

Abstract
Objective:
To discuss the diagnosis and management of primary carcinoid tumors of the liver in light of our experience and a literature review.
Summary Background Data:
Carcinoid tumors of the liver are rare and pose a diagnostic and management dilemma. This series is the largest reported and the only one to include liver transplantation as a treatment option.
Methods:
Between March 1994 and May 2002, we treated 8 patients (4 male, 4 female) with primary hepatic carcinoid tumors. Carcinoid syndrome complicated only 1 of the cases. Treatment was by liver resection in 6 patients and orthotopic liver transplantation in 2.
Results:
The diagnosis was confirmed histologically with light microscopy and immunohistochemistry in the absence of an alternative primary site. Six patients remain alive and disease free after follow-up of more than 3 years: 39, 43, 45, 50, 50, and 95 months. Two patients are recently postoperative.
Conclusions:
Active exclusion of an extrahepatic primary site is essential for the diagnosis of primary carcinoid of the liver. The mainstay of treatment should be liver resection, although liver transplantation may be considered in patients with widespread hepatic involvement. A radical surgical approach is warranted as this disease carries a better prognosis than for other primary hepatic tumors and for secondary hepatic carcinoids.
Primary hepatic carcinoid tumors are rare and difficult to diagnose, with fewer than 60 cases reported elsewhere. The diagnosis is important as long-term disease-free survival can be achieved by a combination of aggressive surgery and vigilant follow-up. Hepatic resection and transplantation are valid treatment options for these tumors.
The term “carcinoid” is a generic name for tumors derived from neuroendocrine cells and was first coined by Oberndorfer in 1907.1 Ninety per cent of carcinoid tumors occur within the gastrointestinal tract,2 most commonly in the appendix and the terminal ileum. The liver is a common site for carcinoid metastases. Conversely, primary hepatic carcinoid tumor is a very rare entity, with fewer than 60 cases reported in the English language literature.3–32 Most of these are single case reports, although 4 series, of 6 patients,7 5 patients,28 4 patients,20 and 3 patients29 have been published.
The diagnosis of primary hepatic carcinoid tumor is based principally on the histopathological confirmation of carcinoid tumor and the exclusion of a nonhepatic primary tumor. This requires preoperative imaging but most importantly a thorough laparotomy and rigorous follow-up. The suggested diagnostic process is outlined in the flow diagram (Fig. 1).

FIGURE 1. Flow diagram representing the suggested diagnostic pathway for primary hepatic carcinoid tumor.
In this paper, we describe the clinical course and management of 8 cases of primary hepatic carcinoid tumor diagnosed in accordance with our protocol. These patients have presented to our center during the past 8 years and form the largest single center report, and the only report to include transplantation as a treatment option.
PATIENTS AND METHODS
Between 1994 and 2002, 8 patients underwent liver resection or transplantation for primary hepatic carcinoid at our Hepatobiliary Unit. Our unit serves a population of approximately 4 million for liver resection (90–110 liver resections each year currently) and 9 million for transplantation (110–130 liver transplants each year currently). These patients form part of our prospective database for liver tumor patients. The case records of these patients were reviewed, and the clinical notes, operative records, and imaging and pathology data were examined. There were 4 females and 4 males, ranging in age from 31 to 65 years. All were white and 7 of the 8 presented from our local catchment area for liver tumors/transplantation. All were assessed and operated upon by the senior author (J.P.A.L.). During this time, our unit also treated 36 patients with secondary hepatic carcinoid: 14 by liver resection, 4 by liver transplantation, 2 by multivisceral transplantation, and 16 by nonsurgical therapies.
Table 1 summarizes the modes of presentation, biochemical findings, and the surgical treatment of these patients. The histologic and immunohistochemical findings of the resected tumors are summarized in Table 2. A summary of the history of each patient is given below.
TABLE 1. Patient Details

TABLE 2. Macroscopic, Microscopic, and Immunohistochemical Findings

Patient One
A 40-year-old woman presented with abdominal distension and pain. Computed tomography (CT) and magnetic resonance imaging (MRI) demonstrated a 12-cm centrally placed liver tumor, of borderline resectability. A percutaneous biopsy was taken and this showed neuroendocrine tumor, evidenced by staining with chromogranin, protein gene product (PGP) 9.5 and neuron-specific enolase (NSE). Contrast-enhanced CT, MRI, and a thorough examination at laparotomy did not reveal evidence of a primary lesion elsewhere. Intraoperative ultrasonography demonstrated that the tumor had extensively involved all 3 hepatic veins, and it was regarded to be unresectable. Celiac lymph nodes, however, were negative for tumor on frozen section examination, and she was listed for urgent liver transplantation. The following day, she underwent total hepatectomy with radical celiac lymphadenectomy and orthotopic liver transplantation. Postoperative recovery was uneventful and she was discharged on day 14. Pathologic examination showed a 12-cm tumor with several satellite nodules. Tumor cells were also seen within the portal and hepatic vein branches, but there was no lymph node disease. The patient remains alive and well at 8 years following surgery, with no evidence of tumor on serial CT and somatostatin scan assessments.
Patient Two
A 37-year-old woman presented with a 2-year history of right upper abdominal pain and intermittent flushing. Past history included an appendicectomy 12 years previously. Liver function tests were normal. CT showed an 8-cm tumor in the left lobe of the liver. An ultrasound-guided biopsy showed features characteristic of a neuroendocrine tumor. Upper and lower gastrointestinal endoscopy, contrast-enhanced CT, bone scan, and mammography were normal. A somatostatin scan showed intense uptake in the region of the tumor only (Fig. 2). At laparotomy, a small nodule was found in the appendix stump and a right hemicolectomy, left hemi-hepatectomy (resection of segments 2, 3, and 4), and celiac lymph node sampling were performed. Histology and immunohistochemistry of the liver tumor confirmed the findings of the biopsy and revealed node-negative disease. The nodule at the appendix stump showed fibrosis but no neoplasia. The histology from the original appendicectomy was reviewed and revealed no features of carcinoid tumor. One year later, she required surgical exploration and adhesiolysis for subacute small bowel obstruction. A small nodule was noted in the ileum and was resected with a 5-cm length of intestine in case it was a primary tumor missed previously. Histopathological analysis showed only fibrous tissue associated with adhesion formation, and there was no evidence of carcinoid tumor. The patient remains alive and disease free 4 years after the initial surgery, evidenced by serial CT and somatostatin scanning.
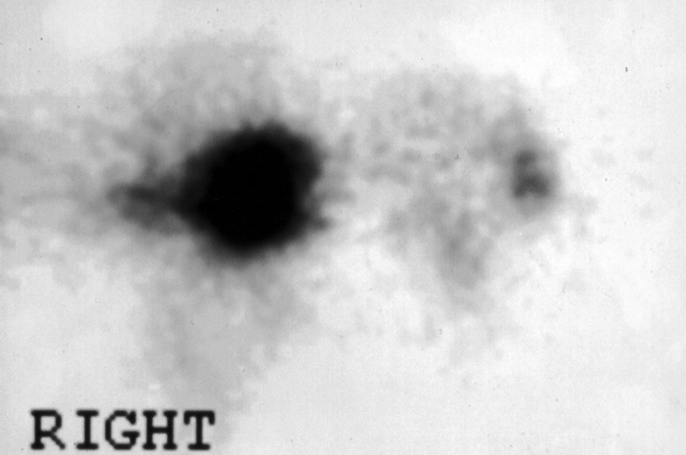
FIGURE 2. A somatostatin scan shows an intense uptake in the region of a large tumor in the left liver (patient 2).
Patient Three
A 31-year-old Croatian woman was referred with a scan-detected hepatic tumor on a background of hepatitis B and C liver disease. Liver function tests, alpha-fetoprotein (AFP), carcinoembryonic antigen (CEA), CA-125, and CA19-9 were all normal. MRI showed a hypervascular lobulated mass in segment 8. Isotope bone scan and mammography were normal. Percutaneous biopsies obtained elsewhere were reported as hepatocellular carcinoma, but on review with immunohistochemistry this was revised to neuroendocrine tumor at our center. At operation, a large tumor was found obliterating segment 1 and much of segments 4, 5, and 8 of the liver. A full laparotomy revealed no other primary site in the abdomen. She underwent a right hepatic trisectionectomy with caudate lobectomy (resection of segments 1, 4, 5, 6, 7, and 8), bile duct excision, celiac lymphadenectomy, and hepaticojejunostomy to the segment 2/3 hepatic duct. Histologic analysis of the liver showed a solitary 8-cm tumor with metastatic spread to the hepatic artery lymph node but not to celiac or peripancreatic lymph nodes. The postoperative recovery was complicated by sepsis and portal vein thrombosis, and she required surgical exploration on day 17. A further laparotomy 2 days later for a marked abdominal pain and distension revealed gross colonic pseudo-obstruction and a cecostomy was performed. She returned to Croatia 4 months post surgery and her cecostomy was closed 2 months later. At 4 years, she is well and disease free.
Patient Four
A 50-year-old man was referred with a 6-week history of right upper abdominal pain and mild hepatomegaly. There were no clinical features of chronic liver disease. Liver function tests showed an obstructed picture and CA19-9 was raised, presumed to be secondary to the biliary obstruction. Serum concentrations of AFP and CEA were normal. Abdominal ultrasonography showed a heterogeneous mass around the porta hepatis extending into the left lobe of liver. A targeted liver biopsy showed features of carcinoma with the possibility of a neuroendocrine lesion; there was insufficient tissue for immunohistochemistry. Imaging (Fig. 3) revealed a 12 × 10-cm tumor enveloping both left and right branches of the portal vein, hepatic artery, and hepatic ducts, but no extrahepatic disease was detected. An octreotide (somatostatin) isotope scan showed a reduced uptake by the tumor compared with the surrounding liver, therefore making a neuroendocrine tumor an unlikely diagnosis. A laparotomy with a view to a left hepatic trisectionectomy revealed an unresectable tumor involving hepatic segments 2–7. There was also a 6-mm nodule in the mid small bowel, which was resected as a possible primary. A tumor biopsy was taken which suggested a neuroendocrine tumor of the liver. The small bowel lesion was a benign stromal tumor. The patient underwent an orthotopic liver transplantation 3 months later with an uneventful recovery. The liver contained a single, solid, white, central tumor displacing structures at the porta hepatis. The patient is currently alive and disease free 45 months posttransplantation, evidenced by CT scans.
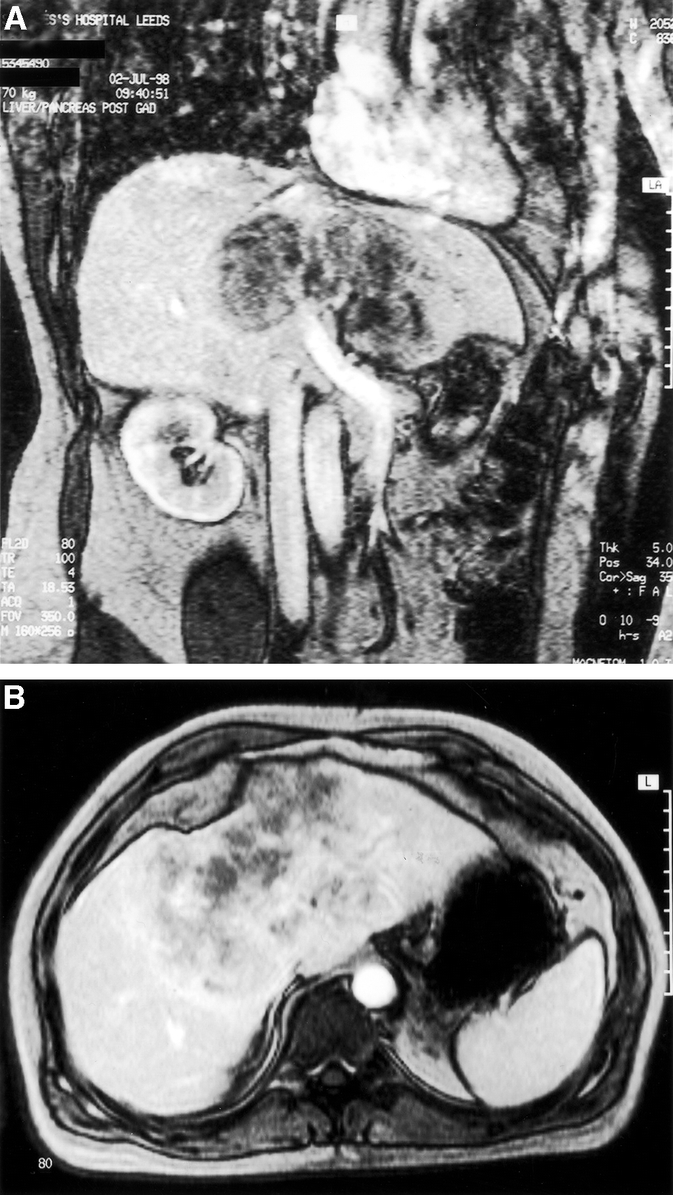
FIGURE 3. Coronal (A) and transverse (B) section magnetic resonance imaging demonstrates a 12 × 10-cm tumor enveloping both left and right branches of the portal vein, hepatic artery, and hepatic ducts (patient 4), for which an orthotopic liver transplant was performed.
Patient Five
A 40-year-old man presented to another hepatobiliary unit in the United Kingdom with a short history of malaise and jaundice. Endoscopic retrograde cholangiopancreatography showed a hilar stricture consistent with a cholangiocarcinoma. He underwent bile duct resection and a Roux-en-Y hepaticojejunostomy over 2 U-tubes. Histology was reported to show a hepatocellular carcinoma with positive hepatic duct resection margins, and he was subsequently treated by endoluminal postoperative brachytherapy with iridium wires inserted via the U-tubes. His follow-up was uneventful until 6 years later when serum AFP concentration increased to 14 kU/L (normal <10.0 kU/L), his liver function deteriorated and a CT scan revealed a 6 × 8-cm mass in the left liver. He was referred to our unit where further scanning did not show any extrahepatic disease. He underwent a modified left hepatic trisectionectomy with caudate lobectomy (resection of hepatic segments 1, 2, 3, 4, 5, 8, and much of segment 7) with en bloc resection of the majority of segment 7 and a Roux-en-Y hepaticojejunostomy to the residual right posterior sectional hepatic duct. Postoperative recovery was uneventful. The resection specimen contained a 6.5-cm tumor extending into the hepatic resection margin, and into the small bowel wall at the site of the previous hepaticojejunostomy. The tumor from the first resection 6 years previously was reexamined and found to be a neuroendocrine tumor on immunohistochemical analysis. At 26 months post surgery, a routine follow-up CT scan showed a lesion in the liver remnant. This was resected; however, histology showed only a secondary sclerosing cholangitis with no evidence of malignancy. The patient remains alive and disease free (serial CT scanning) 43 months following surgery.
Patient Six
A 63-year-old man was admitted acutely with a 2-week history of diarrhea, jaundice, lethargy, and pruritis. Liver function tests showed a serum bilirubin of 87 mmol/L (normal 3–15 mmol/L), ALT of 407 IU/L (normal <35 IU/L), and alkaline phosphatase of 2002 IU/L (normal 70–300 IU/L). The serum concentration of CA19-9 was markedly elevated at 1589 IU/mL (normal <37 IU/mL), presumed to be secondary to the biliary obstruction. CEA and AFP were normal. MRI scan showed a 3-cm hilar mass adjacent to the portal vein, suggestive of a cholangiocarcinoma. At operation, a polypoid tumor was identified at the common hepatic duct bifurcation extending up the right and left hepatic ducts. There was no evidence of extrahepatic disease. The bile duct was divided at the upper border of the pancreas and removed en bloc with a right trisectionectomy and caudate lobectomy (segments 1, 4, 5, 6, 7, and 8). A celiac lymphadenectomy was also performed. A Roux-en-Y hepaticojejunostomy to the segment 2/3 hepatic duct was created. Postoperative recovery was uneventful. This tumor was sausage shaped, solid and yellow, ramifying within and distending the right hepatic duct tributaries, with neither invasive component nor nodal metastases. This patient remains alive and disease free 39 months from surgery, evidenced by serial CT and somatostatin scans.
Patient Seven
A 56-year-old man was admitted with a short history of jaundice, in a poor clinical state with impending liver failure. His history dated back 11 years, in that in 1991 he had undergone surgery for a sigmoid volvulus and a 5-cm centrally located liver mass had been detected. There was no evidence of a primary tumor in the resected large bowel, and a subsequent liver biopsy suggested the tumor was carcinoid. At that time, he visited a number of hepatic surgeons and it was felt that the tumor was inoperable and he was lost to follow-up. Liver MRI demonstrated that the tumor had enlarged to 15 cm in diameter, primarily involving the right liver, but with significant extension into the left liver and there was bilateral biliary obstruction (Fig. 4). A possible solitary spinal metastasis in the transverse process of T7 was identified, but there was no other evidence of extrahepatic disease (CT head, chest, abdomen, pelvis, and isotope bone scan). His clinical condition spontaneously improved and the patient underwent urgent laparotomy. At surgery, the tumor appeared to be virtually inoperable as hepatic segments 2 and 3 were very small (with distorted anatomy and segment 4 hypertrophy), there was tumor invading the portal hilum, and the mass was largely overhanging the hilum making dissection hazardous. A 2.8-kg mass was removed by right hemihepatectomy (segments 5–8) with resection of much of segment 4 and the external biliary tree, with reconstruction by Roux-en-Y hepaticojejunostomy. Anesthesia was complicated by profound vasodilatation, requiring the use of norepinephrine and massive transfusion despite minimal blood loss (with recovery of vasoactive tone within 4 hours of completion of surgery). Postoperative recovery was complicated by recurrent sepsis and portosystemic encephalopathy. A laparotomy was required on the 10th postoperative day for unexplained coma, which resolved within hours after decompression of distended small intestine, and we attribute this to bacterial translocation or to cholangitis as a result of reflux up the Roux. A prolonged paralytic ileus resulted in ascending cholangitis at 20 days, again causing encephalopathy and depressed consciousness but responding to antibiotics and enemas. The patient is currently disease free on CT scanning at 4 months. He awaits spinal surgery.
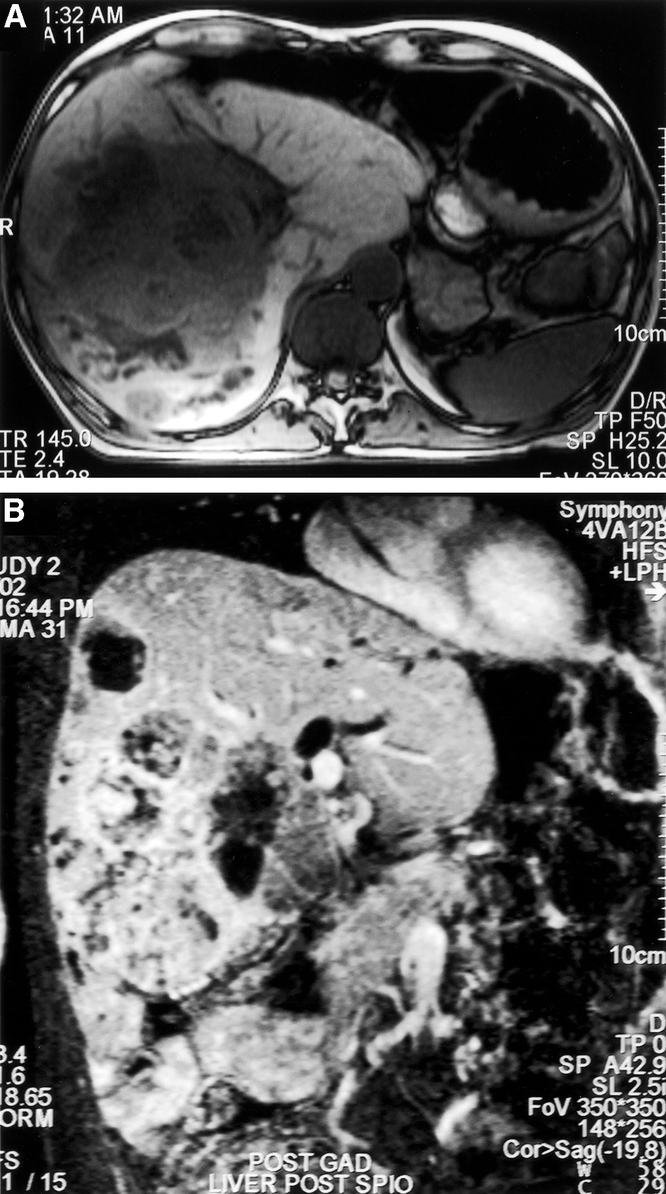
FIGURE 4. Transverse (A) and coronal oblique (B) section magnetic resonance imaging demonstrating a 15-cm diameter tumor, primarily involving the right liver, but with significant extension into the left liver. There is evident biliary obstruction (patient 7). Hepatic resection was possible by a modified right hemihepatectomy technique.
Patient Eight
A 65-year-old woman was admitted with a 3-month history of right-sided upper abdominal pain. Liver function tests were mildly deranged. Serum CEA was elevated at 137 μg/L. Liver MRI demonstrated an extensive intrahepatic tumor replacing segments 2, 3, and 4 and crossing the middle hepatic vein into segment 8. The findings were in keeping with an extensive intrahepatic cholangiocarcinoma. Otherwise, abdominal, pelvic, and thoracic imaging was normal. There was no evidence of bony metastases. At laparotomy, the tumor was found to extend behind the portal hilar structures, into segment 1, and up to the right hepatic vein. One lymph node at the celiac trifurcation was resected. There was otherwise no extrahepatic disease and no evidence of a primary tumor. The liver tumor was resected by means of a left hepatic trisectionectomy with caudate lobectomy (segments 1, 2, 3, 4, 5, and 8). The patient made a slow but uneventful recovery from surgery and was discharged home on day 13. The liver contained a single 16-cm diameter tumor, extending up to the resection margin. The celiac node was involved. At 2 months, the patient appears disease free.
DISCUSSION
Primary carcinoid tumors of the liver are rare. The largest series previously reported was of 6 patients, accumulated over an 8-year period at the National Cancer Institute in Milan.5 While we have treated 8 patients in 8 years, there is no direct evidence of a cluster effect. Six of the patients came from within our region, which serves a population of about 4 million people for liver tumors, while 2 others were referred from elsewhere (1 within our liver transplant zone and 1 from abroad). The 2 transplanted patients accounted for 0.3% of liver transplants, and the 6 patients who underwent liver resection accounted for 1.3% of hepatic resections carried out in our unit during 1994–2002.
The origin of primary hepatic carcinoid tumors is not clear. It has been proposed that they arise from scattered neuroendocrine cells in the intrahepatic biliary epithelium,33 an observation based on animal studies as such cells have only been found in the rat liver but not in humans.34 The scarcity of such cells may explain the rarity of primary neuroendocrine tumors of the liver. It is suggested that chronic inflammation in the biliary system may predispose to the development of neuroendocrine tumor by initiating intestinal metaplasia in which neuroendocrine cells are more numerous.35 Biliary obstruction was a major feature in our series; however, neither intestinal metaplasia nor previous biliary disease was found in our cases. Another theory is that these tumors originate from heterotopic pancreatic or adrenal tissue in the liver, and this is in keeping with their preponderance in the central part or perihilar areas of the liver, where heterotopic pancreatic tissue is most often located.14 It must, however, be considered that an apparent primary hepatic carcinoid tumor may be metastatic disease where, despite extensive assessment, the primary tumor remains obscure.
Terminology is constantly being revised, and most recently it has been suggested that “carcinoid” should be replaced by “well differentiated neuroendocrine tumor.”36 The presentation of primary hepatic neuroendocrine tumor tends to be nonspecific, and this was the case with our patients. Only in 1 patient (patient 2) were there symptoms attributable to the carcinoid syndrome, and this patient's tumor was the only 1 showing argentaffin positivity (Grimelius positivity) characteristic of tumors producing serotonin. The carcinoid syndrome, characterized by flushing, diarrhea, and less commonly wheezing and right heart failure,2 occurs as a result of the release of neurosecretory products, principally serotonin but also histamine, bradykinin, and prostaglandins, into the systemic circulation.37 It is typically seen in patients with carcinoid hepatic metastases as the portal circulation is bypassed. It appears that most primary liver neuroendocrine tumors are endocrinologically silent. It has not been possible to test our cases for specific gut hormone products by immunohistochemistry.
Preoperative diagnosis of primary hepatic carcinoid tumor is difficult as the radiologic appearance on ultrasound, CT, and MRI can mimic hepatocellular carcinoma, cholangiocarcinoma, and metastases, as illustrated by our series. Tumor markers are unhelpful. While not usually advocating preoperative liver biopsies for liver tumors, we did find it useful in this small group. A prominent feature in all of the cases presented was biliary involvement, evidenced by obstructive pattern liver function tests and/or radiologic features (Fig. 4B; Table 1). The close association of the tumor to the portal hilar structures, with biliary involvement at laparotomy, necessitated excision of the external biliary tree in combination with hepatic resection in 4 cases and progression to liver transplantation in 2 (Table 1). Six of the tumors required major hepatic resection, using modified resection methods, as the tumors presented late and liver anatomy was distorted. In the other 2 cases, an attempt at surgical resection failed and transplantation was offered. While an untested therapy for this disease at that time, these 2 cases suggest a favorable outlook after 95 and 45 months of follow-up. We currently recommend hepatic resection as the primary treatment modality for primary hepatic carcinoid tumors, but the complexity of this approach should not be underestimated, and recourse to transplantation in case of difficulty appears to be reasonable.
The histologic diagnosis of neuroendocrine tumors may be difficult as highlighted in 2 of our patients (patients 3 and 5) in whom the tumor analyzed elsewhere was labeled as a hepatocellular carcinoma but was found to have features of neuroendocrine tumor on immunohistochemistry when examined at our center. The macroscopic appearance is usually of a solid white tumor that may show areas of cystic degeneration.20 At microscopy, the architecture of the tumor may be insular, trabecular, papillary, or mixed, with a vascular stroma and the cells being monomorphic, having round or oval nuclei, pink granular cytoplasm, and showing few mitoses (Fig. 5A). Areas comprised of much larger cells but without evidence of necrosis or mitotic activity were present in 3 of our cases, and seem to be a feature of hepatic carcinoids (Fig. 5B). Several immunohistochemical markers can be used to confirm the diagnosis of neuroendocrine tumor; PGP 9.5, NSE, chromogranin, and synaptophysin were used in our series (Table 2). The use of electron microscopy to detect neurosecretory vesicles is also well documented, especially prior to the availability of immunohistochemistry.8,9,13,14,17,20,23 However, ultrastructural preservation was too poor after reprocessing tissue from paraffin blocks to allow identification of cytoplasmic organelles in our cases.
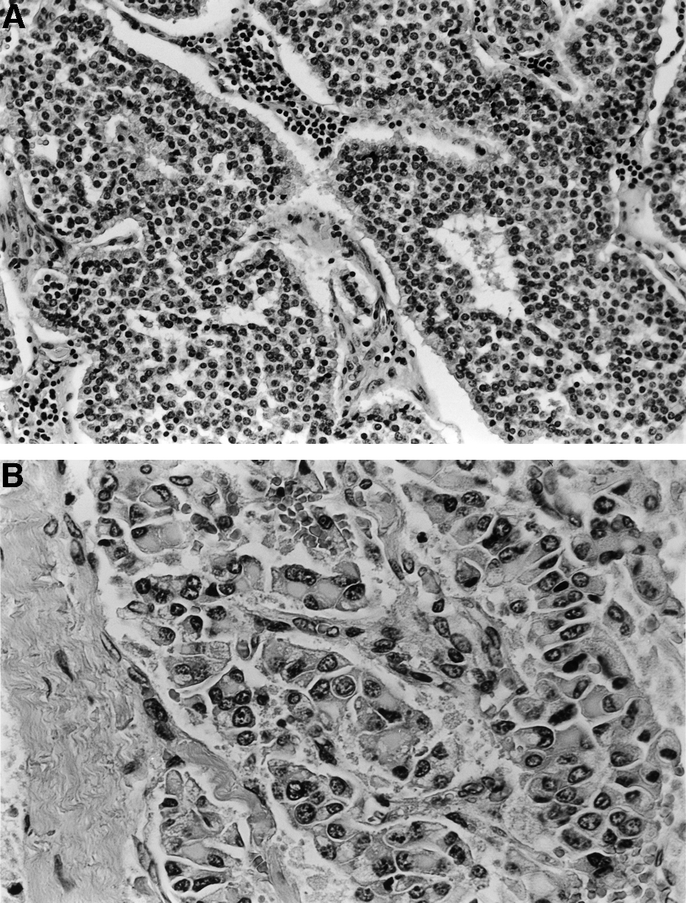
FIGURE 5. A: Photomicrograph showing typical morphology of low-grade endocrine tumor, showing trabecular arrangement of cells with regular round nuclei, peripheral pallisading, and highly vascular stroma. (patient 2; original magnification ×200). B: Photomicrograph of an area of tumor in which the cells show larger, pleomorphic nuclei and distinctive perinuclear eosinophilic inclusions (patient 3; original magnification ×400).
Histologically, all of the tumors showed morphologic characteristics of carcinoid tumors (ie, low-grade neuroendocrine tumors), including trabecular or insular architecture, with peripheral palisading of cells, abundant vascularity of the stroma, uniform cell size with generally rounded nuclei, very infrequent mitotic figures, and no evidence of tumor necrosis. Tumor cells often appeared to lie within vascular spaces. In patients 4 and 6, these features were less marked; however, there were no features to suggest cholangiocarcinoma (eg, mucin production, dense fibrous stroma, ductal morphology) or hepatocellular carcinoma. Immunohistochemistry showed positivity for cytokeratin and also for at least 2 of the neuroendocrine markers NSE, synaptophysin, PGP 9.5, and chromogranin. Three of the tumors (patients 1, 3, and 5) also showed areas of much larger cells (Fig. 5B), either forming sheets or contained within more typical islands of carcinoid tumor with peripheral palisades of smaller cells.
The differentiation between primary and secondary neuroendocrine tumors of the liver is not possible by histology alone, although the position of the tumor, if central and solitary, may suggest a primary tumor. The diagnosis of a primary tumor must follow a careful search for any other possible site, and this requires thorough investigations, operative inspection, and long-term follow-up. In 2 of our patients (patients 2 and 4), lesions that could have been a primary tumor were excised for histologic analysis, although none proved to be the culprit. It is important to review the histology of any previous surgical resection, particularly if the patient has undergone an appendicectomy.
The mainstay of treatment of primary hepatic neuroendocrine tumor is liver resection. In our series, 6 cases were treated with resection and the 4 with adequate follow-up are all currently tumor free. Two had tumors that were unresectable and the patients proceeded to liver transplantation. Although liver transplantation is a well-recognized option for some secondary neuroendocrine tumors of the liver,38,39 this is the first report of its use in the treatment of primary hepatic neuroendocrine tumors. Thus far, the outcome of liver transplantation in our patients remains favorable, with no disease recurrence at 45 and 95 months. These limited data suggest a better overall prognosis for patients with primary hepatic neuroendocrine disease, when compared with secondary tumors of this type, whether treated by resection or transplantation. Furthermore, the use of a percutaneous biopsy to establish a diagnosis is justified as the discovery of a carcinoid tumor should promote a more aggressive approach to treatment.
Patient 7 was known to have a small solitary spinal metastasis, so we decided not to offer transplantation, as bone metastasis has been the most common tumor recurrence following transplantation for secondary hepatic carcinoids in our experience (most usually occurring at between 2 and 5 years from transplantation). We recognize that we may be criticized for offering hepatic resection to this patient, but this was carried out after full surgical and medical oncology counseling. The long-term outlook for this patient is currently unknown. At presentation this year, the patient was moribund. The rate of tumor growth (11-year history) suggests that he may now gain several years of survival. Spinal surgery is planned.
The data on the role of chemotherapy in the treatment of primary hepatic neuroendocrine tumors are scarce. In the series of Andreola et al,7 1 patient with inoperable disease responded to intensive systemic 5-fluorouracil, which down-staged the disease and enabled subsequent resection. However, the disease progressed in 2 other patients despite systemic and intra-abdominal chemotherapy.7 Selective embolization of the hepatic artery may also be used for vascular neuroendocrine tumors, although data remain limited. Krishnamurthy et al reported a 40% reduction in tumor bulk in 1 patient treated with hepatic arterial embolization.20 While we commonly treat secondary neuroendocrine tumors of the liver with chemoembolization, we have not applied this technique to patients with primary hepatic carcinoid disease.
Octreotide, a 5-hydroxytryptamine inhibitor, is the mainstay of treatment in patients with metastatic disease from a primary hepatic neuroendocrine tumor. In addition to achieving a satisfactory symptomatic control, there are in vitro data to support an antiproliferative role for octreotide in cell culture systems,40 although there are few data to support an effect in vivo.41 Future therapies are likely to be receptor-targeted with radiolabeled octreotide. Yttrium-90-labeled octreotide is reported to have some therapeutic value.42
Regular clinical review and CT imaging are essential following resection of primary hepatic neuroendocrine tumors to detect disease recurrence in the liver that may be treated by re-resection or liver transplantation, if possible, as was exemplified by patient 5 in our series. In addition, regular follow-up may enable the detection of a previously unrecognized extrahepatic primary site, and it is our practice to use radiolabeled somatostatin scanning techniques following tumor excision as a primary follow-up investigation. The overall prognosis for patients with these tumors is considerably better than for other hepatic carcinomas.5,13,21,22 All of our patients attend an active follow-up program, and the 6 with adequate follow-up remain well and disease free. Hepatic resection and transplantation can be recommended as treatment of primary hepatic carcinoid tumors, with expected long-term survival.
Footnotes
Reprints: J. Peter A. Lodge, MD, FRCS, Hepatobiliary and Transplantation Surgery, Hepatobiliary Unit, St. James's University Hospital, Leeds LS9 7TF, UK. E-mail: PeterLodge@aol.com.
REFERENCES
- 1.Oberndorfer S. Karzinoide tumoren des dunndarms. Frank Z Pathol. 1907;1:426–429. [Google Scholar]
- 2.Caplin ME, Buscombe JR, Hilson AJ, et al. Carcinoid tumor. Lancet. 1998;352:799–805. [DOI] [PubMed] [Google Scholar]
- 3.Yamashita Y, Takahashi M, Tsuji A, et al. Primary carcinoid of the liver: a case report. J Comput Assist Tomogr. 1986;10:313–317. [DOI] [PubMed] [Google Scholar]
- 4.Xi YP, Yu JY. Primary neuroendocrine carcinoma of the liver. Ultrastruct Pathol. 1986;10:331–336. [DOI] [PubMed] [Google Scholar]
- 5.Larsen LG, Billesbolle P. Primary liver apudoma. Ann Chir Gynaecol. 1987;76:280–282. [PubMed] [Google Scholar]
- 6.Miura K, Shirasawa H. Primary carcinoid tumor of the liver. Am J Clin Pathol. 1988;89:561–564. [DOI] [PubMed] [Google Scholar]
- 7.Andreola S, Lombardi L, Audisio RA, et al. A clinicopathologic study of primary hepatic carcinoid tumors. Cancer. 1990;65:1211–1218. [DOI] [PubMed] [Google Scholar]
- 8.Sioutos N, Virta S, Kessimian N. Primary hepatic carcinoid tumor: an electron microscopic and immunohistochemical study. Am J Clin Pathol. 1991;95:172–175. [DOI] [PubMed] [Google Scholar]
- 9.Aoki K, Sakamoto M, Mukai K, et al. Signet-ring cell carcinoid: a primary hepatic carcinoid tumor with cytoplasmic inclusions comprising of aggregates of keratin. Jpn J Clin Oncol. 1992;22:54–59. [PubMed] [Google Scholar]
- 10.Mathur SK, Supe AN, Nagral SS, et al. Primary hepatic carcinoid (a case report). Indian J Gastroenterol. 1992;11:3–8.1551711 [Google Scholar]
- 11.Pi ZM. 20 rare primary hepatic malignant tumors. Chin J Oncol. 1992;14:210–220. [PubMed] [Google Scholar]
- 12.Takayasu K, Muramatsu Y, Sakamoto M, et al. Findings in primary hepatic carcinoid tumor: US, CT, MRI, and angiography. J Comput Assist Tomogr. 1992;16:99–102. [DOI] [PubMed] [Google Scholar]
- 13.Wang CY, Chen A, Tseng HH, et al. Carcinoid tumor localised in the liver-two case reports: immunohistochemical and ultrastructural studies. Chin Med J. 1992;49:365–372. [PubMed] [Google Scholar]
- 14.Hsueh C, Tan XD, Gonzalez-Crussi F. Primary hepatic neuroendocrine carcinoma in a child: morphological, immunocytological, and molecular biologic studies. Cancer. 1993;71:2600–2605. [DOI] [PubMed] [Google Scholar]
- 15.Imaoka I, Sugimura K, Tamura K. Case report: MR imaging of a carcinoid tumor of the liver. Clin Radiol. 1993;47:287–289. [DOI] [PubMed] [Google Scholar]
- 16.Inoue Y, Nakamura H, Mizumoto S, et al. Primary hepatic carcinoid with production of gastrin: a case report. Radiat Med. 1993;11:102–106. [PubMed] [Google Scholar]
- 17.Yasoshima H, Uematsu K, Sakurai K, et al. Primary hepatic carcinoid tumor. Acta Pathol Jpn. 1993;43:783–789. [DOI] [PubMed] [Google Scholar]
- 18.Artopoulos JG, Destuni C. Primary mixed hepatocellular carcinoma with carcinoid characteristics: a case report. Hepatogastroenterology. 1994;41:442–444. [PubMed] [Google Scholar]
- 19.Holbrook RF, Koo K, Ryan JA. Resection of malignant primary liver tumors. Am J Surg. 1996;171:453–455. [DOI] [PubMed] [Google Scholar]
- 20.Krishnamurthy SC, Dutta V, Pai SA, et al. Primary carcinoid tumor of the liver: report of four resected cases including one with gastrin production. J Surg Oncol. 1996;62:218–221. [DOI] [PubMed] [Google Scholar]
- 21.Mehta DC, Warner RR, Parnes I, et al. An 18-year follow-up of primary hepatic carcinoid with carcinoid syndrome. J Clin Gastrenterol. 1996;23:60–62. [DOI] [PubMed] [Google Scholar]
- 22.Cianchi F, Carassale G, Palomba A, et al. A case report of primary hepatic carcinoid: a report of its surgical resolution. Minerva Chir. 1997;52:433–437. [PubMed] [Google Scholar]
- 23.Oh YH, Kang GH, Kim OJ. Primary hepatic carcinoid tumor with a paranuclear clear zone: a case report. J Korean Med Sci. 1998;13:317–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Asakawa T, Tomioka T, Abe K, et al. Primary hepatic carcinoid tumor. J Gastroenterol. 1999;34:123–127. [DOI] [PubMed] [Google Scholar]
- 25.Fujino K, Koito K, Sano S, et al. A primary hepatic carcinoid tumor: evaluation by computed tomography and magnetic resonance imaging. Radiat Med. 1998;16:371–373. [PubMed] [Google Scholar]
- 26.Kehagias D, Moulopoulos L, Smirniotis V, et al. Imaging findings in primary carcinoid tumor of the liver with gastrin production. Br J Radiol. 1999;72:207–209. [DOI] [PubMed] [Google Scholar]
- 27.Nemes B, Podder H, Jaray J, et al. Primary hepatic carcinoid in a renal transplant patient. Pathol Oncol Res. 1999;5:67–69. [DOI] [PubMed] [Google Scholar]
- 28.Pilichowska M, Kimura N, Ouchi A, et al. Primary hepatic carcinoid and neuroendocrine carcinoma: clinicopathological and immunohistochemical study of five cases. Pathol Int. 1999;49:318–324. [DOI] [PubMed] [Google Scholar]
- 29.Sano K, Kosuge T, Yamomoto J, et al. Primary hepatic carcinoid tumors confirmed with long-term follow-up after resection. Hepatogastroenterology. 1999;46:2547–2550. [PubMed] [Google Scholar]
- 30.Mizuno Y, Ohkohchi N, Fujimori K, et al. Primary hepatic carcinoid tumor: a case report. Hepatogastroenterology. 2000;47:528–530. [PubMed] [Google Scholar]
- 31.Iwao M, Nakamuta M, Enjoji M, et al. Primary hepatic carcinoid tumor: case report and review of 53 cases. Med Sci Monit. 2001;7:746–750. [PubMed] [Google Scholar]
- 32.Kim SR, Imoto S, Maekawa Y, et al. CEA producing primary hepatic carcinoid. Hepatol Res. 2002;22:313–321. [DOI] [PubMed] [Google Scholar]
- 33.Alpert LI, Zak FG, Wethamer S, et al. Cholangiocarcinoma: a clinicopathologic study of five cases with ultrastructural observations. Hum Pathol. 1974;5:709–728. [DOI] [PubMed] [Google Scholar]
- 34.Payne CM, Noyle RB, Paplanus SH, et al. Fibrolamellar carcinoma of liver: a primary malignant oncocytic carcinoid. Ultrastruct Pathol. 1986;10:539–552. [DOI] [PubMed] [Google Scholar]
- 35.Brown WM, Henderson JM, Kennedy JC. Carcinoid tumor of the bile duct: a case report and literature review. Am Surg. 1990;56:343–346. [PubMed] [Google Scholar]
- 36.Wick MR, Ritter JH. Neuroendocrine neoplasms: evolving concepts and terminology. Curr Diagn Pathol. 2002;8:102–112. [Google Scholar]
- 37.Creutzfeld W, Stockmann F. Carcinoids and carcinoid syndrome. Am J Med. 1987;82:4–12. [DOI] [PubMed] [Google Scholar]
- 38.Le Treut YP, Delperro JR, Dousset B, et al. Results of liver transplantation in the treatment of metastatic neuroendocrine tumors. Ann Surg. 1997;225:355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lehnert T. Liver transplantation for metastatic neuroendocrine carcinoma. Transplantation. 1998;66:1307–1312. [DOI] [PubMed] [Google Scholar]
- 40.Wanberg B, Nilsson O, Johansson V, et al. Somatostatin receptors in the diagnosis and therapy of neuroendocrine tumors. Oncologist. 1997;2:50–58. [PubMed] [Google Scholar]
- 41.Arnold R, Trautmann ME, Creutzfeldt W, et al. Somatostatin analogue octreotide and inhibition of tumour growth in metastatic endocrine gastroenteropancreatic tumours. Gut. 1996;38:430–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Otte A, Muller-Brand J, Dellas S, et al. Yttrium-90-labelled somatostatin analogue for cancer treatment. Lancet. 1998;351:417–418. [DOI] [PubMed] [Google Scholar]
- 43.Strasberg SM, Belghiti J, Clavien P-A, et al. The Brisbane 2000 terminology of liver anatomy and resections. HPB Surg. 2000;2:333–339. [Google Scholar]