

Abstract
Secondary metabolites are a diverse set of plant compounds believed to have numerous functions in plant–environment interactions. The large chemical diversity of secondary metabolites undoubtedly arises from an equally diverse set of enzymes responsible for their biosynthesis. However, little is known about the evolution of enzymes involved in secondary metabolism. We are studying the biosynthesis of glucosinolates, a large group of secondary metabolites, in Arabidopsis to investigate the evolution of enzymes involved in secondary metabolism. Arabidopsis contains natural variations in the presence of methylsulfinylalkyl, alkenyl, and hydroxyalkyl glucosinolates. In this article, we report the identification of genes encoding two 2-oxoglutarate–dependent dioxygenases that are responsible for this variation. These genes, AOP2 and AOP3, which map to the same position on chromosome IV, result from an apparent gene duplication and control the conversion of methylsulfinylalkyl glucosinolate to either the alkenyl or the hydroxyalkyl form. By heterologous expression in Escherichia and the correlation of gene expression patterns to the glucosinolate phenotype, we show that AOP2 catalyzes the conversion of methylsulfinylalkyl glucosinolates to alkenyl glucosinolates. Conversely, AOP3 directs the formation of hydroxyalkyl glucosinolates from methylsulfinylalkyl glucosinolates. No ecotype coexpressed both genes. Furthermore, the absence of functional AOP2 and AOP3 leads to the accumulation of the precursor methylsulfinylalkyl glucosinolates. A third member of this gene family, AOP1, is present in at least two forms and found in all ecotypes examined. However, its catalytic role is still uncertain.
INTRODUCTION
The sessile nature of plants forces them to cope directly with environmental stresses, which include insect herbivory, pathogen attack, UV light radiation, and drought. Secondary metabolites are believed to be an important mechanism that allows plants to respond to these environmental challenges. There are more than 100,000 known plant secondary metabolites, which probably represent less than 10% of the actual total in nature (Wink, 1988). A large fraction of this diversity is derived from differential modification of common backbone structures, which requires the evolution of numerous enzymes with different product specificities. There are several mechanisms by which this can occur. First, as is the case with terpene synthases, a single protein can produce numerous products from a single substrate (Steele et al., 1998). A few amino acid substitutions can alter the ratio of products generated by a terpene synthase (Back and Chappell, 1996). The second mechanism is the use of gene duplication. In this model, a gene encoding an enzyme is duplicated, allowing one copy to evolve a new product profile while the other copy maintains its original function (Lynch and Force, 2000). To examine the mechanisms responsible for the evolution of diversity in plant secondary metabolites, we are studying glucosinolate biosynthesis in Arabidopsis.
The largest known group of secondary metabolites in Arabidopsis is the glucosinolates, with more than 30 different structures described (Hogge et al., 1988; D. Kliebenstein and T. Mitchell-Olds, unpublished data). In Arabidopsis, glucosinolates are biosynthesized from a variety of typical protein amino acids (methionine, tryptophan, and phenylalanine) and their chain-elongated analogs. The first step in glucosinolate biosynthesis is the oxidation of the amino group to an oxime moiety by amino acid–specific cytochrome P450 monooxygenases (Du et al., 1995; Wittstock and Halkier, 2000). After a thiohydroximate intermediate is formed, sequential transfer of glucose and sulfate residues creates the basic glucosinolate skeleton (Halkier and Du, 1997). The initially formed glucosinolate can undergo a variety of subsequent transformations that modify the side chain. For example, the C4 dihomomethionine-derived glucosinolate backbone can be modified into seven different glucosinolates in Arabidopsis (Figure 1). Because most of these modifications are sequential, each step probably is catalyzed by a different enzyme that likely had to evolve independently.
Figure 1.

Side Chain Modifications of Methionine-Derived Glucosinolates in Arabidopsis.
Potential side chain modifications for the elongated methionine derivative C4 dihomomethionine are shown. Steps with natural variation in Arabidopsis are shown in boldface to the right or left of each enzymatic arrow with the name of the corresponding locus.
When tissue disruption brings the glucosinolate into contact with myrosinase, a thioglucosidase, numerous compounds (isothiocyanates, nitriles, epithiocyanates, and thiocyanates) with diverse biological activities are formed. The glucosinolates and their derivatives can act as feeding deterrents or attractants for different insect species (Giamoustaris and Mithen, 1995). In addition, they can have nutritional effects that range from being goitrogenic to potential anticarcinogens (Faulkner et al., 1998). This assortment of biological activities associated with glucosinolates varies with the chemical nature of the glucosinolate side chain. For example, if 4-methylthiobutyl glucosinolate is oxidized, it generates the anticarcinogenic 4-methylsulfinylbutyl glucosinolate (Figure 1). However, if this product undergoes two additional reactions to generate the 2-hydroxy-3-butenyl glucosinolate, the product is goitrogenic without significant anticancer activity (Faulkner et al., 1998). Thus, it will be advantageous to manipulate glucosinolate structures to alter the nutritional and economic value of a food crop. This variation in glucosinolate structure and corresponding activity provides a useful system in which to investigate both the evolutionary and biological aspects of secondary metabolism.
In this article, we report the molecular basis of the natural variations for the presence of the GS-ALK (alkenyl producing) and GS-OHP (hydroxyalkyl producing) reactions in Arabidopsis (Figure 1). We have identified a single region that controls both reactions. In this region, there was a small gene family of 2-oxoglutarate–dependent dioxygenases that result from a gene duplication that apparently led to the evolution of two different enzymatic reactions. We present data showing that one member of this gene family controls the GS-ALK reaction and a second member controls the GS-OHP reaction. Importantly, natural variations at these genes predict glucosinolate concentration and type in Arabidopsis ecotypes.
RESULTS
Cosegregation of GS-ALK, GS-OHP, and GS-AOPnull
Previous reports found variations within Arabidopsis and within the genera Thlaspi and Malcolmia, also in the family Brassicaceae, for the presence of alkenyl, methylsulfinylalkyl, and hydroxyalkyl glucosinolates (Figure 1). These differences have been ascribed to the GS-ALK and GS-OHP loci (Giamoustaris and Mithen, 1996), which are alleles of the same locus or tightly linked loci. Both the GS-ALK and GS-OHP variants map to the top of chromosome IV; thus, we refer to them jointly as GS-AOP (Mithen et al., 1995). To map the GS-AOP region, we used 300 lines of the Landsberg erecta (Ler) × Columbia (Col) recombinant inbred lines (RILs) and 162 lines of the Ler × Cape Verde Islands (Cvi) RILs (Lister and Dean, 1993; Alonso-Blanco et al., 1998b). Ler contains the GS-OHP product 3-hydroxypropyl glucosinolate as the predominant short-chain aliphatic glucosinolate, and Cvi contains the GS-ALK products allyl glucosinolate and 3-butenyl glucosinolate (Figure 2). The Col ecotype accumulates the 4-methylsulfinylbutyl glucosinolate, which is the presumed substrate of the enzyme activities represented by GS-OHP and GS-ALK, suggesting that this ecotype is null for both GS-OHP and GS-ALK (GS-AOPnull; Figure 2). In addition to the variations at GS-AOP, Ler, Col, and Cvi have variations at the previously described GS-Elong locus (J. Kroymann and T. Mitchell-Olds, unpublished data). Variation at GS-Elong leads to the production of aliphatic glucosinolates with either a three-carbon-atom (Ler) or a four-carbon-atom (Cvi and Col) side chain.
Figure 2.

HPLC Results of Ler, Cvi, and Col Glucosinolate Profiles.
Shown are HPLC results monitored at 229 nm of samples from Ler, Cvi, and Col prepared from equal amounts of 2-week-old rosette tissue. The major methionine-derived glucosinolate peaks are labeled for each ecotype. The large peak at ∼18.5 min in Ler and Col is a tryptophan-derived glucosinolate. The large peak at ∼2.5 min is due to solvent mixing from the injected sample. mAu, milliabsorbance units.
Of the 300 Ler × Col RILs, 152 lines produced principally C3 glucosinolates, with 79 accumulating 3-hydroxypropyl glucosinolate, indicative of GS-OHP, and 73 containing the GS-AOPnull 3-methylsulfinylpropyl glucosinolate. All of the 148 Ler × Col RILs with predominantly C4 glucosinolates accumulated 4-methylsulfinylbutyl glucosinolate, with no lines containing 4-hydroxybutyl glucosinolate instead of the 1:1 mix of 4-methylsulfinylbutyl to 4-hydroxybutyl glucosinolate, as expected. This finding suggests that the enzyme encoded by the GS-OHP locus is unable to catalyze the conversion of 4-methylsulfinylbutyl glucosinolate to 4-hydroxybutyl glucosinolate and has specificity for substrates with side chains of three carbon atoms (Giamoustaris and Mithen, 1996). This further supports the theory that methylsulfinylalkyl accumulation is indicative of a nonfunctional GS-AOP variant. In addition to containing mostly C4 glucosinolates, many of these 148 RILs also contained minor levels of C3 glucosinolates, making it possible to score GS-OHP. Analysis of C3 glucosinolates showed that 65 lines contained 3-methylsulfinylalkyl and 58 lines accumulated 3-hydroxypropyl. The other 25 lines contained no detectable C3 glucosinolates; therefore, we were unable to score them for GS-OHP. In total, 138 of the Ler × Col RILs scored as GS-AOPnull and 137 scored as GS-OHP. This 1:1 segregation further supports the theory that GS-OHP and GS-AOPnull are two alleles of the same gene or are in close proximity in the genome.
To test the linkage of the GS-ALK and GS-OHP variants in Arabidopsis, we analyzed the 162 Ler × Cvi RILs. Like Ler × Col, this population segregated for different alleles of GS-AOP and GS-Elong. In lines that generated C3 glucosinolates, 42 produced 3-hydroxypropyl glucosinolate and 24 produced allyl glucosinolate. In the C4 glucosinolate–producing lines, 72 produced butenyl and allyl glucosinolates, and 28 produced primarily 4-methylsulfinylbutyl glucosinolate, with minor amounts of 3-hydroxypropyl glucosinolate. This finding is in agreement with the previous observation that GS-OHP is C3 specific and unable to convert the 4-methylsulfinylbutyl to 4-hydroxybutyl glucosinolate. In contrast, GS-ALK was able to convert at least the C3 and C4 methylsulfinylalkyl glucosinolates to alkenyl glucosinolates. In total, 96 of the Ler × Cvi RILs scored as GS-ALK and 70 lines scored as GS-OHP. This nearly 1:1 segregation pattern and the lack of recombinant individuals containing both the GS-OHP and GS-ALK phenotypes suggest that they are alleles of the same locus or map within close proximity. Furthermore, the GS-AOP region in the Ler × Cvi recombinant inbred population had a significant effect on the accumulation of aliphatic glucosinolates such that the GS-ALK lines had threefold higher glucosinolate levels than did the GS-OHP lines (P < 0.00001; 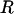 2
2  ). This suggests that GS-AOP is a major determinant of glucosinolate concentration in these lines. Together, the two populations of RILs suggest that the methylsulfinyl, hydroxyl, and alkenyl GS-AOP variants are alleles of the same locus or within a small genetic region in Arabidopsis, because there was no recombination between the variants in 462 lines. Furthermore, we were unable to identify any recombinant progeny in 5000 F2 progeny from a Ler × Col cross. If these variants represent alleles at two different loci, they are very tightly linked.
). This suggests that GS-AOP is a major determinant of glucosinolate concentration in these lines. Together, the two populations of RILs suggest that the methylsulfinyl, hydroxyl, and alkenyl GS-AOP variants are alleles of the same locus or within a small genetic region in Arabidopsis, because there was no recombination between the variants in 462 lines. Furthermore, we were unable to identify any recombinant progeny in 5000 F2 progeny from a Ler × Col cross. If these variants represent alleles at two different loci, they are very tightly linked.
Fine-Scale Mapping of GS-AOP
Comparison of markers mapped previously in the Ler × Col recombinant inbred population with the GS-AOP phenotype placed GS-ALK and GS-AOPnull at the top of chromosome IV, as described previously (Mithen et al., 1995). As shown in Figure 3, we fine-scale mapped GS-AOP to the region between microsatellite markers T5J8 and F4C21. This placed GS-AOP on three possible bacterial artificial chromosomes (BACs): F4C21, T4I9, and T5J8. Analysis of the Ler × Cvi RILs amplified fragment length polymorphism (AFLP) map also placed GS-ALK and GS-OHP at the top of chromosome IV (5-centimorgan centromeric from GS.250C and 17- centimorgan telomeric of CH.169.C; Alonso-Blanco et al., 1998b). Fine-scale mapping in this cross placed GS-AOP on the same BACs as those identified in the Ler × Col cross (Figure 3). In this region, the Col sequence from BAC T4I9 contains a small family of candidate genes. We identified three putative functional and one pseudo 2-oxoglutarate–dependent dioxygenase (2-ODD) genes (Figure 3). 2-ODDs are non-heme-iron–containing dioxygenases that typically use molecular oxygen, 2-oxoglutarate, ascorbate, and ferrous ions to catalyze substrate hydroxylations and other oxidations (Prescott, 1993). The reactions catalyzed by the GS-OHP and GS-ALK gene products, the conversion of methylsulfinylalkyl glucosinolates to alkenyl or hydroxypropyl glucosinolates, could plausibly be catalyzed by 2-ODDS. We have named these genes AOP1, AOP2, and AOP3 (Figure 3). The three AOP genes appear to result from a gene duplication event, because a BlastN search on GenBank produced no other homologs of significant similarity. Amino acid comparison showed that the three genes are ∼60 to 70% similar to each other and ∼25% similar to other 2-ODD genes (data not shown). The pseudogene is a partial open reading frame that contains several frameshift mutations. The presence of a small gene family in a restricted region could explain why two different enzymatic activities map to the same region.
Figure 3.

Fine-Scale Map of GS-AOP.
A fine-scale genomic map of GS-AOP is shown for reference. Horizontal solid lines represent the four BACs used, and the vertical lines represent the positions of the microsatellites on each BAC. The number to the right of each recombinant inbred population name is the number of recombinations between the given marker and the GS-AOP phenotype. The vertical black bar represents the genomic location of the three 2-ODD genes. The three 2-ODD genes and one related 2-ODD pseudogene are labeled AOP1, AOP2, AOP3, and Pseu, respectively. The F9H3 and T5J8 microsatellite markers are separated by ∼300 kb. The dotted line indicates the region between the two microsatellite markers for the Ler × Cvi cross (top line) or the Ler × Col cross (bottom line).
Differential Expression of AOP2 and AOP3 Is Correlated with GS-ALK and GS-OHP
To investigate the association of the AOP genes with the already documented variation in glucosinolate phenotype, we developed primers that specifically recognize the corresponding full-length cDNAs for each gene. These primers first were tested on genomic DNA from Ler, Col, and Cvi and shown by gel analysis and sequencing to amplify the expected fragment from each ecotype (data not shown). This demonstrates that the primers will recognize the corresponding cDNA from each ecotype. We then used these primers for quantitative reverse transcription–polymerase chain reaction (RT-PCR) detection of the different cDNAs. As shown in Figure 4, the AOP1 and Ran (control) genes are expressed at similar levels in all three ecotypes. However, we detected AOP3 cDNA only in the GS-OHP–containing Ler and not in the GS-ALK–containing Cvi or GS-AOPnull–containing Col (Figure 4). This finding suggests that AOP3 may be responsible for the GS-OHP reaction. In contrast with AOP3, AOP2 expression was detected only in the GS-ALK Cvi ecotype and the GS-AOPnull Col ecotype, with Cvi containing higher levels than Col (Figure 4). Sequencing of the AOP2 cDNA from Cvi and Col showed that the Cvi sequence contains a full-length 2-ODD, whereas the Col cDNA contains a 5-bp deletion that leads to a nonfunctional protein (data not shown). Thus, functional AOP2 is present only in the GS-ALK–containing ecotype. Furthermore, this agrees with the observation that the GS-AOPnull allele in Col is a null allele of both GS-ALK and GS-OHP, because Col does not express AOP3 or contain functional AOP2.
Figure 4.

Ecotype-Specific Expression of the AOP Genes.
Shown are the ethidium bromide–stained gels from a quantitative RT-PCR experiment with the Ler, Col, and Cvi parental ecotypes. The Ran gene was used as a loading control. Numbers above the gels (1, 0.3, and 0.1) indicate the amounts of cDNA (μL) used for the RT-PCRs.
Ecotype Association Study
To further test the relation between the AOP genes and natural variations in glucosinolate biosynthesis, we analyzed 21 Arabidopsis ecotypes for AOP gene expression and glucosinolate phenotype. As shown in Table 1, this analysis revealed a complete association between glucosinolate profiles and AOP gene expression. All ecotypes that expressed functional AOP2 accumulated the alkenyl glucosinolate and did not express AOP3 (Table 1). These observations support the hypothesis that AOP2 is responsible for GS-ALK. The hypothesis that AOP3 is responsible for the hydroxylation reaction is supported by the observation that AOP3 is expressed only in those ecotypes that produce the hydroxyalkyl glucosinolates (Table 1). Thus, expression of functional AOP3 is completely associated with the GS-OHP variant, and expression of functional AOP2 is completely associated with GS-ALK. Furthermore, both ecotypes that accumulate methylsulfinylalkyl glucosinolate, Col and Per-1, do not express AOP3 and contain the AOP2 copy that has a 5-bp frameshift mutation that destroys its function (Table 1). Therefore, the absence of AOP2 and AOP3 leads to the accumulation of the methylsulfinylalkyl precursor.
Table 1.
Genetic Association of GS-AOP Phenotype with AOP Expressiona
|
AOP2
|
||||||
|---|---|---|---|---|---|---|
| Ecotype | GS-AOP | Expb | Fxnlc | AOP3 Exp | AOP1.1 Exp | AOP1.2 Exp |
| Cal-0 | ALK | + | + | − | + | − |
| Cnt-1 | ALK | + | + | − | + | − |
| Kondara | ALK | + | + | − | + | − |
| Cvi | ALK | + | + | − | + | − |
| Ema-1 | ALK | + | + | − | + | − |
| Hodja | ALK | + | + | − | + | − |
| Kas-1 | ALK | + | + | − | + | − |
| Mrk-0 | ALK | + | + | − | + | − |
| Sorbo | ALK | + | + | − | + | − |
| Su-0 | ALK | + | + | − | + | − |
| Tac | ALK | + | + | − | + | − |
| Yo-0 | ALK | + | + | − | + | − |
| Per-1 | Null | + | − | − | + | − |
| Col | Null | + | − | − | + | − |
| Bl-1 | OHP | − | + | − | + | |
| Ka-0 | OHP | − | + | − | + | |
| Ler | OHP | − | + | − | + | |
| Lip-0 | OHP | − | + | − | + | |
| Pet | OHP | − | + | − | + | |
| Pi-0 | OHP | − | + | − | + | |
| Wei-0 | OHP | − | + | − | + | |
(+), the presence of mRNA expression and/or functional protein; (−), the absence of mRNA expression or nonfunctional protein.
Exp, expression.
The sequence contains no premature stop codons or frameshift mutations.
Biosynthetic Capacity of Heterologously Expressed AOP2 and AOP3
To test the hypothesis that AOP2 is responsible for the GS-ALK reaction and AOP3 is responsible for the GS-OHP reaction, we heterologously expressed thioredoxin fusion proteins for AOP2 and AOP3 in Escherichia. Each gene was cloned into the vector as a full-length cDNA by using the native stop codon, and the corresponding fusion protein was expressed in an arabinose-dependent manner. Crude extracts from induced and uninduced bacterial cultures were incubated with 3-methylsulfinylpropyl glucosinolate and 4-methylsulfinylbutyl glucosinolate to test the activity of the induced fusion proteins. As shown in Figure 5, induced AOP2 bacterial extracts catalyzed the conversion of the 3-methylsulfinylpropyl to the allyl glucosinolate and the 4-methylsulfinylbutyl to the 3-butenyl glucosinolate (cf. Figures 5A with 5B and 5C with 5D). The identities of the allyl and 3-butenyl products were confirmed by coincidence of retention time and UV light spectra with those of authentic standards and by liquid chromatography–mass spectrometry. The uninduced bacterial extract produced neither of these products (data not shown). The two peaks at 6.2 and 6.8 min are bacterial contaminants that copurified with the glucosinolates. This finding shows that AOP2 is capable of catalyzing the GS-ALK reaction with both the 3-methylsulfinylalkyl and 4-methylsulfinylalkyl glucosinolates.
Figure 5.

Enzymatic Activity of Heterologously Expressed AOP2.
HPLC results (monitored at 229 nm) of purified desulfoglucosinolates from bacterial extracts containing heterologously expressed AOP2 fusion proteins are shown. All compound identities were confirmed by comparison of both retention times and UV light absorption profiles with those of authentic standards. 3-MSO, 3-methylsulfinylalkyl glucosinolate; 4-MSO, 4-methylsulfinylalkyl glucosinolate.
(A) Extract from 3-methylsulfinylalkyl glucosinolate treated with uninduced AOP2 bacterial extract.
(B) Extract from 3-methylsulfinylalkyl glucosinolate treated with AOP2 enzyme.
(C) Extract from 4-methylsulfinylalkyl glucosinolate treated with uninduced AOP2 bacterial extract.
(D) Extract from 4-methylsulfinylalkyl glucosinolate treated with AOP2 enzyme.
In contrast with AOP2, the induced bacterial extracts with the AOP3 constructs displayed a weak conversion of 3-methylsulfinylpropyl glucosinolate to 3-hydroxypropyl glucosinolate, as determined by retention time and diode array spectral comparisons with purified 3-hydroxypropyl glucosinolate (Figure 6). Insufficient material was available to confirm the identity of 3-hydroxypropyl glucosinolate by liquid chromatography–mass spectrometry. We were unable to show any conversion of 4-methylsulfinylbutyl glucosinolate to 4-hydroxybutyl glucosinolate (data not shown). Analysis of the bacterial extracts by SDS-PAGE showed that the AOP3 fusion protein was insoluble, which could explain the very low activity of AOP3. These data support the idea that the natural variation in Arabidopsis for the production of hydroxyl and alkenyl glucosinolates can be explained by AOP2 encoding the GS-ALK enzyme and AOP3 controlling the GS-OHP reaction.
Figure 6.

Enzymatic Activity of AOP3 Heterologously Expressed in Escherichia coli.
E. coli extract was assayed with 3-methylsulfinylpropyl glucosinolate, and products were extracted and analyzed by HPLC as described in Methods.
(A) Sample of authentic 3-hydroxypropyl glucosinolate (monitored at 229 nm).
(B) Extract from AOP3 enzyme assay with 3-methylsulfinylpropyl glucosinolate as substrate (monitored at 229 nm).
(C) Diode array spectrum of standard 3-hydroxypropyl desulfoglucosinolate in (A).
(D) Diode array spectrum of 3-hydroxypropyl desulfoglucosinolate produced by AOP3 in (B).
Analysis of AOP Genes
To analyze the effect of gene duplication and independent evolution on secondary metabolism, we sequenced the AOP1, AOP2, and AOP3 genes from the Ler, Cvi, and Col ecotypes. The cDNA primers were used to amplify both genomic DNA and cDNA from all three ecotypes. The AOP2 cDNA from the GS-ALK–containing Cvi is 1296 nucleotides in length and encodes a protein of 432 amino acids. As mentioned above, the cDNA and genomic DNA for AOP2 in the GS-AOPnull–containing Col contains a 5-bp deletion at base 484, which generates a frameshift mutation and results in a severely truncated protein. Interestingly, the AOP2 cDNA sequence obtained from Ler genomic DNA shows that the open reading frame is intact and that the sequence is similar to that of Cvi (Figure 7). This finding suggests that the functional difference in phenotype between Ler and Cvi is due to an expression polymorphism and that if AOP2 were expressed in Ler it would accumulate alkenyl glucosinolates (Figure 4).
Figure 7.

Tree of AOP cDNAs from Ler, Cvi, and Col.
Cladogram of the relations of AOP1, AOP2, and AOP3 from the Ler, Cvi, and Col reference ecotypes. Either experimental cDNA sequences or predicted cDNA sequences from genomic sequences were used for the alignments shown. The scale was reduced between 0.02 and 0.16, and midpoint branching was used for this tree. Each sequence is listed with the ecotype from which it was obtained. AOP1 A. lyrata was obtained from A. lyrata.
We next analyzed variation in AOP3. The AOP3 cDNA from the hydroxyalkyl glucosinolate–forming Ler is 1230 nucleotides in length and encodes a 410–amino acid protein. AOP3 cDNA sequences obtained from both Col and Cvi genomic sequences show that these genes are highly related to the Ler AOP3 sequence. AOP1 cDNA has a sequence of 960 to 966 nucleotides that predicts a protein of 320 to 322 amino acids. The difference in length is due to variation in a GAT trinucleotide microsatellite within the AOP1 open reading frame. In addition, we obtained two different sequences from the Ler AOP1 genomic DNA. One sequence corresponded to the expressed AOP1 cDNA, whereas the other sequence was more closely related to AOP1 from Cvi and Col (Figure 7). This finding suggests that Ler contains a duplication of AOP1 and that the original AOP1 has somehow been silenced. We have named the original AOP1 gene AOP1.1 and the duplicated Ler copy AOP1.2. Mapping of AOP1.2 showed that the gene was tightly linked to the AOP region, but we were unable to obtain a precise genomic location (data not shown).
Interestingly, analysis of AOP1 showed that AOP1.1 is expressed only in the GS-ALK and GS-AOPnull ecotypes and that AOP1.2 is expressed only in GS-OHP lines (Table 1). The presence of functional AOP1.1 in the methylsulfinylalkyl lines suggests that AOP1 is not responsible for the formation of alkenyl glucosinolates. Furthermore, heterologously expressed AOP1.1 or AOP1.2 did not convert purified methylsulfinylalkyl or methylthioalkyl to either alkenyl or hydroxyalkyl glucosinolates (data not shown). In addition, the AOP1.1/AOP1.2 duplication is younger than the speciation event between Arabidopsis thaliana and A. lyrata (Figure 7). Thus, this duplication is much too young to explain the presence of GS-ALK/GS-OHP variation in Arabidopsis and the genera Thlaspi and Malcolmia, also in the family Brassicaceae (Daxenbichler et al., 1991). Consequently, AOP1 probably plays no role in GS-AOP variation. However, it is possible that AOP1 plays another role in the production of aliphatic glucosinolates. Additional work is required to determine if AOP1 has a role in aliphatic glucosinolate biosynthesis or if the relation with GS-AOP variation is due to evolutionary history.
DISCUSSION
AOP2 and AOP3 Control the Formation of Alkenyl and Hydroxyalkyl Glucosinolates
Arabidopsis contains natural variation for the production of methylsulfinylalkyl, hydroxyalkyl, and alkenyl glucosinolates. The locus controlling this variation mapped to the top of chromosome IV near a small gene family of 2-ODD genes, which could catalyze the conversion of methylsulfinylalkyl glucosinolate to hydroxyalkyl or alkenyl glucosinolate. Enzyme assays of heterologously expressed fusion protein showed that the AOP2 enzyme catalyzes the conversion of methylsulfinylalkyl glucosinolates to alkenyl glucosinolates and that the AOP3 enzyme catalyzes the formation of hydroxyalkyl glucosinolates (Figures 5 and 6). Analysis of the 2-ODD cDNAs from 21 ecotypes showed that expression of functional AOP2 is perfectly correlated with the products of the GS-ALK reaction and that expression of AOP3 is completely associated with the products of the GS-OHP reaction. This finding was supported further by the presence of a frameshift mutation in the Col and Per-1 AOP2 genes, which evidently caused a GS-AOPnull mutant and accumulation of the GS-AOP precursor, methylsulfinylalkyl glucosinolate. Thus, these experiments show that AOP2 is responsible for the GS-ALK reaction and that AOP3 is responsible for the GS-OHP reaction (Figure 8). In addition, the absence of either enzyme leads to the accumulation of the precursor methylsulfinylalkyl glucosinolate.
Figure 8.

Putative Enzymatic Reactions for the 2-ODD Genes in the GS-AOP Region.
The molecular structures and putative enzymatic reactions catalyzed by the AOP2 and AOP3 enzymes are shown. Side chains of up to eight carbon atoms (n 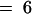 ) are known in Arabidopsis. Heterologous expression studies demonstrated the conversion of methylsulfinylalkyl glucosinolates to their alkenyl and hydroxyalkyl derivatives for three-carbon-atom side chains (propyl; n
) are known in Arabidopsis. Heterologous expression studies demonstrated the conversion of methylsulfinylalkyl glucosinolates to their alkenyl and hydroxyalkyl derivatives for three-carbon-atom side chains (propyl; n 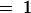 ). For glucosinolates with four-carbon-atom side chains (butyl; n
). For glucosinolates with four-carbon-atom side chains (butyl; n 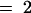 ), only the methylsulfinylalkyl-to-alkenyl conversion was demonstrated.
), only the methylsulfinylalkyl-to-alkenyl conversion was demonstrated.
Role of Gene Duplication in the Evolution of AOP2 and AOP3
BlastN searches with AOP1, AOP2, and AOP3 showed that they are their own closest homologs and that they appear to be the result of two different gene duplication events. The first event separated AOP1 from the AOP2/3 progenitor, and the second event led to the formation of AOP2 and AOP3. After the gene duplications, the separate copies were able to evolve new enzymatic functions. In this case, AOP2 evolved the GS-ALK function, AOP3 evolved the GS-OHP function, and AOP1 appears to have a third, unidentified function. The length of the branches linking AOP2 and AOP3 suggests that the duplication occurred long before the separation of A. lyrata from A. thaliana (Figure 7). This finding agrees with previous data showing that GS-ALK/GS-OHP variation occurs in the distantly related Arabidopsis and in the genera Thlaspi and Malcolmia (Daxenbichler et al., 1991). The identification of this gene duplication that apparently led to two different enzymatic reactions using the same precursor will allow us to study the biochemical and structural mechanisms leading to these different reactions. It will be interesting as well to identify the reaction catalyzed by AOP1, which should increase our understanding of the evolutionary change involved in generating the new product/substrate specificities present in AOP2 and AOP3.
Role of Amino Acid Evolution versus Promoter Mutation in the Evolution of GS-AOP
An unresolved question in evolutionary genetics concerns the importance of amino acid substitution versus promoter mutation in generating natural phenotypic variations. We found that the natural variation for the GS-ALK and GS-OHP reactions is due primarily to changes in the expression of the AOP2 and AOP3 genes (Figure 4). In the GS-ALK ecotypes, AOP2 is expressed but AOP3 is apparently silent. Conversely, in the GS-OHP ecotypes, AOP3 is expressed and AOP2 is silent (Table 2). Because the genomes of all ecotypes tested contain the genes for both AOP2 and AOP3 (data not shown), the major mechanism leading to the natural variation between GS-ALK and GS-OHP is promoter changes in the corresponding genes. Hence, analysis of the AOP2 and AOP3 promoter sequences in the different ecotypes will help elucidate the mechanisms that control the expression of these glucosinolate biosynthetic genes. In contrast with the GS-ALK versus GS-OHP variation, the observed variation between GS-AOPnull and GS-ALK/GS-OHP is the result of a 5-bp deletion that generates a truncated AOP2 open reading frame and presumably generates an inactive protein. Therefore, the natural variation between GS-ALK and GS-AOPnull is due to the functionality of the protein. Thus, both variation in promoters and protein function have strong effects on phenotypic variation in Arabidopsis glucosinolates.
Table 2.
AOP Primers
| Name | Repeat | Sequence | Ler | Col | Cvi |
|---|---|---|---|---|---|
| F9H3-F | TA | TTGCCAAAATTTCTGTAGCA | 126 | 130 | 122 |
| F9H3-R | CTCGACGACTTGTTGTTGGT | ||||
| T5J8-F1 | GAA | GCCAAGACGCAGAAGAAGAG | 125 | 150 | 225 |
| T5J8-R1 | TCTCATTATTCCCCACAATGC | ||||
| F4C21-F | TA | GCGCTTCATCTAGTTACGCTTT | 165 | 171 | 167 |
| F4C21-R | CCCGGACTGAACCAAACTAA | ||||
| T5J8-F2 | TA | CGATCATCGGTGTTCACCTT | 160 | 125 | 140 |
| T5J8-R2 | GAAAATAAATCGTCATATGGTGTACTG | ||||
| AOP1-F | ATGGATTCAGACTTTGTTCCT | ||||
| AOP1-R | AAAGGCAGCGAAAGCATGG | ||||
| AOP2-F | ATGGGTTCATGCAGTCTTCA | ||||
| AOP2-R | TGCTTCGGAGACGGCACAAT | ||||
| AOP3-F | ATGGGTTCATGCAGTCCTC | ||||
| AOP3-R | TTTCCCAGCAGAGACGCC |
GS-AOP Variation and Speciation
Published reports have shown that other Brassicaceae species contain variation for GS-ALK versus GS-OHP. For example, species within the genera Thlaspi and Malcolmia, both within the Brassicaceae, vary for the presence of the two reactions (Daxenbichler et al., 1991). Thus, the variation for the two different classes of glucosinolate is found in species that diverged more than 10 million years ago (Koch et al., 1999). One possible explanation for this wide variation in the GS-ALK and GS-OHP reactions is that AOP2 and AOP3 arose before these species diverged. The presence of one gene or the other within a given species may lead to the natural variation. This presents the possibility that the presence of an environmental pressure, such as heterogeneous selection, maintains the variation. The role of AOP2/3 in controlling this variation in other species will be tested by correlating the presence of AOP2 or AOP3 with the GS-ALK or GS-OHP reactions in numerous species.
GS-AOP Variation Controls the Level of Leaf Aliphatic Glucosinolates
Analysis of the Ler × Cvi recombinant inbred line studies showed that the GS-ALK variant increased leaf aliphatic glucosinolates up to threefold over either the GS-OHP or the GS-AOPnull variant (P < 0.00001; 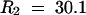 ). This shows that GS-AOP has a significant level of control over the total aliphatic glucosinolate concentration in the leaf. One possible explanation for this is that glucosinolate biosynthesis is regulated via a feedback mechanism in which the alkenyl glucosinolate is not sensed as well as the hydroxyalkyl or methylsulfinylalkyl glucosinolate is sensed. Thus, it may be possible to change glucosinolate concentrations in vegetables and forage crops merely by altering side chain modification. Future quantitative trait loci mapping studies will enable us to discover the mechanisms that control these differences.
). This shows that GS-AOP has a significant level of control over the total aliphatic glucosinolate concentration in the leaf. One possible explanation for this is that glucosinolate biosynthesis is regulated via a feedback mechanism in which the alkenyl glucosinolate is not sensed as well as the hydroxyalkyl or methylsulfinylalkyl glucosinolate is sensed. Thus, it may be possible to change glucosinolate concentrations in vegetables and forage crops merely by altering side chain modification. Future quantitative trait loci mapping studies will enable us to discover the mechanisms that control these differences.
Seed versus Leaf AOP Phenotype
Our data suggest that AOP2 and AOP3 control the decision between producing alkenyl and hydroxypropyl glucosinolates in the leaves of Arabidopsis. However, as has been described previously, the seeds of Col-0 contain 4-hydroxybutyl and 4-methylsulfinylbutyl glucosinolate (Hogge et al., 1988). Analysis of segregation in the Ler × Col-0 RILs suggests that the production of 4-hydroxybutyl glucosinolate in the seed is regulated by a locus other than GS-AOP (D.J. Kliebenstein and T. Mitchell-Olds, unpublished data). This locus could possibly encode one of the numerous other 2-ODDs present in the Arabidopsis genome. This second locus suggests that there may be multiple loci controlling similar side chain modifications in Arabidopsis. However, this does not diminish the observation that GS-AOP as a major determinant of glucosinolate side chain structure in Arabidopsis.
Future
The identification of these genes from Arabidopsis provides an opportunity to modify the composition of glucosinolate side chains in other species. For example, identification of the homologous genes in some brassicaceous food crops could allow modification of the nutritional aspects of these crops. Analysis of the evolution of the different enzymatic activities of AOP2 and AOP3 also will suggest a means to study the generation of novel enzymatic activities involved in secondary metabolism through gene duplication.
METHODS
Ninety-Six–Well Glucosinolate Extraction and Purification
The glucosinolate extraction and purification technique follows the basic sephadex/sulfatase Arabidopsis protocol previously described (Hogge et al., 1988). Samples were harvested into deep-well microtiter tubes (either 10 fresh leaf discs frozen in liquid nitrogen, 10 mg of freeze-dried leaf material, or 5 mg of dried seeds). Four 2.3-mm ball bearings were added, and the samples were ground into a fine powder in a paint shaker by high-speed agitation. To extract glucosinolates, we added 400 μL of methanol, 10 μL of 0.3 M lead acetate, and either 120 μL of water for seeds and freeze-dried material or 80 μL of water for fresh tissue. The samples were mixed for 1 min in the paint shaker and allowed to incubate for 60 min at 180 rpm on a rotary shaker. The tissue and protein were pelleted by centrifuging at 2500g for 10 min, and the supernatant was used for anion-exchange chromatography. Ninety-six–well filter plates from Millipore (model MAHVN4550) were loaded with 45 μL of DEAE Sephadex A-25 by using the Millipore multiscreen column loader. Then, 300 μL of water was added and allowed to equilibrate for 2 to 4 hr. After water was removed with 2 to 4 sec of vacuum on the Qiagen vacuum manifold, 150 μL of the supernatant was added to the 96-well columns, and the liquid was removed by 2 to 4 sec of vacuum. This step was repeated once to bring the total volume of plant extract to 300 μL. The columns were washed four times with 150 μL of 67% methanol, three times with 150 μL of water, and three times with 150 μL of 1 M sodium acetate. To desulfate glucosinolates on the column, we added 10 μL of water and 10 μL of sulfatase solution to each column, and the plates were incubated overnight at room temperature (Hogge et al., 1988). Desulfoglucosinolates were eluted by placing a deep-well 2-mL 96-well plate in the bottom of the 96-well vacuum manifold and aligning the 96-well column plate. The DEAE Sephadex then was washed twice with 100 μL of 60% methanol and twice with 100 μL of water.
HPLC
Forty microliters of the glucosinolate extract was run on a Hewlett-Packard Lichrocart 250-4 RP18e 5-μm column on a Hewlett-Packard 1100 series HPLC. Compounds were detected at 229 nm and separated using either of the two following programs with aqueous acetonitrile. For seeds, the program was an 8-min gradient from 1.5 to 5.0% acetonitrile, a 2-min gradient from 5 to 7% acetonitrile, a 32-min gradient from 7 to 52% acetonitrile, a 2-min gradient from 52 to 92% acetonitrile, 5 min at 92% acetonitrile, a 3-min gradient from 92 to 1.5% acetonitrile, and a final 8 min at 1.5% acetonitrile. For leaf material, the program was a 6-min gradient from 1.5 to 5.0% acetonitrile, a 2-min gradient from 5 to 7% acetonitrile, a 7-min gradient from 7 to 25% acetonitrile, a 2-min gradient from 25 to 92% acetonitrile, 6 min at 92% acetonitrile, a 1-min gradient from 92 to 1.5% acetonitrile, and a final 5 min at 1.5% acetonitrile.
Experimental Design
Five plants from each of the 162 Landsberg erecta (Ler) × Cape Verde Islands (Cvi) recombinant inbred lines (RILs) were planted in 4-inch pots with one Ler and one Cvi parental control pot per flat of 36 pots. The planting order was randomized for each of three experiments. The plants were grown for 3 weeks after planting under an 11-hr-light/13-hr-dark photoperiod, at which time 10 1.0-cm2 leaf discs were harvested for glucosinolate extraction. The plants were allowed to mature, and the seed from the five plants were pooled. This procedure was repeated for three independent replicates for each line and 15 replicates for each parent.
The Ler × Columbia (Col) RILs were given 3 days of cold treatment in the dark and then allowed to grow in a 16-hr-light/8-hr-dark photoperiod for 3 weeks. At 3 weeks, they were harvested and lyophilized. Glucosinolates were extracted from each leaf and seed sample three times, and the results are reported as the arithmetic mean of each ecotype or line.
Ninety-Six–Well DNA Extraction
Plants were grown for 3 weeks under a 9-hr-light/15-hr-dark photoperiod. At 3 weeks of age, one large leaf was harvested into a deep-well microtiter tube from Integra Bioscience (Fernwald, Germany). This procedure was repeated for 95 other samples until a full plate of 96 samples was obtained. The samples were frozen in liquid nitrogen and lyophilized overnight. Four 2.3-mm ball bearings from VandP Scientific (San Diego, CA) were added, and the samples were ground into a fine powder in a paint shaker. Three hundred fifty microliters of extraction buffer (200 mM Tris-HCl, pH 7.5, 250 mM NaCl, 25 mM EDTA, and 0.5% SDS) was added to each tube and incubated at 65°C for 1 hr. The tubes were removed and allowed to cool for at least 10 min. One hundred microliters of 5 M potassium acetate, pH 7.5, was added, and the samples were centrifuged for 15 min at 2500g to precipitate the protein and insoluble material. Two hundred twenty microliters of the supernatant was transferred to a 96-well microtiter plate (wells were 2 mL deep) that contained 220 μL of ice-cold isopropanol. The DNA was precipitated by centrifugation at 1800g for 15 min. The supernatant was removed by tipping the plate upside down, and the pellets were washed with 200 μL of 75% ethanol and centrifuged for 5 min at 1800g. The supernatant again was removed by tipping the plate upside down, and the plate was left overnight to dry at room temperature. Four hundred microliters of 100 mM Tris-HCl, pH 8.0, was added, and the pellet was dissolved for 10 min at 65°C. Forty microliters then was divided into aliquots on two to four microtiter plates, depending on the desired number of reactions. If the DNA was to be used for multiplex microsatellite reactions, it was reprecipitated and resuspended in one-tenth the initial volume of 100 mM Tris-HCl, pH 8.0.
Microsatellites
Previously published microsatellite primer sequences were obtained from the Arabidopsis Information Resource (www.arabidopsis.org). All other microsatellites were identified by scanning bacterial artificial chromosome (BAC) sequences obtained from the Arabidopsis Information Resource with RepeatMasker-Repetitive Element Filter (http://www.genome.washington.edu/UWGC/analysistools/repeatmask.htm; A.F.A. Smit and P. Green, unpublished data). The primers listed in Table 2 were used for fine-scale mapping. Five microliters of the resuspended 96-well DNA extraction DNA was added to 20 μL of polymerase chain reaction (PCR) mixture (2.5 mM MgCl2, 200 picomoles primers, and 0.5 units of Taq). These then were run with the following cycle program: 95°C for 3 min; 40 cycles of 95°C for 20 sec, 56°C for 20 sec, and 72°C for 1 sec; 72°C for 3 min; and 4°C final. The polymorphisms were scored on 4% agarose. Mapping was done using Mapmaker version 3 (Lander et al., 1987).
Quantitative Reverse Transcription–PCR and cDNA Sequencing
Total RNA was isolated from ∼100 mg of leaf tissue using the Trizol reagent. Approximately 1 μg of total RNA was used for cDNA synthesis, as described (Frohman et al., 1988). For quantitative reverse transcription (RT)–PCR, 0.1, 0.3, or 1 μL of the resulting total cDNA was added to 25-μL PCRs with the gene-specific primers listed in Table 2. The resulting product was visualized on a 1.5% agarose gel stained with ethidium bromide. For cDNA sequencing, 1 to 8 μL of the resulting total cDNA then was added to 25-μL PCRs with the gene-specific primers listed in Table 2 to amplify the corresponding specific cDNA. The resulting product then was separated on a 1.5% agarose gel, the band was removed, and the cDNA was purified with Qiagen (Valencia, CA) gel purification columns. The resulting cDNA was sequenced with the primers listed in Table 2 by using Perkin-Elmer Big Dye terminator chemistry on a Perkin-Elmer 310 sequencer. The sequences then were analyzed with the DNAstar analysis package (DNAstar, Madison, WI). Trees were generated using TREECON for Windows version 1.3b with 100 reiterations (Van de Peer and De Wachter, 1994).
Generation of Thioredoxin Fusion Proteins and Enzyme Assays
The cDNA primers listed in Table 2 were used to amplify the full-length cDNAs from AOP1.1, AOP1.2, AOP2, and AOP3. These cDNAs were used with the pBAD/Thio-E Echo Cloning System from Invitrogen (Carlsbad, CA). This places the cDNAs in a fusion protein with thioredoxin under the control of an arabinose-induced promoter. After cloning, the inserted cDNA was sequenced to verify the junctions and to verify that there had been no mutations due to PCR. The proteins were induced as described by the manufacturer (Invitrogen). The bacteria were pelleted and resuspended in 1 M Tris-HCl, pH 7.5, and lysed by five repetitive freeze–thaw cycles. Four hundred microliters of the crude lysate was used in reactions with the purified intact glucosinolate, as described, with the addition of 200 mM sucrose and the use of 10 mM oxoglutarate and 10 mM ascorbate instead of 40 mM of each of these (Hedden, 1991). The reactions were allowed to proceed for 4 hr at 28°C, and the glucosinolates then were purified and analyzed by HPLC or HPLC–mass spectrometry. We attempted to solubilize and/or stabilize the AOP3 fusion protein with 20% sorbitol, 10% glycerol, Triton X-100 (0.01, 0.05, or 0.1%), and Tween 20 (0.01, 0.05, or 0.1%). These were attempted individually and in all possible combinations.
Preparation of Intact Glucosinolates
Extracts were made using methods adapted from previously published protocols (Thies, 1979; Zrybko et al., 1997). Lyophilized samples (1 g) were immersed in boiling deionized water (40 mL) containing 100 μL of 0.3 M Pb(OAc)2, and 0.3 M Ba(OAc)2 solution for 10 min. After 20 min of gentle shaking at room temperature, samples were centrifuged at 4000g for 10 min. The supernatant fraction (extract) was cleaned by modification of the procedure of Thies (1988). The extract was loaded onto a DEAE Sephadex A25 column (1 g dry weight). The column was rinsed with 5 mL of water followed by 2 × 5 mL of formic acid:isopropanol:water (3:2:5 [v/v/v]) and 4 × 5 mL of water. Glucosinolates were eluted with 25 mL of 0.5 M K2SO4/3% isopropanol dropped into 25 mL of ethanol. After centrifugation (4000g for 10 min), the supernatant was evaporated, and the residue was transferred with 4 × 3 mL of methanol into a centrifuge tube. It was stored for 15 min at −20°C and centrifuged (4000g for 10 min), and the supernatant was evaporated. The residue was transferred with 2 × 0.5 mL of water into autosampler vials.
Separation of intact glucosinolates was achieved on a Hewlett-Packard 1100 series system with autosampler, column oven, and diode array detector as described (Zrybko et al., 1997) with some modifications. The procedure used a C-18, fully endcapped, reverse-phase column (LiChrospher RP-18; 250 × 4.6 mm i.d., 5-μm particle size; Chrompack, Raritan, NJ) operated at 1 mL/min and 25°C. Injection volume was 30 μL. Elution was accomplished with a gradient (solvent A, 0.075% triethylamine and 0.05% trifluoroacetic acid in water; solvent B, methylcyanide) of 1% solvent B (6-min hold) and 1 to 30% solvent B (24 min) followed by a cleaning cycle (30 to 95% solvent B in 1 min, 3-min hold, 95 to 1% solvent B in 1 min, 10-min hold). Eluting compounds were monitored at 229 nm and collected with a fraction collector. The fractions containing single intact glucosinolates were evaporated nearly to dryness and reconstituted with 2 × 0.5 mL of 1 M Tris-HCl, pH 7.5.
Mass Spectrometry
Analysis of glucosinolates was performed on a Quattro II (Micromass Ltd. Altrincham, Cheshire, UK) tandem quadrupole mass spectrometer equipped with an atmospheric pressure chemical ionization source. The corona pin was operated at 3.5 kV, and the sample cone was operated at 16 V. HPLC was performed as described previously. Vaporization was achieved with a nitrogen sheath gas (300 L/hr) and drying gas (150 L/hr) at 400°C. Mass spectra of 3-methylsulfinylpropyl, 4-methylsulfinylbutyl, allyl, 3-butenyl, and 3-hydroxypropyl glucosinolates from the enzymatic reactions were analyzed with the first quadrupole set to detect a single M+H+ specific for the glucosinolate being tested.
Statistics
Statistical analysis was performed using Systat version 7 (SPSS Inc., Chicago, IL).
References
- Alonso-Blanco, C., Peeters, A.J.M., Koornneef, M., Lister, C., Dean, C., Vandenbosch, N., Pot, J., and Kuiper, M.T.R. (1998. b). Development of an AFLP based linkage map of Ler, Col and Cvi Arabidopsis thaliana ecotypes and construction of a Ler/Cvi recombinant inbred line population. Plant J. 14, 259–271. [DOI] [PubMed] [Google Scholar]
- Back, K., and Chappell, J. (1996). Identifying functional domains within terpene cyclases using a domain-swapping strategy. Proc. Natl. Acad. Sci. USA 93, 6841–6845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daxenbichler, M.E., Spencer, G.F., Carlson, D.G., Rose, G.B., Brinkler, A.M., and Powell, R.G. (1991). Glucosinolate composition of seeds from 297 species of wild plants. Phytochemistry 30, 2623–2638. [Google Scholar]
- Du, L., Lykkesfeldt, J., Olsen, C., and Halkier, B. (1995). Involvement of cytochrome P450 in oxime production in glucosinolate biosynthesis as demonstrated by an in vitro microsomal enzyme system isolated from jasmonic acid-induced seedlings of Sinapis alba L. Proc. Natl. Acad. Sci. USA 92, 12505–12509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faulkner, K., Mithen, R., and Williamson, G. (1998). Selective increase of the potential anticarcinogen 4-methylsulphinylbutyl glucosinolate in broccoli. Carcinogenesis 19, 605–609. [DOI] [PubMed] [Google Scholar]
- Frohman, M.A., Dush, M.K., and Martin, G.R. (1988). Rapid production of full-length cDNAs from rare transcripts: Amplification using a single gene-specific oligonucleotide primer. Proc. Natl. Acad. Sci. USA 85, 8998–9002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giamoustaris, A., and Mithen, R. (1995). The effect of modifying the glucosinolate content of leaves of oilseed rape (Brassica napus ssp oleifera) on its interaction with specialist and generalist pests. Ann. Appl. Biol. 126, 347–363. [Google Scholar]
- Giamoustaris, A., and Mithen, R. (1996). Genetics of aliphatic glucosinolates. IV. Side-chain modification in Brassica oleracea. Theor. Appl. Genet. 93, 1006–1010. [DOI] [PubMed] [Google Scholar]
- Halkier, B.A., and Du, L. (1997). The biosynthesis of glucosinolates. Trends Plant Sci. 2, 425–431. [DOI] [PubMed] [Google Scholar]
- Hedden, P. (1991). Gibberellin biosynthetic enzymes and the regulation of gibberellin concentration. In Gibberellins; 1989 July 20–23; Tokyo, Japan, N. Takahashi, B.O. Phinney, and J. MacMillan, eds (New York: Springer-Verlag).
- Hogge, L.R., Reed, D.W., Underhill, E.W., and Haughn, G.W. (1988). HPLC separation of glucosinolates from leaves and seeds of Arabidopsis thaliana and their identification using thermospray liquid chromatography-mass spectrometry. J. Chromatogr. Sci. 26, 551–556. [Google Scholar]
- Koch, M., Bishop, J., and Mitchell-Olds, T. (1999). Molecular systematics of Arabidopsis and Arabis. Plant Biol. 1, 529–537. [Google Scholar]
- Lander, E.S., Green, P., Abrahamson, J., Barlow, A., Daly, M.J., Lincoln, S.E., and Newburg, L. (1987). Mapmaker: An interactive computer package for constructing primary genetic linkage maps of experimental and natural populations. Genomics 1, 174–181. [DOI] [PubMed] [Google Scholar]
- Lister, C., and Dean, C. (1993). Recombinant inbred lines for mapping RFLP and phenotypic markers in Arabidopsis thaliana. Plant J. 4, 745–750. [DOI] [PubMed] [Google Scholar]
- Lynch, M., and Force, A. (2000). The probability of duplicate gene preservation by subfunctionalization. Genetics 154, 459–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mithen, R., Clarke, J., Lister, C., and Dean, C. (1995). Genetics of aliphatic glucosinolates. III. Side chain structure of aliphatic glucosinolates in Arabidopsis thaliana. Heredity 74, 210–215. [Google Scholar]
- Prescott, A.G. (1993). A dilemma of dioxygenases (or where biochemistry and molecular biology fail to meet). J. Exp. Bot. 44, 849–861. [Google Scholar]
- Steele, C.L., Crock, J., Bohlmann, J., and Croteau, R. (1998). Sesquiterpene synthases from grand fir (Abies grandis): Comparison of constitutive and wound-induced activities, and cDNA isolation, characterization and bacterial expression of delta-selinene synthase and gamma-humulene synthase. J. Biol. Chem. 273, 2078–2089. [DOI] [PubMed] [Google Scholar]
- Thies, W. (1979). Detection and utilization of a glucosinolate sulfohydrolase in the edible snail Helix pomatia. Naturwissenschaften 66, 364–365. [Google Scholar]
- Thies, W. (1988). Isolation of sinigrin and glucotropaeolin from cruciferous seeds. Fat Sci. Technol. 8, 311–314. [Google Scholar]
- Van de Peer, Y., and De Wachter, R. (1994). TREECON for Windows: A software package for the construction and drawing of evolutionary trees for the Microsoft Windows environment. Comput. Appl. Biosci. 10, 569–570. [DOI] [PubMed] [Google Scholar]
- Wink, M. (1988). Plant breeding: Importance of plant secondary metabolites for protection against pathogens and herbivores. Theor. Appl. Genet. 75, 225–233. [Google Scholar]
- Wittstock, U., and Halkier, B.A. (2000). Cytochrome P450 CYP79A2 from Arabidopsis thaliana L. catalyzes the conversion of l-phenylalanine to phenylacetaldoxime in the biosynthesis of benzylglucosinolate. J. Biol. Chem. 275, 14659–14666. [DOI] [PubMed] [Google Scholar]
- Zrybko, C.L., Fukuda, E.K., and Rosen, R.T. (1997). Determination of glucosinolates in domestic and wild mustard by high-performance liquid chromatography with confirmation by electrospray mass spectrometry and photodiode-array detection. J. Chromatogr. 767, 43–52. [Google Scholar]