemistry 37:10173-10180. [DOI] [PubMed] [Google Scholar]
- 33.Porter, D. J. T., and E. A. Austin. 1993. Cytosine deaminase. J. Biol. Chem. 268:24005-24011. [PubMed] [Google Scholar]
- 34.Radosevich, M., S. J. Traina, Y.-L. Hao, and O. H. Tuovinen. 1995. Degradation and mineralization of atrazine by a soil bacterial isolate. Appl. Environ. Microbiol. 61:297-302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Raushel, F. M., and H. M. Holden. 2000. Phosphotriesterase: an enzyme in search of its natural substrate. Adv. Enzymol. Relat. Areas Mol. Biol. 74:51-93. [DOI] [PubMed] [Google Scholar]
- 36.Rousseaux, S., A. Hartmann, and G. Soulas. 2001. Isolation and characterization of new gram-negative and gram-positive atrazine degrading bacteria from different French soils. FEMS Microbiol. Ecol. 36:211-222. [DOI] [PubMed] [Google Scholar]
- 37.Rupp, H., K. K. Rao, D. O. Hall, and R. Cammack. 1978. Electron spin relaxation of iron-sulphur proteins studies by microwave power saturation. Biochim. Biophys. Acta 537:255-269. [DOI] [PubMed] [Google Scholar]
- 38.Sadowsky, M. J., Z. Tong, M. de Souza, and L. P. Wackett. 1998. AtzC is a new member of the amidohydrolase protein superfamily and is homologous to other atrazine-metabolizing enzymes. J. Bacteriol. 180:152-158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sahlin, M., A. Graeslund, and A. Ehrenberg. 1986. Determination of relaxation times for a free radical from microwave saturation studies. J. Magn. Reson. 67:135-137. [Google Scholar]
- 40.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 41.Seffernick, J. L., and L. P. Wackett. 2001. Rapid evaluation of bacterial catabolic enzymes: a case study with atrazine chlorohydrolase. Biochemistry 40:12747-12753. [DOI] [PubMed] [Google Scholar]
- 42.Seffernick, J. L., M. L. de Souza, M. J. Sadowsky, and L. P. Wackett. 2001. Melamine deaminase and atrazine chlorohydrolase: 98 percent identical but functionally different. J. Bacteriol. 183:2405-2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Segel, I. H. 1975. Other methods of plotting enzyme kinetics data, p. 208-224. In I. H. Segal (ed.), Enzyme kinetics: behavior and analysis of rapid equilibrium and steady-state enzyme systems. Wiley-Interscience, New York, N.Y.
- 44.Shim, H., and F. M. Raushel. 2000. Self-assembly of the binuclear metal center of phosphotriesterase. Biochemistry 39:7357-7364. [DOI] [PubMed] [Google Scholar]
- 45.Smolin, E. M., and L. Rapoport. 1959. s-Triazines and derivatives, p. 269-308. In A. Weissberger (ed.), The chemistry of heterocyclic compounds. Interscience Publishers, Inc., New York, N.Y.
- 46.Struthers, J. K., K. Jayachandran, and T. B. Moorman. 1998. Biodegradation of atrazine by Agrobacterium radiobacter J14a and use of this strain in bioremediation of contaminated soil. Appl. Environ. Microbiol. 64:3368-3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thurston, J. T., J. R. Dudley, D. W. Kaiser, I. Hechenbleikner, F. C. Schaefer, and D. -H. Hansen. 1951. Cyanuric chloride derivatives. J. Am. Chem. Soc. 73:2981-2983. [Google Scholar]
- 48.Topp, E., W. M. Mulbry, H. Zhu, S. M. Nour, and D. Cuppels. 2000. Characterization of S-triazine herbicide metabolism by a Nocardioides sp. isolated from agricultural soils. Appl. Environ. Microbiol. 66:3134-3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Topp, E., H. Zhu, S. M. Nour, S. Houot, M. Lewis, and D. Cuppels. 2000. Characterization of an atrazine-degrading Pseudoaminobacter sp. isolated from Canadian and French agricultural soils. Appl. Environ. Microbiol. 66:2773-2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.U.S. Department of Agriculture. 2001. Agricultural chemical usage 2000 restricted use summary, p. 5. Agricultural Statistics Board. National Agricultural Statistics Service, U.S. Department of Agriculture, Washington, D.C.
- 51.Wilson, D. K., and F. A. Quiocho. 1993. A pre-transition-state mimic of an enzyme: X-ray structure of adenosine deaminase with bound 1-deazaadenosine and zinc-activated water. Biochemistry 32:1689-1694. [DOI] [PubMed] [Google Scholar]
- 52.Yanze-Kontchou, C., and N. Gschwind. 1994. Mineralization of the herbicide atrazine as a carbon source by a Pseudomonas strain. Appl. Environ. Microbiol. 60:4297-4302. [DOI] [PMC free article] [PubMed] [Google Scholar]