

Abstract
Protein-protein interactions, particularly weak and transient ones, are often mediated by peptide recognition domains, such as Src Homology 2 and 3 (SH2 and SH3) domains, which bind to specific sequence and structural motifs. It is important but challenging to determine the binding specificity of these domains accurately and to predict their physiological interacting partners. In this study, the interactions between 35 peptide ligands (15 binders and 20 non-binders) and the Abl SH3 domain were analyzed using molecular dynamics simulation and the Molecular Mechanics/Poisson-Boltzmann Solvent Area method. The calculated binding free energies correlated well with the rank order of the binding peptides and clearly distinguished binders from non-binders. Free energy component analysis revealed that the van der Waals interactions dictate the binding strength of peptides, whereas the binding specificity is determined by the electrostatic interaction and the polar contribution of desolvation. The binding motif of the Abl SH3 domain was then determined by a virtual mutagenesis method, which mutates the residue at each position of the template peptide relative to all other 19 amino acids and calculates the binding free energy difference between the template and the mutated peptides using the Molecular Mechanics/Poisson-Boltzmann Solvent Area method. A single position mutation free energy profile was thus established and used as a scoring matrix to search peptides recognized by the Abl SH3 domain in the human genome. Our approach successfully picked ten out of 13 experimentally determined binding partners of the Abl SH3 domain among the top 600 candidates from the 218,540 decapeptides with the PXXP motif in the SWISS-PROT database. We expect that this physical-principle based method can be applied to other protein domains as well.
Synopsis
One of the central questions of molecular biology is to understand how signals are transduced in the cell. Intracellular signal transduction is mainly achieved through cascades of protein-protein interactions, which are often mediated by peptide-binding modular domains, such as Src Homology 2 and 3 (SH2 and SH3). Each family of these domains binds to peptides with specific sequence and structural characteristics. To reconstruct the protein-protein interaction networks mediated by modular domains, one must identify the peptide motifs recognized by these domains and understand the mechanism of binding specificity. These questions are challenging because the domain-peptide interactions are usually weak and transient. Here, the authors took a physical-principles approach to address these difficult questions for the SH3 domain of human protein Abl, which binds to peptides containing the PXXP motif (where P is proline and X is any amino acid). They generated a position-specific scoring matrix to represent the binding motif of the Abl SH3 domain. Analysis on the binding free energy components suggested insights into how the binding specificity is achieved. Most known protein interacting partners of the Abl SH3 domain were correctly identified using the position-specific scoring matrix, and other potential interacting partners were also suggested.
Introduction
The interactions between protein domains and their peptide ligands play critical roles in signal transduction and many other key biological processes. Because domain-peptide interactions are usually weak and transient, and often depend upon post-translational modification, they tend to be under-represented in high-throughput and computational studies [1], thus highlighting the need to develop new methods to identify these interactions. The Src Homology 3 (SH3) domain is the most abundant modular domain in the human proteome and presents in a wide variety of proteins, such as kinases, lipases, GTPases, and adaptor proteins, to orchestrate diverse cellular processes [2–6]. SH3 domains are 50–70 amino acids long and consist of five β-strands arranged into two sheets packed at right angles. They recognize the proline-rich peptides with the consensus motif PXXP (where P is proline and X is any amino acid) [7, 8] that forms a left-handed poly-proline type II (PPII) helix [9]. Depending on the position of the positive residue in the peptide sequence, the majority of SH3 ligands fall into two classes that bind to the protein in opposite orientations [10]: N-terminal to C-terminal (class I) or C-terminal to N-terminal (class II). Class I peptides typically consist of a core motif of RXLPX#P (where # is usually a hydrophobic residue), whereas the class II peptides contain a core motif of PX#PXR. In class I peptides, the proline residues in bold occupy the sites in the hydrophobic pocket that are normally referred as position P0 and P3, while the Arg residue occupies position P−3 (the positions are often dubbed as P−3, P−2, P−1, P0, P1, P2, and P3 from N-terminal to C-terminal, namely from R to P, in the motif RXLPX#P [2]). A notable variance of this motif is the one recognized by the Abl SH3 domain, which contains a tyrosine or a large hydrophobic residue at position P−3.
The binding specificity of a specific SH3 domain is determined by the flanking residues around the core motif PXXP [10]. Understanding the molecular basis of the specificity for each SH3 domain and identifying the sequence motif it recognizes are crucial to reconstruct the complete protein-protein interaction networks mediated by SH3 domains. Both experimental and computational methods have been developed to tackle this problem. Peptide library screening is often used to determine the binding motif of a SH3 domain, in which the binding peptides are sequenced and aligned to generate a frequency matrix representing the amino acid preference at each position [11, 12]. Bias may be introduced by not completely sampling all possible peptides, not quantitatively weighting the contribution of peptides to the matrix based on their binding strength and/or not distinguishing peptides bound to the SH3 domain in different binding modes. Computational methods such as Scansite [13], SPOT [14], and VIP [15] methods have been developed to predict interacting proteins of a domain. The performance of Scansite totally depends on the accuracy of the frequency matrix determined by the peptide library experiments. SPOT is limited by the relatively small number of residue contact pairs between SH3 domains and peptides. The performance of the VIP method can be improved if more conformational sampling is done and more rigorous binding energy prediction method, including the conformational energy change and the desolvation contribution upon peptide binding, is applied.
In this study, we analyzed the binding specificity of the SH3 domain of the human protein Abl. The Molecular Mechanics/Poisson-Boltzmann Solvent Area (MM/PBSA) method [16] was first applied to calculate the binding free energies between the Abl SH3 domains and 35 ten-residue-long peptides (15 binders and 20 non-binders). As a validation of the MM/PBSA method on the domain-peptide system, the calculated binding free energies of the 15 known binders correlated well with the experimental values [17] and were distinct from those of the non-binders. Analysis of the molecular dynamics (MD) trajectories and binding free energy components shed light into understanding the mechanism of the binding specificity of the Abl SH3 domain. The residue preference at each position of the peptide ligand was then studied systematically by single position mutation and MM/PBSA calculations, which we call the virtual mutagenesis (VM) method [18, 19]. A single position mutation free energy profile (SPMFEP) was established from such analysis to quantitatively represent the binding motif and was in good agreement with the experimental measurements. We used SPMFEP as a scoring matrix to search the SWISS-PROT database for potential binding partners of the Abl SH3 domain. Most experimentally determined binding proteins of the Abl SH3 domain were ranked in the top 600 candidates among about 6.2 × 107 decapeptides in the database and many promising candidates were also suggested.
Results
Molecular Basis of the Binding Specificity of the Abl SH3 Domain
The calculated binding free energies correlate well with the experimental values.
We first evaluated the performance of the MM/PBSA method on calculating the binding free energies of the Abl SH3 domain and its peptide ligands (15 binders and 20 non-binders). As shown in Table 1 and Figure 1, the calculated relative binding free energies of the 15 known binders show good correlation with the experimental values (the correlation coefficient r and standard deviation [SD] are 0.82 and 1.7, respectively). We then analyzed the free energy components to search for the dominant factor that dictates the binding specificity (Table 1). As the favorable electrostatic interaction between the peptide and the SH3 domain ΔE ele is canceled by the unfavorable electrostatic contribution to desolvation ΔG PB, the van der Waals interaction ΔE vdw is the most favorable component of the binding free energy. The favorable ΔE vdw is mainly from the interactions between the conserved proline residues of the PXXP motif and the hydrophobic surface that is formed by Tyr7, Phe9, Trp36, Tyr52, and Pro49 of the Abl SH3 domain and conserved in almost all SH3 domains. To investigate which energetic factor determines the relative binding affinities of these 15 binders, we compared the correlations between the measured binding free energies and each of the four free energy components, ΔE ele, ΔE vdw, ΔG PB, and ΔG SA. None of these components shows good correlation with the experimental values and the largest correlation coefficient is only 0.43 for ΔE ele, which suggests that no individual free energy component dominates the binding specificity. We then analyzed the non-polar (ΔE vdw + ΔG SA) and the electrostatic contribution (ΔE ele + ΔG PB) to the binding free energy. The electrostatic contribution correlates well with the binding free energies (r = 0.73, SD = 1.6) while the non-polar contribution does not show any correlation (r = −0.32, SD = 2.0). It suggests that the binding preference of these peptides is mainly determined by the electrostatic contribution to binding.
Table 1.
Energetic Components and Binding Affinities for the 15 Peptide Ligands of the Abl SH3 Domain (kcal/mol)

Figure 1. The Correlations between Experimental and Predicted Relative Binding Free Energies of the 15 Binders of the Abl SH3 Domain.

We also found that the conformational change of the peptide upon binding ΔE conf_pep was important (r = 0.54, SD =1.8). The sum of ΔE ele, ΔE vdw, ΔG PB, ΔG SA, and ΔE conf_pep correlates well with the binding free energies (r = 0.79, SD =1.8) whereas the sum of the first four terms does not (r = 0.52). Our calculations highlight the crucial effect of the change of the conformational energy of peptide to the affinities, which is not always appropriately considered [20, 21]. Inclusion of the conformational entropy −TΔS in the free energy calculation only slightly improves the correlation coefficient from 0.79–0.82, which suggests that the conformational entropy is not the determinant factor of the binding specificity of the binders.
The binding free energies and the free energy components for the 20 non-binders of the Abl SH3 domain were also calculated (Table S1). Two distributions of the binding free energies for binders and non-binders are distinct (Figure 2), which indicates that the MM/PBSA method can distinguish binders from non-binders. We also found that most non-binders preferred unbound conformations. First, the average conformational entropy change upon binding −TΔS for binders and non-binders are 32.1 kcal/mol and 35.9 kcal/mol, respectively, indicating that most non-binders lost more entropy upon binding than did binders. Second, the change of conformational energy for binders and non-binders are also significantly different: the average value for binders and non-binders are 2.0 kcal/mol and 4.3 kcal/mol, respectively.
Figure 2. The Distributions of the Predicted Binding Free Energies for Binders and Non-Binders of the Abl SH3 Domain.

The binding motif can be revealed by the VM method.
To understand the mechanism of the binding specificity, we need to determine the binding motif of the domain. We analyzed the amino acid preference at each position of the peptide using the VM method (see Materials and Methods). We compared our results with the available experimental measurements at positions P3, P0, P−3, and P−5 (Tables S3–S6) [17, 22]. These four positions are particularly important for the peptide binding: the two usually conserved Pro residues at P3 and P0 ensure strong binding affinity and residues at P−3, and P−5 are essential to the binding specificity [9, 10]. To determine the residues of the Abl SH3 domain that are important for peptide binding, the contribution of each SH3 domain residue to binding with the template peptide APSYSPPPPP (the two conserved Pro residues are in bold) was analyzed (the polar contribution to desolvation was calculated using the GB/SA method implemented in the mm_pbsa module of AMBER 8 for the sake of efficiency.) (Table S7) [19].
In the crystal structure of APSYSPPPPP complexed with the Abl SH3 domain, the residue at position P−5 in the peptide occupies a hydrophobic pocket of the SH3 domain. Proline at this position has relatively strong van der Waals interactions with Trp36 (−1.3 kcal/mol) and Trp47 (−0.9 kcal/mol) in the Abl SH3 domain (Table S7). Several other residues, Asn, Leu, Met, Phe, Tyr, and Val, are also strongly favored at this position (Figure 3A). It is worth pointing out that the mutation of Pro to Tyr or Phe, does not significantly impair the van der Waals interaction between the peptide and the SH3 domain due to the conformational change of the peptide backbone. Our predicted preference based on free energy calculations is consistent with the experimental results: Pro is the most preferred, whereas other residues especially hydrophobic ones (Phe, Leu, Met, Val, and Trp) are also favored [22].
Figure 3. The Preference of Residue Based on the Binding Free Energy Difference between the Mutated Peptide and the Template Peptide.

The preference of residue at positions (A) P−5, (B) P−3, (C) P0, and (D) P3 based on the binding free energy difference between the mutated peptide and the template peptide APSYSPPPPP. In (D), the conformational entropy was included in the binding free energies.
It should be noted that the preference of residue at P−5 are closely related to the residues at the adjacent positions. For example, the known binder FGTYPPPLPP (A4 in Table 1) has a Gly at P−5, which is not favored at this position based on the VM result. By analyzing the MD trajectory on A4-SH3 complex, we found that Phe at P−6 in A4 can occupy the binding pocket that is occupied by Pro at P−5 in the template peptide APSYSPPPPP to form favorable van der Waals interactions with Trp36 and Trp47. Moreover, the benzyl ring of Phe at P−6 is parallel to the aromatic ring of Trp47 to form strong π-π stacking interactions. Therefore, if there is a small residue (Ala, Gly, or Ser) at P−5, an aromatic residue (Tyr, Phe, or Trp) may be preferred at P−6. This suggests that the repertoire of SH3 domain-binding peptides may be much larger than previously thought.
Our analysis showed that Trp, Phe, Tyr, Met, and Pro are favored at P−3 (Figure 3B), which is in good agreement with the study of Villanueva's et al. that the most favorable residues are Trp, Tyr, Phe, and Met (ordered based on the binding free energies) [22]. This observation is consistent with findings that an aromatic residue is favored at P−3 of mouse protein 3BP1, a known binder of the Abl SH3 domain [12, 23, 24]. The energy component analysis (Table S4) suggests that the strong preference of these four residues at this position is mainly due to the favorable non-polar contribution (ΔEvdw + ΔGSA) upon peptide binding: ΔE vdw + ΔG SA for Trp, Tyr, Met, and Phe are −55.9, −55.4, −54.4, and −53.8 kcal/mol, respectively, which are stronger than the other 16 residues at this position. Interestingly, positive charged residues, Arg and Lys, are not favored at this position, which is in contrast to most SH3 domain-binding peptides. To investigate the reason, we have compared the electrostatic surfaces of four SH3 domains, Abl tyrosine kinase SH3 domain (PDB entry 1bbz) [25], c-Crk N-terminal SH3 domain (PDB entry 1cka) [26], Grb2 N-terminal SH3 domain (PDB entry 1gbq) [27], and rat amphiphysin-2 SH3 domain (PDB entry 1bb9) [28] (Figure 4). Electrostatic potentials were calculated by solving the Poisson-Boltzmann equation using the Delphi program [29] in Insight II [30]. We find that the rat amphiphysin-2 SH3 domain has the largest areas of negative electrostatic potentials, which is mainly due to the acidic residues in Arg-Thr and extended n-Src loops of the domain. The large patch of negative electrostatic potential explains why the amphiphysin SH3 domain specifically recognizes the PXRPXR motif with two positively charged Arg residues. The c-Crk N-terminal and the Grb2 N-terminal SH3 domain have distinct but relatively small negative potential near the Arg-Thr and n-Src loop, which may explain why these two SH3 domains bind to peptides only possessing a single positively charged residue. Compared with the other three SH3 domains, the Abl SH3 domain does not possess remarkable and continuous distribution of the negative electrostatic potentials and therefore positively charged residues are not strongly preferred.
Figure 4. The Electrostatic Potentials of the Peptide-Binding Interfaces for Four SH3 Domains.

(A) 1bbz, (B) 1cka, (C) 1gbq, and (D) 1bb9.
The scale of gradation was from −5 kT/e to +5 kT/e corresponding to red color to blue color. The electrostatic potentials of proteins were calculated using the Delphi module in Insight II. The salt concentration was set to 0.0 M because electrostatic potentials had small changes in the range of the experimental salt concentrations. The internal and external dielectric constants were set to 1 and 80, respectively. Electrostatic potentials were computed using a grid space of 0.5 Å with the focusing technique. The structures of the four SH3 domains were aligned using the Homology module in Insight II. The Tyr residue at P−3 in peptide APSYSPPPPP was shown in stick.
Proline is highly preferred at positions P0 and P3. We find that Pro at P0 has favorable interactions with Phe9 and Tyr52 (−1.0 kcal/mol and −1.0 kcal/mol) (Table S7), as well as favorable van der Waals interaction with Pro49 (−0.8 kcal/mol). Experimental and theoretical studies have focused on understanding how SH3 domains recognize the core motif PXXP [31, 32]. Proline is the most favorable residue at this position based on our free energy calculation (Figure 3C). When only the non-polar contribution (ΔE vdw + ΔG SA) is considered, the peptide with Pro has the strongest interaction with SH3 (−54.8 kcal/mol) (Table S5), which agrees with the study of Wang et al. [31]. In addition to the non-polar contribution, we found the less unfavorable desolvation-free energy is also an important factor for the preference of proline. For example, the residues at P0 in peptides 16, 19, and 20 (Table S5) all have relatively small side chains (Thr, Val, and Pro, respectively) and have similar protein-ligand electrostatic interactions ΔE ele. When the polar contributions of desolvation-free energies ΔG PB were considered, the peptide 20 has the least unfavorable contribution (ΔE ele + ΔG PB = + 20.5 kcal/mol). We believe that upon peptide binding the desolvation cost for the nitrogen-substituted atom in Pro may be less than that of the non-substituted nitrogen atom in the other amino acids, because the non-substituted nitrogen atom can be easily polarized by the solvent [32].
Proline is also strongly selected at P3 in almost all SH3 domain-binding peptides. From the calculated binding free energies (Figure 3D), it is interesting to find that, although Pro is preferred at this position by the Abl SH3 domain, the preference is not very strong. Proline at this position can be mutated to several other residues including Trp, Phe, Met, His, Leu, Gln, and Tyr, and the mutated peptides still have relatively strong binding free energies. Our calculation seems to contrast with what has been suggested about the critical role of Pro at P3 in SH3 ligands [10]. However, in the mutation experiments reported by Pisabarro et al. [17], Pro at P3 in peptide APTYPPPLPP was mutated to His, Leu, and Tyr, and the binding affinities of the mutated peptides were only slightly decreased from −7.1 kcal/mol to −6.2, −6.2, and −6.2 kcal/mol, respectively. These three favorable residues reported by Pisabarro et al. were also relatively favored in our predictions.
Identifying Physiological Interacting Partners of the Abl SH3 Domain
Based on the comparison between the experimental and calculated results, we have shown that the VM method can determine the binding motif of the Abl SH3 domain. The difference between the binding free energies of the mutated peptide at each position and the template peptide APSYSPPPPP, called SPMFEP, can be used as a position specific scoring matrix to predict the binding affinities of peptides (Table 2). To evaluate the performance of SPMFEP, the 15 binders and 20 non-binders were first scored using SPMFEP. Two obvious distributions for binders and non-binders can be observed (Figure 5), indicating that binders and non-binders can be successfully distinguished by SPMFEP. The binding affinity of peptide A4 was under-estimated. From our analysis (see above), we know that Phe at P−6 in the peptide A4 is favorable in the hydrophobic binding pocket that is originally occupied by Pro at P−5 in the template peptide. Consequently, the residues at P−6 and P−5 may interact with each other. In SPMFEP, the inter-dependence between positions is not considered. In fact, all methods using a position specific scoring matrix, such as Scansite [13], have the same limitations. Overall, SPMFEP performs well on the selected 35 peptides.
Table 2.
SPMFEPs of the Abl SH3 Domain
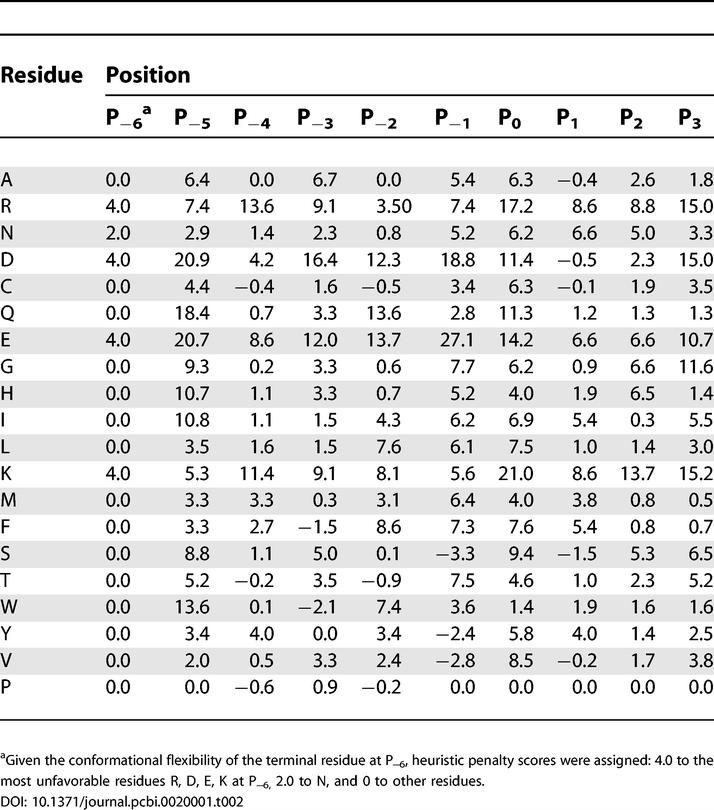
Figure 5. The Distribution of the Predicted Binding Free Energies Based on SPMFEP for Binders and Non-Binders.

We next scanned the SWISS-PROT using SPMFEP to predict interacting partners of the Abl SH3 domain. There are about 6.2 × 107 ten-residue-long peptides in the current SWISS-PROT database (May 2005), in which about 218,540 ten-residue-long peptides have the PXXP motif. Only about 2,600 peptides have scores smaller than two, which are in the top 0.005% (the top 600 peptide sequences in 353 unique human proteins are listed in Table S9). We first carefully examined the top ten candidates in the human proteome (Table 3), among which WASF1 and EVL are known interacting partners of the Abl SH3 domain [33, 34]. WASF4 is a homology of WASF1 and is in the same protein family. SEM6A is a homology of the mouse protein SEM6D, a known Abl SH3 domain-binding protein, and is therefore likely to be a true binder [35]. In total, we have identified two known binders and two candidates highly supported by experimental evidence, among the top ten peptides, which is a surprisingly good result. As a comparison, the top ten human peptides in the Scansite search are [13] 3BP2, RX, RBMG, TACT, PRL3, SCA3, AT19, AD08, DYN2, and SEP4, among which only 3BP2, the homology of a known binder (mouse protein 3BP2), is likely to be a true binder [36] but no binding information is found for all other candidates in BIND [36] and MINT [37] databases to interact with Abl protein or the Abl SH3 domain. If only considering the top ten candidates, the SPMFEP method based on VM performs better than Scansite on identifying the interacting partners of the Abl SH3 domain.
Table 3.
Top Ten Binding Partner Candidates of the Abl SH3 Domain Identified by SPMFEP

In MINT [37] and BIND [36], 44 non-redundant proteins have been identified to directly interact with the protein Abl, and 13 of them, including five mouse proteins and eight human proteins, bind to the Abl SH3 domain. We compared the performance of SPMFEP, Scansite [13], and iSPOT [14], to identify these 13 proteins (Table 4). The top 600 candidates found in human proteins and the top 2,000 candidates found in all proteins were saved for further analyses.
Table 4.
Comparison of Predictions of SPMFEP, Scansite, and iSPOT on 13 Known Interacting Proteins of the Abl SH3 Domain

SPMFEP can successfully identify ten known binders: seven of the eight human proteins and three of the five mouse proteins (Table 4 and Table S8). 3BP1_mouse and 3BP2_mouse are not ranked highly, considering all proteins (1,393 and 1,895) but they are in the top 500 if only mouse proteins are considered. The human homologies of 3BP1_mouse and 3BP2_mouse are in the top 600 candidates (249 for 3BP1_human and 502 for 3BP2_human) (Table S9) and it is reasonable to believe that they are true binders of the Abl SH3 domain. Overall, we have successfully identified most of the known binders of the Abl SH3 domain.
Scansite can identify eight known binders of the Abl SH3 domain (Table 4). P73_human, not identified by SPMFEP, is ranked 321 in the Scansite result. Agami et al. [38] reported that a P73 mutant P338A could not form stable P73-Abl complexes. If P73 interacts with Abl by ten-residue-long peptide segment, this peptide segment should be AFKQSPPAVP, which is the same as the peptide identified by Scansite. Based on our VM analysis and mutation experiments by Pisabarro and Serrano [17], Phe at P−5 and Lys at P−4 are not favored. Because Scansite considers 15-residue-long peptides rather than ten-residue-long peptides in SPMFEP, it is likely that the five additional residues may contribute favorably to binding. It is not surprising that the longer the peptide, the more specific, but less sensitive, are the predictions.
Using iSPOT, we can only correctly identify five binders (Table 4). In iSPOT, the scoring matrix was derived from position-specific contacts based on six SH3-peptide or SH3-protein complex structures [14]. The accuracy of the matrix is limited by the relatively small number of residue-resident contacts found between SH3 domains and their binding peptides to fill the 27 × 10 × 10 × 10 position-specific contact matrix.
CABL2_human and SEM6D_mouse (Table 4) cannot be identified by all the three methods. Experiments [35, 39] have shown that their interactions with Abl are mediated by the interaction between the proline-rich region and the Abl SH3 domain. Since the scoring matrix used by all the three methods does not consider dependence between positions, we suspect that synergistic interactions may exist between positions within or beyond the proline-rich regions of the two proteins.
Discussion
In this study, we have demonstrated that the MM/PBSA method can accurately calculate the binding free energies between the Abl SH3 domain and its peptide ligands. Examination of each component of the binding free energy shows that, besides the non-bonded interactions and desolvation effect, the change of the conformational energies of the peptides upon binding is also crucial to determine the binding specificity of the domain. These results are encouraging to apply MD simulation and free energy calculation to understand the molecular mechanism of other domain-peptide and protein-protein interactions.
We have also shown that the VM method can precisely determine the sequence motif recognized by the Abl SH3 domain. The experimental scheme of the VM method is totally different from those of the current peptide library experiments in the following ways: 1) produce all possible peptides that are one amino acid different from the template peptide; (2) measure the “binding” affinities of all these peptides; and, (3) generate a scoring matrix based on the binding affinity differences between the mutated and the template peptides to determine the binding motif.
There are advantages of this scheme. First, the preference of an amino acid is quantitatively measured, based on the binding affinity of the peptide, which at least partially overcomes the sampling difficulty in the current peptide library experiment. Ideally, to determine the binding motif of a domain, one should examine the binding between the domain and all possible peptides of a given length, and align all binding peptides to calculate the frequency of each amino acid occurring at each position. In reality, there are usually only 107–1010 peptides in the library, due to the limit of time and cost. If the length of the binding peptide is ten, there are 1010–1013 possible peptides and the coverage of the peptide sequence space by the peptide library is about 10−6–10−3. If 15-residue-long peptides are considered as in Scansite, the coverage drops dramatically to 10−12–10−9. By measuring the relative preference of every amino acid based on the peptide-binding affinities, one overcomes this insufficient sampling issue and mimics the ideal procedure given the position-independence assumption. Second, the VM method can evaluate the penalties of unfavorable amino acids even the peptide do not really bind to the protein, which is very hard if not impossible in the experimental approaches. Third, since the scoring matrix is obtained by taking the difference between the template and the mutated peptides that are only one amino acid different from the template peptide, some errors due to insufficient sampling of conformational space and/or inaccurate free energy calculation can be cancelled.
There are two major hurdles of applying the VM method in a high-throughput manner. First, the MD simulation and free energy calculations are time-consuming. Second, a domain-peptide complex structure is required. Given the fast pace of advancement of computer power and structural genomics/homology modeling, we believe that the VM method will become more and more useful.
Materials and Methods
MD simulations.
MD simulations were performed on the 15 binders and 20 non-binders of the Abl SH3 domain using the AMBER 8 simulation package [19] and AMBER03 force field [40]. The amino acid sequences and the experimentally determined binding affinities of the 15 binders are shown in Table 1 [17]. Ten peptides, B1 to B10, were randomly selected from the human proteome and are considered as non-binders (Table S1). Ten peptides, C1 to C10, do not bind to the Abl SH3 domain but are Class I binders of other SH3 domains [12]: C1 and C2 bind to the Src SH3 domains, C3 and C4 bind to the Yes SH3 domain, and C5 to C10 bind to the Grb2 N-terminal SH3 domain (Table S1). We chose the crystal structure of the peptide APSYSPPPPP complexed with the Abl SH3 domain (Class I binder and the PDB entry is 1bbz) [25] as the template and mutated it to other peptides using the scap program [41]. The complex was solvated in a rectangular box of about 3,000 TIP3P water molecules so that the boundary of the box is at least 9 Å away from any solute atom. Counter-ions of Na+ were placed based on the Columbic potential to keep the whole system neutral. Particle Mesh Ewald (PME) was employed to consider the long-range electrostatic interactions [42]. Following 2,000 steps of minimization, a 1.2 ns (30 ps temperature increase from 10 °K to 300 °K and 1.17 ns equilibration and data collection) MD simulation with a 2.0 fs time step was performed on each complex. The SHAKE procedure was employed to constrain hydrogen atoms [43] during MD and all heavy atoms of SH3 were restrained using a 5 kcal·mol−1·Å−2 harmonic force (see discussion in Supporting Information).
To determine the conformational energy of unbound peptide in solvent, 2.0 ns MD simulation was conducted on each peptide. Each peptide was solvated in a water box of about 1,600 water molecules, which extended 10 Å away from any peptide atom. 1,000 steps of minimization were followed by a 2.0 ns MD simulation for equilibration and data collection using the same set-up as described above.
Free energy calculations using the MM/PBSA method.
The binding free energy is calculated as:
where ΔEMM is the molecular mechanics interaction energy between the SH3 domain and the peptide, ΔGPB and ΔGSA are the electrostatic and non-polar contributions to desolvation upon peptide binding, respectively, and −TΔS is the conformational entropy change. To consider the conformational flexibility of the peptide, we ran two separate MD simulations on the complex and the free peptide to calculate the binding free energy [16].
ΔEMM was calculated using the sander program in AMBER 8 [19]. ΔGPB was calculated using the pbsa program in AMBER 8. The grid size used to solve the Poisson-Boltzmann equation was 0.5 Å, and the values of interior dielectric constant and exterior dielectric constant were set to 1 and 80, respectively (the influence of the interior dielectric constant value to the free energy calculation is discussed in Supporting Information). ΔGSA was estimated from the surface area [16, 44]. The peptide-SH3 interaction energies were calculated from 150 snapshots taken from 300 ps to 1.2 ns MD simulation trajectories of the complex. 160 snapshots taken from 400 ps to 2.0 ns MD simulations on the unbound peptides were used to calculate the conformational energy change for the peptides.
The normal mode analysis was performed to estimate the vibrational component of the entropy using the nmode program in AMBER 8 [19]. In the absence of solvent, the structures (complex, SH3, and peptide) were minimized with no cutoff for non-bonded interactions, by using conjugate gradient and then Newton-Raphson minimizations until the root mean square of the elements of gradient vector was less than 5 × 10−5 kcal/mol. Then, normal mode calculations were carried out with no cutoff for non-bonded interactions. A distance-dependent dielectric constant (ɛ = 4R ij) was used to mimic solvent screening. Frequencies of the vibrational modes were computed at 300K for these minimized structures and using a harmonic approximation of the energies. Due to the high computational demand, only 25 snapshots taken from MD were used to estimate −TΔS.
The VM method.
To investigate the preference of residues at each position, systematic single point mutation was performed on the peptide. The peptide APSYSPPPPP in the crystal structure 1bbz was used as the template. Each residue of the peptide was mutated to the other 19 residues using the scap program [41]. Minimization, MD simulations, and MM/PBSA calculations were performed on all 190 mutated complexes, as well as on the free peptides using the same set-up described above. Assuming mutating a single residue of the peptide did not significantly change the peptide conformation, we did not include the conformational entropy in the comparison of 20 residues at each position.
Single point mutation free energy profile and database scan.
The SPMFEP is a 10 × 20 matrix, which represents the difference between the binding free energies of the mutated peptides and the template peptide APSYSPPPPP (Table 2). SPMFEP can be used as a position specific scoring matrix. The score of each peptide is calculated as: 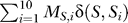 , where MS,i is the score of the amino acid S at ith position in the SPMFEP and Si is the amino acid at the ith position of the peptide. All ten-residue-long peptides in the SWISS-PROT database (release 46.4) were scored using the SPMFEP. The Perl script used for the database scan is available upon request.
, where MS,i is the score of the amino acid S at ith position in the SPMFEP and Si is the amino acid at the ith position of the peptide. All ten-residue-long peptides in the SWISS-PROT database (release 46.4) were scored using the SPMFEP. The Perl script used for the database scan is available upon request.
Supporting Information
(249 KB TIF)
The structure of SH3 shown here was extracted from the snapshot at 0.1 ns. The residues at P3, P−3, P−4, P−5, and P−6 of the peptide are colored in red and other residues are colored according to residue type defined in Insight II.
(2.9 MB TIF)
PB represents the polar contribution to the solvation free energy (ΔGPB). Electrostatic means the electrostatic interaction between the peptide and the SH3 domain (ΔEele), and van de Waals means the van de Waals interactions between the peptide and the SH3 domain (ΔEvdw).
(358 KB TIF)
Two residues, Trp36 and Trp47, are shown in stick, and the peptide is shown in ball-and-stick.
(942 KB TIF)
(42 KB DOC)
(66 KB DOC)
(56 KB DOC)
(63 KB DOC)
(62 KB DOC)
(61 KB DOC)
(65 KB DOC)
(57 KB DOC)
(54 KB DOC)
(643 KB DOC)
Accession Numbers
The Swiss-Prot and TrEMBL (http://www.expasy.org/sprot) accession numbers for the genes and gene products discussed in this paper are: 1bb9 (1bb9), 1bbz (CAB04591), 1cka (CAA18266), 1gbq (1gbqa), 3BP1_human (Q9Y3L3), 3BP1_mouse (P55194), 3BP2 (P78314), 3BP2_mouse (Q06649), AD08 (P78325), AT19 (Q8TE59), CABL2_human (NP_665850), DYN2 (P50570), EVL (Q9UI08), P73_human (O15350), PRL3 (P02814), RBMG (Q9UPN6), RX (Q9Y2V3), SCA3 (O14828), SEM6A (Q9H2E6), SEM6D (Q76KF3), SEM6D_mouse (NP_954711), SEP4 (O43236), TACT (P40200), WASF1 (Q92558), and WASF4 (Q8IV90).
Acknowledgments
The MD simulations were performed on the Linux cluster in the Center for Theoretical Biological Physics (CTBP) at UCSD. This work has been supported by the National Science Foundation PFC-sponsored Center for Theoretical Biological Physics (grants PHY-0216576 and PHY-0225630). TH is supported by a CTBP postdoctoral scholarship. BL is supported by a National Institutes of Health grant to the McCammon group (GM31749). WAM is supported by a NIH training grant (5-T32DK07233). We thank Prof. J. Andrew McCammon for providing access to computer software such as Insight II.
Abbreviations
- MD
molecular dynamics
- MM/PBSA
Molecular Mechanics/Poisson-Boltzmann Solvent Area
- SD
standard deviation
- SH2
Src Homology 2 domain
- SH3
Src Homology 3 domain
- SPMFEP
single position mutation free energy profile
- VM
virtual mutagenesis
Footnotes
Author contributions. TH, KC, and WW conceived and designed the experiments. TH performed the experiments. TH, KC, WAM, and BL analyzed the data. TH and WW wrote the paper.
Competing interests. The authors have declared that no competing interests exist.
References
- Russell RB, Alber F, Aloy P, Davis FP, Korkin D, et al. A structural perspective on protein-protein interactions. Curr Opin Struct Biol. 2004;14:313–324. doi: 10.1016/j.sbi.2004.04.006. [DOI] [PubMed] [Google Scholar]
- Lim WA, Richards FM, Fox RO. Structural determinants of peptide-binding orientation and of sequence specificity in SH3 domains. Nature. 1994;372:375–379. doi: 10.1038/372375a0. [DOI] [PubMed] [Google Scholar]
- Feng SB, Chen JK, Yu H, Simon JA, Schreiber SL. Two binding orientations for peptides to the src SH3 domain: Development of a general model for SH3-ligand interactions. Science. 1994;266:1241–1247. doi: 10.1126/science.7526465. [DOI] [PubMed] [Google Scholar]
- Macias MJ, Hyvonen M, Baraldi E, Schultz J, Sudol M, et al. Structure of the WW domain of a kinase-associated protein complexed with a proline-rich peptide. Nature. 1996;382:646–649. doi: 10.1038/382646a0. [DOI] [PubMed] [Google Scholar]
- Kuriyan J, Cowburn D. Modular peptide recognition domains in eukaryotic signaling. Annu Rev Biophys Biomol Struct. 1997;26:259–288. doi: 10.1146/annurev.biophys.26.1.259. [DOI] [PubMed] [Google Scholar]
- Mahoney NM, Rozwarski DA, Fedorov E, Federov AA, Almo SC. Profilin binds proline-rich ligands in two distinct amide backbone orientations. Nat Struct Biol. 1999;6:666–671. doi: 10.1038/10722. [DOI] [PubMed] [Google Scholar]
- Zhou SY. Recognition and regulation of primary-sequence motifs by signaling modular domains. Prog Biophys Mol Biol. 1999;71:359–372. doi: 10.1016/s0079-6107(98)00045-5. [DOI] [PubMed] [Google Scholar]
- Dalgarno DC, Botfield MC, Rickles RJ. SH3 domains and drug design: Ligands, structure, and biological function. Biopolymers. 1998;43:383–400. doi: 10.1002/(SICI)1097-0282(1997)43:5<383::AID-BIP4>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Cesareni G, Panni S, Nardelli G, Castagnoli L. Can we infer peptide recognition specificity mediated by SH3 domains? FEBS Lett. 2002;513:38–44. doi: 10.1016/s0014-5793(01)03307-5. [DOI] [PubMed] [Google Scholar]
- Zarrinpar A, Bhattacharyya RP, Lim WA. The structure and function of proline recognition domains. Sci STKE. 2003;203:RE8. doi: 10.1126/stke.2003.179.re8. [DOI] [PubMed] [Google Scholar]
- Rickles RJ, Botfield MC, Zhou XM, Henry PA, Brugge JS, et al. Phage display selection of ligand residues important for Src homology 3 domain-binding specificity. Proc Natl Acad Sci USA. 1995;92:10909–10913. doi: 10.1073/pnas.92.24.10909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparks AB, Rider JE, Hoffman NG, Fowlkes DM, Quillam LA, et al. Distinct ligand preferences of Src homology 3 domain from Src, Yes, Abl, Cortactin, p53bp2, Plc-g, Crk, and Grb2. Proc Natl Acad Sci USA. 1996;93:1540–1544. doi: 10.1073/pnas.93.4.1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obenauer JC, Cantley LC, Yaffe MB. Scansite 2.0: Proteome-wide prediction of cell signaling interactions using short sequence motifs. Nucleic Acids Res. 2003;31:3635–3641. doi: 10.1093/nar/gkg584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brannetti B, Via A, Cestra G, Cesareni G, Helmer Citterich M. SH3-SPOT: An algorithm to predict preferred ligands to different members of the SH3 gene family. J Mol Biol. 2000;298:313–328. doi: 10.1006/jmbi.2000.3670. [DOI] [PubMed] [Google Scholar]
- Wollacott AM, Desjarlais JR. Virtual interaction profiles of proteins. J Mol Biol. 2001;313:317–342. doi: 10.1006/jmbi.2001.5035. [DOI] [PubMed] [Google Scholar]
- Kollman PA, Massova I, Reyes C, Kuhn B, Huo SH, et al. Calculating structures and free energies of complex molecules: Combining molecular mechanics and continuum models. Acc Chem Res. 2000;33:889–897. doi: 10.1021/ar000033j. [DOI] [PubMed] [Google Scholar]
- Pisabarro MT, Serrano L. Rational design of specific high-affinity peptide ligands for the Abl-SH3 domain. Biochemistry. 1996;35:10634–10640. doi: 10.1021/bi960203t. [DOI] [PubMed] [Google Scholar]
- Wang W, Kollman PA. Free energy calculations on dimer stability of the HIV protease using molecular dynamics and a continuum solvent model. J Mol Biol. 2000;303:567–582. doi: 10.1006/jmbi.2000.4057. [DOI] [PubMed] [Google Scholar]
- Case DA, Darden TA, Cheatham IIITE, Simmerling CL, Wang J, et al. AMBER: Assisted model building with energy refinement, version 8 [computer program] San Francisco: University of California San Francisco; 2004. [Google Scholar]
- Froloff N, Windemuth A, Honig B. On the calculation of binding free energies using continuum methods: Application to MHC class I protein-peptide interactions. Protein Sci. 1997;6:1293–1301. doi: 10.1002/pro.5560060617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnini S, Juffer AH. Calculation of affinities of peptides for proteins. J Comput Chem. 2003;25:393–411. doi: 10.1002/jcc.10387. [DOI] [PubMed] [Google Scholar]
- Villanueva J, Fernández-Ballester G, Querol E, Aviles FX, Serrano L. Ligand screening by exoproteolysis and mass spectrometry in combination with computer modeling. J Mol Biol. 2003;330:1039–1048. doi: 10.1016/s0022-2836(03)00664-8. [DOI] [PubMed] [Google Scholar]
- Rickles RJ, Botfield MC, Weng Z, Taylor JA, Green OM, et al. Identification of Src, Fyn, Lyn, PI3K, and Abl SH3 domain ligands by screening a random phage display library. EMBO J. 1994;13:5598–5604. doi: 10.1002/j.1460-2075.1994.tb06897.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng ZG, Rickles RJ, Feng SB, Richard S, Shaw AS, et al. Structure-function analysis of SH3 domains: SH3 binding-specificity altered by single amino acid substitutions. Mol Cell Biol. 1995;15:5627–5634. doi: 10.1128/mcb.15.10.5627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisabarro MT, Serrano L, Wilmanns M. Crystal structure of the Abl-SH3 domain complexed with a designed high-affinity peptide ligand: Implications for SH3-ligand interactions. J Mol Biol. 1998;281:513–521. doi: 10.1006/jmbi.1998.1932. [DOI] [PubMed] [Google Scholar]
- Wu XD, Knudsen B, Feller SM, Zheng J, Sali A, et al. Structural basis for the specific interaction of lysine-containing proline-rich peptides with the n-terminal SH3 domain of c-Crk. Structure. 1995;3:215–226. doi: 10.1016/s0969-2126(01)00151-4. [DOI] [PubMed] [Google Scholar]
- Wittekind M, Mapelli C, Lee V, Goldfarb V, Friedrichs MS, et al. Solution structure of the Grb2 N-terminal SH3 domain complexed with a ten-residue peptide derived from SOS: Direct refinement against NOEs, J-couplings, and 1H and 13C chemical shifts. J Mol Biol. 1997;267:933–952. doi: 10.1006/jmbi.1996.0886. [DOI] [PubMed] [Google Scholar]
- Owen DJ, Wigge P, Vallis Y, Moore JD, Evans PR, et al. Crystal structure of the amphiphysin-2 SH3 domain and its role in the prevention of dynamin ring formation. EMBO J. 1998;17:5273–5285. doi: 10.1093/emboj/17.18.5273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honig B, Nicholls A. Classical electrostatics in biology and chemistry. Science. 1995;268:1144–1149. doi: 10.1126/science.7761829. [DOI] [PubMed] [Google Scholar]
- Accelrys. Insight II 2000 User Guide [computer program] San Diego (California): Accelrys; 2000. [Google Scholar]
- Wang W, Lim WA, Jakalian A, Wang J, Wang JM, et al. An analysis of the interactions between the Sem-5 SH3 domain and its ligands using molecular dynamics, free energy calculations, and sequence analysis. J Am Chem Soc. 2001;123:3986–3994. doi: 10.1021/ja003164o. [DOI] [PubMed] [Google Scholar]
- Nguyen JT, Turck CW, Cohen FE, Zuckermann RN, Lim WA. Exploiting the basis of proline recognition by SH3 and WW domains: Design of N-substituted inhibitors. Science. 1998;292:2088–2092. doi: 10.1126/science.282.5396.2088. [DOI] [PubMed] [Google Scholar]
- Westphal RS, Soderling SH, Alto NM, Langeberg LK, Scott JD. Scar/WAVE-1, a Wiskott-Aldrich syndrome protein, assembles an actin-associated multi-kinase scaffold. EMBO J. 2000;19:4589–4600. doi: 10.1093/emboj/19.17.4589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambrechts A, Kwiatkowski AV, Lanier LM, Bear JE, Vandekerckhove J, et al. cAMP-dependent protein kinase phosphorylation of EVL, a mena/VASP relative, regulates its interaction with actin and SH3 domains. J Biol Chem. 2000;275:36143–36151. doi: 10.1074/jbc.M006274200. [DOI] [PubMed] [Google Scholar]
- Toyofuku T, Zhang H, Kumanogoh A, Takegahara N, Yabuki M, et al. Guidance of myocardial patterning in cardiac development by Sema6D reverse signaling. Nat Cell Biol. 2004;6:1204–1211. doi: 10.1038/ncb1193. [DOI] [PubMed] [Google Scholar]
- Bader GD, Betel D, Hogue CWV. BIND: The Biomolecular Interaction Network Database. Nucleic Acids Res. 2003;31:248–250. doi: 10.1093/nar/gkg056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanzoni A, Montecchi-Palazzi L, Quondam M, Ausiello G, Helmer-Citterich M, et al. MINT: A Molecular INTeraction database. FEBS Lett. 2002;513:135–140. doi: 10.1016/s0014-5793(01)03293-8. [DOI] [PubMed] [Google Scholar]
- Agami R, Blandino G, Oren M, Shaul Y. Interaction of c-Abl and p73 alpha and their collaboration to induce apoptosis. Nature. 1999;399:809–813. doi: 10.1038/21697. [DOI] [PubMed] [Google Scholar]
- Sato H, Nishimoto I, Matsuoka M. ik3–2, a relative to ik3–1/Cables, is associated with cdk3, cdk5, and c-abl. Biochim Biophys Acta. 2002;19:157–163. doi: 10.1016/s0167-4781(01)00367-0. [DOI] [PubMed] [Google Scholar]
- Duan Y, Wu C, Chowdhury S, Lee MC, Xiong G, et al. A point-charge force field for molecular mechanics simulations of proteins. J Comput Chem. 2003;24:1999–2012. doi: 10.1002/jcc.10349. [DOI] [PubMed] [Google Scholar]
- Xiang Z, Honig B. Extending the accuracy limits of prediction of side-chain conformations. J Mol Biol. 2001;311:421–430. doi: 10.1006/jmbi.2001.4865. [DOI] [PubMed] [Google Scholar]
- Darden T, York D, Pedersen L. Particale mesh Ewald and N.log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
- Ryckaert J, Ciccotti G, Berendsen HJC. Numerical-integration of cartesian equations of motion of a system with constraints: Molecular-dynamics of n-alkanes. J Comput Phys. 1977;23:327–341. [Google Scholar]
- Wang W, Donini O, Reyes CM, Kollman PA. Biomolecular simulations: Recent developments in force fields, simulations of enzyme catalysis, protein-ligand, protein-protein, and protein-nucleic acid noncovalent interactions. Annu Rev Biophys Biomol Struct. 2001;30:211–243. doi: 10.1146/annurev.biophys.30.1.211. [DOI] [PubMed] [Google Scholar]
- Cicchetti P, Mayer BJ, Thiel G, Baltimore D. Identification of a protein that binds to the SH3 region of abi and is similar to bcr and gap-rho. Science. 1992;257:803–806. doi: 10.1126/science.1379745. [DOI] [PubMed] [Google Scholar]
- Ren R, Mayer BJ, Cicchetti P, Baltimore D. Identification of a 10-amino acid proline-rich SH3 binding-site. Science. 1993;259:1157–1161. doi: 10.1126/science.8438166. [DOI] [PubMed] [Google Scholar]
- Glover RT, Angiolieri M, Kelly S, Monaghan DT, Wang JY, et al. Interaction of the N-methyl-D-aspartic acid receptor NR2D subunit with the c-Abl tyrosine kinase. J Biol Chem. 2000;275:12725–12729. doi: 10.1074/jbc.275.17.12725. [DOI] [PubMed] [Google Scholar]
- Sun J, Zhao J, Schwartz MA, Wang JY, Wiedmer T, et al. c-Abl tyrosine kinase binds and phosphorylates phospholipid scramblase 1. J Biol Chem. 2001;276:28984–18990. doi: 10.1074/jbc.M102505200. [DOI] [PubMed] [Google Scholar]
- Beard MB, O'Connell JC, Bolger GB, Houslay MD. The unique N-terminal domain of the cAMP phosphodiesterase PDE4D4 allows for interaction with specific SH3 domains. FEBS Lett. 1999;22:173–177. doi: 10.1016/s0014-5793(99)01335-6. [DOI] [PubMed] [Google Scholar]
- Yamamoto A, Suzuki T, Sakaki Y. Isolation of hNap1BP which interacts with human Nap1 (NCKAP1) whose expression is down-regulated in Alzheimer's disease. Gene. 2001;271:159–169. doi: 10.1016/s0378-1119(01)00521-2. [DOI] [PubMed] [Google Scholar]
- Gertler FB, Niebuhr K, Reinhard M, Wehland J, Soriano P. Mena, a relative of VASP and Drosophila enabled, is implicated in the control of microfilament dynamics. Cell. 1996;87:227–239. doi: 10.1016/s0092-8674(00)81341-0. [DOI] [PubMed] [Google Scholar]
- Donaldson LW, Gish G, Pawson T, Kay LE, Forman-Kay JD. Structure of a regulatory complex involving the Abl SH3 domain, the Crk SH2 domain, and a Crk-derived phosphopeptide. Proc Natl Acad Sci USA. 2002;99:14053–14058. doi: 10.1073/pnas.212518799. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(249 KB TIF)
The structure of SH3 shown here was extracted from the snapshot at 0.1 ns. The residues at P3, P−3, P−4, P−5, and P−6 of the peptide are colored in red and other residues are colored according to residue type defined in Insight II.
(2.9 MB TIF)
PB represents the polar contribution to the solvation free energy (ΔGPB). Electrostatic means the electrostatic interaction between the peptide and the SH3 domain (ΔEele), and van de Waals means the van de Waals interactions between the peptide and the SH3 domain (ΔEvdw).
(358 KB TIF)
Two residues, Trp36 and Trp47, are shown in stick, and the peptide is shown in ball-and-stick.
(942 KB TIF)
(42 KB DOC)
(66 KB DOC)
(56 KB DOC)
(63 KB DOC)
(62 KB DOC)
(61 KB DOC)
(65 KB DOC)
(57 KB DOC)
(54 KB DOC)
(643 KB DOC)