

Abstract
Intraclonal genome diversity of Pseudomonas aeruginosa was studied in one of the most diverse mosaic regions of the P. aeruginosa chromosome. The ca. 110-kb large hypervariable region located near the lipH gene in two members of the predominant P. aeruginosa clone C, strain C and strain SG17M, was sequenced. In both strains the region consists of an individual strain-specific gene island of 111 (strain C) or 106 (SG17M) open reading frames (ORFs) and of a 7-kb stretch of clone C-specific sequence of 9 ORFs. The gene islands are integrated into conserved tRNAGly genes and have a bipartite structure. The first part adjacent to the tRNA gene consists of strain-specific ORFs encoding metabolic functions and transporters, the majority of which have homologs of known function in other eubacteria, such as hemophores, cytochrome c biosynthesis, or mercury resistance. The second part is made up mostly of ORFs of yet-unknown function. Forty-seven of these ORFs are mutual homologs with a pairwise amino acid sequence identity of 35 to 88% and are arranged in the same order in the two gene islands. We hypothesize that this novel type of gene island derives from mobile elements which, upon integration, endow the recipient with strain-specific metabolic properties, thus possibly conferring on it a selective advantage in its specific habitat.
Genetic variability within bacterial species can be the result of nucleotide substitutions, intragenomic reshuffling, and acquisition of DNA sequences from another organism (3). The considerable impact of the last strategy, termed horizontal gene transfer, on microbial evolution and its integral role in the diversification and speciation of the bacteria has become apparent from recent analyses based on the growing pool of genomic sequence information (7, 18, 23, 28). Prominent examples are the pathogenicity islands of many obligatory pathogens (14). These chromosomally encoded regions typically contain large clusters of virulence genes not present in closely related nonpathogenic strains and can, upon integration, transform a benign organism into a pathogen. Whereas the molecular mechanism of chromosomal integration has been resolved for some conjugative transposons and bacteriophages and details about the transmissibility of conjugative plasmids are well known, the evolution and mobility of gene islands remain obscure (14). Often these DNA blocks are integrated adjacent to or within tRNA genes, and some contain a phage-related integrase gene near one end, suggesting that gene islands may have been generated by a phage or by a plasmid with integrative functions (14, 42). Nevertheless, the comparative sequence analysis of gene islands so far have not pointed to any common genetic repertoire that confers transmission and acquisition.
The gram-negative bacterium Pseudomonas aeruginosa is ubiquitously distributed in aquatic and soil habitats, and it is an opportunistic pathogen for plants, animals, and humans (38). No correlation between certain P. aeruginosa clones and disease habitats or environmental niches could be detected (1, 9). Although the genome sequence of the reference strain PAO1 provides insights into the versatility and intrinsic drug resistance of P. aeruginosa (48), the genetic origin of the broad range of metabolic capacities and the evolutionary history of chromosome organization have not been determined in sufficient depth for this phenotypically and genetically diverse species. Our previous analyses have shown that the P. aeruginosa chromosome possesses three regions with pronounced genomic variability (15, 33). These three so-called hypervariable regions close to the pilA, phnAB, and lipH loci could even be found at the intraclonal level (35). Comparative genome mapping was used to unambiguously identify the chromosomal difference regions of the two related strains C and SG17M, both belonging to the predominant P. aeruginosa clone C but recovered from different habitats (40).
In order to resolve the chromosomal structure and the genetic makeup of one of the hypervariable areas of the P. aeruginosa genome, we determined the sequence of the region located near the lipH gene for strains C and SG17M. The annotation revealed that the hypervariable region resembles a mosaic of species-, clone-, and strain-specific DNA segments in both strains. The two identified strain-specific gene islands have been integrated into tRNAGly genes and probably originated from mobile circular elements. They are composed of strain-specific open reading frames (ORFs) encoding metabolic functions, of phage- and plasmid-like genes, and of a set of previously unknown genes which display a very high degree of homology between the two islands.
MATERIALS AND METHODS
Bacterial strains, plasmids, and culture conditions.
The P. aeruginosa strains C and SG17M selected for this study both belong to the major P. aeruginosa clone C (35). P. aeruginosa strain C was isolated from the lung of a cystic fibrosis (CF) patient, while strain SG17M was recovered from the aquatic environment (37). Cloning was done in E. coli strain DH5α or XL1-Blue MR (Stratagene) by using the broad-host-range vector pLAFR3 (tetracycline resistance) (47), the cosmid SuperCos-1 (ampicillin resistance) (Stratagene), and the plasmid pTZ19R-Δbla-cat (chloramphenicol resistance) (this study). To construct the plasmid pTZ19R-Δbla-cat, we replaced the ampR gene-containing 0.7-kb DraI fragment in pTZ19R (MBI Fermentas) with a chloramphenicol acetyltransferase-encoding BssHII fragment from pHK (22). It was necessary to use chloramphenicol rather than ampicillin resistance because the plasmid vector was used for subcloning of the SuperCos-1 cosmids, which also carry the ampR gene. Bacteria were routinely grown at 37°C in Luria-Bertani medium (39). For maintenance of pLAFR3 cosmids in Escherichia coli DH5α, the media were supplemented with 20 μg of tetracycline per ml. For E. coli XL1-Blue carrying SuperCos-1 cosmids, 2YT medium (17) supplemented with 100 μg of ampicillin per ml was used, and E. coli DH5α with pTZ19R-Δbla-cat plasmids was propagated in TB broth (39) containing 25 μg of chloramphenicol per ml.
DNA techniques.
DNA manipulations were by standard procedures (5). High-molecular-weight chromosomal DNA of P. aeruginosa was prepared by the protocol of Goldberg and Ohman (11). Small-scale isolations of plasmid and cosmid DNAs were performed by using QIAprep spin miniprep kits (Qiagen), while larger amounts of cosmid DNA were purified by using QIAtip100 columns (Qiagen) according to the instructions of the supplier.
Construction of cosmid libraries.
A genome-wide cosmid library was constructed for each P. aeruginosa strain according to the protocols described previously (52). Chromosomal DNA, partially Sau3AI digested and size fractionated by preparative sucrose gradient ultracentrifugation (11), was cloned into the BamHI sites of pLAFR3 for strain SG17M and of SuperCos-1 for strain C. The ligated DNA was packaged into phage λ particles in vitro by using the λ-DNA in vitro packaging module (Amersham). For strain SG17M, E. coli DH5α was transfected with the λ particles containing the pLAFR3 cosmid DNA. After selection for tetracycline resistance, 768 recombinant clones were transferred to 96-well plates; the resulting cosmid library was named pKSCS. The packaged SuperCos-1 cosmids with DNA of P. aeruginosa C were introduced into E. coli XL1-Blue MR. The corresponding cosmid library pKSCC was made by picking 960 recombinant clones resistant to ampicillin into 96-well plates. A further 20,000 colonies were recovered and stored as a pool.
Southern hybridization.
For colony blots, cell suspensions were inoculated on Hybond N membranes (Amersham) by using a 96-needle replication device and grown either on Luria-Bertani medium-tetracycline plates or on 2YT-ampicillin plates. Alternatively, colony lifts were performed directly from agar plates onto Hybond N membranes. The cells were lysed, and the DNA was fixed (52). Blotting of chromosomal or cosmid DNA digested with appropriate restriction enzymes to nylon membranes, hybridization, and immunological detection of probe signals were performed by previously described protocols (34).
Probe preparation.
The following probes were used for Southern hybridization: strain-specific subtraction clones generated by reciprocal subtractive hybridization (40), cloned gene probes as described previously (35), a selection of P. aeruginosa PAO1-derived SpeI linking clones (36), and insert DNAs from the cosmids themselves. The probes were prepared from gel-purified restriction fragments of cosmids or plasmids by using a digoxigenin labeling kit (Roche Diagnostics) (34). For the pKSCC library, single-stranded probes specific for the ends of a cosmid insert were obtained by using asymmetric PCR with a T3 (5′-AATTAACCCTCACTAAAGGG) or T7 (5′-CATAATACGACTCACTATAGGG) primer and a digoxigenin PCR labeling mixture (Roche Diagnostics); asymmetric PCR was performed in a volume of 50 μl containing 0.5 μg of cosmid DNA as a template, 1 μM primer, 5 μl of digoxigenin PCR labeling mix, 5% dimethyl sulfoxide, 1.5 mM MgCl2, and 2.5 U of Taq polymerase (InViTec) in 1× reaction buffer (InViTec). Extension of the T3 or T7 primer was performed in a Thermo-Cycler (Landgraf) with the following program: 420 s at 95°C and 60 cycles of 120 s at the annealing temperature, 120 s at 72°C, and 120 s at 92°C. The annealing temperatures were 54°C for the T7 primer and 46°C for the T3 primer. After amplification, the reaction mixture was purified as described previously (34).
Construction of ordered cosmid contigs.
To identify the cosmids at the borders of the hypervariable genomic region in P. aeruginosa strains C and SG17M, the corresponding libraries were both screened with the lipH gene probe and a PAO1-derived linking clone covering the SpeI junction SpV-SpAK in strain PAO1, SpV-SpX in strain C, and SpAF′-SpX in SG17M (35, 41). To obtain cosmids covering the strain-specific inserts, both libraries were screened with selected subtraction clones (40). The DNA of each cosmid clone identified in this screen was prepared, and probes specific for the whole insert or only for the ends were generated. These probes derived from the insert ends were used for further hybridization experiments in order to identify overlapping cosmids. All cosmids identified in the walk were individually controlled by hybridization to Southern blots of SpeI digests of PAO1, C, and SG17M chromosomal DNAs to verify their genomic localization and to exclude chimeric cosmids or false-positive signals associated with repeated regions. Comparison of the EcoRI and HindIII restriction fragment patterns and hybridization with the aforementioned probes were used to order the cosmids and to establish the minimal tilting path for the strain-specific regions. Altogether, 27 pKSCC and 34 pKSCS cosmids were identified for P. aeruginosa C and SG17M, respectively, located within the region of interest from the lipH gene to the SpeI junction SpV-SpX in strain C or SpAF′-SpX in strain SG17M. In strain SG17M the following cosmids were selected for sequence analysis: pKSCS 572, 052, 149, 427, 795, and 282. A remaining gap of about 9 kb between pKSCS 572 and 052 was closed by long-range PCR using the Proofsprinter kit (Hybaid). For strain C it was necessary to use an alternative strategy because extensive cross-hybridization prevented the generation of an unequivocal cosmid contig. In order to obtain unique tags, BamHI, HindIII, and EcoRI sublibraries of the pulsed-field gel electrophoresis gel-eluted SpeI fragment SpV were generated. In parallel, the restriction map of the SpV fragment was constructed for the same enzymes by Smith-Birnstiel mapping (16). Thus, the subcloned fragments could be mapped. Subclones carrying unique sequence located within the gap were used as probes for further colony hybridization. More than 3,000 additional pKSCC cosmids had to be screened to gain a contiguous order of cosmids, of which the following five cosmids were selected for sequencing: pKSCC 323, 022, 1064, 1065, and 273.
Sequencing.
To determine the DNA sequence of the entire cosmid inserts, separate plasmid libraries were constructed for each cosmid. DNA from each cosmid was sheared by hydrodynamic cleavage (29), size fractionated, and subcloned into the SmaI site of pTZ19R-Δbla-cat. DNA sequencing of the resulting plasmid libraries was performed on a LICOR 4200 sequencer (MWG Biotech) or on an ABI 377 sequencer (Applied Biosystems). For each cosmid, the individual reads were assembled into contigs by using the base-caller program Phred (8) and the Staden package (46) with the Phrap algorithm integrated (12). Sequencing gaps were closed by primer walking, while combinatorial PCR was used to span physical gaps. The sequence of the 9.8-kb long-range PCR product was determined by primer walking. Finally, the sequences of the individual cosmids and the PCR product were assembled into one contig for each P. aeruginosa strain.
Annotation.
Putative ORFs were identified by using GeneMark.HMM and GeneMark (6, 26). Public databases were searched for similar sequences with the BlastN, BlastX, and BlastP algorithms (2). Predicted ORFs were reviewed individually for start codon assignment based on additional contextual information such as the proximity of ribosome binding sequence motifs. tRNA genes were identified by the program tRNA-scan-SE (25). Pairwise sequence comparisons and multiple alignments were generated using Clustal W (50). Long-range restriction maps were constructed with the in-house program MasterMap (51). Codon usage patterns were analyzed using the in-house programs and the program CodonW (written by John Peden and available at ftp://molbiol.ox.ac.uk/cu). The relative synonymous codon usage (RSCU) was determined for each gene; the RSCU is the observed frequency of a particular codon divided by its expected frequency under the assumption of equal usage of the synonymous codons for an amino acid (43). The genomic codon index (GCI) (21) is a quantitative measure for the synonymous codon bias of a particular gene compared to the average codon usage in the genome. It is defined as the geometric mean of the RSCU values corresponding to each of the codons used in that gene, divided by the maximum possible GCI for a gene of the same amino acid composition:
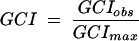 |
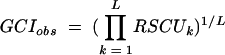 |
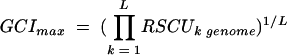 |
where RSCUk is the RSCU value for the kth codon in the gene, RSCUkgenome is the maximal genomic RSCU value for the amino acid encoded by the kth codon in the gene, and L is the number of codons in the gene. The GCI was defined in analogy to the codon adaptation index (43).
For comparison with the P. aeruginosa PAO1 genome sequence, the information at http://www.pseudomonas.com was used (48). Preliminary sequence data were obtained from the Department of Energy Joint Genome Institute at http://www.jgi.doe.gov/tempweb/JGI_microbial/html/index.html.
Nucleotide sequence accession numbers.
The nucleotide sequences reported in this paper have been deposited in the GenBank database (accession no. AF440523 for P. aeruginosa C and AF440524 for SG17M).
RESULTS AND DISCUSSION
A mosaic of species-, clone-, and strain-specific DNA makes up one of the most diverse regions of the P. aeruginosa chromosome. Among the three hypervariable regions in the P. aeruginosa clone C genome (35, 41), the most diverse region near the lipH gene was selected for comparative sequencing of the two P. aeruginosa strains C and SG17M. Both strains belong to clone C, but they were recovered from different habitats. An ordered cosmid contig covering this hypervariable region was constructed for each strain. A contiguous set of cosmids was selected for each strain and sequenced by a shotgun approach. The final contig was 158,230 bp in size for strain C and 128,136 bp for strain SG17M. Sequence comparison revealed that each strain contains an individual large, novel gene cluster flanked by species-specific DNA known from the P. aeruginosa PAO1 genome sequencing project (48). Both insertions are composed of a minor portion of 6,872 bp of DNA, identical in both clone C strains, and a major portion of strain-specific DNA sequence [104,955 bp for strain C, designated PAGI-2(C), and 103,304 bp for SG17M, designated PAGI-3(SG)] (Table 1). (PAGI stands for P. aeruginosa genomic island, in accordance with the nomenclature introduced by Liang et al. [24]). Instead of the 6,872-bp clone C-specific DNA, the genome of P. aeruginosa PAO1 carries a 2,001-bp individual sequence from bp 3173531 to 3171531 at this chromosomal position (Fig. 2). The alignment of the strain C and PAO1 sequences revealed that the analyzed portion of 46.4-kb species-specific DNA shows a very high degree of conservation characterized by identical gene order and a very low nucleotide substitution rate of 0.39%, in agreement with published data of 0.3% sequence diversity in housekeeping genes of P. aeruginosa (20). In total, 184 nucleotide substitutions without any frameshifts or nonsense mutations were identified in this 46.4 kb of DNA. Fewer than 20% of these are nonsynonymous substitutions, resulting in a protein with an altered amino acid composition. Furthermore, no nucleotide alterations could be detected between strains C and SG17M in the analyzed portion of 24.8 kb of shared DNA sequence.
TABLE 1.
Comparison of general features of the sequenced gene islands and the PAO1 genome
| Genomic region | Size (bp) | % G+C | % Coding regions | No. of ORFs
|
Mean GCI | |
|---|---|---|---|---|---|---|
| Total | Per 10 kb | |||||
| PAGI-2(C) | 104,955 | 64.7 | 90.4 | 113 | 10.7 | 0.537 |
| PAGI-3(SG) | 103,304 | 59.2 | 82.7 | 105 | 10.2 | 0.448 |
| C-specific DNA | 6,872 | 66.1 | 83.8 | 9 | 13.1 | 0.65 |
| PAO genomea | 6,264,403 | 66.6 | 89.4 | 5,570 | 8.9 | 0.678 |
FIG. 2.

Gene maps of the P. aeruginosa strain PAO1, C, and SG17M hypervariable genome regions. Predicted coding regions are shown by arrows indicating the direction of transcription. The tRNA genes and attachment sites are depicted by rectangles. Vertical lines and their connections represent the borders of the gene islands and their sites of integration in comparison to the PAO1 genome. Genes are color coded according to their functional category (adapted from http://www.pseudomonas.com). All genes carry identification numbers (C1 to C111 and SG1 to SG105 in the two strain-specific gene islands and C112 to C120 in the clone C specific region [highlighted in pink]), but some have been omitted because of space limitations. In cases of a high degree of homology to already-characterized proteins, three-letter designations are provided for individual genes. ORFs with mutual homologs in both gene islands are shown with a light-blue background. Additionally, ORFs with equivalents in the detected gene island of X. fastidiosa are marked with blue boxes and the corresponding gene identification numbers of the sequencing project (44). IS elements and transposons are shaded in gray.
Strain-specific gene islands integrated into tRNAGly genes.
Comparison of the P. aeruginosa C, SG17M, and PAO1 sequences showed that the two large strain-specific gene islands are inserted into one tRNAGly gene within a cluster comprising one tRNAGlu gene followed by two identical tRNAGly genes (Fig. 1). Within the PAO sequence these tRNA genes are located from bp 3173912 to 3173599. In strain SG17M, the first tRNAGly gene was used for integration of PAGI-3(SG), whereas in strain C, the PAGI-2(C) DNA was incorporated into the second tRNAGly gene. Upon integration, the entire tRNAGly gene was reconstructed at the left end of the gene island, designated attL, whereas in strain C the terminal 16 nucleotides and in strain SG17M the terminal 24 nucleotides of the 3′ end of the tRNAGly gene were present as direct repeat at the right end, designated attR (Fig. 1). Alignment of the attachment sites attL and attR showed a high degree of sequence homology at both junctions (data not shown). The attL sites of both integrated gene islands and the attB2 chromosomal target sites following the second tRNAGly gene share similar AT-rich inverted repeat sequences. Interestingly, similar genomic structures were found by analyzing the chromosomal insertions of the 105-kb clc element in Pseudomonas putida (30, 31) and of a 67-kb gene island in the plant pathogen Xylella fastidiosa (reference 44 and this study). In both cases, the complete tRNAGly gene was reconstructed at the left border, whereas the 18-bp 3′ end of the tRNAGly gene was repeated at the right border of the integrated element (Fig. 1). All four gene islands possess similarly structured attachment sites and surrounding sequences including the conserved inverted repeats (Fig. 1). Only the length of attR varies between the different gene islands (Fig. 1). At the left junction the four gene islands share not only the attL sites but also a highly homologous intergenic spacer (228 bp in strain C, 225 bp in strain SG17M, 226 bp in the P. putida clc element, and 226 bp in X. fastidiosa) and the first ORF, encoding very similar site-specific integrases of the bacteriophage P4 integrase subfamily (the sequence alignment is at our website, http://www.mh-hannover.de/kliniken/kinderheilkunde/kfg/index.htm). The three highly related integrases of strain C, P. putida, and X. fastidiosa are of considerably higher molecular weight than the typical phage P4-related integrases and possess an unusual C terminus showing homology to a putative transposase of Pseudomonas sp. strain B4 (accession no. emb/CAB93963).
FIG. 1.

Organization of the boundaries of the gene islands. The structure of the genomic region around a cluster of three tRNA genes is shown for P. aeruginosa strains PAO1, C, and SG17M. In P. putida F1 (structure adapted from references 30 and 31) and X. fastidiosa (sequence taken from reference 44), the gene islands integrated into a single tRNAGly gene. Map positions in the genome sequence are indicated for P. aeruginosa PAO1 and X. fastidiosa. Large inverted repeats (IRs) are shown as loop structures. Numbers above the maps indicate the lengths (in base pairs) of the corresponding sequences. The 84-bp spacer s1 separating the two tRNAGly genes differs by only two nucleotide substitutions between P. aeruginosa PAO1 and the two clone C strains. The localization of attachment sites attB, attL, and attR (see text for explanation) is indicated. All sequences flanking inverted repeats were named (s2, s2c, and s2c∗, etc.) and aligned to visualize the high degree of homology among the different gene islands and strains. Additionally, the sequences of the depicted tRNAGly genes, highlighted in black, are shown for the three species.
The integrase int-B13 of P. putida has been shown to be responsible for site-specific integrative recombination between the clc element's attachment site (attP) and chromosomal attachment (attB) genes (30, 31, 45). The 105-kb self-transmissible clc element, encoding the degradation of 3-chlorobenzoate, is capable of integrating site and sequence specifically into a tRNAGly gene of its host. The clc element is transferred in plate matings with a frequency of about 10−7 per recipient cell (27). Despite these low frequencies, transfer of the clc element to endogeneous bacteria seems to readily occur in complex microbial communities, such as sludges from soil or wastewater treatment plants (49, 53). When the clc-carrying P. putida strain BN210 was inoculated into a bacterial population in 3-chlorobenzoate-contaminated wastewater, the clc element was taken up by P. aeruginosa strains or by strains belonging to the genus Ralstonia or related β-proteobacteria such as Comamonas (45). Although PAGI-2(C) and PAGI-3(SG) have been stably kept by strains C and SG17M in vitro and in the lungs of the affected CF patient for more than 17 years now with no evidence for loss of the island, these data on the clc element suggest that PAGI-2(C) and PAGI-3(SG) could potentially be mobilized and transferred to other strains, even across species barriers. Hence, gene islands of this type may be widely distributed in terms of species, geographical region, and habitat. This hypothesis is supported by the fact that a copy of PAGI-2(C) with 99.972% nucleotide sequence identity was identified in the Ralstonia metallidurans CH34 chromosome (preliminary sequence data were obtained at http://www.jgi.doe.gov/tempweb/JGI_microbial/html/index.html). P. aeruginosa strain C was isolated in 1986 from a patient in northern Germany, whereas the sequenced R. metallidurans strain was isolated 1976 from the sludge of a zinc decantation tank in Belgium that was polluted with high concentrations of several heavy metals.
Sequence analysis and annotation of PAGI-2(C) and PAGI-3(SG).
The organization of predicted ORFs within the hypervariable region is displayed in Fig. 2. The G+C content and the proportion of coding sequence of PAGI-2(C) are closer to those of the PAO genome than are those of PAGI-3(SG) (Table 1). The mean GCI is significantly lower in PAGI-2(C) and PAGI-3(SG) than in the P. aeruginosa PAO1 genome, indicating that in these islands codon usage is different from that of a typical P. aeruginosa gene. The 6,872-bp region of clone C-specific DNA, however, exhibits a G+C content and GCI values characteristic of P. aeruginosa.
The annotation revealed 111 ORFs in PAGI-2(C) (Table 2) and 106 ORFs in PAGI-3(SG) (Table 3). Tables 2 and 3 show for each ORF its coordinates within the gene island, direction of transcription, size of the gene product, G+C content, and GCI value. Furthermore, the accession number and the name of the homolog that was chosen to assign the function of the gene product are given, together with the corresponding E value from the Blast search. More than 60% of the genes are either conserved hypothetical genes of unknown function or genes with no apparent homology to any reported sequences (Fig. 2; Table 4). Interestingly, these hypothetical ORFs are clustered in the gene islands.
TABLE 2.
Annotation of all ORFs located within the gene island PAGI-2(C) in P. aeruginosa strain C
| Gene identifi- cation | Coordinates
|
Direc- tion | Length (amino acids) | G+C (%) | GCI | Gene name | Homolog product | GenBank accession no. | E value (Blast search) | |
|---|---|---|---|---|---|---|---|---|---|---|
| Left | Right | |||||||||
| C1a | 229 | 2160 | → | 644 | 63.8 | 0.571 | int | Phage-related integrase XF1718 (X. fastidiosa) | AAF84527 | 0E + 00 |
| C2a | 2360 | 3016 | ← | 219 | 59.1 | 0.341 | Hypothetical protein XF1719 (X. fastidiosa) | AAF84528 | 1E − 71 | |
| C3a | 3136 | 3429 | → | 98 | 58.8 | 0.280 | Hypothetical protein XF1720 (X. fastidiosa) | AAF84529 | 9E − 30 | |
| C4a | 3451 | 4341 | ← | 297 | 61.6 | 0.427 | bphR | BphR regulatory protein (R. eutropha) | CAB72138 | 9E − 83 |
| C5 | 4379 | 4702 | ← | 108 | 63.0 | 0.396 | No significant similarity | |||
| C6 | 4734 | 6110 | ← | 459 | 67.4 | 0.537 | Pyridine nucleotide-disulfide oxidoreductase, class I, VC2638 (Vibrio cholerae) | AAF95779 | 1E − 73 | |
| C7 | 6153 | 6959 | ← | 269 | 65.6 | 0.508 | Conserved hypothetical protein str1262 (Synechocystis sp. strain PCC 6803) | BAA17856 | 5E − 25 | |
| C8 | 7050 | 7823 | ← | 258 | 64.0 | 0.541 | dsbG | Thiol:disulfide interchange protein DsbG (PA2476) (P. aeruginosa) | AAG05864 | 2E − 51 |
| C9 | 7826 | 8662 | ← | 279 | 63.2 | 0.437 | Probable thiol:disulfide interchange protein (PA2477) (P. aeruginosa) | AAG05865 | 6E − 48 | |
| C10 | 8662 | 10515 | ← | 618 | 64.9 | 0.464 | dsbD | Probable thiol:disulfide interchange protein (PA2478) (P. aeruginosa) | AAG05866 | 1E − 135 |
| C11 | 10598 | 11479 | ← | 294 | 61.8 | 0.359 | cycH | Cytochrome c-type biogenesis protein CycH (Sinorhizobium meliloti) | P45400 | 5E − 17 |
| C12 | 11476 | 11931 | ← | 152 | 57.5 | 0.392 | cycL | Cytochrome c-type biogenesis protein CycL precursor (S. meliloti) | P45406 | 3E − 25 |
| C13 | 11928 | 12452 | ← | 175 | 60.4 | 0.473 | ccmG | Cytochrome c biogenesis protein CcmG (PA1481) (P. aeruginosa) | AAG04870 | 3E − 37 |
| C14 | 12449 | 14410 | ← | 654 | 62.3 | 0.431 | ccmF | Cytochrome c-type biogenesis protein CcmF (PA1480) (P. aeruginosa) | AAG04869 | 0E + 00 |
| C15 | 14414 | 14860 | ← | 149 | 60.6 | 0.427 | cycJI ccmE | Cytochrome c-type biogenesis protein CycJ (P. fluorescens) | AAC44225 | 6E − 35 |
| C15b | 14844 | 15035 | ← | 64 | 62.5 | 0.430 | ccmD | Heme exporter protein D (cytochrome c-type biogenesis protein CcmD) (V. cholerae) | AAF95200 | 2E − 03 |
| C16 | 15032 | 15769 | ← | 246 | 62.3 | 0.519 | ccmC | Heme exporter protein C (cytochrome c-type biogenesis protein CcmC) (V. cholerae) | AAF95201 | 9E − 67 |
| C17 | 15782 | 16468 | ← | 229 | 63.2 | 0.415 | ccmB | Cytochrome c maturation protein B (Shewanella putrefaciens) | AAC02694 | 6E − 65 |
| C18 | 16465 | 17076 | ← | 204 | 59.6 | 0.354 | ccmA | Heme exporter protein A (cytochrome c-type biogenesis ATP/binding protein CcmA) (V. cholerae) | AAF95203 | 7E − 39 |
| C19 | 17257 | 17925 | → | 223 | 65.0 | 0.525 | armR | Response regulator ArmR (two-component transcriptional regulator) (Pseudomonas sp. strain JR1) | AAF80268 | 4E − 59 |
| C20 | 17922 | 19307 | → | 462 | 63.1 | 0.424 | armS | Sensor kinase ArmS (two-component sensor protein) (Pseudomonas sp. strain JR1) | AAF80269 | 5E − 77 |
| C21 | 19461 | 21059 | ← | 532 | 64.5 | 0.459 | cutE | Apolipoprotein N-acyltransferase (copper homeostasis protein CutE homolog) (P. aeruginosa) | AAC97167 | 3E − 67 |
| C22 | 21084 | 23399 | ← | 772 | 65.5 | 0.459 | Putative metal transporter ATPase [Streptomyces coelicolor A3(2)] | CAB96031 | 9E − 87 | |
| C23 | 23323 | 24420 | ← | 366 | 61.7 | 0.441 | Hypothetical protein PA2481 (P. aeruginosa) (probable cytochrome c) | AAG05869 | 4E − 70 | |
| C24 | 24404 | 25222 | ← | 273 | 64.3 | 0.476 | ORF21 (Moritella marina) (probable cytochrome c4) | BAA89395 | 4E − 23 | |
| C25 | 25219 | 25860 | ← | 214 | 64.0 | 0.490 | fixO/ccoO | ORF20 (M. marina) (cytochrome c oxidase, monoheme subunit, membrane-bound) | BAA89394 | 3E − 17 |
| C26 | 25857 | 27413 | ← | 519 | 60.4 | 0.498 | fixN/ccoN | ORF20 (M. marina) (cytochrome c oxidase, heme b- and copper-binding subunit, membrane bound) | BAA89393 | 6E − 68 |
| C27 | 27932 | 29602 | ← | 557 | 67.6 | 0.651 | Conserved hypothetical protein PA2345 (P. aeruginosa) | AAG05733 | 1E − 125 | |
| C28 | 29651 | 30610 | ← | 320 | 64.8 | 0.564 | Hypothetical protein PA2915 (P. aeruginosa) | AAG06303 | 2E − 99 | |
| C29 | 30717 | 31244 | ← | 176 | 68.2 | 0.544 | Hypothetical protein (E. coli) | AAC75715 | 1E − 47 | |
| C30 | 31267 | 31578 | ← | 104 | 61.9 | 0.611 | Transcriptional activator HlyU (V. cholerae) | AAF93843 | 6E − 16 | |
| C31 | 31728 | 32954 | → | 409 | 70.2 | 0.646 | Similar to metabolite transport protein (Bacillus subtilis) | CAB12326 | 2E − 34 | |
| C32 | 33031 | 33408 | → | 126 | 65.9 | 0.535 | Hypothetical protein Rv1767 (Mycobacterium tuberculosis) | CAB09310 | 3E − 22 | |
| C33 | 33519 | 33890 | → | 124 | 58.1 | 0.491 | No significant similarity | |||
| C34 | 33950 | 34744 | ← | 265 | 64.9 | 0.619 | fenO | Hydroxybutyryl dehydratase (B. subtilis) (probable enoyl coenzyme A hydratase/isomerase) | AAF32340 | 2E − 24/PICK> |
| C35 | 35156 | 36151 | → | 332 | 63.6 | 0.476 | Probable transcriptional regulator (PA1182) (P. aeruginosa) | AAG04571 | 2E − 42 | |
| C36a | 36199 | 38091 | ← | 631 | 65.0 | 0.489 | Hypothetical protein XF1753 (X. fastidiosa) | AAF84562 | 0E − 00 | |
| C37a | 38407 | 39033 | → | 209 | 65.6 | 0.603 | Conserved hypothetical protein XF1754 (X. fastidiosa) | AAF84563 | 1E − 105 | |
| C38a | 39046 | 39678 | → | 211 | 64.0 | 0.588 | Conserved hypothetical protein XF1755 (X. fastidiosa) | AAF84564 | 1E − 112 | |
| C39a | 39752 | 40111 | → | 120 | 65.6 | 0.466 | Hypothetical protein XF1756 (X. fastidiosa) | AAF84565 | 1E − 15 | |
| C40a | 40127 | 41674 | ← | 516 | 62.2 | 0.536 | No significant similarity | |||
| C41a | 41690 | 42046 | ← | 119 | 70.0 | 0.611 | No significant similarity | |||
| C42a | 42043 | 43437 | ← | 465 | 67.5 | 0.611 | No significant similarity | |||
| C43a | 43447 | 44397 | ← | 317 | 66.7 | 0.689 | No significant similarity | |||
| C44a | 44394 | 44840 | ← | 149 | 68.0 | 0.512 | No significant similarity | |||
| C45a | 45005 | 45499 | ← | 165 | 63.0 | 0.516 | radC | DNA repair protein (XF0148) (X. fastidiosa) | AAF82961 | 9E − 34 |
| C46a | 45675 | 46439 | ← | 255 | 67.5 | 0.508 | Hypothetical protein PA0982 (P. aeruginosa) | AAG04371 | 4E − 28 | |
| C47a | 46464 | 49295 | ← | 944 | 66.6 | 0.606 | Low homology at the N terminus to sex pilus assembly and synthesis protein (Sphingomonas aromaticivorans); origin of replication binding domain | AAD03958 | 1E − 07 | |
| C48a | 49356 | 49796 | ← | 147 | 68.9 | 0.704 | No significant similarity | |||
| C49a | 49777 | 51195 | ← | 473 | 68.7 | 0.672 | No significant similarity | |||
| C50a | 51185 | 52096 | ← | 304 | 71.5 | 0.631 | No significant similarity | |||
| C51a | 52093 | 52785 | ← | 231 | 68.1 | 0.659 | No significant similarity | |||
| C52a | 52782 | 53180 | ← | 133 | 69.9 | 0.618 | No significant similarity | |||
| C53a | 53193 | 53552 | ← | 120 | 66.7 | 0.733 | No significant similarity | |||
| C54a | 53569 | 53802 | ← | 78 | 64.5 | 0.628 | No significant similarity | |||
| C55a | 53799 | 54182 | ← | 128 | 72.4 | 0.599 | No significant similarity | |||
| C56 | 54386 | 54856 | → | 157 | 59.7 | 0.442 | Putative excisionase ORF277 (S. aromaticivorans plasmid pNL1) | AAD03880 | 2E − 16 | |
| C57 | 54853 | 55428 | → | 192 | 61.1 | 0.467 | Hypothetical protein ORF271 (S. aromaticivorans plasmid pNL1) | AAD03879 | 2E − 23 | |
| C58 | 55446 | 56360 | → | 305 | 59.1 | 0.405 | CG11743 gene product (Drosophila melanogaster) | AAF54250 | 7E − 26 | |
| C59 | 56357 | 56827 | → | 157 | 61.4 | 0.407 | No significant similarity | |||
| C60 | 56824 | 57324 | → | 167 | 56.7 | 0.449 | No significant similarity | |||
| C61 | 57324 | 58226 | → | 301 | 58.7 | 0.536 | No significant similarity | |||
| C62 | 58031 | 58990 | → | 320 | 60.4 | 0.430 | No significant similarity | |||
| C63 | 59000 | 61624 | → | 875 | 66.7 | 0.534 | No significant similarity | |||
| C64a | 61665 | 62414 | ← | 250 | 66.0 | 0.649 | No significant similarity | |||
| C65a | 62411 | 64600 | ← | 730 | 65.6 | 0.612 | Hypothetical protein (Salmonella enterica serovar Typhi) | AAF69957 | 7E − 30 | |
| C66a | 64605 | 65153 | ← | 183 | 72.9 | 0.583 | No significant similarity | |||
| C67a | 65150 | 65740 | ← | 197 | 73.1 | 0.622 | Hypothetical protein RP457 (Rickettsia prowazekii) | CAA14913 | 4E − 12 | |
| C68a | 65722 | 66459 | ← | 246 | 70.6 | 0.684 | No significant similarity | |||
| C69a | 66472 | 67116 | ← | 215 | 71.0 | 0.616 | No significant similarity | |||
| C70a | 67113 | 67712 | ← | 200 | 68.8 | 0.514 | PilL (type IV pili) (Salmonella serovar Typhi) | AAF14812 | 3E − 19 | |
| C71a | 67851 | 70130 | ← | 760 | 64.9 | 0.615 | Hypothetical protein pXO1-08 (Bacillus anthracis virulence plasmid pXO1) (with helicase domain) | AAD32312 | 9E − 43 | |
| C72a | 70267 | 70572 | ← | 102 | 64.1 | 0.576 | No significant similarity | |||
| C73a | 70662 | 70982 | ← | 107 | 61.7 | 0.556 | No significant similarity | |||
| C74a | 71033 | 72142 | ← | 370 | 66.2 | 0.597 | Hypothetical protein pXO1-10 (B. anthracis virulence plasmid pXO1) | AAD32314 | 5E − 11 | |
| C75a | 72207 | 72854 | ← | 216 | 67.3 | 0.646 | No significant similarity | |||
| C76a | 72931 | 73191 | ← | 87 | 60.5 | 0.573 | Hypothetical protein XF1757 (X. fastidiosa) | AAF84566 | 1E − 39 | |
| C77a | 73208 | 73615 | ← | 136 | 65.7 | 0.577 | Hypothetical protein XF1758 (X. fastidiosa) | AAF84567 | 6E − 68 | |
| C78a | 73720 | 74061 | ← | 114 | 61.7 | 0.460 | Conserved plasmid protein XF1759 (X. fastidiosa) | AAF84568 | 3E − 50 | |
| C79a | 74156 | 74845 | ← | 230 | 67.2 | 0.566 | Hypothetical protein XF1760 (X. fastidiosa) | AAF84569 | 1E − 106 | |
| C80a | 74940 | 75767 | ← | 276 | 63.4 | 0.567 | Hypothetical protein ORF273 (oriT 5′ region) (E. coli plasmid F) | AAA99218 | 2E − 88 | |
| C81a | 75913 | 76911 | ← | 333 | 64.9 | 0.570 | Hypothetical protein XF1761 (X. fastidiosa) | AAF84570 | 1E − 156 | |
| C82a | 77129 | 77413 | ← | 95 | 75.1 | 0.638 | Conserved hypothetical protein XF1762 (X. fastidiosa) | AAF84571 | 2E − 41 | |
| C83a | 77721 | 77981 | ← | 87 | 69.0 | 0.570 | Hypothetical protein XF1764 (X. fastidiosa) | AAF84573 | 7E − 36 | |
| C84 | 78051 | 78692 | ← | 214 | 65.3 | 0.652 | tnp* | Transposase (P. fluorescens) | CAA70408 | 2E − 90 |
| C84b | 78533 | 79048 | ← | 172 | 66.3 | 0.458 | tnp* | TnpA transposase (Tn21) (E. coli) | AAC33926 | 6E − 51 |
| C85 | 79067 | 80755 | ← | 563 | 69.0 | 0.620 | merA | Mercuric [Hg(II)] reductase (Thiobacillus sp.) | CAA72398 | 0E + 00 |
| C86 | 80766 | 81053 | ← | 96 | 66.0 | 0.671 | merP | Periplasmic mercuric ion binding protein (Sphingomonas paucimobilis) | AAD23805 | 5E − 32 |
| C87 | 81066 | 81416 | → | 117 | 67.2 | 0.626 | merT | MerT protein (mercuric transport protein) (E. coli plasmid pDU1358) | AAA98222 | 2E − 55 |
| C88 | 81488 | 81895 | → | 136 | 63.5 | 0.517 | merR | Organomercurial resistance regulatory protein (P. stutzen) | AAC38229 | 9E − 52 |
| C89a | 82157 | 82981 | ← | 275 | 64.7 | 0.611 | No significant similarity | |||
| C90a | 83270 | 83548 | ← | 93 | 60.2 | 0.655 | No significant similarity | |||
| C91a | 83646 | 84383 | ← | 246 | 64.8 | 0.585 | No significant similarity | |||
| C92a | 84467 | 85204 | ← | 246 | 63.7 | 0.594 | No significant similarity | |||
| C93a | 85336 | 85728 | ← | 131 | 62.1 | 0.629 | Hypothetical protein XF1771 (X. fastidiosa) | AAF84580 | 2E − 66 | |
| C94a | 85750 | 86163 | ← | 138 | 63.5 | 0.442 | Hypothetical protein XF1772 (X. fastidiosa) | AAF84581 | 2E − 35 | |
| C95a | 86300 | 86611 | ← | 104 | 62.2 | 0.423 | Hypothetical protein XF1773 (X. fastidiosa) | AAF84582 | 1E − 17 | |
| C96 | 86948 | 87448 | ← | 167 | 61.5 | 0.444 | lspA | Lipoprotein signal peptidase LspA (Serratia marcescens) | AAC82524 | 1E − 32 |
| C97 | 87452 | 90364 | ← | 971 | 65.7 | 0.584 | Probable metal-transporting P-type ATPase (PA3690) (P. aeruginosa) | AAG07078 | 0E +00 | |
| C98 | 90456 | 90854 | → | 133 | 60.2 | 0.476 | Probable transcriptional regulator (PA3689) (P. aeruginosa) | AAG07077 | 5E − 37 | |
| C99 | 91309 | 91545 | ← | 79 | 61.2 | 0.496 | No significant similarity | |||
| C100 | 91930 | 92565 | → | 212 | 61.5 | 0.417 | Putative integral membrane protein/transporter (Neisseria meningitidis) | AAF42077 | 8E − 26 | |
| C101a | 93289 | 95319 | ← | 677 | 66.4 | 0.580 | topB | DNA topoisomerase III (XF1776) (X. fastidiosa) | AAF84584 | 0E + 00 |
| C102a | 95603 | 96043 | ← | 147 | 65.5 | 0.644 | ssb | Single-stranded-DNA binding protein (XF1778) (X. fastidiosa) | AAF84586 | 1E − 71 |
| C103a | 96117 | 96644 | ← | 176 | 64.2 | 0.533 | Hypothetical protein XF1779 (X. fastidiosa) | AAF84587 | 9E − 78 | |
| C104a | 96641 | 97432 | ← | 264 | 66.4 | 0.601 | Hypothetical protein XF1780 (X. fastidiosa) | AAF84588 | 1E − 123 | |
| C105a | 97862 | 99100 | ← | 413 | 67.7 | 0.513 | Hypothetical protein XF1781 (X. fastidiosa) | AAF84589 | 0E + 00 | |
| C106a | 99104 | 99664 | ← | 187 | 65.6 | 0.661 | Conserved hypothetical protein (XF1782) (X. fastidiosa) | AAF84590 | 1E − 96 | |
| C107a | 99679 | 101358 | ← | 560 | 69.9 | 0.590 | Protein fused from two hypothetical proteins (XF1783 and XF1784) (X. fastidiosa) | AAF84591, AAF84592 | 1E − 111, 1E − 117 | |
| C108a | 101604 | 102479 | ← | 292 | 68.7 | 0.551 | soj | Chromosome partitioning-related protein (XF1785) (X. fastidiosa) | AAF84593 | 1E − 150 |
| C109a | 102522 | 102743 | ← | 74 | 64.0 | 0.703 | Phage-related protein (XF1786) (X. fastidiosa) | AAF84594 | 9E − 35 | |
| C110a | 102853 | 103599 | ← | 249 | 57.6 | 0.402 | Hypothetical protein XF1787 (X. fastidiosa) | AAF84595 | 1E − 101 | |
| C111a | 104050 | 104550 | → | 167 | 55.7 | 0.399 | No significant similarity | |||
ORF defined as noncargo in the text (including the homologs).
TABLE 3.
Annotation of all ORFs located within the gene island PAGI-3(SG) in P. aeruginosa strain SG17M
| Gene identifi- cation | Coordinates
|
Direc- tion | Length (amino acids) | G+C (%) | GCI | Gene name | Homolog product | GenBank accession no. | E value (Blast search) | |
|---|---|---|---|---|---|---|---|---|---|---|
| Left | Right | |||||||||
| SG1a | 226 | 1635 | → | 470 | 61.1 | 0.458 | int | Phage-related integrase (XF1718) (X. fastidiosa) | AAF84527 | 1E − 178 |
| SG2 | 1909 | 2970 | ← | 354 | 56.0 | 0.352 | hemE | Uroporphyrinogen decarboxylase (E. coli K-12) | AAC76971 | 1E − 149 |
| SG3 | 3360 | 3815 | → | 152 | 55.7 | 0.350 | Conserved hypothetical protein (Paracoccus denitrificans) | AAC44549 | 2E − 14 | |
| SG4 | 4145 | 5131 | → | 329 | 46.1 | 0.215 | Methyl-accepting domain of probable chemotaxis transducer PA4844 (P. aeruginosa) | AAG08229 | 1E − 17 | |
| SG5 | 5201 | 5953 | ← | 251 | 46.1 | 0.205 | Domain of conserved hypothetical protein PA4601 (P. aeruginosa) | AAG07989 | 1E − 51 | |
| IS element | 6212 | 8612 | → | [2,401 bp] | IS with inverted repeats and two ORFs (transposase and nucleoside triphosphate-binding protein); upon insertion into ORF (5 + 8), a sequence of 7 bp (CCTTAGT) was repeated | No homology at the nucleotide level | ||||
| SG6 | 6318 | 7823 | → | 502 | 56.4 | 0.339 | istA | Transposase IstA (IS1326) | AAA79725 | 1E − 109 |
| SG7 | 7813 | 8556 | → | 248 | 55.4 | 0.370 | istB | Nucleoside triphosphate-binding protein IstB (IS1326) (Ralstonia eutropha) | AAA79726 | 9E − 72 |
| SG8 | 8596 | 9831 | ← | 412 | 53.2 | 0.255 | Conserved hypothetical protein ORF1 (Rhizobium etli) | AAC64871 | 9E − 30 | |
| SG5 + SG8 | 5201 | 9831 | ← | 740 | 50.6 | 0.232 | Conserved hypothetical protein PA4601 (P. aeruginosa) (after deletion of the IS element) | AAG07989 | 6E − 88 | |
| SG9 | 10249 | 11025 | ← | 259 | 53.9 | 0.363 | Conserved hypothetical protein Orf3 (Methylobacterium extorquens) | AAB66495 | 5E − 23 | |
| SG10 | 11025 | 12479 | ← | 485 | 55.3 | 0.296 | gabD | Succinate semialdehyde dehydrogenase (Pseudonocardia sp. strain K1) | CAC10505 | 5E − 56 |
| IS element | 13380 | 15209 | ← | [1,830 bp] | IS containing three ORFs (two fragments of a putative transposase and a hypothetical protein); no flanking repeats could be detected | No homology at the nucleotide level | ||||
| SG11 | 13380 | 14258 | ← | 293 | 58.4 | 0.382 | Similar to domain of conserved hypothetical protein (Wolbachia sp. strain wKue) (putative transposase) | BAA89629 | 8E − 49 | |
| SG12 | 14280 | 14723 | ← | 148 | 59.2 | 0.436 | Similar to domain of conserved hypothetical protein (Wolbachia sp. strain wKue) (putative transposase) | BAA89629 | 5E − 31 | |
| SG11 + SG12 | 13380 | 14723 | ← | 448 | 58.9 | 0.402 | Fusion of ORFs SG11 and SG12 (change of the stop codon TAG to TCG); full-length similarity to conserved hypothetical protein (Wolbachia sp. strain wKue) (putative transposase) | BAA89629 | 7E − 90 | |
| SG13 | 14892 | 15209 | ← | 106 | 58.8 | 0.353 | Conserved hypothetical protein PA0979 (P. aeruginosa); in other species often associated with IS elements | AAG05325 | 8E − 13 | |
| SG14 | 15612 | 16592 | → | 327 | 56.0 | 0.371 | yumC | Thioredoxin reductase (Bacillus halodurans) | BAB07127 | 2.E − 71 |
| SG15 | 16993 | 18375 | → | 461 | 51.4 | 0.326 | glnA4 | Putative glutamine-synthetase GlnA4 (Mycobacterium tuberculosis) | CAA15522 | 2E − 73 |
| SG16 | 18447 | 19607 | → | 387 | 51.8 | 0.320 | Cytochrome P450 (monooxygenase) (Rhizobium sp. strain NGR234) | AAB91895 | 2E − 45 | |
| SG17 | 19840 | 20724 | → | 295 | 50.3 | 0.251 | Vng2501c (Halobacterium sp. strain NRC-1) putative glutamine amidotransferase | AAG20565 | 1E − 11 | |
| SG18 | 20789 | 22219 | → | 477 | 48.6 | 0.241 | Putative amino acid permease [Streptomyces coelicolor A3(2)] | CAB46781 | 3E − 68 | |
| SG19 | 22330 | 23838 | → | 503 | 54.5 | 0.285 | Aldehyde dehydrogenase PA5312 (P. aeruginosa) | AAG08697 | 1E − 162 | |
| SG20 | 24412 | 25527 | → | 372 | 57.9 | 0.391 | Enoyl coenzyme A hydratase (P. putida) | AAB62303 | 1E − 120 | |
| SG21 | 25509 | 25970 | → | 154 | 58.2 | 0.391 | Acyl coenzyme A dehydrogenase (Bacillus subtilis) | CAB14346 | 1E − 12 | |
| SG22 | 26463 | 27677 | → | 405 | 67.7 | 0.656 | pntAA | Proton-translocating NAD(P) transhydrogenase, alpha subunit, PntAA (Rhodospirillum rubrum) | AAA62493 | 4E − 93 |
| SG23 | 27689 | 28006 | → | 106 | 62.6 | 0.756 | pntAB | Proton-translocating NAD(P) transhydrogenase, alpha2 subunit, PntAB (R. rubrum) | AAA62494 | 1E − 23 |
| SG24 | 28006 | 29469 | → | 488 | 64.9 | 0.744 | pntB | Pyridine nucleotide transhydrogenase, beta subunit, PA0196 (P. aeruginosa) | AAG03585 | 0E + 00 |
| SG25 | 29816 | 29914 | ← | 33 | 58.6 | 0.398 | Only fragment of transposase (Agrobacterium tumefaciens) | CAA79150 | 0.033 | |
| SG26 | 30368 | 30913 | ← | 182 | 50.4 | 0.274 | Transcriptional regulator, HTH_3 family (Vibrio cholerae) | AAF96189 | 1E − 16 | |
| SG27 | 31278 | 32030 | ← | 251 | 57.8 | 0.462 | Putative short-chain type dehydrogenase/reductase [S. coelicolor A3(2)] | CAA20822 | 9E − 38 | |
| SG28 | 32892 | 33644 | → | 251 | 57.2 | 0.353 | Probable glutamine amidotransferase PA0297 (P. aeruginosa) | AAG03686 | 1E − 51 | |
| SG29 | 33730 | 34770 | → | 347 | 57.9 | 0.383 | adh | Alcohol dehydrogenase PA5427 (P. aeruginosa) | AAG08812 | 7E − 45 |
| SG30 | 35076 | 36383 | → | 436 | 56.6 | 0.335 | Aminotransferase class III (adenosylmethionine-8-amino-7-oxononanoate) (B. halodurans) | BAB05979 | 2E − 94 | |
| SG31 | 36446 | 36931 | ← | 162 | 60.5 | 0.349 | Fragment of transposase-like protein TnpA1 (P. stutzeri) | AAD02143 | 3E − 28 | |
| SG32 | 37018 | 37281 | ← | 88 | 53.4 | 0.345 | Fragment of transposase-like protein TnpA1 (P. stutzeri) | AAD02143 | 9E − 08 | |
| SG33 | 37736 | 38716 | ← | 327 | 58.9 | 0.565 | tnp | Transposase (P. putida) | AAC98743 | 0E + 00 |
| SG34 | 39004 | 39186 | ← | 61 | 42.6 | 0.182 | No significant similarity | |||
| IS element | 39545 | 41180 | → | [1,636 bp] | IS with inverted repeats and two ORFs (transposase and hypothetical protein) | No homology at the nucleotide level | ||||
| SG35 | 39645 | 40109 | → | 155 | 63.9 | 0.545 | ORF within IS1240 (P. syringae) | AAB81643 | 7E − 35 | |
| SG36 | 40106 | 41155 | → | 350 | 67.0 | 0.642 | tnp | Transposase within IS1240 (P. syringae) | AAB81642 | 1E − 100 |
| SG37 | 41450 | 41629 | → | 60 | 38.3 | 0.181 | No significant similarity | |||
| SG38 | 41634 | 42404 | → | 257 | 49.8 | 0.318 | Conserved hypothetical protein (B. subtilis) | BAA19344 | 1E − 64 | |
| SG39 | 42455 | 42865 | → | 137 | 45.3 | 0.261 | No significant similarity | |||
| SG40 | 43008 | 44006 | ← | 333 | 50.4 | 0.258 | Probable transcriptional regulator (AraC family) PA3782 (P. aeruginosa) | AAG07169 | 2E − 87 | |
| SG41 | 44594 | 45082 | → | 163 | 57.5 | 0.476 | No significant similarity | |||
| SG42 | 45079 | 45471 | → | 131 | 60.3 | 0.404 | Monophosphatase (Synechocystis sp.) | BAA18648 | 2E − 08 | |
| SG43 | 45732 | 46247 | ← | 172 | 61.6 | 0.550 | Hypothetical protein jhp0584 (Helicobacter pylori strain J99) | AAD06175 | 1E − 34 | |
| SG44a | 46405 | 48207 | ← | 601 | 62.3 | 0.437 | Hypothetical protein XF1753 (X. fastidiosa) | AAF84562 | 0E + 00 | |
| SG45a | 48517 | 48834 | → | 106 | 61.7 | 0.416 | HtaR suppressor protein slr0724 (Synechocystis sp. strain PCC 6803) | BAA16671 | 4E − 06 | |
| SG46a | 48834 | 49292 | → | 153 | 61.7 | 0.502 | Conserved hypothetical protein slr0725 (Synechocystis sp. strain PCC 6803) | BAA16672 | 2E − 28 | |
| SG47a | 49322 | 49675 | → | 118 | 61.2 | 0.401 | Hypothetical protein XF1756 (X. fastidiosa) | AAF84565 | 6E − 03 | |
| SG48a | 49672 | 51174 | ← | 501 | 62.4 | 0.588 | No significant similarity | |||
| SG49a | 51187 | 51516 | ← | 110 | 70.3 | 0.700 | No significant similarity | |||
| SG50a | 51513 | 52916 | ← | 468 | 66.1 | 0.609 | No significant similarity | |||
| SG51a | 52925 | 53860 | ← | 312 | 66.1 | 0.638 | No significant similarity | |||
| SG52a | 53857 | 54294 | ← | 146 | 65.8 | 0.442 | No significant similarity | |||
| SG53a | 54459 | 54959 | ← | 167 | 60.7 | 0.496 | radC | Probable DNA repair protein RadC VC1786 (V. cholerae) | AAF94935 | 3E − 30 |
| SG54a | 55313 | 55684 | → | 124 | 61.8 | 0.451 | Hypothetical protein (similar to spdB3 gene in pSG5) (A. rhizogenes) | BAB16262 | 4E − 20 | |
| SG55a | 55748 | 55993 | → | 82 | 71.1 | 0.637 | No significant similarity | |||
| SG56a | 56002 | 56388 | ← | 129 | 62.8 | 0.484 | No significant similarity | |||
| SG57a | 56401 | 59268 | ← | 956 | 67.4 | 0.670 | Low homology at the N terminus to sex pilus assembly and synthesis protein (Sphingomonas aromaticivorans); origin of replication binding domain | AAD03958 | 4E − 10 | |
| SG58a | 59268 | 59687 | ← | 140 | 67.6 | 0.654 | No significant similarity | |||
| SG59a | 59668 | 61089 | ← | 474 | 66.5 | 0.608 | No significant similarity | |||
| SG60a | 61079 | 61954 | ← | 292 | 71.1 | 0.649 | No significant similarity | |||
| SG61a | 61951 | 62625 | ← | 225 | 67.3 | 0.658 | No significant similarity | |||
| SG62a | 62622 | 63017 | ← | 132 | 67.4 | 0.535 | No significant similarity | |||
| SG63a | 63034 | 63396 | ← | 121 | 67.2 | 0.666 | No significant similarity | |||
| SG64a | 63409 | 63642 | ← | 78 | 63.2 | 0.630 | No significant similarity | |||
| SG65a | 63639 | 64010 | ← | 124 | 67.7 | 0.443 | No significant similarity | |||
| SG66 | 64314 | 65819 | → | 502 | 62.0 | 0.512 | Domain of hypothetical protein ORF261 [S. aromaticivorans plasmid pNL1] | AAD03878 | 6E − 08 | |
| SG67a | 65838 | 66587 | ← | 250 | 64.7 | 0.641 | No significant similarity | |||
| SG68a | 66584 | 68758 | ← | 725 | 65.8 | 0.622 | Hypothetical protein (Salmonella enterica serovar Typhi) | AAF69957 | 1E − 31 | |
| SG69a | 68769 | 69296 | ← | 176 | 69.3 | 0.552 | No significant similarity | |||
| SG70a | 69293 | 69856 | ← | 188 | 70.9 | 0.574 | Hypothetical protein RP457 (Rickettsia prowazekii) | CAA14913 | 1E − 12 | |
| SG71a | 69856 | 70581 | ← | 242 | 70.0 | 0.571 | No significant similarity | |||
| SG72a | 70591 | 71244 | ← | 218 | 68.0 | 0.567 | No significant similarity | |||
| SG73a | 71241 | 71768 | ← | 176 | 68.8 | 0.471 | PilL (type IV pili) (Salmonella serovar Typhi) | AAF14812 | 7E − 20 | |
| SG74 | 72311 | 73741 | ← | 477 | 59.6 | 0.502 | Conserved hypothetical protein PA1368 (P. aeruginosa), putative transposase | AAG04757 | 0E + 00 | |
| SG75 | 73871 | 74323 | ← | 151 | 53.2 | 0.270 | No significant similarity | |||
| SG76 | 74592 | 75101 | → | 170 | 56.3 | 0.345 | Conserved hypothetical protein PA2582 (P. aeruginosa) | AAG05970 | 4E − 45 | |
| SG77 | 75509 | 76585 | → | 359 | 43.8 | 0.217 | No significant similarity | |||
| SG78 | 76585 | 77550 | → | 322 | 44.0 | 0.209 | Domain of conserved hypothetical protein (Deinococcus radiodurans) | AAF11191 | 3E − 12 | |
| SG79 | 77705 | 78451 | ← | 249 | 49.9 | 0.197 | Domain of hypothetical protein Y4jT (Rhizobium sp. strain NGR234) plasmid pNGR234a | AAB91732 | 2E − 23 | |
| SG80 | 78232 | 78501 | ← | 90 | Hypothetical ORF, no significant similarity | |||||
| SG81a | 78843 | 81116 | ← | 758 | 62.7 | 0.556 | Hypothetical protein pXO1-08 (Bacillus anthracis virulence plasmid pXO1) (with helicase domain) | AAD32312 | 3E − 42 | |
| SG82a | 81203 | 81499 | ← | 99 | 63.0 | 0.504 | No significant similarity | |||
| SG83a | 81718 | 82827 | ← | 370 | 64.2 | 0.557 | Hypothetical protein pXO1-10 (B. anthracis virulence plasmid pXO1) | AAD32314 | 2E − 09 | |
| SG84a | 82892 | 83548 | ← | 219 | 63.2 | 0.465 | No significant similarity | |||
| SG85a | 83683 | 84354 | ← | 224 | 64.3 | 0.466 | Hypothetical protein XF1760 (X. fastidiosa) | AAF84569 | 1E − 87 | |
| SG86a | 84444 | 85271 | ← | 276 | 62.8 | 0.590 | Hypothetical protein, ORF273 plasmid protein (E. coli K-12) | AAC75681 | 1E − 83 | |
| SG87a | 85460 | 86344 | ← | 295 | 61.8 | 0.510 | Hypothetical protein XF1761 (X. fastidiosa) | AAF84570 | 4E − 97 | |
| SG88a | 86670 | 86894 | → | 75 | 54.7 | 0.252 | No significant similarity | |||
| SG89a | 87095 | 87262 | ← | 56 | 61.3 | 0.398 | Hypothetical protein XF1764 (X. fastidiosa) | AAF84573 | 2E − 06 | |
| SG90a | 87280 | 88077 | ← | 266 | 60.3 | 0.481 | No significant similarity | |||
| SG91a | 88389 | 89102 | ← | 238 | 60.4 | 0.491 | No significant similarity | |||
| SG92a | 89199 | 89591 | ← | 131 | 60.3 | 0.443 | Hypothetical protein XF1771 (X. fastidiosa) | AAF84580 | 4E − 51 | |
| SG93a | 89615 | 90022 | ← | 136 | 66.7 | 0.408 | Hypothetical protein XF1772 (X. fastidiosa) | AAF84581 | 3E − 19 | |
| SG94 | 90168 | 91730 | ← | 521 | 60.1 | 0.452 | Domain of hypothetical protein ORF299 (Sphingomonas aromaticivorans plasmid pNL1) | AAD03882 | 3E − 08 | |
| SG95 | 92316 | 92435 | ← | 40 | 59.2 | 0.297 | No significant similarity | |||
| SG96a | 92619 | 94637 | ← | 673 | 66.5 | 0.573 | topB | DNA topoisomerase III (XF1776) (X. fastidiosa) | AAF84584 | 0E +00 |
| SG97a | 94881 | 95276 | ← | 132 | 62.6 | 0.569 | ssb | Single-stranded-DNA binding protein (XF1778) (X. fastidiosa) | AAF84586 | 3E − 50 |
| SG98a | 95273 | 95824 | ← | 184 | 63.4 | 0.460 | Hypothetical protein XF1779 (X. fastidiosa) | AAF84587 | 4E − 56 | |
| SG99a | 95821 | 96612 | ← | 264 | 66.3 | 0.617 | Hypothetical protein XF1780 (X. fastidiosa) | AAF84588 | 1E − 102 | |
| SG100a | 96782 | 97945 | ← | 388 | 64.1 | 0.483 | Hypothetical protein XF1781 (X. fastidiosa) | AAF84589 | 1E − 127 | |
| SG101a | 97950 | 98510 | ← | 187 | 61.0 | 0.537 | Conserved hypothetical protein (XF1782) (X. fastidiosa) | AAF84590 | 6E − 73 | |
| SG102a | 98529 | 100196 | ← | 556 | 62.5 | 0.471 | Protein fused from two hypothetical proteins (XF1783 and XF1784) (X. fastidiosa) | AAF84591 + AAF84592 | 3E − 62 + 1E − 100 | |
| SG103a | 100376 | 101239 | ← | 288 | 64.2 | 0.501 | soj | Chromosome partitioning-related protein (XF1785) (X. fastidiosa) | AAF84593 | 1E − 109 |
| SG104a | 101270 | 101488 | ← | 73 | 56.2 | 0.399 | Phage-related protein (XF1786) (X. fastidiosa) | AAF84594 | 3E −20 | |
| SG105a | 101939 | 102838 | ← | 300 | 57.6 | 0.384 | bphR | LysR-type regulatory protein BphR (Pseudomonas sp. strain KKS102) | BAA07613 | 1E −56 |
| SG106a | 102979 | 103197 | → | 73 | 57.5 | 0.269 | No significant similarity | |||
ORF defined as noncargo in the text (including the homologs).
TABLE 4.
Features of coding sequences within the strain-specific gene islands
| Categorya | No. of ORFs in:
|
|||||
|---|---|---|---|---|---|---|
| PAGI-2(C)
|
PAGI-3(SG)
|
|||||
| All | Cargob | Noncargoc (all/XF/SG17M) | All | Cargod | Noncargoc (all/XF/C) | |
| Strong homologs of genes with demonstrated function | 30 | 24 | 6/5/6 | 18 | 12 | 6/5/6 |
| Genes with proposed function based on motif searches or limited homology | 14 | 12 | 2/2/1 | 19 | 18 | 1/1/1 |
| Homologs of reported genes of unknown function | 36 | 7 | 29/21/19 | 36 | 13 | 22/12/19 |
| No homology to any reported sequences | 33 | 8 | 25/0/21 | 32 | 7 | 25/0/21 |
| Total | 113 | 51 | 62/28/47 | 105 | 51 | 54/18/47 |
Definitions are as for the PAO1 genome (48).
Cargo ORFs in strain C are C5 to C35, C56 to C63, C84 to C88, and C96 to C100.
All ORFs of the gene island except the cargo ORF. Subgroup XF, ORFs with homologs in the X. fastidiosa gene island, subgroup SG17M or C, ORFs with mutual homologs in SG17M and C, respectively. Compare with Fig. 2 for the exact gene identifications within the subgroups.
Cargo ORFs in strain SG17M are SG2 to SG43, SG66, SG74 to SG79, SG94 and SG95.
In both strains the gene islands are partitioned into two blocks (Fig. 2). The cluster adjacent to the attL site consists of genes that are specific for each strain. The encoded function could be attributed to most of these so-called strain-specific genes (termed cargo ORFs in Table 4). The other cluster predominantly contains hypothetical ORFs, of which 47 are mutual homologs in both gene islands. Of these 47 ORFs, 28 ORFs in strain C and 18 ORFs in strain SG17M have homologs in the tRNAGly-associated island of X. fastidiosa mentioned above (Table 4; Fig. 2). The putative function could be recognized for a few homologs (Tables 2 to 4). Three genes encode elements of DNA recombination or repair (ssb [single-strand binding protein], C102 and SG97 [accession number XF1778]; topB [topoisomerase B], C101 and SG96 [XF1776]; and radC [DNA repair protein], C45 and SG53). One gene product is associated with the partitioning of chromosomal or extrachromosomal elements in the cell (soj, C108 and SG103 [XF1785]), and another gene product is associated with site-specific integration into the chromosome (int [phage-type P4 integrase], C1 and SG1 [XF1718]) (see above). Additionally, a few conserved hypothetical genes show strong homology to already identified plasmid (C71 and SG81, C74 and SG83, C78, and C80 and SG86) or phage (C109 and SG104) genes.
The cargo ORFs, of which 51 each were found in PAGI-2(C) and PAGI-3(SG), build up the individual part of the gene island. Of these 102 ORFs, the closest homolog identified from BLAST searches was frequently found in other P. aeruginosa strains [12 in PAGI-2(C) and 10 in PAGI-3(SG)]; in other type I pseudomonads, such as P. fluorescens, P. syringae, P. putida, or P. stutzeri [3 in PAGI-2(C) and 6 in PAGI-3(SG)]; or in “honorary” pseudomonads that had been removed from the Pseudomonas genus after introduction of the ribosomal DNA-based phylogeny [3 in PAGI-2(C) and 1 in PAGI-3(SG)]. Hence, a substantial portion of the genes have homologs in other pseudomonads.
The cargo genes endow the strains with some extra metabolic features and transport and resistance capacities (Tables 2 and 3). PAGI-3(SG) of the environmental isolate SG17M is a metabolic island of complex architecture that encodes a broad variety of enzymes, the majority of which are encoded by single genes. The strain-specific portion of PAGI-3(SG) contains genes related to the metabolism and transport of amino acids (SG15, SG17, SG18, and SG28), coenzymes (SG22 to SG24), and porphyrins (SG2), and other putative enzymes (SG10, SG14, SG16,. SG19, SG20, SG21, SG27, SG29, SG30, and SG42). Various small transposable elements such as insertion sequences (ISs) are integrated into this part of the gene island, sometimes disrupting the encoded genes (e.g., ORFs SG5 and SG8 in Table 3). Future functional studies will determine to what extent this set of enzymes strengthens the metabolic versatility of strain SG17M.
The cargo genes of PAGI-2(C) encode proteins for the complexation and transport of heavy metal ions. Gene clusters encoding all nine essential proteins for the cytochrome c biogenesis system I (C11 to C18) and related thiol-disulfide exchange proteins (C8 to C10) could be identified. Additionally, proteins associated with the transport of cations (C22 and C97), a two-component regulatory system (C19 and C20), several transcriptional regulators (C30, C35, and C98), a transposon conferring mercuric resistance (C84 to C88), and several other transporters are located on PAGI-2(C). Strain C is a disease isolate from the airways of a patient with CF. The expression of the genes for cytochrome c biogenesis encoded by PAGI-2(C) could facilitate iron uptake and inactivation of peroxides (10) and thus may confer an advantage for the bacteria to persist in the CF lung, where they are exposed to iron limitation and oxidative stress (13, 32). However, it is not obvious why the presence of a copper homeostasis protein (C21) or a mercuric resistance operon (C84 to C88) could be of advantage for survival in the CF host. These genes should be highly relevant in an environment with high concentration of heavy metal ions. A copy of PAGI-2(C) was identified in the unfinished sequence of the R. metallidurans CH34 genome. The R. metallidurans island is also integrated into a tRNAGly gene and differs from PAGI-2(C) by only 29 nucleotide substitutions in a stretch of 105,049 bp (PAO coordinates 3173676 to 3173597) (Fig. 2). R. metallidurans flourishes in millimolar concentrations of toxic heavy metals, and all cargo genes of PAGI-2(C) can add to the bacterial fitness against heavy metal stress.
Comparison of gene islands.
Table 5 displays the distribution of G+C contents and GCI values in PAGI-2(C), PAGI-3(SG), and the small clone C-specific segment compared to those in the PAO1 genome. Whereas the G+C content of most noncargo genes with their many mutual homologs comes quite close to typical values of the GC-rich P. aeruginosa, the strain-specific cargo genes are less GC rich, which is more pronounced in PAGI-3(SG) than in PAGI-2(C). The plot of the GC content in Fig. 3, with its broad range and numerous shifts, visually shows this mosaicism between cargo and noncargo genes. As indicated by their low GCI values, the codon usages of the majority of PAGI-3(SG) and PAGI-2(C) genes are significantly different from those in the PAO1 genome. The P. aeruginosa PAO1 genes are characterized by consistently high GCI values, which do not vary with the chromosomal localization of the respective gene (21). The only exceptions are 15 islands that carry five or more consecutive genes with low GCI values (21). Hence, we conclude that PAGI-2(C) and PAGI-3(SG), with their more than 100 genes, represent a very large island with atypical codon usage in P. aeruginosa C, where the cargo genes are more atypical in their codon usage than the noncargo genes and PAGI-3(SG) is more atypical than PAGI-2(C).
TABLE 5.
Distribution of G+C contents and GCI values of PAGI-2(C) and PAGI-3(SG) compared to those in the PAO1 genome
| Genomic region | ORFsa (n) | G+C content (%)
|
GCI
|
||
|---|---|---|---|---|---|
| Avg | Median (inner quartiles; range) | Avg | Median (inner quartiles; range) | ||
| PAGI-2(C) | All (113) | 64.6 | 64.8 (61.9-66.7; 55.7-75.1) | 0.537 | 0.541 (0.460-0.611; 0.280-0.733) |
| Cargo (51) | 63.2 | 63.2 (61.1-65.4; 56.7-70.2) | 0.495 | 0.476 (0.434-0.537; 0.354-0.671) | |
| Noncargo (62) | 65.8 | 65.6 (63.7-68.1; 55.7-75.1) | 0.573 | 0.589 (0.534-0.627; 0.280-0.733) | |
| PAGI-3(SG) | All (105) | 59.8 | 61.0 (56.2-64.3; 38.3-71.1) | 0.448 | 0.452 (0.349-0.557; 0.181-0.756) |
| Cargo (51) | 55.2 | 56.3 (50.9-59.0; 38.3-67.6) | 0.371 | 0.350 (0.272-0.420; 0.181-0.756) | |
| Noncargo (54) | 64.2 | 63.8 (61.7-66.7; 54.7-71.1) | 0.521 | 0.507 (0.459-0.590; 0.252-0.700) | |
| Clone C DNA | All (9) | 65.4 | 65.4 (63.3-66.5; 62.6-70.0) | 0.645 | 0.639 (0.629-0.667; 0.539-0.724) |
| PAO genome | All (5,570) | 66.7 | 67.3 (64.9-69.3; 29.9-76.2) | 0.678 | 0.697 (0.638-0.741; 0.139-0.896) |
For definitions of subgroups, see Table 2, footnote a.
FIG. 3.

Comparison of the strain-specific gene islands in P. aeruginosa SG17M (upper line) and C (lower line). Genes are represented by arrows as in Fig. 2. Homologous ORFs are linked by light blue bars. A slightly darker blue line connects the corresponding bphR genes located at the right border of the SG17M gene island and at the left border of the C-specific insertion. Genes with homologs in the X. fastidiosa gene island are highlighted with a dark blue background. Gray boxes above and below the gene maps mark all ORFs that are presumably associated with the mobilization and transfer of the gene islands (called noncargo ORFs in the text; compare with Tables 2 and 3 for the corresponding gene identification numbers). Additionally, a 500-bp sliding window plot of G+C content is displayed for each gene island.
The homologous proteins in the gene islands of strain C, strain SG17M, R. metallidurans, and X. fastidiosa exhibit high levels of amino acid identity and similarity. The pairwise comparison revealed the highest values between the corresponding genes of strain C, R. metallidurans, and X. fastidiosa. The average amino acid identity between C and R. metallidurans was 100%, that between C and X. fastidiosa was 79.8%, that between C and SG17M was 64.8%, and that between SG17M and X. fastidiosa was 62.6%. In other words, the homologs of strain C are more related to those in the gene islands of phylogenetically unrelated species than to those found in a member of the same P. aeruginosa clone. This statement is corroborated by the finding that the X. fastidiosa gene island shares 28 homologs with PAGI-2(G) but only 18 homologs with PAGI-3(SG) (Table 4).
The order of the homologs is conserved in PAGI-2(C) and PAGI-3(SG) for 46 of the 47 genes. The exception encodes the transcriptional regulator BphR (C4 and SG105). The gene contig, however, is disrupted several times by the insertion of strain-specific ORFs (Fig. 3).
PAGI-2(C) and PAGI-3(SG) are not the only gene islands that are known in P. aeruginosa. We have previously described 100-kb large gene islands that were derived from episomal plasmids and reversibly recombined with either of the two tRNALys genes of clone C and K chromosomes (19). The tRNALys- and tRNAGly-associated gene islands share P4-type int and homologous soj genes adjacent to the recombination breakpoints, but otherwise their genetic contents are different (unpublished data). Gene islands, however, are not necessarily inserted into tRNA genes. So far, two islands that are not integrated into a tRNA gene have been identified in P. aeruginosa. The first example is the 48.9-kb PAGI-1, which has been found in 85% of tested P. aeruginosa clinical isolates from sepsis and urinary tract infections and hence has been suggested to confer virulence traits (24). The other example is a ca. 16-kb large DNA segment in strain PAK that carries genes for the glycosylation of a-flagellin, among others (4).
PAGI-2(C) and PAGI-3(SG) have a bipartite structure: a set of strain-specific ORFs encoding metabolic functions and transporters and a set of conserved hypothetical genes and unknown genes, of which most genes are homologs with high sequence similarity. The conserved order of the homologs (many of which are also found in a tRNAGly-associated island in X. fastidiosa), the similar global structures of PAGI-2(C) and PAGI-3(SG), and the role of the few homologs with a recognized function in DNA recombination or repair (ssb, topB, and radC) are three striking features that point to important and conserved roles of the large cassette of homologous genes. We hypothesize that besides the int and soj genes, at least some of the homologs are responsible for the mobilization, transfer, and stabilization of the island (Fig. 3). In other words, genes of the cassette of conserved homologs should mediate lateral gene transfer, whereas the other half of the island would represent the individual cargo that endows the recipient with strain-specific metabolic properties. The forthcoming genome projects will resolve whether or not this peculiar type of gene island with its mosaic structure of individual cargo and of conserved homologs is obligatorily associated with tRNAGly genes. These potentially transmissible islands seem to be rather common among metabolically versatile proteobacteria that initially had been classified as pseudomonads by physiology-oriented taxonomists. We have preliminary evidence from ongoing Southern and in silico analyses that homologs of PAGI-2 or PAGI-3 or conserved ORFs thereof exist not only in R. metallidurans CH34 and X. fastidiosa but also in other P. aeruginosa strains, type I pseudomonads, and Burkholderia spp.
Acknowledgments
We cordially thank C. Weinel and C. Kiewitz for support in computer-assisted calculations and sequence analysis. We are indebted to U. Bode, M. Bömeke, S. Schlenczek, S. Steckel, and I. Kovolik for their expert technical assistance in sequencing.
Financial support by the Deutsche Forschungsgemeinschaft (Tu 40/5-1, 5-2) is gratefully acknowledged. K.D.L. has been a recipient of a postgraduate stipend and J.K. is a recipient of a graduate stipend of the European Graduate College (“Pseudomonas: Pathogenicity and Biotechnology”).
REFERENCES
- 1.Alonso, A., F. Rojo, and J. L. Martínez. 1999. Environmental and clinical isolates of Pseudomonas aeruginosa show pathogenic and biodegradative properties irrespective of their origin. Environ. Microbiol. 1:421-430. [DOI] [PubMed] [Google Scholar]
- 2.Altschul, S. F., T. L. Madden, A. A. Schäffer, J. Zhang, Z. Zhang, W. Miller, and D. J. Lipman. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arber, W. 2000. Genetic variation: molecular mechanisms and impact on microbial evolution. FEMS Microbiol. Rev. 24:1-7. [DOI] [PubMed] [Google Scholar]
- 4.Arora, S. K., M. Bangera, S. Lory, and R. Ramphal. 2001. A genomic island in Pseudomonas aeruginosa carries the determinants of flagellin glycosylation. Proc. Natl. Acad. Sci. USA 98: 9342-9347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ausubel, F. M., R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidmann, J. A. Smith, and K. Struhl (ed.). 1994. Current protocols in molecular biology. Wiley, New York, N.Y.
- 6.Besemer, J., and M. Borodovsky. 1999. Heuristic approach to deriving models for gene finding. Nucleic Acids Res. 27:3911-3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de la Cruz, F., and J. Davies. 2000. Horizontal gene transfer and the origin of species: lessons from bacteria. Trends Microbiol. 8:128-133. [DOI] [PubMed] [Google Scholar]
- 8.Ewing, B., L. Hillier, M. Wendl, and P. Green. 1998. Base-calling of automated sequencer traces using Phred. I. Accuracy assessment. Genome Res. 8:175-185. [DOI] [PubMed] [Google Scholar]
- 9.Foght, J. M., D. W. Westlake, W. M. Johnson, and H. F. Ridgway. 1996. Environmental gasoline-utilizing isolates and clinical isolates of Pseudomonas aeruginosa are taxonomically indistinguishable by chemotaxonomic and molecular techniques. Microbiology 142:2333-2340. [DOI] [PubMed] [Google Scholar]
- 10.Gaballa, A., C. Baysse, N. Koedam, S. Muyldermans, and P. Cornelis. 1998. Different residues in periplasmic domains of the CcmC inner membrane protein of Pseudomonas fluorescens ATCC 17400 are critical for cytochrome c biogenesis and pyoverdine-mediated iron uptake. Mol. Microbiol. 30:547-555. [DOI] [PubMed] [Google Scholar]
- 11.Goldberg, J. B., and D. E. Ohman. 1984. Cloning and expression in Pseudomonas aeruginosa of a gene involved in the production of alginate. J. Bacteriol. 158:1115-1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gordon, D., C. Abajian, and P. Green. 1998. Consed: a graphical tool for sequence finishing. Genome Res. 8:195-202. [DOI] [PubMed] [Google Scholar]
- 13.Govan, J. R., and V. Deretic. 1996. Microbial pathogenesis in cystic fibrosis: mucoid Pseudomonas aeruginosa and Burkholderia cepacia. Microbiol. Rev. 60:539-574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hacker, J., and J. B. Kaper (ed.). 2002. Pathogenicity islands and the evolution of pathogenic microbes. Curr. Top. Microbiol. Immunol. 264/I:1-211. [Google Scholar]
- 15.Heuer, T., C. Bürger, G. Maaβ, and B. Tümmler. 1998. Cloning of prokaryotic genomes in yeast artificial chromosomes: application to the population genetics of Pseudomonas aeruginosa. Electrophoresis 19:486-494. [DOI] [PubMed] [Google Scholar]
- 16.Heuer, T., C. Bürger, and B. Tümmler. 1998. Smith/Birnstiel mapping of genome rearrangments in Pseudomonas aeruginosa. Electrophoresis 19:495-499. [DOI] [PubMed] [Google Scholar]
- 17.Hoheisel, J. D., E. Maier, R. Mott, and H. Lehrach. 1996. Integrated genome mapping by hybridization techniques, p. 319-346. In B. Birren and E. Lai (ed.) Nonmammalian genomic analysis: a practical guide. Academic Press., San Diego, Calif.
- 18.Karlin, S. 2001. Detecting anomalous gene clusters and pathogenicity islands in diverse bacterial genomes. Trends Microbiol. 9:335-343. [DOI] [PubMed] [Google Scholar]
- 19.Kiewitz, C., K. Larbig, J. Klockgether, C. Weinel, and B. Tümmler. 2000. Monitoring genome evolution ex vivo: reversible chromosomal integration of a 106 kb plasmid at two tRNALys gene loci in sequential Pseudomonas aeruginosa airway isolates. Microbiology 146:2365-2373. [DOI] [PubMed] [Google Scholar]
- 20.Kiewitz, C., and B. Tümmler. 2000. Sequence diversity of Pseudomonas aeruginosa: impact on population structure and genome evolution. J. Bacteriol. 182:3125-3135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kiewitz, C., C. Weinel, and B. Tümmler. 2002. Genome codon index of Pseudomonas aeruginosa, a codon index that utilizes whole genome sequence data. Genome Lett. 1:61-70. [Google Scholar]
- 22.Kolmar, H., E. Ferrando, F. Hennecke, J. Wippler, and H. J. Fritz. 1992. General mutagenesis/gene expression procedure for the construction of variant immunoglobulin domains in Escherichia coli. J. Mol. Biol. 228:359-365. [DOI] [PubMed] [Google Scholar]
- 23.Lan, R., and P. R. Reeves. 2000. Intraspecies variation in bacterial genomes: the need for a species concept. Trends Microbiol. 8:396-401. [DOI] [PubMed] [Google Scholar]
- 24.Liang, X., X.-Q. T. Pham, M. V. Olson, and S. Lory. 2001. Identification of a genomic island present in the majority of pathogenic isolates of Pseudomonas aeruginosa. J. Bacteriol. 183:843-853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lowe, T. M., and S. R. Eddy. 1997. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25:955-964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lukashin, A., and M. Borodovsky. 1998. GeneMark.hmm: new solutions for gene finding. Nucleic Acids Res. 26:1107-1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mokross, H., E. Schmidt, and W. Reineke. 1990. Degradation of 3-chlorobiphenyl by in vivo constructed hybrid pseudomonads. FEMS Microbiol. Lett. 59:179-185. [DOI] [PubMed] [Google Scholar]
- 28.Ochman, H., J. G. Lawrence, and E. A. Groisman. 2000. Lateral gene transfer and the nature of bacterial innovation. Nature 405:299-304. [DOI] [PubMed] [Google Scholar]
- 29.Oefner, P. J., S. P. Hunicke-Smith, L. Chiang, F. Dietrich, J. Mulligan, and R. W. Davis. 1996. Efficient random subcloning of DNA sheared in a recirculating point-sink flow system. Nucleic Acids Res. 24:3879-3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ravatn, R., S. Studer, D. Springael, A. J. B. Zehnder, and J. R. van der Meer. 1998. Chromosomal integration, tandem amplification, and deamplification in Pseudomonas putida F1 of a 105-kilobase genetic element containing the chlorocatechol degradative genes from Pseudomonas sp. strain B13. J. Bacteriol. 180:4360-4369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ravatn, R., S. Studer, A. J. B. Zehnder, and J. R. van der Meer. 1998. Int-B13, an unusual site-specific recombinase of the bacteriophage P4 integrase family, is responsible for chromosomal insertion of the 105-kilobase clc element of Pseudomonas sp. strain B13. J. Bacteriol. 180:5505-5514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Regelmann, W. E., C. M. Siefferman, J. M. Herron, G. R. Elliott, C. C. G. Clawson, and B. H. Gray. 1995. Sputum peroxidase activity correlates with the severity of lung disease in cystic fibrosis. Pediatr. Pulmonol. 19:1-9. [DOI] [PubMed] [Google Scholar]
- 33.Römling, U., J. Greipel, and B. Tümmler. 1995. Gradient of genomic diversity in the Pseudomonas aeruginosa chromosome. Mol. Microbiol. 17:323-332. [DOI] [PubMed] [Google Scholar]
- 34.Römling, U., T. Heuer, and B. Tümmler. 1994. Bacterial genome analysis by pulsed field gel electrophoresis techniques. Adv. Electrophoresis 7:353-406. [Google Scholar]
- 35.Römling, U., K. D. Schmidt, and B. Tümmler. 1997. Large genome rearrangements discovered by the detailed analysis of 21 Pseudomonas aeruginosa clone C isolates found in environment and disease habitats. J. Mol. Biol. 271:386-404. [DOI] [PubMed] [Google Scholar]
- 36.Römling, U., and B. Tümmler. 1992. Comparative mapping of the Pseudomonas aeruginosa PAO genome with rare-cutter linking clones or two-dimensional pulsed-field gel electrophoresis protocols. Electrophoresis 14:283-289. [DOI] [PubMed] [Google Scholar]
- 37.Römling, U., J. Wingender, H. Müller, and B. Tümmler. 1994. A major Pseudomonas aeruginosa clone common to patients and aquatic habitats. Appl. Environ. Microbiol. 60:1734-1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sabath, C. D. (ed.). 1980. Pseudomonas aeruginosa: the organism, diseases it causes, and their treatment. Hans Huber Publishers, Berne, Switzerland.
- 39.Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Plainview, N.Y.
- 40.Schmidt, K. D., T. Schmidt-Rose, U. Römling, and B. Tümmler. 1998. Differential genome analysis of bacteria by genomic subtractive hybridization and pulsed-field gel electrophoresis. Electrophoresis 19:509-514. [DOI] [PubMed] [Google Scholar]
- 41.Schmidt, K. D., B. Tümmler, and U. Römling. 1996. Comparative mapping of Pseudomonas aeruginosa PAO with P. aeruginosa C, which belongs to a major clone in cystic fibrosis patients and aquatic habitats. J. Bacteriol. 178:85-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Semsey, S., B. Blaha, K. Köles, L. Orosz, and P. P. Papp. 2002. Site-specific integrative elements of rhizobiophage 16-3 can integrate into proline tRNA (CGG) genes in different bacterial genera. J. Bacteriol. 184:177-182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sharp, P. M., and W.-H. Li. 1987. The codon adaptation index—a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 15:1281-1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Simpson, A. J., F. C. Reinach, P. Arruda, F. A. Abreu, M. Acencio, R. Alvarenga, L. M. Alves, J. E. Araya, G. S. Baia, C. S. Baptista, M. H. Barros, E. D. Bonaccorsi, S. Bordin, J. M. Bove, M. R. S. Briones, M. R. P. Bueno, A. A. Camargo, L. E. A. Camargo, D. M. Carraro, H. Carrer, N. B. Colauto, C. Colombo, F. F. Costa, M. C. R. Costa, C. M. Costa-Neto, L. L. Coutinho, M. Cristofani, E. Dias-Neto, C. Docena, H. El-Dorry, A. P. Facincani, A. J. S. Ferreira, V. C. A. Ferreira, J. A. Ferro, J. S. Fraga, S. C. Franca, M. C. Franco, M. Frohme, L. R. Furlan, M. Garnier, G. H. Goldman, M. H. S. Goldman, S. L. Gomes, A. Gruber, P. L. Ho, J. D. Hoheisel, M. L. Junqueira, E. L. Kemper, J. P. Kitajima, J. E. Krieger, E. E. Kuramae, F. Laigret, M. R. Lambais, L. C. C. Leite, E. G. M. Lemos, M. V. F. Lemos, S. A. Lopes, C. R. Lopes, J. A. Machado, M. A. Machado, A. M. B. N. Madeira, H. M. F. Madeira, C. L. Marino, M. V. Marques, E. A. L. Martins, E. M. F. Martins, A. Y. Matsukuma, C. F. M. Menck, E. C. Miracca, C. Y. Miyaki, C. B. Monteiro-Vitorello, D. H. Moon, M. A. Nagai, A. L. T. O. Nascimento, L. E. S. Netto, A. Nhani, Jr., F. G. Nobrega, L. R. Nunes, M. A. Oliveira, M. C. de Oliveira, R. C. de Oliveira, D. A. Plamieri, A. Paris, B. R. Peixoto, G. A. G. Pereira, H. A. Pereira, Jr., J. B. Pesquero, R. B. Quaggio, P. G. Roberto, V. Rodrigues, A. J. de M. Rosa, V. E. de Rosa, Jr., R. G. de Sa, R. V. Santelli, H. E. Sawasaki, A. C. R. de Silva, F. R. de Silva, W. A. Silva, Jr., J. F. de Silveira, M. L. Z. Silvestri, W. J. Sequeira, A. A. de Souza, A. P. de Souza, M. F. Terenzi, D. Truffi, S. M. Tsai, M. H. Tsuhako, H. Vallada, M. A. van Sluys, S. Verjovski-Almeida, A. L. Vettore, M. A. Zago, M. Zatz, J. Meidanis, and J. C. Setubal. 2000. The genome sequence of the plant pathogen Xylella fastidiosa. Nature 406:151-157. [DOI] [PubMed] [Google Scholar]
- 45.Springael, D., K. Peys, A. Ryngaert, S. V. Roy, L. Hooyberghs, R. Ravatn, M. Heyndrickx, J. R. Meer, C. Vandecasteele, M. Mergeay, and L. Diels. 2002. Community shifts in a seeded 3-chlorobenzoate degrading membrane biofilm reactor: indications for involvement of in situ horizontal transfer of the clc-element from inoculum to contaminant bacteria. Environ. Microbiol. 4:70-80. [DOI] [PubMed] [Google Scholar]
- 46.Staden, R., K. F. Beal, and J. K. Bonfield. 2000. The Staden package. Methods Mol. Biol. 132:115-130. [DOI] [PubMed] [Google Scholar]
- 47.Staskawicz, B., D. Dahlbeck, N. Keen, and C. Napoli. 1987. Molecular characterization of cloned avirulence genes from race 0 and race 1 of Pseudomonas syringae pv. glycinea. J. Bacteriol. 169:5789-5794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stover, C. K., X. Q. Pham, A. L. Erwin, S. D. Mizoguchi, P. Warrener, M. J. Hickey, F. S. Brinkman, W. O. Hufnagle, D. J. Kowalik, M. Lagrou, R. L. Garber, L. Goltry, E. Tolentino, S. Westbrock-Wadman, Y. Yuan, L. L. Brody, S. N. Coulter, K. R. Folger, A. Kas, K. Larbig, R. Lim, K. Smith, D. Spencer, G. K. Wong, Z. Wu, I. T. Paulsen, J. Reizer, M. H. Saier, R. E. W. Hancock, S. Lory, and M. V. Olson. 2000. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 406:959-964. [DOI] [PubMed] [Google Scholar]
- 49.Thiem, S. M., M. L. Krumme, R. L. Smith, and J. M. Tiedje. 1994. Use of molecular techniques to evaluate the survival of a microorganism injected into an aquifer. Appl. Environ. Microbiol. 60:1059-1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thompson, J. D., D. G. Higgins, and T. J. Gibson. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, positions-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673-4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Weinel, C., B. Tümmler, H. Hilbert, K. E. Nelson, and C. Kiewitz. 2001. General method of rapid Smith/Birnstiel mapping adds for gap closure in shotgun microbial genome sequencing projects: application to Pseudomonas putida KT2440. Nucleic Acids Res. 29:E110.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wenzel, R., and R. Herrmann. 1996. Cosmid cloning with small genomes, p. 197-222. In B. Birren and E. Lai (ed.), Nonmammalian genomic analysis: a practical guide. Academic Press, San Diego, Calif.
- 53.Zhou, J. Z., and J. M. Tiedje. 1995. Gene transfer from a bacterium injected into an aquifer to an indigenous bacterium. Mol. Ecol. 4:613-618. [DOI] [PubMed] [Google Scholar]