

Abstract
The leucine-responsive regulatory protein Lrp regulates the expression of a number of operons in Escherichia coli, including the ilvIH operon. Earlier in vitro experiments showed purified Lrp binding to two regions of DNA proximal to the ilvIH promoter, an upstream region (−260 to −190) and a downstream region (−150 to −40). The effect of mutations in these regions on ilvIH promoter expression in vivo led to the proposal that activation of transcription required Lrp binding to downstream sites 3, 4, 5, and 6. Binding of Lrp to upstream sites 1 and 2 seemed to enhance promoter expression but was not absolutely required (Q. Wang and J. M. Calvo, J. Mol. Biol. 229:306-318, 1993). Here we present data that require a reevaluation of the above conclusion. Constructs having either a deletion of DNA or a 100-bp substitution of DNA upstream of position −160 showed no ilvIH promoter activity in vivo. These results unambiguously establish that DNA at or upstream of position −160 is required for ilvIH promoter expression. Together with previous results, we conclude that Lrp bound at downstream sites is necessary but not sufficient for promoter activation. In addition, insertion of 4, 6, 8, or 10 bp between the upstream and downstream regions also resulted in a very strong reduction of in vivo promoter expression, even though the binding of Lrp in vitro was not greatly affected by these mutations. Closer inspection showed that the affinity of Lrp for the upstream region of all of these constructs was about the same but that Lrp bound to the downstream region of the wild-type construct with a higher degree of cooperativity than in the case of the others. These mutations may have reduced promoter activity in vivo by eliminating a binding site for some transcription factor other than Lrp. Alternatively, the small-addition mutations may have affected the geometry of these complexes, preventing either an interaction between Lrps bound at upstream and downstream sites (which might be necessary for promoter expression) or preventing the positioning of Lrp bound at upstream sites for productive interaction with the promoter.
The leucine-responsive regulatory protein Lrp of Escherichia coli controls the expression of a large number of genes, including some involved in amino acid biosynthesis, amino acid degradation, transport of metabolites, pilus formation, and one-carbon metabolism (2, 14). The most striking feature of the Lrp regulon is the variety of ways in which Lrp and leucine affect gene expression. Genes may either be activated or repressed by Lrp, and leucine may in each case antagonize, potentiate, or have no effect on this regulation (2, 14).
Lrp has a pI above 9, has a monomer molecular mass of 18.8 kDa (164 amino acids), and is a stable dimer in vitro in solutions containing nanomolar concentrations of Lrp (23). The total Lrp monomer concentration in E. coli cells is in the micromolar range (about 6,000 monomers per cell in cells grown in a minimal medium) (23). At that concentration in vitro, Lrp exists mainly as an octamer or hexadecamer, and millimolar concentrations of leucine promote conversion of hexadecamer to octamer. For E. coli grown in a minimal medium, about 60% of the total Lrp is bound nonspecifically to DNA, but the concentration of free Lrp is high enough that some mixture of octamers and hexadecamers is expected in vivo (4, 5).
A consensus sequence for binding Lrp has been proposed (7), but it is not always readily discernible in some known members of the regulon. For members of the regulon that have been studied in detail, Lrp interacts with multiple sites upstream of the promoter region, and binding to groups of sites is highly cooperative (2). The binding seems to perturb the structure of a 100- to 200-bp region of DNA, as observed in DNase I footprinting experiments, and the pattern of bands is reminiscent of similar experiments with nucleosomes (2). Furthermore, multiple Lrps seem to bind to the same face of the DNA (22). These results together suggest a structure in which DNA is wrapped around a core composed of multiple Lrp units. Contributing to DNA wrapping is a propensity of Lrp to bend DNA at sites to which it binds specifically (21). For PutR, an Lrp-like protein from Agrobacterium tumefaciens, atomic force microscopy pictures show DNA loops induced by this protein (11). DNA wrapping around an Lrp core is also supported by the three-dimensional structure of an Lrp-like protein from Pyrococcus furiosus that was published recently (12). That structure shows an octamer composed of four dimers, with four symmetrically placed DNA binding sites on the outside (12).
The ilvIH operon, one of the best-studied members of the Lrp regulon, encodes an acetohydroxy acid synthase that is involved in the biosynthesis of branched-chain amino acids (8). Lrp directly activates the ilvIH promoter, and strains that have an inactive lrp gene have only very low levels of ilvIH expression (16). Addition of leucine to the growth medium of wild-type strains causes a 5- to 10-fold reduction in ilvIH promoter activity (9). In vitro, Lrp binds to six sites within a 200-bp sequence of DNA upstream of the ilvIH promoter (20). Binding of Lrp to sites 1 and 2 (upstream region) is highly cooperative, as is binding to sites 3, 4, 5, and 6 (downstream sites). Mutations in each of the individual binding sites have previously been characterized; most of them cause reduced Lrp binding in vitro and reduced ilvIH expression in vivo (20).
The exact mechanism by which Lrp controls gene expression is not well understood. In a model put forward by Wang and Calvo for regulation of ilvIH expression, Lrp bound at downstream sites 3, 4, 5, and 6 was postulated to interact directly with RNA polymerase at the promoter (20). In this model, Lrp bound at upstream sites was not essential for promoter expression but contributed to more efficient expression, perhaps by interacting with Lrp bound at downstream sites. Here we present new data that force us to reevaluate this model. Taken together with other data, our new data indicate that occupancy by Lrp at both upstream and downstream sites is required for ilvIH promoter expression.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
All strains used in this study are derivatives of E. coli K-12 and are described in Table 1. Strain P90C [ara (lac-pro) thi] was the host for plasmid pRS415 and for phage lambda and their derivatives (19). Luria broth (13) and Vogel and Bonner minimal salts supplemented with 0.4% glucose, 50 μg of l-proline per ml, and 1 μg of thiamine per ml were used as rich and minimal media, respectively. Media were sometimes supplemented with l-leucine (50 μg/ml), ampicillin (100 μg/ml), or 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal, 50 μg/ml).
TABLE 1.
Bacterial strains, phages, and plasmids used in this study
| Strain, plasmid, or phage | Relevant genotype | Source or reference |
|---|---|---|
| E. coli K-12 strains | ||
| CV981a | ara (lac-pro) thi | 13 |
| CV1042 | CV981-λQW156 | 20 |
| CV1327 | CV981-λQW223 | 20 |
| CV1555 | CV981-λSJ315 | This study |
| CV1556 | CV981-λSJ316 | This study |
| CV1557 | CV981-λSJ317 | This study |
| CV1558 | CV981-λSJ318 | This study |
| CV1559 | CV981-λSJ319 | This study |
| CV1560 | CV981-λSJ320 | This study |
| CV1561 | CV981-λSJ321 | This study |
| CV1562 | CV981-λSJ322 | This study |
| Plasmids | ||
| pRS415 | Contains promoterless lacZYA | 19 |
| pCV156 | pRS415 containing −332 to +29 of ilvIH region fused to lacZYA | 20 |
| pCV223 | pCV156 but Δ1, 2, 3, 4, 5, 6 | 20 |
| pCV315 | pCV156 but Δ1&2 | This study |
| pCV316 | pCV156 but 100-bp insertion | This study |
| pCV317 | pCV156 but Δ1&2, 100-bp insertion | This study |
| pCV318 | pCV156 but 2-bp insertion | This study |
| pCV319 | pCV156 but 4-bp insertion | This study |
| pCV320 | pCV156 but 6-bp insertion | This study |
| pCV321 | pCV156 but 8-bp insertion | This study |
| pCV322 | pCV156 but 10-bp insertion | This study |
| Phages | ||
| λRS45 | Contains promoterless lacZYA | 19 |
| λQW156 | λRS415 containing −332 to +29 of ilvIH region fused to lacZYA | 20 |
| λQW223 | λQW156 but Δ1, 2, 3, 4, 5, 6 | 20 |
| λSJ315 | λQW156 but Δ1&2 | This study |
| λSJ316 | λQW156 but 100-bp insertion | This study |
| λSJ317 | λQW156 but Δ1&2, 100-bp insertion | This study |
| λSJ318 | λQW156 but 2-bp insertion | This study |
| λSJ319 | λQW156 but 4-bp insertion | This study |
| λSJ320 | λQW156 but 6-bp insertion | This study |
| λSJ321 | λQW156 but 8-bp insertion | This study |
| λSJ322 | λQW156 but 10-bp insertion | This study |
Strain CV981 is strain P90C (CSH26) of Miller (13).
In vitro mutagenesis.
Insertion and deletion mutations were generated by PCR mutagenesis (1). Plasmid pCV156 containing the region from −332 to +29 of the ilvIH operon (20) was used as the template, and primers for PCR were designed to create addition mutations between bases −160 and −161 of the ilvIH operon. To prepare construct +2, two PCR products were generated with primers 1 and 2 and, separately, primers 3 and 4 (Table 2). The two products were purified by electrophoresis through 1% agarose, mixed, and used in a second PCR with primers 1 and 4, yielding the desired PCR product. For PCR, a 100-μl volume contained 1 ng of template DNA, 1 μg of each primer, 50 mM KCl, 10 mM Tris-HCl (pH 8.8), 2.5 mM MgCl2, 0.01% gelatin, 0.5 mM each of the four deoxynucleoside triphosphates, and 5 U of Pfu DNA polymerase (Stratagene). For each of 30 cycles, samples were denatured at 94°C for 1 min, annealed at 55°C for 1 min, and extended at 72°C for 2 min. Constructs +4, +6, +8, and +10 were prepared similarly with the primers identified in Table 2.
TABLE 2.
Primers used in creating mutationsa
| Mutation created | Primer | Position of 3′ end of primer | Sequence of primer (5′ → 3′) |
|---|---|---|---|
| 1 | −320 | GCAGAATTCCCCCATCAGTGGATG | |
| 4 | —b | CCAGGGTTTTCCCAGTCACGACG | |
| +2 | 2 | −172 | CCACAACTTAGCTAGCAATTTCTC |
| 3 | −151 | GAGAAATTGCTAGCTAAGTTGTGG | |
| +4 | 5 | −172 | CCACAACTTGCTAGCAGCAATTTCTC |
| 6 | −151 | GAGAAATTGCTGCTAGCAAGTTGTGG | |
| +6 | 7 | −172 | CCACAACTTAGCTAGCCAGCAATTTCTC |
| 8 | −151 | GAGAAATTGCTGGCTAGCTAAGTTGTGG | |
| +8 | 9 | −172 | CCACAACTTAAGCTAGCTCAGCAATTTCTC |
| 10 | −151 | GAGAAATTGCTGAGCTAGCTTAAGTTGTGG | |
| +10 | 11 | −172 | CCACAACTTAAGCTAGATCTCAGCAATTTCTC |
| 12 | −151 | GAGAAATTGCTGAGATCTAGCTTAAGTTGTGG | |
| +100 | 13 | —c | TAGAGAAATTGCTGGTTGTCCCAATTCTTGTTG |
| 14 | —c | GGCTGAATCCCACAACTTAGGGTAAGTTTTCCGTATG | |
| 15 | −142 | CATACGGAAAACTTACCCTAAGTTGTGGGATTCAGCC | |
| 16 | −174 | CAACAAGAATTGGGACAACCAGCAATTTCTCTA | |
| Δ1&2 | 17 | −142 | CGAGAATTCTAAGTTGTGGGATTCAGCC |
| Δ1&2, +100 | 18 | —c | CGAGAATTCGTTGTCCCAATTCTTGTT |
Inserted bases are shown in bold, and ensuing restriction sites are underlined. The numbering refers to the startpoint of transcription of the ilvIH operon (20).
This primer corresponds to sequences in lacZ in plasmid pCV156. Plasmid pCV156 contains the region from −332 to +29 of the ilvIH operon, followed by trp and lac sequences.
These primers correspond to sequences in the gfp gene, encoding green fluorescent protein.
The insertion at position −160 of 100 bp of unrelated DNA from the gfp gene of Aequoria victoria was carried out in a similar way but with additional steps to create intermediate fragments. Fragments 1 and 2 were created with primers 13 and 14 and the gfp template and separately with primers 4 and 15 and plasmid pCV156 as the template, respectively. Fragment 3 was created with primers 4 and 13 and fragments 1 and 2 as the template. Fragment 4 was created with primers 1 and 16 and plasmid pCV156 as the template. The desired construct was formed by PCR with primers 1 and 4 and fragments 3 and 4 as the template.
A deletion of all bases upstream of −160 in the ilvIH operon was generated by a one-step amplification process in which the desired region containing the downstream sites was amplified with primers 4 and 17 and plasmid pCV156 as the template. A similar construct having the 100-bp insertion was prepared with primers 4 and 18 and pCV316 as the template.
The EcoRI-BamHI fragment of plasmid pCV156 was replaced with the mutation-containing fragments described above by cleaving with restriction enzymes and ligation, placing the ilvIH promoter and upstream region in front of the lacZYA reporter gene in plasmid pRS415. The constructs were introduced into strain P90C (CV981) by transformation (18), transferred to a derivative of phage lambda by homologous recombination, and then introduced into the E. coli chromosome of strain P90C in single copy by forming lysogens (19). Single-copy lysogens were identified by assaying five colonies for β-galactosidase activity.
β-Galactosidase assays.
The specific activity of β-galactosidase was measured in cells in log phase (A600, about 0.5) with the chloroform-sodium dodecyl sulfate lysis procedure described by Miller (13). Results are averages from at least two separate samples in which the activity of each sample was determined in triplicate. The results obtained from two separate samples varied by less than 10%.
Gel mobility shift experiments.
The 375-bp EcoRI-BamHI fragment from pCV156 and derivative plasmids was excised by treatment with EcoRI and BamHI, end labeled at the 3′ end with [32P]dATP (Amersham) and the Klenow fragment of E. coli DNA polymerase, and purified by electrophoresis through 5% polyacrylamide. Binding reactions contained 1 ng of labeled DNA and Lrp in 20 μl of a buffer containing 20 mM Tris-hydrochloride (pH 8.0), 0.4 mM EDTA, 0.1 mM dithiothreitol, 50 mM NaCl, 1 mM MgCl2, 12.5% glycerol, 50 μg of calf thymus DNA per ml, and 100 μg of bovine serum albumin per ml. After 20 min at room temperature, samples were fractionated by electrophoresis at 10 V/cm in 5% polyacrylamide gels in TBE buffer (pH 8.0) containing 89 mM Tris, 89 mM borate, and 2 mM EDTA. Gels were transferred to 3MM Whatman paper, dried, and autoradiographed. Radioactivity was quantitated with a Storm B840 Phosphorimager (Molecular Dynamics).
Analysis of Lrp-DNA complexes formed from a mixture of fragments.
The wild-type, +4, +6, and +10 fragments were each end labeled with 32P and purified by electrophoresis through 5% polyacrylamide. Approximately equal amounts of each fragment (in terms of radioactivity) were mixed, and gel shift mobility experiments were performed as described above except that reaction components and volumes were scaled up by a factor of 10 (to ensure enough counts for analysis) and each sample was loaded into two large wells. After electrophoresis, gels were wrapped in Saran wrap and placed on X-ray film overnight.
The faster- and slower-moving bands (each containing four different complexes) were excised and eluted with 0.5 M ammonium acetate containing 1 mM EDTA. The eluate was passed through Ultrafree-Probind filters (Millipore) to remove protein, and the DNA in the samples was precipitated and resuspended in a solution containing 80% (vol/vol) formamide, 10 mM NaOH, 1 mM EDTA, 0.1% xylene cyanol, and 0.1% bromophenol blue. Samples were fractionated by electrophoresis through 5% polyacrylamide containing 8 M urea at 2,000 constant volts for 4 h (movement of xylene cyanol was about two gel lengths). After drying, the relative intensities of the four bands in each lane were determined with a phosphorimager. Another lane in the same gel contained the input mixture of constructs. The mole fraction of each construct in the input DNAs and in the slower- and faster-moving complexes was calculated from the data.
A computational method was used to determine the likelihood of obtaining the results of these experiments by chance. A computer program generated random input numbers around four experimentally determined input means. To do this, random numbers between zero and one were generated, and those numbers were transformed into random numbers about a mean with a standard deviation value of 4% and the equations provided by R. Strawderman (the average standard deviation for 20 experimentally determined means, each mean being the average of four measurements, was 3%). Ten thousand sets of four input values were generated in this way each time the program was run, but in groups of 2,500. For each group of 2,500 values, three of the input means were generated as described above, and the fourth was calculated from 100 minus the sum of the others. Each set of four values (randomized around the experimentally derived input values) was used to generate a set of 16 values equivalent to those in the table in Fig. 3D; thus, in each operation of the program, 10,000 sets of numbers having the appearance of that in Fig. 3D were generated. Each line in each table had input values randomized around experimentally derived input means and four output values that were randomly generated around those input values, assuming a 4% standard deviation. Finally, the computer program analyzed each of 10,000 tables for hits, a hit being a situation in which, on any line in the table, values 1 and 3 were less than the input by fraction P and fraction Q, respectively, and values 2 and 4 were greater than the input by fraction R and fraction S, respectively. For analysis of the data in Fig. 3D, values corresponding to the wild type (Fig. 3D, line 4) were chosen: P = 0.2, Q = 0.1, R = 0.1, and S = 0.05. In running the program multiple times, only 15 hits were observed for 250,000 tables analyzed.
FIG. 3.

Analysis of Lrp binding to a mixture of wild-type and mutant constructs in vitro. (A) Diagrammatic representation of free DNAs and complexes formed after Lrp is incubated with a mixture containing the wild-type, +4, +6, and +10 constructs. Solid inverted arrowheads represent addition mutations, and solid circles represent Lrp. (B) Gel mobility shift experiments performed with the indicated amounts of Lrp and a mixture of constructs. Lines identify bands as containing either a mixture of free DNAs or a mixture of Lrp-DNA complexes. (C) Samples from panel B were excised from the gel and, after removal of protein, fractionated by electrophoresis under denaturing conditions. The four bands in each lane are the wild-type (highest mobility), +4, +6, and +10 constructs. (D) Quantitation of data from panel C. Numbers reflect the radioactivity in a band as a percentage of the total radioactivity in all four bands in a lane.
Determination of association constants for binding of Lrp to ilvIH DNA.
The relatively stable complexes formed from Lrp binding to ilvIH DNA are here called complex 1 (Lrp bound to sites 1 and 2) and complex 2 (Lrp bound to sites 1, 2, 3, 4, 5, and 6).
For complex 1:
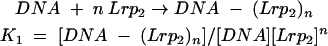 |
(1) |
For complex 2:
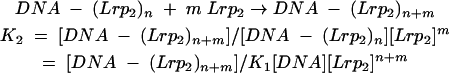 |
(2) |
where n and (n + m) are numbers of Lrp dimers in complexes 1 and 2, respectively, K1 and K2 are association constants for complexes 1 and 2, respectively, Lrp2 represents Lrp dimer, and brackets indicate concentration of Lrp as a dimer. The fraction of the total DNA that is free (F0), in complex 1 (F1), or in complex 2 (F2) is given by
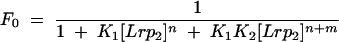 |
(3) |
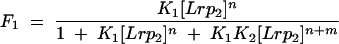 |
(4) |
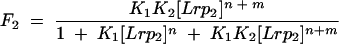 |
(5) |
To estimate K1 and K2, data from gel mobility shift experiments at different Lrp concentrations were globally fit to equations 3, 4, and 5 with a nonlinear least-square regression method from Origin (Microcal Software). Values for n and m were set at 2 or 4, and the total protein concentration was used in place of free protein concentration because protein is in large excess over DNA in these experiments.
RESULTS
Effects of upstream deletions and insertions on ilvIH promoter expression in vivo.
The constructs used in this study (Fig. 1) contain the ilvIH promoter together with various amounts of upstream DNA as part of a transcriptional fusion to lacZ. These constructs were transferred to bacteriophage lambda and introduced as single lysogens into strain CV981 by the procedures of Simons et al. (19). Strains were grown in a minimal medium in the presence or absence of leucine, and the strength of the ilvIH promoter was assessed by measuring the specific activity of β-galactosidase.
FIG. 1.

Expression from the ilvIH promoter in vivo in strains having addition and deletion mutations. The specific activity of β-galactosidase (Miller units) (13) was measured in the indicated strains in cells grown in minimal medium lacking (−Leu) or containing (+Leu) leucine (50 μg/ml). Sites at which Lrp binds upstream of the promoter are represented in the cartoon as solid circles (dimer assumed to bind to one site). Arrow, start point for transcription; wt, wild type; Δ, deletion.
As previously observed (20), a strain having the wild-type construct had approximately 450 U of β-galactosidase activity, and that activity was reduced about 10-fold in cells grown in the presence of leucine. By contrast, a strain in which the entire region upstream of the ilvIH promoter was deleted (strain CV1327) had low β-galactosidase activity when grown in the presence or absence of leucine. Similar results were obtained with a strain that contained Lrp binding sites 3, 4, 5, and 6 but lacked binding sites 1 and 2 (Fig. 1, compare lines 1, 2, and 3).
An insertion of 100 bp between the upstream and downstream sites at position −160 also resulted in a loss of Lrp-mediated activation in vivo, indicating that the correct spacing and/or phasing of the upstream region with respect to the downstream sites or the promoter is important. A strain containing the 100-bp insertion but from which the upstream region had been deleted had the expected null phenotype (Fig. 1, compare lines 1, 4, and 5). These results indicate that the presence of the two most upstream Lrp binding sites, centered at positions −246 and −221, is crucial for Lrp-activated ilvIH promoter expression and that the spacing and/or phasing of these sites relative to the remainder of the operon is important.
Expression of ilvIH promoter in strains containing differentially phased DNA constructs.
To investigate potential phase relationships between upstream and downstream Lrp binding sites, we used PCR mutagenesis to insert up to 10 additional base pairs between the two regions in increments of 2 bp. These mutations were not expected to affect the strength of Lrp binding to these six binding sites in a major way, and in vitro gel retardation experiments suggested that this was indeed the case (see below). Strains containing single-copy lambda prophage with these mutations were constructed, and β-galactosidase levels were measured in cells grown in the absence or presence of leucine.
Strain CV1558 having an insertion of 2 bp between the upstream and the downstream regions had approximately fourfold less β-galactosidase activity than the wild-type strain, and the residual activity was repressed still further by the presence of leucine in the medium (Fig. 1). Insertions of 4 or more base pairs resulted in a further decrease in ilvIH expression, and for the +6 and +8 constructs, the promoter activity was as low as that for a construct lacking all six Lrp binding sites (Fig. 1, compare lines 1, 2, 6, 7, 8, 9, and 10). These results show that a change as small as a 2-bp addition (expected change in the phase of about 72o) can have a significant effect on promoter function. It is interesting that the +10 mutation, in which the upstream and the downstream regions remained in the same phase relationship as in the wild-type operon, was still a strong down mutation for ilvIH expression in vivo. Thus, the precise distance between the upstream and downstream regions may also be important for Lrp-mediated activation of the ilvIH promoter.
Binding of Lrp to differentially phased DNA constructs.
We performed gel mobility shift assays to measure the affinity of Lrp for wild-type and differentially phased construct DNAs in vitro. DNA fragments approximately 375 bp in length cut from plasmid pCV316 and mutant derivatives pCV318 to pCV322 were labeled and used for this analysis (nucleotides −332 to +29 of ilvIH DNA). Figure 2 shows the results of titration experiments performed with the wild-type and +10 constructs and increasing concentrations of purified Lrp. As previously shown by Ricca et al. (17) and Wang and Calvo (20), two major bands were observed in these titrations, representing a faster-moving species containing Lrp bound to sites 1 and 2 and a slower-moving species containing Lrp bound to sites 1, 2, 3, 4, 5, and 6. Trace amounts of other complexes seen in the figure represent DNAs containing Lrp bound to one, three, four, or five sites (20).
FIG. 2.

Binding of Lrp to wild-type and +10 constructs in vitro, as measured by gel mobility shift experiments. Lrp (0, 2, 4, 8, 17, 33, 66, and 133 nM) was incubated with a labeled 375-bp fragment containing wild-type DNA (A) or the same fragment having a 10-bp addition at −160 (B). Samples were fractionated by electrophoresis through polyacrylamide, and radioactivity was measured with a phosphorimager. Arrows denote the positions of free DNA, complex 1 (Lrp bound to sites 1 and 2), and complex 2 (Lrp bound to sites 1, 2, 3, 4, 5, and 6).
Visual inspection of Fig. 2 shows the patterns of binding of Lrp to the wild-type and +10 constructs to be very similar, if not identical. A similar conclusion was reached by comparing the Lrp concentrations that reduced the free DNA concentration by 50%; at a molar concentration of Lrp monomers that was more than 20-fold higher than the concentration of DNA, a concentration of about 3.6 × 10−9 M Lrp was required to shift 50% of free DNA into complexes for both the wild-type and +10 constructs. Note that under the conditions of these gel mobility shift experiments, Lrp is predominantly a dimer in solution. Similar results were obtained in comparisons of the wild type to the +2, +4, +6, +8, and +100 constructs (data not shown).
A more detailed analysis of the binding data was carried out with a simple model of Lrp binding to two regions of DNA. The experimental data were fit to equations derived from the model, and estimates of K1 and K2, association constants for binding to each of the two regions, were derived by global curve fitting (see Materials and Methods for derivation of equations). This analysis indicated that Lrp binds strongly and with about equal avidity to one region of these constructs, at least within a factor of about 2 (Table 3, compare K1 values). Previous work showed that Lrp binds with highest avidity to the upstream sites (20), and we presume that K1 reflects binding to upstream sites for all of these constructs. Values for K2, by contrast, were more variable, reflecting some degree of cooperativity in binding to the two regions. The absolute value of the cooperativity cannot be deduced from these data because the two association constants are not identical, but the relative cooperativity can be deduced by comparing the K1/K2 ratios. This comparison shows that cooperativity associated with Lrp binding was 3- to 9-fold higher for the wild-type and +2 constructs than for the others.
TABLE 3.
Association constants for Lrp binding to upstream and downstream sitesa
| Construct | K1 (10−3 nM−4) | K2 (10−5 nM−4) | K2/K1 (10−2) |
|---|---|---|---|
| Wild type | 5.3 (±1.1) | 16 (±3) | 3.0 (±0.6) |
| +2 | 4.9 (±0.8) | 13 (±3) | 2.7 (±0.6) |
| +4 | 7.6 (±1.9) | 8 (±2) | 1.1 (±0.3) |
| +6 | 4.6 (±0.9) | 6 (±1) | 1.3 (±0.2) |
| +8 | 3.6 (±0.6) | 0.95 (±0.16) | 0.26 (±0.04) |
| +10 | 2.9 (±0.2) | 1.0 (±0.3) | 0.34 (±0.10) |
Data from gel mobility shift experiments of the type shown in Fig. 2 were globally fit to equations 3, 4, and 5 by using a nonlinear least-square regression method from Origin software (Microcal). For the data shown, values for n and m were set at 2 and 4, respectively. When n and m were assumed to be 4 and 4, respectively, the K1 and K2 values were different but the same trend of decreasing cooperativity was observed. Values are means (±standard deviation).
Since the differences in binding described above were relatively small and derived from a comparison of gel mobility patterns of different samples, we decided to repeat the above experiment but in a way that allowed comparisons of complexes in the same lane to be made. To do this, different amounts of Lrp were incubated with a mixture of constructs. At equilibrium, one expects a mixture of free DNA, DNAs of each type with Lrp bound to upstream sites, and DNAs of each type with Lrp bound at both upstream and downstream sites (represented in Fig. 3A). Upon fractionation of these DNAs and complexes, a typical gel mobility shift pattern of bands was observed (Fig. 3B), but each of the major bands was actually a mixture of four different DNAs or DNA-protein complexes. We isolated both groups of complexes and, after removing protein and electrophoresis under denaturing conditions, determined the mole fraction of each construct in the mixture. Figure 3C shows the separation of complexes after deproteinization and electrophoresis. Note that the +2 and +6 constructs were omitted from this analysis so as to achieve good resolution of the other constructs.
A comparison of the mole fraction values determined from Fig. 3C is given in Fig. 3D. In multiple measurements of the mole fractions of the input DNAs used in these experiments, we observed a variation of about 3%. This variation, caused mainly by slight differences in defining bands produced by the phosphorimager, accounts for some of the variation in the rest of the table, but inspection uncovered a trend in the data that was unique to the wild-type construct. For each of the equilibrium mixtures formed at three different Lrp concentrations, the mole fraction of the wild-type construct in complex 1 (DNA bound to sites 1 and 2) was lower than its mole fraction in the input mixture of DNAs, whereas the reverse was true for complex 2 (DNA bound to all six sites). In evaluating the significance of this result, it may be helpful to consider a hypothetical case in which the input mole fractions were each 0.25 for four DNA constructs. If the avidities of Lrp for binding sites on these constructs were identical and there were no cooperative interactions during binding, then the output mole fractions should be identical to the input values for both types of complexes. If, by contrast, one of the constructs bound Lrp to downstream sites with higher avidity than the others, then the mole fraction of that construct should be lower in the first complex and higher in the second complex. Also, by necessity, the mole fraction of at least one of the other constructs must be higher in the first complex and lower in the second. This is exactly what was observed in Fig. 3D. The chance that the observed data resulted from normal variation associated with these measurements is less than 1 in 16,666. A repetition of this experiment with three different Lrp concentrations gave the same pattern of results: only the wild-type construct behaved as described above (data not shown).
These results demonstrate that Lrp binds to the downstream region of the wild-type construct with higher affinity than it does to the downstream regions of constructs +4, +6, and +10.
DISCUSSION
There are three significant findings in this work: DNA located at or upstream of position −160 is absolutely required for expression from the ilvIH promoter; addition mutations that are expected to change both the phasing and the distance between upstream and downstream binding sites substantially reduce promoter expression; and good ilvIH promoter activity is correlated with cooperative interactions between Lrp binding to upstream and downstream sites and not so much with the fact that all sites contain bound Lrp. We discuss these findings below in the light of new findings by us and others.
DNA at or upstream of position −160 is required for ilvIH promoter expression.
In a model for activation of ilvIH expression, Wang and Calvo postulated that Lrp bound to downstream sites 3, 4, and 5 was required for RNA polymerase action at the promoter and that binding of Lrp to sites further upstream improved but was not absolutely required for promoter function (20). The reasoning underlying this model derived mainly from an analysis of the effects of deletion (10) and substitution (20) mutations on promoter action in vivo: mutations in sites 3, 4, or 5 almost completely eliminated expression, whereas mutations in sites 1 and/or 2 reduced but did not eliminate expression.
In reevaluating the earlier work, it seems that the evidence was substantial but not compelling. For example, for the deletion analysis (10), the endpoints of the deletions within the vector were not identical, and it is conceivable that vector sequences had some effect on ilvIH expression. For example, a weak promoter might have been present within the vector sequences of some constructs but not others. In the experiments of Wang and Calvo (20), 6-bp substitution mutations in sites 1 and 2 and a 16-bp substitution mutation affecting both sites 1 and 2 all reduced promoter expression by about 50%. Based on an analysis of the contribution of each of the consensus base pairs to the energetics of binding (7), it was predicted that mutant sites 1 and 2 would not be recognized by Lrp. Binding was indeed severely reduced, but some residual binding was still evident for these mutated constructs (20), highlighting the fact that the rules by which Lrp recognizes binding sites have not yet been established.
The analysis above calls into question our earlier interpretation of the importance of upstream binding sites and our focus on Lrp bound at downstream sites as the most significant factor in transcription activation. Here we show that constructs having either a deletion of DNA or a 100-bp substitution of DNA upstream of position −160 showed no ilvIH promoter activity in vivo, nor did constructs having from 4- to 10-bp additions at position −160. These results unambiguously establish that DNA at or upstream of position −160 is required for ilvIH promoter expression. Together with our previous results, we conclude that Lrp bound at downstream sites is necessary but not sufficient for promoter activation.
Addition mutations located between upstream and downstream binding sites substantially reduce promoter expression.
As few as 2 bp inserted between positions −160 and −161 caused an approximately 60% decrease in expression from the ilvIH promoter, and expression dropped to near background levels with additions of 4, 6, 8, and 10 bp. One possibility consistent with these results is that expression from the ilvIH promoter may require transcription factors in addition to Lrp and the binding site for one of them may include position −160. For example, transcription of the gltBDF operon requires binding of integration host factor to a site between the promoter and upstream Lrp binding sites (15).
Two other possibilities that emphasize phasing and potential interaction between the upstream and downstream Lrp binding sites are discussed below.
Correlation between ilvIH promoter activity in vivo and cooperative interactions between Lrp binding to upstream and downstream sites in vitro.
Here we showed that Lrp binds in vitro to the wild-type and +2 constructs with slightly higher affinity than to the +4, +6, +8, and +10 constructs and that the increased affinity is due to increased cooperativity of binding. In addition, we confirmed the higher affinity of the wild-type construct by showing that, within the same reaction mixture with the +4, +6, and +10 constructs, it tended to be more fully occupied with Lrp than did the others. The increased cooperativity is most likely the result of an interaction between Lrp bound at upstream and downstream sites, as represented diagrammatically in Fig. 4. However, the extent of cooperativity is relatively small, and for that reason we have represented the complex in Fig. 4 as an equilibrium between two conformers that differ in their extents of Lrp interaction. One or the other of these two conformers might support transcription initiation, and we deal with each possibility separately below.
FIG. 4.

Cartoon representing Lrp-DNA and Lrp-Lrp interactions. Lrp (cylinders) is shown binding to upstream (hatched) and downstream (stippled) sites near the ilvIH promoter (P→). Two conformations are represented, the one on the right highlighting an interaction between Lrps bound at upstream and downstream sites. The relative sizes of DNA and Lrp, the positions of binding sites, and the 2-, 4-, 6-, 8-, and 10-bp addition site (triangle) are drawn roughly to scale, but the path of DNA relative to Lrp is imaginary.
How might promoter function depend on an interaction between Lrps bound at upstream and downstream sites? The simplest possibility is that the stronger binding resulting from cooperative interactions ensures Lrp occupancy. However, a cooperativity difference of about 3 between the wild type and construct +4 is not large enough to result in a major difference in Lrp occupancy of these constructs in vivo. Another possibility is that the higher oligomer formed through cooperative interactions, per se, is required for activity of the ilvIH promoter. This possibility is interesting in view of recent studies from our laboratory demonstrating that Lrp exists at micromolar concentrations as a hexadecamer and/or an octamer and that leucine shifts the equilibrium in favor of the octamer (5).
In another study, we measured the equilibrium constants for the hexadecamer-octamer transition and for binding of leucine to hexadecamer and octamer (3). Furthermore, we measured the amount of free Lrp and Lrp bound to DNA in cells grown in minimal medium and in minimal medium containing leucine (4). These measurements allowed us to calculate that in cells grown in a minimal medium, about 40% of the total Lrp is free and that free protein is distributed as 187 hexadecamer molecules, 99 octamers, and 65 leucine-bound octamers (with almost no leucine-bound hexadecamers). At present, we have no evidence bearing on the question of whether hexadecamers and/or octamers are active in stimulating ilvIH promoter expression.
If cooperative interactions between Lrp bound at upstream and downstream sites are required for promoter activity (and not just correlated with it), then this would provide some support for the idea that hexadecamers but not octamers activate the ilvIH promoter. This is so because all of the constructs presumably have bound octamer but only the wild-type and +2 constructs have elevated cooperative interactions. For the +4, +6, and +8 constructs, interaction of Lrps bound at upstream and downstream sites might be precluded because of a change in phase angle of 144, 216, and 288°, respectively, associated with these additions. For the +10 construct, the phase should be restored to that of the wild-type construct, but the positioning of the two Lrp constructs might be such that interaction was precluded.
Of course, the correlation between cooperative interactions between Lrp bound at upstream and downstream sites and promoter activity may have no significance, and a conformer related to the one on the left of Fig. 4 may be active in transcription initiation. In this model, Lrp bound to upstream sites directly stimulates transcription, whereas Lrp bound to downstream sites is necessary to bend the DNA so that the upstream Lrp is positioned properly with respect to the promoter. Thus, all of the constructs can have Lrp positioned at upstream sites, but only the wild-type and +2 constructs have an overall conformation that is consistent with promoter activity. If this model is correct, then this case is one of the exceptions to the general situation summarized by Collado-Villes et al. (6) that sigma 70 promoters are activated by proteins that bind at or very near the promoter; here, activation would be from protein bound to a region several hundred base pairs upstream of the promoter. Another likely exception is gltBDF, in which Lrp bound at sites upstream of −128 stimulates transcription from a downstream promoter (15).
Whether any of these models is correct remains to be determined. What is clear from this work, however, is that binding of Lrp to two distinct regions is required for transcription from the ilvIH promoter and that the phasing and distance between the two regions may also be important.
Acknowledgments
We thank Zhiqi Hao for writing the computer program used to analyze the data in Fig. 3 and Robert Strawderman for help with the design of this analysis.
This work was supported by National Institute of Health grant GM39496.
REFERENCES
- 1.Blomfield, I. C., V. Vaughn, R. R. Rest, and B. I. Eisenstein. 1991. Allelic exchange in Escherichia coli with the Bacillus subtilis sacB gene and a temperature-sensitive pSC101 replicon. Mol. Microbiol. 5:1447-1457. [DOI] [PubMed] [Google Scholar]
- 2.Calvo, J. M., and R. G. Matthews. 1994. The leucine-responsive regulatory protein (Lrp), a global regulator of metabolism in Escherichia coli. Microbiol. Rev. 58:466-490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen, S., and J. M. Calvo. 2002. Leucine-induced dissociation of Escherichia coli Lrp hexadecamers to octamers. J. Mol. Biol. 318:1031-1042. [DOI] [PubMed] [Google Scholar]
- 4.Chen, S., Z. Hao, E. Bieniek, and J. M. Calvo. 2001. Modulation of Lrp action in Escherichia coli by leucine: effects on non-specific binding of Lrp to DNA. J. Mol. Biol. 314:1067-1075. [DOI] [PubMed] [Google Scholar]
- 5.Chen, S., M. H. Rosner, and J. M. Calvo. 2001. Leucine-regulated self-association of leucine-responsive regulatory protein (Lrp) from Escherichia coli. J. Mol. Biol. 312:625-635. [DOI] [PubMed] [Google Scholar]
- 6.Collado-Vides, J., B. Magasanik, and J. D. Gralla. 1991. Control site location and transcriptional regulation in Escherichia coli. Microbiol. Rev. 55:371-394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cui, Y., Q. Wang, G. D. Stormo, and J. M. Calvo. 1995. A consensus sequence for binding of Lrp to DNA. J. Bacteriol. 177:4872-4880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.DeFelice, M., C. T. Lago, C. H. Squires, and J. M. Calvo. 1982. Acetohydroxy acid synthase isoenzymes of Escherichia coli K12 and Salmonella typhimurium. Ann. Microbiol. Inst. Pasteur 133A:251-256. [PubMed]
- 9.DeFelice, M., and M. Levinthal. 1977. The acetohydroxy acid synthase III isoenzyme of Escherichia coli K-12: regulation of synthesis by leucine. Biochem. Biophys. Res. Commun. 79:82-87. [DOI] [PubMed] [Google Scholar]
- 10.Haughn, G. W., C. H. Squires, M. DeFelice, C. T. Lago, and J. M. Calvo. 1985. Unusual organization of the ilvIH promoter of Escherichia coli. J. Bacteriol. 163:186-198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jafri, S., S. Evoy, K. Cho, H. G. Craighead, and S. C. Winans. 1999. An Lrp-type transcriptional regulator from Agrobacterium tumefaciens condenses more than 100 nucleotides of DNA into globular nucleoprotein complexes. J. Mol. Biol. 288:811-824. [DOI] [PubMed] [Google Scholar]
- 12.Leonard, P. M., H. J. Smits, S. E. Sedelnikova, A. B. Brinkman, W. M. deVos, J. van der Oost, D. W. Rice, and J. B. Rafferty. 2001. Crystal structure of the Lrp-like transcriptional regulator from the archaeon Pyrococcus furiosus. EMBO J. 20:990-997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miller, J. H. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
- 14.Newman, E. B., T. T. Lin, and R. D'Ari. 1996. The leucine/Lrp regulon, p. 1513-1525. In F. C. Neidhardt et al. (ed.), Escherichia coli and Salmonella: cellular and molecular biology, 2nd ed., vol. 1. ASM Press, Washington, D.C.
- 15.Paul, L., R. M. Blumenthal, and R. G. Matthews. 2001. Activation from a distance: roles of Lrp and integration host factor in transcriptional activation of gltBDF. J. Bacteriol. 183:3910-3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Platko, J. V., D. A. Willins, and J. M. Calvo. 1990. The ilvIH operon of Escherichia coli is positively regulated. J. Bacteriol. 172:4563-4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ricca, E., D. A. Aker, and J. M. Calvo. 1989. A protein that binds to the regulatory region of the ilvIH operon of Escherichia coli. J. Bacteriol. 171:1658-1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, p. 1.4, vol. 1. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
- 19.Simons, R. W., F. Houman, and N. Kleckner. 1987. Improved single and multicopy lac-based cloning vectors for protein and operon fusions. Gene 53:85-96. [DOI] [PubMed] [Google Scholar]
- 20.Wang, Q., and J. M. Calvo. 1993. Lrp, a global regulatory protein of E. coli, binds cooperatively to multiple sites and activates transcription of ilvIH. J. Mol. Biol. 229:306-318. [DOI] [PubMed] [Google Scholar]
- 21.Wang, Q., and J. M. Calvo. 1993. Lrp, a major regulatory protein in Escherichia coli, bends DNA and can organize the assembly of a higher-order nucleoprotein structure. EMBO J. 12:2495-2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weise, D. E., B. R. Ernsting, R. M. Blumenthal, and R. G. Matthews. 1997. A nucleoprotein activation complex between the leucine-responsive regulatory protein and DNA upstream of the gltBDF operon in Escherichia coli. J. Mol. Biol. 270:152-168. [DOI] [PubMed] [Google Scholar]
- 23.Willins, D. A., C. W. Ryan, J. V. Platko, and J. M. Calvo. 1991. Characterization of Lrp, an Escherichia coli regulatory protein that mediates a global response to leucine. J. Biol. Chem. 266:10768-10774. [PubMed] [Google Scholar]