

Abstract
Two of the four human FGF8 splice isoforms, FGF8a and FGF8b, are expressed in the mid-hindbrain region during development. Although the only difference between these isoforms is the presence of an additional 11 amino acids at the N terminus of FGF8b, these isoforms possess remarkably different abilities to pattern the midbrain and anterior hindbrain. To reveal the structural basis by which alternative splicing modulates the organizing activity of FGF8, we solved the crystal structure of FGF8b in complex with the “c” splice isoform of FGF receptor 2 (FGFR2c). Using surface plasmon resonance (SPR), we also characterized the receptor-binding specificity of FGF8a and FGF8b, the “b” isoform of FGF17 (FGF17b), and FGF18. The FGF8b-FGFR2c structure shows that alternative splicing permits a single additional contact between phenylalanine 32 (F32) of FGF8b and a hydrophobic groove within Ig domain 3 of the receptor that is also present in FGFR1c, FGFR3c, and FGFR4. Consistent with the structure, mutation of F32 to alanine reduces the affinity of FGF8b toward all these receptors to levels characteristic of FGF8a. More importantly, analysis of the mid-hindbrain patterning ability of the FGF8bF32A mutant in chick embryos and murine midbrain explants shows that this mutation functionally converts FGF8b to FGF8a. Moreover, our data suggest that the intermediate receptor-binding affinities of FGF17b and FGF18, relative to FGF8a and FGF8b, also account for the distinct patterning abilities of these two ligands. We also show that the mode of FGF8 receptor-binding specificity is distinct from that of other FGFs and provide the first biochemical evidence for a physiological FGF8b-FGFR1c interaction during mid-hindbrain development. Consistent with the indispensable role of FGF8 in embryonic development, we show that the FGF8 mode of receptor binding appeared as early as in nematodes and has been preserved throughout evolution.
Keywords: FGF8 subfamily, FGF receptors, alternative splicing, mid-hindbrain organizer, surface plasmon resonance
FGF8 is expressed in many organizing centers during embryogenesis and regulates patterning of the brain, limbs, heart, ear, and eye (Crossley et al. 1996a,b; Meyers et al. 1998; Sun et al. 1999). Unlike other FGFs, the biological activity of FGF8 is regulated by alternative splicing (Crossley and Martin 1995), which occurs at the N termini of these ligands and gives rise to four FGF8 isoforms (a, b, e, f) in humans (Gemel et al. 1996). The smallest FGF8 isoform, FGF8a, represents the common core region of all FGF8 isoforms, and the remaining isoforms differ by the presence of additional N-terminal amino acid sequences of variable length and sequence (Fig. 2B, below). A similar alternative splicing event occurs in FGF17 and results in two isoforms, FGF17a and FGF17b, which correspond to FGF8a and FGF8b, respectively. In contrast, FGF18 is not subject to alternative splicing.
Figure 2.

Structure-based sequence analysis of the FGF8-FGFR mode of receptor-binding specificity. (A) Structure-based sequence alignment of selected FGFs. Predicted signal sequences have been omitted from the alignment. Residue numbers are in bold print to the left of the sequence alignment. FGF8 and FGF17 are numbered according to the “b” isoform of these ligands. The alternatively spliced N-terminal regions of FGF8b and FGF17b that are absent in FGF8a and FGF17a, respectively, are enclosed in a black box. The location of the secondary structural elements of FGF8b are shown on top of the sequence alignment. The N-terminal boundary of the β1 strands of other structurally characterized FGFs are marked by a red line. The N-terminal boundaries of FGF1, FGF2, and FGF10 have been assigned using the receptor-bound form of these ligands (PDB identification codes 1DJS, 1EVT, and 1NUN, respectively). The cysteine residues in FGF8b that form the disulfide bridge are marked with a violet double-headed arrow. A dash represents a gap introduced to optimize the alignment. FGF residues that interact with receptor D2, linker, the constant region of D3, and the alternatively spliced region of D3, are colored green, red, cyan, and purple, respectively. The six FGF8b residues that play a particularly important role in determining the unique mode of FGF8b receptor-binding specificity are marked with an asterisk. The FGF8b residues that correspond to the FGF1, FGF2, and FGF10 residues that form the βC′-βE loop-interacting hydrophobic patch of these ligands are indicated by black triangles. The sequence of Danio rerio FGF8b (drfgf8) and C. elegans EGL17 are also shown to highlight the evolutionary conservation of the unique mode FGF8b-FGFR-binding specificity. (B) Sequence alignment of the alternatively spliced N-terminal region of the human FGF8 and FGF17 isoforms. The amino acid sequences in the FGF8 and FGF17 proteins have been boxed according to the exons from which they are encoded: FGF8 exons are boxed in black and FGF17 exons are boxed in red. Note that the “a” isoforms of FGF8 and FGF17 are generated by the use of an internal splice-acceptor site in exon 1D (indicated by asterisk). The exon boundaries for FGF8 and FGF17 were obtained from Gemel et al. (1996) and Xu et al. (1999), respectively. The predicted signal peptide cleavage sites for FGF8 and FGF17 (Tanaka et al. 1992; Hoshikawa et al. 1998) are indicated by a black and red arrow, respectively. (C) Structure-based sequence alignment of the alternatively spliced, C-terminal half of D3 from human FGFRs. The location and length of the β strands are shown on top of the sequence alignment. A period indicates sequence identity to FGFR2c. A dash represents a gap introduced to optimize the alignment. FGFR2c residues that form the D3 hydrophobic groove and that interact with FGF8b are colored purple. Two key constituents of the groove are indicated with asterisks. The βC′ and βE strands of FGFR1c, FGFR2c, FGFR3c, and FGFR2b from previous FGF-FGFR structures (PDB identification codes: 1EVT, 1DJS, 1RY7, and 1NUN, respectively) are indicated by red boxes.
The biological significance of alternative splicing of FGF8 subfamily members is most extensively studied in the context of mid-hindbrain development. All three members of the FGF8 subfamily (FGF8, FGF17, and FGF18), including the “a” and “b” isoforms of FGF8 and FGF17, are expressed by the isthmic organizer (Maruoka et al. 1998; Sato et al. 2001) and play distinct roles in early patterning of the vertebrate midbrain and anterior hindbrain (Liu and Joyner 2001a; Sato et al. 2004). Ectopic expression of FGF8a or FGF8b in the midbrain of transgenic mice shows that FGF8a expands the midbrain (Lee et al. 1997), whereas FGF8b transforms midbrain into cerebellum (Liu et al. 1999). Similar phenotypes are also produced when FGF8a or FGF8b cDNAs are electroporated into embryonic chick midbrain (Sato et al. 2001). Interestingly, electroporation of FGF17b or FGF18 into embryonic chick midbrain leads to an FGF8a-like expansion of the midbrain (Liu et al. 2003). The distinct mid-hindbrain patterning abilities of these FGFs correlate with differences in the ability of these ligands to alter the expression patterns of genes implicated in mid-hindbrain development, including the homeobox transcription factors Gbx2, Otx2, and Engrailed (En) 1 and 2, and the cytoplasmic negative regulators of FGF signaling Sprouty (Spry) 1 and 2 (for review, see Joyner et al. 2000).
Cumulative genetic data indicate that FGFR1 mediates the majority of FGF8b signaling in mid-hindbrain development. Conditional inactivation of FGFR1 in the midbrain and rhombomere 1 of mice leads to a phenotype that is similar to that of conditional FGF8 knockouts (Chi et al. 2003; Trokovic et al. 2003). Furthermore, knock-down of FGFR1in zebrafish phenocopies many aspects of acerebellar mutant zebrafish, which harbor a deletion of a major portion of the FGF8b core region (Reifers et al. 1998; Scholpp et al. 2004). However, mitogenic studies using transfected BaF3 cell lines overexpressing the seven principal FGFRs showed that FGF8b activates FGFR4 and the “c” isoforms of FGFR2 (FGFR2c) and FGFR3 (FGFR3c), but not the “c” isoform of FGFR1 (FGFR1c) or the “b” isoforms of FGFR1-3 (MacArthur et al. 1995b). Moreover, in vitro binding studies found that FGF8b does not compete with FGF1 for binding to FGFR1c (Chellaiah et al. 1999). Hence, there is controversy as to whether FGF8-FGFR1 forms a signaling axis during neuronal development.
The molecular basis by which FGF8a and FGF8b differ in their organizing abilities is also currently unknown. Ectopic expression of low amounts of FGF8b leads to morphological changes that are characteristic of those induced by high levels of FGF8a expression (Sato et al. 2001). Furthermore, only FGF8b was shown to activate the Ras-Erk pathway in chick midbrain (Sato and Nakamura 2004). These data have led to the hypothesis that differences in the organizing abilities of FGF8a and FGF8b are due to differences in the intensity of the signal generated by these isoforms (Sato et al. 2001; Sato and Nakamura 2004). Consistent with this hypothesis, electroporation of constitutively active forms of the “c” isoforms of FGFR1c or FGFR2c into chick midbrain leads to similar gene expression changes to those induced by FGF8b (Liu et al. 2003).
To decipher how alternative splicing modulates FGF8 organizing activity during mid-hindbrain development, and to gain further insights into the identity of the FGFRs that mediate the FGF8b signal during mid-hind-brain development, we solved the crystal structure of FGF8b in complex with FGFR2c at 2.2-Å resolution. To complement our structural studies, we characterized the receptor-binding affinity and specificity of FGF8a, FGF8b, FGF17b, and FGF18 using surface plasmon resonance (SPR). Our structural and SPR data demonstrate that the biological role of alternative splicing is to regulate the binding affinity of FGF8 toward the “c” isoforms of FGFR1-3 and FGFR4. Surprisingly, only a single residue, phenylalanine 32 (F32), from the alternatively spliced N-terminal region of FGF8b accounts for the higher receptor-binding affinity of FGF8b relative to FGF8a, and for the unique ability of FGF8b to transform midbrain into cerebellum. We also suggest that the intermediate receptor-binding affinity of FGF17b and FGF18, relative to FGF8a and FGF8b, accounts for the unique mid-hindbrain patterning abilities of these ligands.
Results and Discussion
To provide the molecular basis by which alternative splicing modulates the diverse biological activity of FGF8, including the organizing ability of FGF8 in the brain, we crystallized FGF8b in complex with the extracellular region of FGFR2c encompassing Ig domains 2 (D2) and 3 (D3). The FGF8b-FGFR2c structure was refined to 2.2-Å resolution with working and free R-values of 23.9% and 27.2%, respectively (Table 1). As expected based on previous FGF-FGFR crystal structures (for review, see Mohammadi et al. 2005), FGF8b interacts with D2, D3, and the interconnecting D2-D3 linker of FGFR2c (Fig. 1A). However, the structural basis by which FGF8 attains its unique pattern of FGFR-binding specificity/promiscuity is drastically different from that of other FGFs.
Table 1.
Summary of crystallographic analysis
| Data collection statistics | |||||
|---|---|---|---|---|---|
| Resolution (Å) 30.0-2.2 | Reflections (total/unique) 321931/44825 | Completeness (%) 100 (100)b | Rsyma (%) 8.4 (30.6)b | Signal (<I/σI>) 15.0 |
| Refinement statisticsc | |||||||
|---|---|---|---|---|---|---|---|
| Root-mean-square deviations
|
|||||||
| Resolution (Å) 25.0-2.2 | Reflections 43837 | Rcryst/Rfreed (%) 23.9%/27.2% | Bonds (Å) 0.006 | Angles (°) 1.4 | B-factorse (Å2) 1.3 | ||
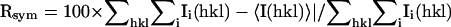 .
.
Value in parentheses is for the highest resolution shell: 2.28-2.2 Å.
Atomic model: 5200 protein atoms.
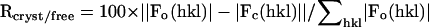 , where Fo (>0) and Fc are the observed and calculated structure factors, respectively. 15% of the reflections were used for calculation of Rfree.
, where Fo (>0) and Fc are the observed and calculated structure factors, respectively. 15% of the reflections were used for calculation of Rfree.
For bonded protein atoms.
Figure 1.

Overall features of the FGF8b-FGFR2c complex. (A) Molecular surface representation of the FGF8b-FGFR2c structure. D2 and the D2-D3 linker of receptor are colored green and gray, respectively. The constant N-terminal half of D3 and the alternatively spliced C-terminal half of D3 of receptor are colored cyan and purple, respectively. FGF8b is colored orange, and is also represented as a ribbon diagram. The only three residues from the alternatively spliced N-terminal region of FGF8b that are ordered are colored blue. Phenylalanine 32 from the alternatively spliced region of FGF8b is shown as sticks. F32 is a key player in the molecular mechanism by which alternative splicing modulates the biological activity of FGF8. In all figures, graphic representation of the FGF8b-FGFR2c complex is made using the copy of the FGF8b-FGFR2c complex in the asymmetric unit in which the βC′-βE loop of FGFR2c is ordered. (B) Ribbon representation of FGF8b from the FGF8b-FGFR2c structure. The β-strands of FGF8b are labeled according to the conventional strand nomenclature for FGF1 and FGF2. (NT) N terminus of FGF8b; (CT) C terminus of FGF8b; (gN) N-terminal helix of FGF8b. Intramolecular interactions that stabilize the unique conformation of the FGF8b N terminus and tether the N-terminal helix to the core are shown. Hydrogen bonds are shown as dashed black lines. The disulfide bridge between cysteines 109 (in β6) and 127 (in β8) is shown as a dashed green line, and serves to stabilize the conformation of the β7-β8 loop, which is one residue longer in FGF8 subfamily members vis-a-vis other FGFs. (C) The spatial positioning of the FGF8b N terminus relative to the β-trefoil core is opposite to that of FGF1 and FGF10 in their receptor-bound forms. This is also the case for the receptor-bound form of FGF2 (PDB identification code 1EV2; not shown for the sake of clarity). The view of FGF8b is the same as in B. The Cα trace of the FGF10 β-trefoil from the FGF10-FGFR2b structure (PDB identification code 1NUN) and of the FGF1 β-trefoil from the FGF1-FGFR2c structure (PDB identification code: 1DJS) were superimposed onto the Cα trace of the FGF8b β-trefoil. The surface of the FGF8b β-trefoil core is shown as gray mesh. The β1 strands and N termini of FGF8 (orange), FGF1 (green), and FGF10 (red) are shown as ribbons. The three ordered residues from the alternatively spliced region of FGF8b are colored blue. The N termini of FGF8, FGF1, and FGF10 are labeled NT.
Structural basis for the receptor-binding specificity/promiscuity of the FGF8 subfamily
The FGF8b core region adopts a β-trefoil fold consisting of 12 anti-parallel β-strands (β1-β12) (Fig. 1B). Compared with previous FGF structures (free or receptor-bound), a much larger portion of the FGF8b N terminus is ordered, which adopts a drastically different conformation (Fig. 1C). The FGF8b β-trefoil core also exhibits several unique structural features, most notably a one-residue insertion (S95) in the β4-β5 loop of FGF8b (Fig. 2A).
From the 11 N-terminal residues of FGF8b that comprise the alternatively spliced region of FGF8b (Fig. 2A), only the last three residues, F32, T33, and Q34, are ordered (Fig. 1B). These three residues, together with the following six residues from the constant region of FGF8, form a g helix (gN; residues 32-40) that is linked to the β-trefoil core by a nine-residue-long loop (Figs. 1B, 2A). The gN helix and the subsequent gN-β1 loop are tethered to the β4 strand and to the β4-β5 loop within the β-trefoil core via numerous hydrophobic contacts and hydrogen bonds (Fig. 1B), implying that the N terminus of FGF8b is rigid and adopts a similar structure in the absence of receptor.
The spatial positioning of the FGF8b N terminus relative to the globular β-trefoil core is completely opposite to that of FGF1, FGF2, and FGF10 in their receptor-bound structures. The unique spatial positioning of the FGF8b N terminus dictates the novel mode of FGF8b-FGFR-binding specificity (Fig. 1C) by placing the gN helix in the vicinity of a hydrophobic groove on receptor D3, into which F32 and V36 from the gN helix are inserted (Fig. 3). This hydrophobic D3 groove is also engaged by F93 at the tip of the β4-β5 loop of FGF8b, which is pushed into close proximity of the hydrophobic groove (Fig. 3) due to the unique one-residue insertion in the loop (Fig. 2A). F93 also engages in intramolecular hydrophobic contacts with V36 in the gN helix, which thereby facilitates optimal packing of the gN helix against the groove (Figs. 1B, 3).
Figure 3.

Contacts between the N-terminal helix and β4-β5 loop of FGF8b with the D3 hydrophobic groove of FGFR2c dictate FGF8-FGFR-binding specificity. Interactions are shown in stereo and coloring is the same as in Figure 1A. The molecular surfaces of the FGF8b residues that interact with the D3 groove are shown. Note that F32 is the only residue from the alternatively spliced region of FGF8b that interacts with FGFR. Note that the βC′-βE loop of FGFR2c (marked by an arrowhead) does not interact with FGF8b.
The hydrophobic D3 groove is located on the bottom β sheet layer of D3 and is formed by I291, L309, L343, I350, and F352 (Fig. 3). The methyl group of T341 also contributes to the hydrophobicity of this groove. I291 and L309 are on the βC and βC′ strands, respectively, in the first half of D3, which is shared between the “b” and “c” isoforms of a given FGFR. In contrast, T341, L343, I350, and F352 are on the βF and βG strands, which are located in the alternatively spliced second half of D3 (Figs. 2C, 3). Sequence alignment of the seven principal FGFRs indicates that the hydrophobic D3 groove will also be present in FGFR1c, FGFR3c, and FGFR4, whereas in the “b” isoforms of FGFR1-3 the hydophobicity of this groove should be significantly reduced (Fig. 2C). This is because two of the key constituents of the hydrophobic groove (L343 and I350) are replaced by polar residues in “b” isoforms of FGFR1-3 (Fig. 2C). Thus, based on the FGF8b-FGFR2c structure and sequence analysis, FGF8b should bind promiscuously to all three FGFRc isoforms and FGFR4, but should exhibit significantly reduced affinity to FGFRb isoforms. Hence, the FGF8b-FGFR2c structure reveals that the unique mode of FGF8 receptor-binding/specificity is mechanistically linked to the mode by which alternative splicing modulates the diverse biological activity of FGF8 isoforms.
A structure-based sequence alignment of FGF8 subfamily members shows that all the salient features of the FGF8b structure, including the conformation of the N terminus and the β4-β5 loop insertion, are conserved in both FGF17 isoforms and FGF18 (Fig. 2A). Thus, the FGF8b structure represents not only the common core structure of all FGF8 isoforms, but also that of all members of the FGF8 subfamily. Furthermore, 17 out of 20 residues in FGF8b that engage FGFR are conserved in FGF17b and FGF18, including the three key residues of FGF8b (F32, V36, and F93) that engage the hydrophobic groove in receptor D3 (Fig. 2A). Hence, FGF17b and FGF18 should exhibit a similar pattern of FGFR-binding specificity/promiscuity as that elucidated for FGF8b by the FGF8b-FGFR2c structure.
Structural basis by which alternative splicing regulates FGF8 biological activity
Of the three key hydrophobic residues of FGF8b (F32, V36, and F93) that engage the D3 groove of FGFR2c, only F32 resides within the alternatively spliced N-terminal region of FGF8b (Figs. 2A, 3). In fact, residues 23-31 of the alternatively spliced N-terminal region of FGF8b are disordered in our crystal structure, implying that these residues are not important for receptor binding. Based on the FGF8b-FGFR2c structure and sequence analysis, FGF8a should not be able to engage in the hydrophobic F32-D3 interaction, and consequently should bind more weakly than FGF8b to FGFR2c. Hence, based on our structural data, the biological role of alternative splicing is to modulate the receptor-binding affinity of FGF8 isoforms to FGFRc isoforms and FGFR4. Furthermore, FGF17 is spliced in a similar manner to FGF8, such that the “a” and “b” isoforms of these ligands correspond to one another (Fig. 2B). Importantly, the residue corresponding to F32 in FGF8b is conserved in FGF17b (Fig. 2A), and thus the structural mechanism by which alternative splicing modulates the biological activity of FGF8 should also apply to FGF17.
SPR analysis of receptor-binding specificity of FGF8 subfamily members confirms the structural data and shows that FGF8 is capable of interacting with FGFR1c
To validate our structural data, we characterized the FGFR-binding affinity/specificity of the three members of the FGF8 subfamily, including the “a” and “b” isoforms of FGF8, using SPR. The SPR data are summarized in Table 2, and representative sensograms are shown in Figure 4. The SPR data show that FGF8b, FGF17b, and FGF18 all bind to FGFR1c, FGFR2c, FGFR3c, and FGFR4, but not to the “b” isoforms of FGFR1-3 (Fig. 4; Table 2). Furthermore, FGF8a binds markedly more weakly than FGF8b to FGFR1c, FGFR2c, FGFR3c, and FGFR4 (Fig. 4; Table 2). The dissociation constants observed for each of these interactions fall within the range of dissociation constants that we have previously measured for other FGF-FGFR interactions (Ibrahimi et al. 2004a,b). Since the FGF8b ligand used to solve the FGF8b-FGFR2c structure lacks the C-terminal residues 186-215 (FGF8bCTR) (see Materials and Methods), we compared the receptor-binding affinities of FGF8bCTR with that of wild-type FGF8b using SPR. The SPR data show that FGF8bCTR binds to FGFR1c, FGFR2c, FGFR3c, and FGFR4 with dissociation constants that are comparable to those measured for FGF8bWT binding to these FGFRs (Table 2). Therefore, the SPR data demonstrate that our crystal structure captures all of the interactions that take place between full-length FGF8b and FGFR2c.
Table 2.
Summary of kinetic data
| FGF | FGFR1c | FGFR2c | FGFR3c | FGFR4 | FGFR2b | FGFR3b | |
|---|---|---|---|---|---|---|---|
| FGF8a | kon (/M/s)a | 1.24 × 104 | 1.67 × 104 | 4.22 × 103 | 1.44 × 104 | NBc | NB |
| koff (/s)a | 1.55 × 10−2 | 1.81 × 10−2 | 1.08 × 10−2 | 1.62 × 10−2 | NB | NB | |
| KD (M)b | 1.25 × 10−6 | 1.08 × 10−6 | 2.45 × 10−6 | 1.13 × 10−6 | NB | NB | |
| FGF8b | kon (/M/s) | 9.35 × 103 | 3.76 × 104 | 2.78 × 104 | 4.77 × 104 | NB | NB |
| koff (/s) | 1.18 × 10−3 | 5.84 × 10−3 | 3.77 × 10−3 | 0.0235 | NB | NB | |
| KD (M) | 1.26 × 10−7 | 1.55 × 10−7 | 1.36 × 10−7 | 4.92 × 10−7 | NB | NB | |
| FGF17 | kon (/M/s) | 1.77 × 104 | 7.34 × 104 | 8.02 × 103 | 2.25 × 104 | NB | NB |
| koff (/s) | 6.8 × 10−3 | 3.2 × 10−2 | 5.5 × 10−3 | 2.74 × 10−2 | NB | NB | |
| KD (M) | 3.82 × 10−7 | 4.32 × 10−7 | 6.87 × 10−7 | 1.22 × 10−6 | NB | NB | |
| FGF18 | kon (/M/s) | 1.39 × 104 | 6.07 × 104 | 1.16 × 104 | 4.45 × 104 | NB | NB |
| koff (/s) | 4.0 × 10−3 | 2.5 × 10−2 | 8.0 × 10−3 | 4.0 × 10−2 | NB | NB | |
| KD (M) | 2.84 × 10−7 | 4.11 × 10−7 | 7.02 × 10−7 | 9.1 × 10−7 | NB | NB | |
| FGF8bF32A | kon (/M/s) | 8.65 × 103 | 8.93 × 103 | 3.93 × 103 | 6.49 × 103 | NB | NB |
| koff (/s) | 9.61 × 10−3 | 9.29 × 10−3 | 5.14 × 10−3 | 2.37 × 10−2 | NB | NB | |
| KD (M) | 1.11 × 10−6 | 1.04 × 10−6 | 1.31 × 10−6 | 3.65 × 10−6 | NB | NB | |
| FGF8bCTR | kon (/M/s) | 9.66 × 103 | 4.13 × 104 | 2.08 × 104 | 2.41 × 104 | NB | NB |
| koff (/s) | 1.0 × 10−3 | 4.2 × 10−3 | 2.4 × 10−3 | 1.5 × 10−2 | NB | NB | |
| KD (M) | 1.05 × 10−7 | 1.03 × 10−7 | 1.14 × 10−7 | 6.12 × 10−7 | NB | NB | |
| FGF8bNTR | kon (/M/s) | 2.47 × 104 | 3.49 × 104 | 2.48 × 104 | 4.05 × 104 | NB | NB |
| koff (/s) | 1.87 × 10−3 | 6.88 × 10−3 | 3.81 × 10−3 | 2.27 × 10−2 | NB | NB | |
| KD (M) | 7.57 × 10−8 | 1.97 × 10−7 | 1.54 × 10−7 | 5.6 × 10−7 | NB | NB |
kon and koff were derived as described in Materials and Methods. ϰ2 was <10% in all cases.
The apparent affinity, KD is equal to koff/kon.
(NB) Negligible binding.
Figure 4.

SPR analysis of the interaction of FGF8 subfamily members with six of the seven principal FGFRs Representative sensorgrams of injections of 200 nM FGFR1 (A), 200 nM FGFR2c (B), 200 nM FGFR3c (C), and 200 nM FGFR4 (D), over a CM5 chip onto which FGF8b (red), FGF17 (green), FGF18 (blue), FGF8a (pink), or FGF8bF32A (cyan) have been immobilized. FGF8 subfamily members do not significantly interact with the “b” isoforms of FGFR2 and FGFR3, and therefore the sensograms for these interactions are not presented. The biosensor chip response is indicated on the Y-axis (ΔRU) as a function of time (X-axis) at 25°C. Kinetic data are summarized in Table 2.
Cumulatively, our SPR data confirm the mode of receptor-binding specificity for FGF8 subfamily members revealed by the FGF8b-FGFR2c structure. Our data also provide the first direct evidence that FGF8b binds to FGFR1c, which is consistent with and supports mounting genetic data indicating that FGF8b signals through FGFR1c during mid-hindbrain development (Chi et al. 2003; Trokovic et al. 2003; Scholpp et al. 2004). With the critical exception of the direct interaction between FGF8 subfamily members and FGFR1c, our SPR data are qualitatively consistent with published mitogenic data pertaining to the receptor-binding specificity of FGF8 subfamily members (MacArthur et al. 1995b; Xu et al. 2000). In addition, our data demonstrating that FGF8a binds with markedly lower affinity than FGF8b to FGFRc isoforms and FGFR4 (Table 2) provide a satisfying molecular explanation for the lower biological activity of FGF8a and FGF17a relative to FGF8b and FGF17b in mitogenic and cell transformation studies (MacArthur et al. 1995a,b; Xu et al. 2000). Specifically, the higher receptor-binding affinity of FGF8b relative to that of FGF8a should cause a higher degree of receptor activation, which translates into a stronger mitogenic or transforming signal.
Only F32 from the alternatively spliced region of FGF8b accounts for the higher receptor-binding affinity of FGF8b relative to FGF8a
The FGF8b-FGFR2c crystal structure suggests that the higher FGFR affinity of FGF8b compared with FGF8a is solely due to the interaction between F32 of FGF8b and the hydrophobic groove of receptor D3. To test this, we replaced F32 with alanine in FGF8b (FGF8bF32A) and analyzed binding of this mutant to FGFRs using SPR. Importantly, the F32A mutation markedly reduced the affinity of FGF8b towards FGFR1c, FGFR2c, FGFR3c, and FGFR4, effectively converting FGF8b to FGF8a (Fig. 4; Table 2). To exclude the possibility that the flexibility of residues 23-28 from the alternatively spliced region of FGF8b contributes to FGF8b-FGFR-binding affinity, we also measured FGFR-binding affinity of an FGF8b deletion mutant (FGF8bNTR), which lacks residues 23-28 from the alternatively spliced region. Our SPR data show that FGF8bNTR binds with the same specificity and affinity as FGF8bWT to FGFR1c, FGFR2c, FGFR3c, and FGFR4 (Fig. 4; Table 2). These data support our conclusions drawn from the crystal structure that differences in the receptor-binding affinity of FGF8a and FGF8b resulting from the alternative splicing-dependent F32-D3 interaction translate into the differential activities of these isoforms in mitogenesis and transformation assays.
Mutation of F32 in FGF8b results in a conversion of the mid-hindbrain patterning activity of FGF8b to that of FGF8a
Based on our data, we reasoned that the differences in receptor-binding affinity/signaling of FGF8a and FGF8b resulting from the alternative splicing-dependent FGF8b F32-D3 interaction are also responsible for the distinct mid-hindbrain organizing abilities of these two isoforms. To test our hypothesis, we studied the effect of the F32A mutation on the ability of FGF8b to induce morphological and gene expression changes in the developing chick midbrain. As demonstrated in previous studies (Sato et al. 2001; Liu et al. 2003) electroporation of mouse FGF8b cDNA at a concentration of 1 μg/mL into chick midbrain led to a morphological transformation of midbrain into cerebellum (n = 9/9; Fig. 5C), and to strong induction of Spry1 expression (n = 4/4) and repression of the midbrain-specific marker Otx2 (n = 4/4). Electroporation of FGF8a cDNA at the same concentration led to an overgrowth of the midbrain (n = 8/9; Fig. 5D), little or no induction of Spry1 expression (n = 2/3 no induction; 1/3 weak induction), and no repression of Otx2 (n = 3/3). Consistent with our structural and SPR data, electroporation of FGF8bCTR led to morphological transformation of midbrain into cerebellum (n = 2/3; Fig. 5E) and gene expression changes similar to FGF8b (repression of Otx2, n = 2/2; induction of Spry1, n = 2/2), demonstrating that deletion of these C-terminal residues does not affect the biological activity of FGF8b.
Figure 5.

Mutation of F32A in FGF8b abolishes the ability of FGF8b to morphologically transform chick midbrain into hindbrain. (A) Schematic of the experimental design of the in ovo electroporation experiments. Wild-type or mutant FGF8 cDNA (green) is injected with a glass needle into the neural tube at the embryonic mid-hindbrain region of chick embryos at the 10- to 11-somite stage. Only the right side of the midbrain is transfected. The left side of the midbrain is left untreated and serves as an internal control. (B) Dorsal view of a wild-type embryonic day 9.5 (E9.5) chicken brain, 30 h post-electroporation of a GFP expression vector. (Inset) Note the restricted expression of GFP fluorescence on the right side of the mid-hindbrain. (C-G) Dorsal views of E9.5 chicken brains after in ovo electroporation of the indicated FGF8 cDNAs. The red dashed line indicates the midline of the brain, and the asterisk indicates the conversion of midbrain to hindbrain on the transfected side (C,E,G), whereas the arrow indicates expansion of the midbrain on the transfected side (D,F). (Tel) Telencephalon; (Di) diencephalons; (Mid) midbrain; (Cb) cerebellum.
In contrast to FGF8b and FGF8bCTR, the FGF8bF32A mutant failed to transform midbrain into cerebellum (n = 4/4; Fig. 5F), and instead caused an overgrowth of the midbrain (n = 3/4; Fig. 5F). Also like FGF8a, FGF8bF32A failed to induce Spry1 (n = 2/2) expression and to repress Otx2 expression (n = 2/2). Thus, the data show that the mutation of F32A in FGF8b abolishes the ability of FGF8b to transform midbrain into hindbrain and to repress midbrain specific genes. Conversely, when the last four amino acids of the alternatively spliced region of FGF8b (31NFTQ34) were added in-frame to the N terminus of FGF8a, the resulting FGF8a chimera (FGF8aNFTQ) gained the ability to transform midbrain into hindbrain in a manner identical to FGF8b (n = 2/3; Fig. 5G). Taken together, these data exclude the possibility that flexibility of the residues preceding F32 in FGF8b contributes to receptor binding, and confirm that the presence or absence of the F32-D3 interaction alone accounts for the distinct morphological and gene expression changes induced by FGF8a and FGF8b.
To further confirm this conclusion, we implanted heparin-conjugated beads coated with FGF8a, FGF8b, or FGF8bF32A into mouse midbrain explants, and used in situ hybridization to assess the ability of these ligands to alter the expression patterns of genes implicated in mid-hindbrain patterning. Consistent with our previous studies, FGF8b robustly induced expression of Gbx2 (n = 23/25), Spry1 (n = 19/19), and En2 (n = 13/13) (Fig. 6G-I; Liu and Joyner 2001b; Liu et al. 2003). On the other hand, FGF8a failed to induce Gbx2 (11/11) and Spry1 (n = 6/6), but induced En2, albeit to a weaker degree than FGF8b (n = 9/14) (Fig. 6D-F). The results of this latter experiment, which is the first to analyze the effect of FGF8a on midbrain gene expression using the mouse brain explant assay, are consistent with the results of earlier studies of FGF8a activity in chick midbrain using electroporation (Sato et al. 2001; Liu et al. 2003). Importantly, introduction of the F32A mutation into FGF8b abolished the ability of FGF8b to induce Gbx2 (n = 6/6) and Spry1 (n = 5/5), without affecting its ability to induce En2 (n = 5/5) (Fig. 6J-L). These data further demonstrate that the F32A mutation functionally converts FGF8b into FGF8a with respect to both morphological and gene expression patterning abilities. Taken together, these data confirm our hypothesis that differences in the receptor-binding affinity of FGF8a and FGF8b resulting from the alternative splicing-dependent F32-D3 interaction are solely responsible for the distinct patterning abilities of these ligands. In addition to mid-hindbrain development, FGF8 activity in gastrulation and limb development is also likely to be modulated by alternative splicing, and our findings should provide a framework for testing the roles of various FGF8 isoforms in these processes.
Figure 6.

Mutation of F32A in FGF8b converts FGF8b into FGF8a with respect to gene expression changes induced by these ligands during mid-hindbrain development. Heparin-conjugated beads soaked in BSA (A-C), FGF8a (D-F), FGF8b (G-I), or FGF8bF32A (J-L) were implanted into midbrain explants and incubated for 48 h. Whole-mount RNA in situ analysis was then performed with probes for En2, Spry1, and Gbx2, as indicated. FGF8b strongly induces En2 (G), Spry1 (H), and Gbx2 (I); FGF8a weakly induces En1 (D) and Spry1 (E), and fails to induce Gbx2 (F). Note that the F32A mutation functionally converts FGF8b to FGF8a, as FGF8bF32A weakly induces En2 (J) and Spry1 (K) and fails to induce Gbx2 (L).
Distinct patterning abilities of FGF17b or FGF18 correlate with the intermediate receptor-binding affinities of FGF17 and FGF18 relative to FGF8a and FGF8b
Our SPR data also show for the first time that FGF8 subfamily members differ in their receptor-binding affinity. Specifically, FGF17b and FGF18 bind more weakly than FGF8b, but stronger than FGF8a, to FGFRc isoforms and FGFR4 (Fig. 4; Table 2). Consequently, these ligands should exhibit mid-hindbrain patterning activities that are intermediate to those of FGF8a and FGF8b. To test this, we compared the abilities of FGF17b and FGF18 to induce Gbx2, Spry1, and En2 in mouse midbrain explants with those of FGF8a and FGF8b. Consistent with our hypothesis, both FGF17b and FGF18 induced expression of Spry1 in some explants (n = 5/11 for FGF18; n = 6/15 for FGF17b), whereas FGF8a consistently failed to induce this gene. Further consistent with our hypothesis, FGF17b and FGF18 induced En2 (n = 12/14 for FGF18; n = 14/15 for FGF17b) with a greater efficiency than FGF8a did (n = 9/16). Importantly, implantation of FGF17b- or FGF18-coated heparin beads into mouse midbrain explants resulted in a low frequency of Gbx2 induction (n = 2/7 for FGF18; n = 2/8 for FGF17b), whereas FGF8a always failed to induce this gene (n =0/11). Collectively, our data demonstrate that members of the FGF8 subfamily and their splice isoforms exhibit a spectrum of receptor-binding affinities that account for the distinct patterning abilities of this biologically indispensable subfamily of FGFs. It is noteworthy that members of other FGF subfamilies also exhibit distinct biological activities, which may reflect subtle differences in the affinities of these subfamily members toward a common subset of FGFRs.
Distinct structural features of FGFs give rise to two mutually exclusive modes by which FGF-FGFR specificity/promiscuity is achieved
In the FGF8b-FGFR2c structure, the βF and βG strands, residing within the C-terminal half of the alternatively spliced region of receptor D3, harbor the major determinants of the FGF8b-binding specificity (Fig. 3). This is in contrast to our current working model for FGF-FGFR specificity, in which the βC′-βE loop located at the N-terminal region of the alternatively spliced region of receptor D3 harbors the major determinants of ligand-binding specificity (Fig. 7B; for review, see Mohammadi et al. 2005). Indeed, superimposition of FGF8b from the FGF8b-FGFR2c structure onto FGF2 in the FGF2-FGFR2c structure clearly shows that the unique path of the FGF8b N terminus results in a major clash with the βC′-βE loop of D3, and thus is incompatible with the previously elucidated mode of receptor-binding specificity/promiscuity of FGF1 and FGF2 (Fig. 7C).
Figure 7.

The structural mechanism by which FGF8b attains its receptor-binding specificity deviates from the more general mode described for FGF1, FGF2, and FGF10. Comparison of the overall mode of the FGF-D3 interaction observed in the FGF8b-FGFR2c (A) and in the FGF2-FGFR2c (PDB identification code 1EV2) (B) structures. The FGF2-FGFR2c structure is representative of the overall mode of receptor binding previously determined for FGF1, FGF2, and FGF10. In each panel, FGFR is colored as in Figure 1A, and the molecular surface of FGF is shown as a gray mesh. In B, unique hydrogen bonds between the βC′-βE loop of receptor and FGF2 that play a key role in determining FGF2-FGFR2c specificity are represented by dashed lines. The hydrophobic patch of FGF2 that interacts with the βC′-βE loop of receptor (B) and the corresponding patch of FGF8b (A) are shown in red mesh. The reduction of the red mesh area in FGF8b reflects the fact that G122 of FGF8b replaces V97 of FGF2 (indicated by green arrows). (C) The FGF8b mode of binding is incompatible with that of other FGFs. FGF8b from the FGF8b-FGFR2c structure was superimposed onto FGF2 in the FGF2-FGFR2c structure. Note that the N terminus of FGF8b runs directly into the βC′-βE loop of D3 due to opposite spatial positioning of the FGF8b N terminus relative to the β-trefoil core, compared with that of other FGFs (cf. the locations of the N termini of FGF8b and FGF2 in A and B; also refer to Fig. 1C).
Consistent with the lack of a role for the βC′-βE loop in determining FGF8b-FGFR-binding specificity/promiscuity, the βC′-βE loop of FGFR2c is disordered in one of the two FGF8-FGFR2c copies in the asymmetric unit, and the ordering of this loop in the other copy is mainly due to crystal lattice contacts. Thus, the FGF8b-FGFR2c structure reinforces our original hypothesis that the alternatively spliced βC′-βE loop of the receptor is highly flexible in the absence of contacts with the ligand (Olsen et al. 2004). We have previously suggested that the extent of a contact between a hydrophobic patch in the β-trefoil core region of FGF and a hydrophobic residue in the βC′-βE loop of receptor D3 is the main determinant of the local fold of the βC′-βE loop (Mohammadi et al. 2005). This patch formed by residues from the β7-β8 loop, β8 strand, and a third location that is ligand-dependent (β4 in FGF10, β5 in FGF1, or β6 in FGF2) (Figs. 2A, 7B). Contacts between the patch and the βC′-βE loop of receptor D3 facilitate the remainder of the specific contacts that take place between the loop and highly divergent regions FGF (including the FGF N terminus, the β4 strand, and the β4-β5 loop) (Fig. 7B). Interestingly, compared with all other FGFs, the hydrophobicity of this patch is dramatically reduced in FGF8 subfamily members. This is due to the fact that a glycine (G122 in FGF8) substitutes for a key constituent of the hydrophobic patch (V97 in FGF2) (Figs. 2A, 7A). The reduced hydrophobicity of the patch in FGF8 subfamily members agrees with the lack of a role for the βC′-βE loop in the determination of the receptor-binding specificity/promiscuity of this ligand (Fig. 7A).
It is noteworthy that in previous FGF-FGFR structures that contain FGFR1c, FGFR2c, or FGFR3c as the receptors, the hydrophobic groove in receptor D3 is also present (data not shown). However, in these structures that feature FGF1 or FGF2 as the ligands, only the upper edge of the groove interacts with hydrophobic residues that are generally present in the β4-β5 loop of these FGFs at the location corresponding to F93 of FGF8b (Fig. 2A). The smaller size of these residues, together with the shorter β4-β5 loop in FGF1 and FGF2, account for why only the upper edge of the groove is engaged in these other structures. Another major factor for why the bulk of the groove remains empty in these previous structures is that the spatial positioning of the N termini of these ligands is opposite to that of the FGF8b N terminus (Fig. 1C; see also Fig. 7, cf. A and B). Reciprocally, the opposite spatial positioning of the N termini of FGF1 and FGF2 relative to that of FGF8b allows for the hydrophobic contact between the patch of these FGFs with the βC′-βE loop of FGFR to take place (Fig. 7, cf. A and B). This interaction is the hallmark of the previously characterized structural mode of receptor-binding specificity of these ligands.
A key determinant of the novel mode by which FGF8b attains its receptor-binding specificity is that the spatial positioning of the FGF8b N terminus relative to the β-trefoil core is opposite to that of other FGFs (Fig. 1C). The FGF8b-FGFR2c structure shows that this is mainly due to the fact that the β1 strand of FGF8b is extended N-terminally, as it strand-pairs with β4 more extensively than in other FGF structures. Importantly, comparison of the FGF8b structure with previous FGF crystal structures reveals a correlation between the presence of a proline or glycine (residues with low propensity for β stranding) preceding β1, and a β1 strand that is aborted earlier than that of FGF8b (Fig. 2A). In addition to defining the N-terminal boundary of the β1 strand, these proline/glycine residues force the N termini of these other FGFs to turn back toward the opposite side of the β-trefoil core relative to FGF8b (Fig. 1C). Structure-based sequence analysis of the entire FGF family shows that proline/glycine residues are present near the predicted β1 strand region of most FGFs. Therefore, we predict that in most FGFs, the spatial positioning of the N terminus relative to the β-trefoil core will be opposite to that observed in FGF8b.
In conclusion, with the insights gained from the FGF8b-FGFR2c structure, it is apparent that structural differences at the N terminus as well as at the hydrophobic patch within the β-trefoil core of FGFs give rise to two mutually exclusive modes by which FGF-FGFR specificity/promiscuity is achieved: one general mode of FGF-FGFR specificity that is achieved via contacts with the βC′-βE loop of receptor, and another mode that is unique to FGF8 subfamily members that is achieved via contacts with the alternatively spliced βF and βG strands of receptor. Structure-based sequence analysis of FGF and FGFR in metazoans demonstrates that the distinct structural mode by which FGF8 attains its receptor binding and specificity is absolutely conserved throughout the vertebrate kingdom, and even appears to be conserved in Caenorhabditis elegans Egl17 and Egl15 (Fig. 2A).
The FGF8b-FGFR2c structure reveals that FGF8b binding induces a distinct rotation in receptor linker/D3: implications for receptor dimerization and signaling
By superimposing the ligands from the six previously elucidated FGF-FGFR structures featuring FGF1, FGF2, and FGF10 as the ligands, we have previously noted that FGF, receptor D3, and D2-D3 linker operate as one rigid body. We have shown that this is due to the formation of a highly conserved network of hydrogen bonds between FGF, D2-D3 linker, and the common βB′-βC loop of D3 in these different FGF-FGFR complexes (Plotnikov et al. 1999, 2000; Yeh et al. 2003; Olsen et al. 2004). As a main consequence of FGF-linker/D3 acting as a rigid body in these complexes, modeling studies show that the distance between the membrane insertion points of two FGFRs is similar (∼50 Å) in the dimers of these complexes. In contrast, superimposition of FGF8b from the FGF8b-FGFR2c structure onto FGFs from the six previous FGF-FGFR structures shows that the receptor D3 and linker region in the FGF8b-FGFR2c structure undergo a novel ligand-induced rotation, which pivots at the bottom end of D2. Our modeling studies show that this FGF8b-induced linker/D3 rotation places the membrane insertion points of individual FGFRs in an FGF8b-FGFR dimer closer to each other, by ∼15 Å relative to other FGF-FGFR dimers (Fig. 8A).
Figure 8.

FGF8b binding induces a unique rotation of receptor linker/D3. (A) A 2:2 FGF8b-FGFR2c dimer (green) was constructed by superimposing FGF8b from the FGF8b-FGFR2c structure onto each of the FGF2 molecules in the 2:2:2 FGF2-FGFR1c-heparin dimer (orange) (Schlessinger et al. 2000; PDB identification code 1FQ9). (B,C) Comparison of the network of hydrogen bonds that takes place between FGF8b and the linker/D3 region of the FGFR2c (B) with the universally conserved network observed in previous FGF-FGFR structures, as exemplified by the FGF2-FGFR2c structure (C). The structures are shown as ribbon diagrams and are colored as in Figure 1A. Two of the key constituents of the conserved network of hydrogen bonds in previous structures (N104 and Y106 in FGF2) are substituted by T139 and L141 in FGF8b (cf. B and C). The four novel hydrogen bonds observed in the FGF8b-FGFR2c structure (see text) are presented as yellow dashed lines. The remaining hydrogen bonds in the FGF8b-FGFR2c structure are colored black in B to indicate their homology to the hydrogen bonds observed in the previous structures, which are also shown in black in C. Note that E131 of FGF8b (indicated by a black triangle) assumes a different rotamer conformation than that observed in previous structures (indicated by a black triangle in FGF2) because of a novel intramolecular hydrogen bond with the FGF8 subfamily-specific K176. The altered composition and geometry of the network of hydrogen bonds in the FGF8b-FGFR2c structure induces the distinct rotation of D3. The FGF8b-D2 interface harbors a novel salt bridge between R177 from the β12 strand of FGF8b and the FGFR-invariant D247 on the βG strand of D2. Over half of the 18 FGFs have an arginine at the homologous position, suggesting that this salt bridge will also occur at the FGF-D2 interfaces of these ligands (Fig. 2A).
Analysis of the FGF8b-FGFR2c structure shows that the distinct FGF8b-induced D3 rotation is due to the mutation of the key residues that participate in the formation of the FGF-linker/D3 network of hydrogen bonds in other FGF-FGFR structures. These mutations in FGF8b alter both the composition and geometry of the aforementioned network of hydrogen bonds in the FGF8b-FGFR2c structure. The substitution of a highly conserved proline in the β12 strand with a lysine (K176), and the insertion of a serine (S95) in the β4-β5 loop of FGF8b appear to play a major role in the observed D3 rotation (Fig. 8B,C). These changes in FGF8b lead to the formation of four hydrogen bonds that are unique to the FGF8b-FGFR2c structure. It is noteworthy that many of the substitutions in FGF8b complement each other, such that these residues engage in intramolecular interactions that serve to reduce the entropy of the interacting side chains in a manner similar to that seen in previous structures (Fig. 8B,C; for review, see Mohammadi et al. 2005). This further underscores the universal role that this network of hydrogen bonds plays in providing general FGF-FGFR-binding affinity. Notably, all of the divergent FGF8b residues that contribute to the altered network of hydrogen bonds at the FGF-D3/linker interface are also conserved in human FGF17b and FGF18 (Fig. 2A), implying that these ligands will also induce linker/D3 rotation upon receptor binding. Moreover, these residues are also conserved throughout members of the vertebrate FGF8 subfamily, and even in Egl17 of C. elegans, suggesting that the FGF8-induced rotation of FGFR D3 is important for signaling by this subfamily of ligands.
Based on sequence and structural analysis, we suggest that subtle ligand-induced variations exist in the actual orientation of D3 among different FGF-FGFR dimers that may propagate to differences in spatial juxtapositioning of cytoplasmic kinase domains and ultimately modulate the efficiency of signaling by various FGF-FGFR pairs. Our proposal is corollary to a proposal by Wilson and coworkers who, based on a comparison of the crystal structures of erythropoietin receptor in complex with agonists and antagonists, suggested that variations in receptor conformation can serve as an on/off switch in receptor signaling (for review, see Wilson and Jolliffe 1999). It is tempting to speculate that the significantly closer positioning of the kinase domains in the FGF8b-FGFR2c dimer, relative to other FGF-FGFR dimers, may enable FGF8 to send the more robust signal that is required for proper patterning of the mid-hindbrain region during development.
Materials and methods
Protein expression and purification
The DNA fragments encoding full-length, mature FGF8b (residues 23-215), full-length FGF8a (residues 34-215), FGF17b (residues 27-216), and full-length FGF18 (residues 28-207) were amplified by PCR and subcloned into the pET-30a bacterial expression vector. PCR was also used to generate the expression constructs for an N-terminally truncated FGF8b (FGF8bNTR, residues 29-215), a C-terminally truncated FGF8bCTR (residues 23-186), and the F32A FGF8b mutant. Each of the FGFs was expressed in Escherichia coli as inclusion bodies, and was refolded and purified as described previously (Plotnikov et al. 2000). The minimal ligand-binding region of FGFRs, consisting of D2 and D3, were expressed in E. coli, refolded in vitro, and purified as previously described (Plotnikov et al. 2000). The FGF8b-FGFR2c complex was prepared by mixing the purified in vitro refolded FGFR2c ectodomain protein with a slight excess of FGF8b protein, and the 1:1 FGF8b-FGFR2c complex was isolated using a Superdex 200 gel filtration column.
Crystallization, structure determination, and refinement
Large, single crystals of FGF8bCTR in complex with P253R FGFR2c (hereafter referred to as FGF8b-FGFR2c) were grown at 20°C from a mixture of 1.5 μL of protein complex (7.5 mg/mL, 25 mM Hepes at pH 7.5, 450 mM NaCl) with 1.5 μL of crystallization buffer (18.5% polyethylene glycol 5000 [monomethyl ether], 0.15 M magnesium chloride, 0.1 M Tris at pH 7.5), using the hanging drop vapor diffusion method. FGFR2c harboring the Apert Syndrome mutation (P253R) exhibits a generalized enhancement in ligand-binding affinity without affecting the conformation of FGFR (Ibrahimi et al. 2004a), and was used to improve crystal quality. The FGF8b-FGFR2c crystals are in the C-centered monoclinic space group C2, with unit cell dimensions of a = 170.84 Å, b = 46.90 Å, c = 109.61 Å, and β = 91.66 Å. The crystals have a solvent content of 51%. The asymmetric unit contains two FGF8b-FGFR2c complexes. Crystals were flash-frozen in a final cryoprotectant solution composed of the mother liquor and 15% glycerol. Diffraction data were measured at NSLS beamline X4A. The data were processed using DENZO and SCALEPACK (Otwinowski and Minor 1997).
A molecular replacement solution for the two copies of FGF8b-FGFR2c complex in the asymmetric unit of the crystals was found with the program AMORE (Navaza 1994), using the FGF1-FGFR1c crystal structure (PDB ID code 1EVT) as the search model (Plotnikov et al. 2000). The program O (Jones et al. 1991) was used for model building into the 2Fo - Fc and Fo - Fc maps, and CNS (Brunger et al. 1998) was used for all refinements. The refined model for the FGF8b-FGFR2c structure is composed of two FGF8 molecules (residues 32-180), two FGFR2c molecules (residues 150-362), and 125 water molecules. In both FGF8 copies, the N-terminal residues 23-31 and the C-terminal residues 180-186 are disordered. Residues 295-306 (corresponding to the βC-βC′ loop in D3) in both copies of FGFR2c, and residues 315-323 (corresponding to the βC′-βE loop in D3) in one copy of FGFR2c are disordered.
SPR analysis
Real-time biomolecular interactions between FGF and FGFR were studied by SPR, using the BIAcore 3000 system (Biosensor AB). The FGF ligands were coupled onto a research-grade CM5 sensor chip (Biosensor AB) through their free amino groups as described previously (Ibrahimi et al. 2004b). FGF homologous factor (FHF) 1b, which is structurally related to FGFs but fails to bind FGFR, was used as a negative control and was immobilized to a density corresponding to 1000 response units (RU). FGFs were immobilized to similar RU levels based on their molecular masses (FGF8a = 1120 RU, FGF8b = 1186 RU, FGF17 = 1090 RU, FGF18 = 1038 RU, FGF8bF32A = 1227 RU, FGF8bCTR = 932 RU, FGF8bNTR = 1072 RU). Different concentrations of analyte (FGFR) were prepared in HBS-EP (BIAcore) and were injected over the FGF/FHF1b chip at a flow rate of 50 μL/min in HBS-EP buffer. Three minutes after each sample injection, running buffer was passed over the chip surface to monitor the dissociation phase (180 sec). The kinetic parameters for each FGF-FGFR interaction were determined by globally fitting the data to a 1:1 interaction using BIAEVALUATION software (Biosensor AB). A minimum of four different sensograms was used for fitting, and each sensogram was manually examined for data quality and closeness of fit. A χ2 of 10% or less was accepted for each fit.
Mouse explant cultures and in situ hybridizations
Mouse brain explant cultures and in situ hybridizations were carried out as previously described (Liu et al. 1999). Briefly, explants were exposed for 40 h to heparin-conjugated acrylic beads soaked in the indicated FGF and subjected to automated in situ hybridization using our whole-mount protocol (http://saturn.med.nyu.edu/research/dg/joynerlab/protocols.html).
Chick electroporation studies
Fgf8 cDNAs (wild-type or mutant) were cloned into a chicken expression vector, pMiwIII, under the control of the chicken β-actin promoter. Chick electroporation assays were performed as described previously (Liu et al. 2003). Briefly, 1.0 μg/μL of the indicated expression vector together with 0.5 μg/μL of pMiwIII-EGFP (to monitor transfection efficiency) were injected into the mesencephalon of stage 9-12 chicken embryos and electroporated into the right side of the neural tube with five rectangular electric pulses of 18 V and 50-msec intervals. Embryos were dissected after 7 d (E9.5) for morphological analysis.
Acknowledgments
We are grateful to Drs. R. Abramowitz and J. Schwanof for synchrotron beamline assistance, Y. Lu (New York University Protein Analysis Facility) for mass spectrometric analysis, and Dr. R. Goetz for critically reading the manuscript. Beamline X4A at the National Synchrotron Light Source is a Department of Energy facility. This work was supported by National Institute of Health grant DE13686 (M.M.). S.K.O. is partially supported by the Training in Pharmacological Sciences (NIH/NIGMS T32 GM066704). A.L.J. is an HHMI investigator. J.Y.H.L. was partially supported by a National Institutes of Health postdoctoral fellowship. The atomic coordinates and structure factors have been deposited into the Protein Data Bank under accession code 2FDB.
Article published online ahead of print. Article and publication date are at http://www.genesdev.org/cgi/doi/10.1101/gad.1365406.
References
- Brunger, A.T., Adams, P.D., Clore, G.M., DeLano, W.L., Gros, P., Grosse-Kunstleve, R.W., Jiang, J.S., Kuszewski, J., Nilges, M., Pannu, N.S., et al. 1998. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54: 905-921. [DOI] [PubMed] [Google Scholar]
- Chellaiah, A., Yuan, W., Chellaiah, M., and Ornitz, D.M. 1999. Mapping ligand binding domains in chimeric fibroblast growth factor receptor molecules. Multiple regions determine ligand binding specificity. J. Biol. Chem. 274: 34785-34794. [DOI] [PubMed] [Google Scholar]
- Chi, C.L., Martinez, S., Wurst, W., and Martin, G.R. 2003. The isthmic organizer signal FGF8 is required for cell survival in the prospective midbrain and cerebellum. Development 130: 2633-2644. [DOI] [PubMed] [Google Scholar]
- Crossley, P.H. and Martin, G.R. 1995. The mouse Fgf8 gene encodes a family of polypeptides and is expressed in regions that direct outgrowth and patterning in the developing embryo. Development 121: 439-451. [DOI] [PubMed] [Google Scholar]
- Crossley, P.H., Martinez, S., and Martin, G.R. 1996a. Midbrain development induced by FGF8 in the chick embryo. Nature 380: 66-68. [DOI] [PubMed] [Google Scholar]
- Crossley, P.H., Minowada, G., MacArthur, C.A., and Martin, G.R. 1996b. Roles for FGF8 in the induction, initiation, and maintenance of chick limb development. Cell 84: 127-136. [DOI] [PubMed] [Google Scholar]
- Gemel, J., Gorry, M., Ehrlich, G.D., and MacArthur, C.A. 1996. Structure and sequence of human FGF8. Genomics 35: 253-257. [DOI] [PubMed] [Google Scholar]
- Hoshikawa, M., Ohbayashi, N., Yonamine, A., Konishi, M., Ozaki, K., Fukui, S., and Itoh, N. 1998. Structure and expression of a novel fibroblast growth factor, FGF-17, preferentially expressed in the embryonic brain. Biochem. Biophys. Res. Commun. 244: 187-191. [DOI] [PubMed] [Google Scholar]
- Ibrahimi, O.A., Zhang, F., Eliseenkova, A.V., Itoh, N., Linhardt, R.J., and Mohammadi, M. 2004a. Biochemical analysis of pathogenic ligand-dependent FGFR2 mutations suggests distinct pathophysiological mechanisms for craniofacial and limb abnormalities. Hum. Mol. Genet. 13: 2313-2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahimi, O.A., Zhang, F., Eliseenkova, A.V., Linhardt, R.J., and Mohammadi, M. 2004b. Proline to arginine mutations in FGF receptors 1 and 3 result in Pfeiffer and Muenke craniosynostosis syndromes through enhancement of FGF binding affinity. Hum. Mol. Genet. 13: 69-78. [DOI] [PubMed] [Google Scholar]
- Jones, T.A., Zou, J.Y., Cowan, S.W., and Kjeldgaard, G. 1991. Improved methods for binding protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47 (Pt 2): 110-119. [DOI] [PubMed] [Google Scholar]
- Joyner, A.L., Liu, A., and Millet, S. 2000. Otx2, Gbx2 and Fgf8 interact to position and maintain a mid-hindbrain organizer. Curr. Opin. Cell Biol. 12: 736-741. [DOI] [PubMed] [Google Scholar]
- Lee, S.M., Danielian, P.S., Fritzsch, B., and McMahon, A.P. 1997. Evidence that FGF8 signalling from the midbrain-hindbrain junction regulates growth and polarity in the developing midbrain. Development 124: 959-969. [DOI] [PubMed] [Google Scholar]
- Liu, A. and Joyner, A.L. 2001a. Early anterior/posterior patterning of the midbrain and cerebellum. Annu. Rev. Neurosci. 24: 869-896. [DOI] [PubMed] [Google Scholar]
- ____. 2001b. EN and GBX2 play essential roles downstream of FGF8 in patterning the mouse mid/hindbrain region. Development 128: 181-191. [DOI] [PubMed] [Google Scholar]
- Liu, A., Losos, K., and Joyner, A.L. 1999. FGF8 can activate Gbx2 and transform regions of the rostral mouse brain into a hindbrain fate. Development 126: 4827-4838. [DOI] [PubMed] [Google Scholar]
- Liu, A., Li, J.Y., Bromleigh, C., Lao, Z., Niswander, L.A., and Joyner, A.L. 2003. FGF17b and FGF18 have different midbrain regulatory properties from FGF8b or activated FGF receptors. Development 130: 6175-6185. [DOI] [PubMed] [Google Scholar]
- MacArthur, C.A., Lawshe, A., Shankar, D.B., Heikinheimo, M., and Shackleford, G.M. 1995a. FGF-8 isoforms differ in NIH3T3 cell transforming potential. Cell Growth Differ. 6: 817-825. [PubMed] [Google Scholar]
- MacArthur, C.A., Lawshe, A., Xu, J., Santos-Ocampo, S., Heikinheimo, M., Chellaiah, A.T., and Ornitz, D.M. 1995b. FGF-8 isoforms activate receptor splice forms that are expressed in mesenchymal regions of mouse development. Development 121: 3603-3613. [DOI] [PubMed] [Google Scholar]
- Maruoka, Y., Ohbayashi, N., Hoshikawa, M., Itoh, N., Hogan, B.L., and Furuta, Y. 1998. Comparison of the expression of three highly related genes, Fgf8, Fgf17 and Fgf18, in the mouse embryo. Mech. Dev. 74: 175-177. [DOI] [PubMed] [Google Scholar]
- Meyers, E.N., Lewandoski, M., and Martin, G.R. 1998. An Fgf8 mutant allelic series generated by Cre- and Flp-mediated recombination. Nat. Genet. 18: 136-141. [DOI] [PubMed] [Google Scholar]
- Mohammadi, M., Olsen, S.K., and Ibrahimi, O.A. 2005. Structural basis for fibroblast growth factor receptor activation. Cytokine Growth Factor Rev. 16: 107-137. [DOI] [PubMed] [Google Scholar]
- Navaza, J. 1994. AMoRe: An automated package for molecular replacement. Acta Crystallogr. A 50: 157-163. [Google Scholar]
- Olsen, S.K., Ibrahimi, O.A., Raucci, A., Zhang, F., Eliseenkova, A.V., Yayon, A., Basilico, C., Linhardt, R.J., Schlessinger, J., and Mohammadi, M. 2004. Insights into the molecular basis for fibroblast growth factor receptor autoinhibition and ligand-binding promiscuity. Proc. Natl. Acad. Sci. 101: 935-940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski, Z. and Minor, W. 1997. Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276: 307-326. [DOI] [PubMed] [Google Scholar]
- Plotnikov, A.N., Schlessinger, J., Hubbard, S.R., and Mohammadi, M. 1999. Structural basis for FGF receptor dimerization and activation. Cell 98: 641-650. [DOI] [PubMed] [Google Scholar]
- Plotnikov, A.N., Hubbard, S.R., Schlessinger, J., and Mohammadi, M. 2000. Crystal structures of two FGF-FGFR complexes reveal the determinants of ligand-receptor specificity. Cell 101: 413-424. [DOI] [PubMed] [Google Scholar]
- Reifers, F., Bohli, H., Walsh, E.C., Crossley, P.H., Stainier, D.Y., and Brand, M. 1998. Fgf8 is mutated in zebrafish acerebellar (ace) mutants and is required for maintenance of midbrain-hindbrain boundary development and somitogenesis. Development 125: 2381-2395. [DOI] [PubMed] [Google Scholar]
- Sato, T. and Nakamura, H. 2004. The Fgf8 signal causes cerebellar differentiation by activating the Ras-ERK signaling pathway. Development 131: 4275-4285. [DOI] [PubMed] [Google Scholar]
- Sato, T., Araki, I., and Nakamura, H. 2001. Inductive signal and tissue responsiveness defining the tectum and the cerebellum. Development 128: 2461-2469. [DOI] [PubMed] [Google Scholar]
- Sato, T., Joyner, A.L., and Nakamura, H. 2004. How does Fgf signaling from the isthmic organizer induce midbrain and cerebellum development? Dev. Growth Differ. 46: 487-494. [DOI] [PubMed] [Google Scholar]
- Schlessinger, J., Plotnikov, A.N., Ibrahimi, O.A., Eliseenkova, A.V., Yeh, B.K., Yayon, A., Linhardt, R.J., and Mohammadi, M. 2000. Crystal structure of a ternary FGF-FGFR-heparin complex reveals a dual role for heparin in FGFR binding and dimerization. Mol. Cell 6: 743-750. [DOI] [PubMed] [Google Scholar]
- Scholpp, S., Groth, C., Lohs, C., Lardelli, M., and Brand, M. 2004. Zebrafish fgfr1 is a member of the fgf8 synexpression group and is required for fgf8 signalling at the midbrain-hindbrain boundary. Dev. Genes Evol. 214: 285-295. [DOI] [PubMed] [Google Scholar]
- Sun, X., Meyers, E.N., Lewandoski, M., and Martin, G.R. 1999. Targeted disruption of Fgf8 causes failure of cell migration in the gastrulating mouse embryo. Genes & Dev. 13: 1834-1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka, A., Miyamoto, K., Minamino, N., Takeda, M., Sato, B., Matsuo, H., and Matsumoto, K. 1992. Cloning and characterization of an androgen-induced growth factor essential for the androgen-dependent growth of mouse mammary carcinoma cells. Proc. Natl. Acad. Sci. 89: 8928-8932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trokovic, R., Trokovic, N., Hernesniemi, S., Pirvola, U., Vogt Weisenhorn, D.M., Rossant, J., McMahon, A.P., Wurst, W., and Partanen, J. 2003. FGFR1 is independently required in both developing mid- and hindbrain for sustained response to isthmic signals. EMBO J. 22: 1811-1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson, I.A. and Jolliffe, L.K. 1999. The structure, organization, activation and plasticity of the erythropoietin receptor. Curr. Opin. Struct. Biol. 9: 696-704. [DOI] [PubMed] [Google Scholar]
- Xu, J., Lawshe, A., MacArthur, C.A., and Ornitz, D.M. 1999. Genomic structure, mapping, activity and expression of fibroblast growth factor 17. Mech. Dev. 83: 165-178. [DOI] [PubMed] [Google Scholar]
- Xu, J., Liu, Z., and Ornitz, D.M. 2000. Temporal and spatial gradients of Fgf8 and Fgf17 regulate proliferation and differentiation of midline cerebellar structures. Development 127: 1833-1843. [DOI] [PubMed] [Google Scholar]
- Yeh, B.K., Igarashi, M., Eliseenkova, A.V., Plotnikov, A.N., Sher, I., Ron, D., Aaronson, S.A., and Mohammadi, M. 2003. Structural basis by which alternative splicing confers specificity in fibroblast growth factor receptors. Proc. Natl. Acad. Sci. 100: 2266-2271. [DOI] [PMC free article] [PubMed] [Google Scholar]