

Abstract
Objective:
To examine the expression of the transcription factor nuclear factor kappa B (NF-κB) in Barrett's epithelium and adenocarcinoma and the impact of NF-κB expression on tumor stage and response to neoadjuvant chemotherapy and radiation therapy.
Summary Background Data:
Progression of Barrett's esophagus to adenocarcinoma is associated with a wide range of cellular and molecular abnormalities. Nuclear factor-kappa B (NF-κB) regulates several genes involved in inflammatory, immune and apoptotic responses, but its role in esophageal inflammation and tumorigenesis has not been reported.
Methods:
Mobility shift assay was used to measure NF-κB activity in nuclear extracts of fresh-frozen biopsies from tumor and uninvolved tissues (n = 30) and esophageal cell lines OE33, SKGT-4, and OE21. RelA expression was assessed by immunohistochemical staining (n = 97). The NF-κB/RelA and IκB protein expressions were also examined by Western blotting.
Results:
NF-κB was not expressed in normal esophageal squamous epithelium, in contrast to increased expression in 40% of patients with Barrett's epithelium. Sixty-one percent of resected tumors (n = 97) displayed NF-κB immunoreactivity, and 87.5% of the NF-κB- positive tumors were Stage IIb and III compared with only 12.5% of patients with Stage I and IIa disease (P < 0.05). The expression of NF-κB inversely correlated with major or complete pathologic responses to neoadjuvant chemotherapy and radiation therapy, with 15/20 (75%) responders in the NF-κB-negative group compared with 7/38 (18%) in the NF-κB-positive group (P < 0.00001). Moreover, incubation of esophageal cell lines OE33, SKGT-4, and OE21 with deoxycholic acid or low pH induced NF-κB expression.
Conclusions:
Bile acids and low pH induce NF-κB expression in esophageal cell lines. NF-κB activation is common in esophageal adenocarcinoma. In patients with Barrett's epithelium and an associated esophageal adenocarcinoma, there is a progressive expression of NF-κB through Barrett's tumorigenesis. The absence of NF-κB expression in esophageal adenocarcinoma correlates with response to neoadjuvant chemoradiotherapy and may be of value in predicting response to neoadjuvant therapy.
Esophageal adenocarcinoma tumor tissues contain high levels of activated nuclear factor kappa-B (NF-κB), and the absence of NF-κB in esophageal cancer correlates with the early stage of the disease and with responsiveness to neoadjuvant therapy. Exposure of esophageal cancer cells to deoxycholic acid or low pH induced NF-κB expression.
There has been a marked increase in the incidence of esophageal adenocarcinoma over the last 2 decades. A 3-fold increase has been reported in the United States and Western Europe, where adenocarcinoma has supplanted squamous cell cancer as the dominant pathologic phenotype.1,2 Esophageal adenocarcinoma may arise in a background of Barrett's esophagus.1,3, specifically specialized intestinal metaplasia (SIM). Barrett's metaplasia is prevalent in the general population, and the risk of progression to dysplasia and cancer is between 0.2–2% per year,3,4 but whether SIM is an essential first step in all cases of esophageal adenocarcinoma remains unclear.
Esophageal adenocarcinoma remains an aggressive disease with a poor prognosis. The prognosis is better when the tumor is resected at an earlier stage and recent studies of radical en-bloc resection with extended lymphadenectomy report impressive survival rates.5,6 Nonetheless, the 5-year survival after surgery alone is rarely above 20%.7 There is increasing evidence from level II and level III trials that chemotherapy and radiotherapy prior to esophagectomy for esophageal adenocarcinoma may improve survival.8 The response of tumor cells to both cytotoxic drugs and radiation relies on their ability to cause programmed cell death or apoptosis in the cells, and therefore alteration in the molecular mechanisms governing this process may impact on sensitivity or resistance to neoadjuvant therapy9. In esophageal adenocarcinoma, approximately 1 in 4 patients have a complete pathologic response to combined neoadjuvant chemotherapy and radiation therapy, yet over 50% of patients demonstrate no response whatsoever.6 It is clear that a greater understanding of the molecular regulation of this increasingly prevalent cancer may permit improved therapeutic outcomes.
Nuclear factor kappa B (NF-κB) is a ubiquitous transcription factor whose activity is controlled at a subcellular level. NF-κB resides in the cytoplasm of most cells in an inactive form as a heterodimer consisting of p50 and RelA subunits complexed to the inhibitory molecule IκB, which prevents the migration of the heterodimer to the nucleus.10 Upon stimulation of NF-κB by a wide variety of stimuli, including cytotoxic agents and irradiation, NF-κB translocates to the nucleus and binds to its specific DNA site and subsequently up-regulates the transcription of a wide variety of genes involved in the inflammatory and immune response. Several reports have shown a role for NF-κB in regulating cell proliferation, tumor development, and cell transformation.11,12 Altered levels of NF-κB expression have been found in a number of tumors.13,14
It has been demonstrated that activation of NF-κB is involved in the protection against apoptosis.15,16 NF-κB has also been implicated in the pathogenesis of multidrug resistance, although the underlying mechanism is unclear. The activation of NF-κB was found to protect from cell killing and its inhibition enhanced apoptotic killing by TNF, ionizing radiation and cancer therapy.16 Several studies have demonstrated an antiapoptotic function for NF-kB in breast cancer and hepatocellular carcinoma, and the inhibition of NF-κB may lead to cellular apoptosis.13,14,16,17
NF-κB plays an important role in the development and progression of tumor cells in several settings, however, its role has not yet been studied in esophageal adenocarcinoma. We aimed in this study to assess the level of expression of NF-κB in esophageal adenocarcinoma and to correlate this with clinicopathological parameters, survival and responsiveness to neoadjuvant therapy. This study demonstrates that esophageal adenocarcinoma tumor tissues contain high levels of activated NF-κB, that NF-κB activation may be observed in SIM, and that neoadjuvant therapy results in the inhibition of constitutive NF-κB activation. Furthermore, exposure of esophageal cancer cells to the bile acid deoxycholic acid (DCA) and low pH induced NF-κB expression.
PATIENTS AND METHODS
Patient Characteristics and Treatment
Paraffin-fixed specimens from 97 patients with esophageal adenocarcinoma managed in St. James's Hospital, Dublin between 1990 and 2000 were included in this study. The study had the approval of the Institutional Review Board and informed consent was obtained from all patients. Fifty-eight received multimodal therapy and 39 had surgery alone. The neoadjuvant regimen was as described previously.18 Briefly, this comprised external beam radiotherapy (40Gy in 15 fractions over 3 weeks) and chemotherapy given in weeks 1 and 6, consisting of 5-fluorouracil 50 mg/kg for 5 days followed by cisplatin 75 mg/m2 on day 7. Surgery was performed during week 8. Data concerning the clinical and pathologic parameters was obtained from a detailed database maintained in Microsoft Excel 97 and SPSS version 8 on IBM compatible PC. The variables analyzed included age, gender and the histopathologic characteristics of the tumors such as grade and stage. Staging was according to the American Joint Committee on Cancer TNM system.19 Response to neoadjuvant therapy was evaluated on detailed histologic examination of the resection specimen. A complete pathologic response was defined as no residual tumor cells identifiable in the resection, a partial response had evidence of microscopic tumor which was of stage pT2pN0 or less and any tumor above this stage was defined as a nonresponder.
Biopsy and Sample Collection
Fresh biopsy specimens (0.5–1 g) were collected from patients with Barrett's esophagus (Barrett's tissue; n=15 and Barrett's adjacent to adenocarcinoma; n = 10), esophageal adenocarcinoma (tumor and adjacent uninvolved tissues of the same patient; n = 30) and from patients who had received neoadjuvant therapy (n = 8). Biopsy specimens were also collected from the normal stomach of patients and used as controls. Samples of endoscopically normal esophageal squamous epithelium above the affected segment were also obtained from each patient and used as a further control. Each sample was immediately processed for experimental use or snap-frozen on dry ice, stored at −70°C until required.
Cell Isolation
Single-cell suspensions were prepared from both esophageal tumor and adjacent normal esophageal tissues using a modified method of Madrigal et al.20 Briefly, sections of tumor and normal mucosa were incubated in 120 U/mL collagenase type 1 (Sigma) solution at 37°C tumbling for 1 hour. Undigested tissues were removed by centrifugation at low speed of 200 rpm for 1 minute. Single cells were collected and used at 1 × 106/mL for preparation of total cell extracts and nuclear extracts.
Cell Culture
The esophageal epithelial cell lines OE33 (derived from the adenocarcinoma of the lower esophagus; Barrett's metaplasia) and OE21 (derived from a squamous carcinoma of mid esophagus) were obtained from the European Collection of Animal Cell Cultures, ECACC (Porton Down, Salisbury, UK). SKGT-4 cells were a gift from Dr. David S. Schrump (Thoracic Oncology Section, Surgery Branch, National Cancer Institute, NIH, Bethesda, MD). OE33, SKGT-4 and OE21 esophageal epithelial cell lines were grown in RPMI 1640 medium supplemented with 10% filtered fetal calf serum, 100 units/mL penicillin, 100 μg/mL streptomycin, and 2mm l-glutamine.
Cell Culture Treatments
OE33, SKGT-4, and OE21 cells were removed from flasks by trypsin/EDTA treatment and seeded at a density of 5x105 cells/ml in 6-well plates. Confluent cells were exposed to low pH media of different pH values. To adjust media to the required pH value, 0.1 M HCl was added to the cell culture medium and titrated to the required pH. The pH of the culture medium was measured before and after the incubation to ensure that the pH remained unchanged. For controls, volumes of deionized water were added. Cells were also treated with deoxycholic acid (DCA); at different concentrations for different periods of time, as indicated in figure legends. Each experiment was performed at least 3 times.
Preparation of Total Cell Extracts
Cells were collected by centrifugation at 1400 rpm for 5 minutes. The pellet of cells was resuspended in lysis buffer containing 20 mm Tris-HCl (pH 7.5), 1% (wt/vol) sodium dodecyl sulfate (SDS), 150 mm NaCl, 1 mm EGTA, 1 mm EDTA, 0.5 mm phenylmethylsulfonylfluoride (PMSF) and leupeptin (10 μg/ml) and then the cells were solubilized by boiling for 5 minutes.
Western Blot Analysis
Equivalent amounts of total cell extracts (50 μg of protein/lane) were resolved by electrophoresis through polyacrylamide gels using 10% separating gels according to the method of Laemlli.21 Proteins were electrotransfered onto PVDF membrane. Blots were blocked with 5% (wt/vol) dried skim milk in phosphate-buffered saline (PBS) for 1 hour at room temperature and then incubated for 1 hour at room temperature with primary appropriate antibody (anti-IκB-α or anti-RelA in a dilution 1/1000). Blots were then incubated with the relevant antirabbit horseradish peroxidase conjugated secondary antibody (1/1000) for 1 hour at room temperature. Immunodetection was performed by enhanced chemiluminescence.
Cell Fractionation and Nuclear Extract Preparation
Nuclear extracts were prepared from the cells as described previously by Osborn et al22 Briefly, the cells were washed twice in ice-cold PBS. The cells were pelleted by centrifugation at 1400 rpm for 5 minutes and washed once in buffer A (10 mm Hepes, pH 7.9, 1.5 mm MgCl2, 10 mm KCl, 0.5 mm PMSF and 0.5 mm DTT and centrifuged at 10,000 rpm for 10 minutes. The pellet of cells was then resuspended in buffer A containing 0.1% (vol/vol) NP-40 for 10 minutes on ice and lysed cells were centrifuged at 10000 rpm for 10 minutes. The supernatant was discarded and the nuclear pellet was extracted with (15 μL) buffer C (20 mm Hepes, pH 7.9, 420 mm NaCl, 1.5 mm MgCl2, 0.2 mm EDTA, 25% (wt/vol) glycerol and 0.5 mm PMSF) for 15 minutes on ice. After incubation, the nuclei were centrifuged at 10000 rpm for 10 minutes and the supernatant was diluted with 4 volumes of buffer D (10 mm Hepes, pH 7.9, 50 mm KCl, 0.2 mm EDTA, 25% (wt/vol) glycerol and 0.5 mm PMSF). The nuclear extracts were used immediately or stored at −70°C until required. The protein concentration was determined on nuclear extracts by the method of Bradford.23
Electrophoretic Mobility-Shift Assays (EMSA)
For binding assays, nuclear extracts (4 μg of protein) were incubated with 10000 cpm of the 32P-labeled oligonucleotide (22 bp) NF-κB that had been previously labeled with (γ32P) ATP (10 mCi/mmol) at the 5′-ends with T4 polynucleotide kinase. The assay was performed in 20 μL of binding buffer (10 mm Tris (pH 7.5), 4% (wt/vol) glycerol, 5 mm DTT, 1 mm EDTA, 100 mm NaCl and 0.1 mg/ml nuclease-free BSA) in the presence of 2 mg poly(dI-dC) as nonspecific competitor. The reaction mixture was then incubated for 30 minutes at room temperature after the addition of the probe DNA. The binding reaction was terminated using a loading dye (0.25% bromophenol, 0.25% xylene cyenol, 30% (wt/vol) glycerol in deionized water) prior to electrophoretic separation of the DNA-protein complexes on 5% polyacrylamide gels that had been pre-electrophoresed for 30 minutes at 80 V. Gels were run at 150 V for 1–2 hours at room temperature. After electrophoresis, the gels were dried and autoradiographed at −70°C for 24–36 hours with intensifying screens.
Supershift and Competition Assays
In supershift and competition assays, 0.5 μL of antip50, anti-RelA, anti- C-rel antibodies, or 100-fold molar excess of unlabeled oligonulcelotide was preincubated with nuclear extract for 30 minutes at room temperature prior to the addition of the labeled probe.
Immunohistochemistry
Paraffin-embedded blocks of formalin fixed tissue from the surgically resected specimens was analyzed immunohistochemically for altered expression of NF-κB using the standard avidin-biotin-peroxidase complex. Diagnostic biopsy samples taken prior to commencement of chemoradiotherapy were also analyzed in the 58 patients in the multimodal therapy group. Sections (4 μm) were cut from each block and dewaxed in xylene and graded alcohol. Endogenous peroxidase activity was blocked by incubation with 0.3% H2O2 in methanol for 30 minutes followed by Antigen unmasking by pressure cooking for 60 seconds with antigen unmasking agent (Vector Laboratories Inc., Burlingame, CA, USA). Each slide was then incubated with the primary monoclonal antibodies to NF-κBp65 for 60 minutes at room temperature. A biotinylated secondary immunoglobulin was applied for 30 minutes followed by the avidin peroxidase reagent for a further 30 minutes. The stable avidin-biotin complexes were identified by the addition of the chromogen 3,3′ diaminobenzidine tetrachloride for 8 minutes. Staining was completed by counterstaining with Meyers Hematoxylin, dehydrating through graded alcohol to xylene and cover slipping. For a negative control the same procedure was carried out with replacement of the primary antibody with PBS. Sections of appendix were used as a positive control. Each slide was then examined for NF-κB/RelA immunoreactivity. P < 0.05 was considered statistically significant.
Statistical Analysis
For parametric data, an unpaired t test was used for comparison of differences between means in 2 groups. Statistical significance was ascribed to a P value of less than 0.05. All data are reported as medians and interquartile ranges unless otherwise stated. Survival curves are analyzed by Kaplan-Meier method.
RESULTS
Patients
Activation of NF-κB in Esophageal Adenocarcinoma
Nuclear extracts from the tumor tissues showed a high expression of NF-κB compared with nontumor tissues. Twenty-three of 30 of esophageal adenocarcinoma specimens demonstrated elevated levels of NF-κB (Fig. 1A). The specificity of this complex was confirmed in supershift assays, which indicated the presence of both p50 and RelA in nuclear extracts from tumor tissues (Fig. 1B). Competition assays also demonstrated a marked reduction in the density of this complex after preincubation with cold probe (Fig. 1B). In some experiments, gastric tissues used as controls and showed no or minimal activated NF-κB expression compared with tumor tissues from the same patients.
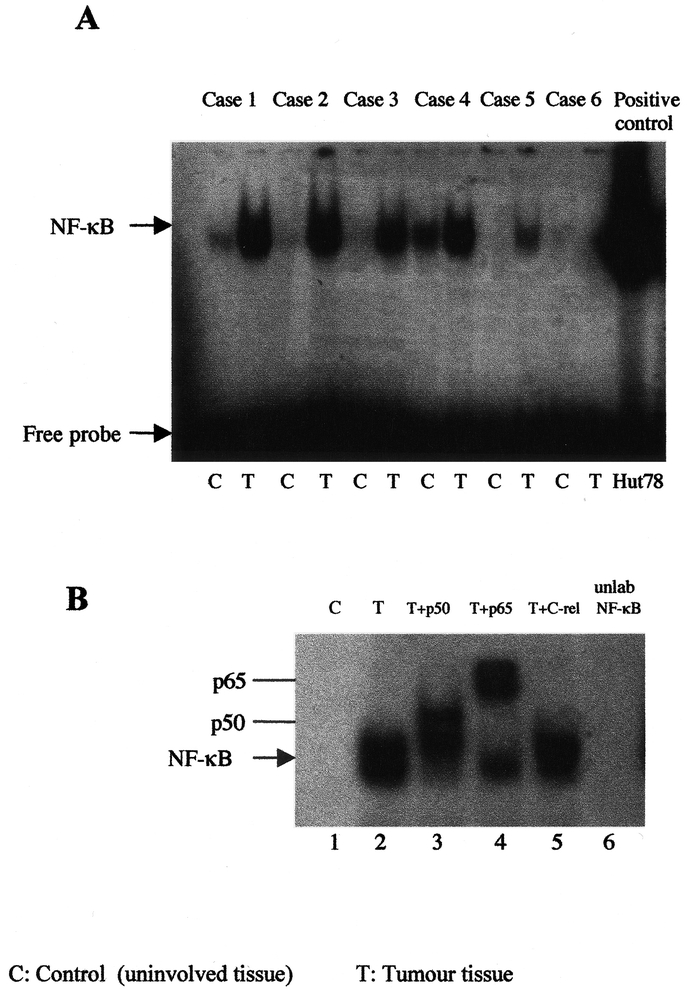
FIGURE 1. Gel mobility shift assay for NF-κB binding activity in esophageal biopsy tissue. A, Expression of NF-κB in nuclear extracts from esophageal tumor tissue, showing a strong band of NF-κB activation in tumor tissues (T) compared with non tumor tissues (C) from the same patient. B, Supershift assay was also performed on the same nuclear extracts using 0.5 μL of rabbit antisera to p50, lane 2, RelA, lane 3, and c-Rel, lane 4 antibodies in esophageal tumor tissue. Competition assay for NF-κB was also performed using molar excess (1 μL) of cold probe, lane 5. Each experiment was repeated 3 times and a representative gel is shown.
IκB-α and RelA Protein Expressions in Esophageal Adenocarcinoma
The translocation of NF-κB to the nucleus is preceded by the phosphorylation and proteolytic degradation of IκB-α. To determine whether NF-κB activation was due to IκB-α degradation, the cytoplasmic levels of IκB-α protein were examined by Western blot analysis. Tumor tissues showed lower IκB-α levels compared with nontumor tissues from the same patients (Fig. 2A), suggesting that the reduction of IκB-α levels in tumor tissue is associated with NF-κB activation. To further examine the presence of NF-κB in tumor tissue, immunoblotting with antibody against RelA indicated the presence of RelA in nuclear extracts from tumor tissue compared with nontumor tissue (Fig. 2B).
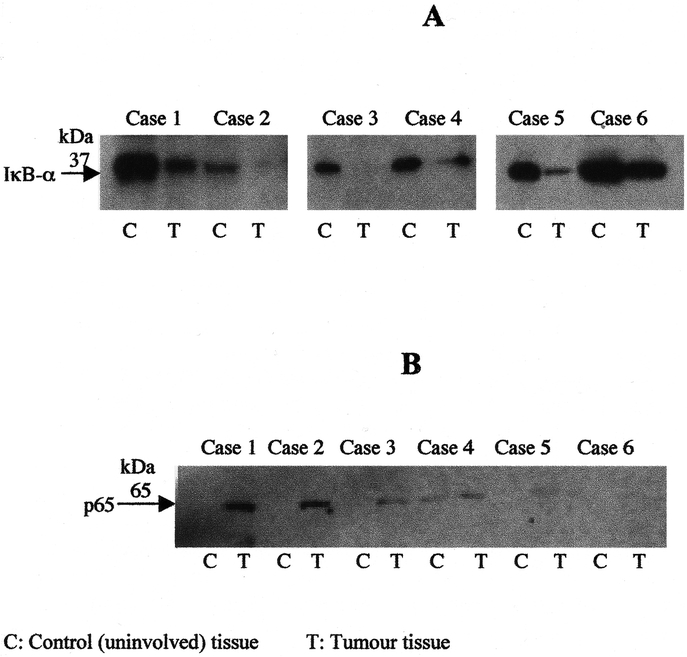
FIGURE 2. Western blot analysis for IκB-α (A) and RelA (B) protein expressions were performed on total and nuclear cell extracts (50 μg protein/lane), respectively, from both esophageal tumor tissues and non tumor tissues and separated by 10% acrylamide SDS-PAGE, blotted onto PVDF membrane and probed with specific antisera. Experiments were performed 3 times and results of 6 independent patients are shown.
Correlation of NF-κB Status With Tumor Characteristics and Pathologic Stage
Adequate pathologic material for immunocytochemistry was available on all 97 patients. In 58 patients undergoing multimodal therapy pretreatment biopsies were available in addition to resection specimens of all patients in this group except the 13 who had a complete pathologic response to neoadjuvant therapy.
Sixty-one percent (59/97) of resected tumors displayed NF-κB immunoreactivity. Eighty of the patients were male (73%), with a median age of 62.1 year ± 12.8. Immunohistochemical staining of RelA also indicated a high expression of RelA in tumor tissues compared with control tissues, as shown in Figure 3. The majority of tumors were moderately or poorly differentiated but there was no correlation with NF-κB status. Thirty-one percent of the NF-κB negative tumors were Stage I or II compared with 12% of the NF-κB positive tumors (P < 0.05).
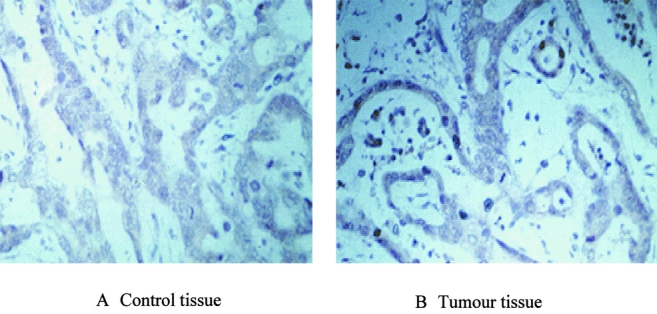
FIGURE 3. Immunohistochemical stains of RelA expression in esophageal adenocarcinoma. Immunohistochemical staining was performed as described under the experimental procedures section on normal and tumor tissues using anti-RelA polyclonal antibody. A, Normal esophageal tissues showed no immunostaining for RelA expression. B, Esophageal tumor tissues showed increased RelA expression, as indicated by brown staining. (Magnification ×400).
There was clear evidence of SIM in association with adenocarcinoma in 22 of the 59 patients who were NF-κB positive, compared with 13 of 38 in the NF-κB negative group.
Response to Chemoradiotherapy
Thirteen of the 58 patients (22%), who received chemoradiotherapy, had a complete pathologic response to multimodal therapy. A partial response was achieved in 9 (16%) and no response in 36 (62%). In patients who were NF-κB-negative (n = 20) on the pretreatment biopsy, 12 (60%) achieved a complete pathologic response, 3 (15%) a partial response, and 5 (25%) had no response to chemoradiotherapy. In the NF-κB-positive group (n = 38), there was 1 (3%) complete pathologic response, 6 (16%) with a partial response, and 31 (82%) with no evidence of a response (P < 0.0001 NF-κB-negative versus NF-κB-positive group)
Correlation of NF-κB Levels to Response to Neoadjuvant Therapy in EMSA Assays
We examined the correlation of NF-κB levels to response rates to neoadjuvant therapy using gel shift assays. Nuclear extracts before and after neoadjuvant therapy from the same patients were used to detect NF-κB levels. The results indicated that 3 of 8 specimens, which showed elevated NF-κB levels before neoadjuvant therapy, had no or little activated NF-κB after treatment (Fig. 4A), whereas 2 of 8 specimens showed partial inhibition of NF-κB (Fig. 4B). No difference in NF-κB levels was seen in 3 of 8 specimens before and after neoadjuvant therapy (Fig. 4C).
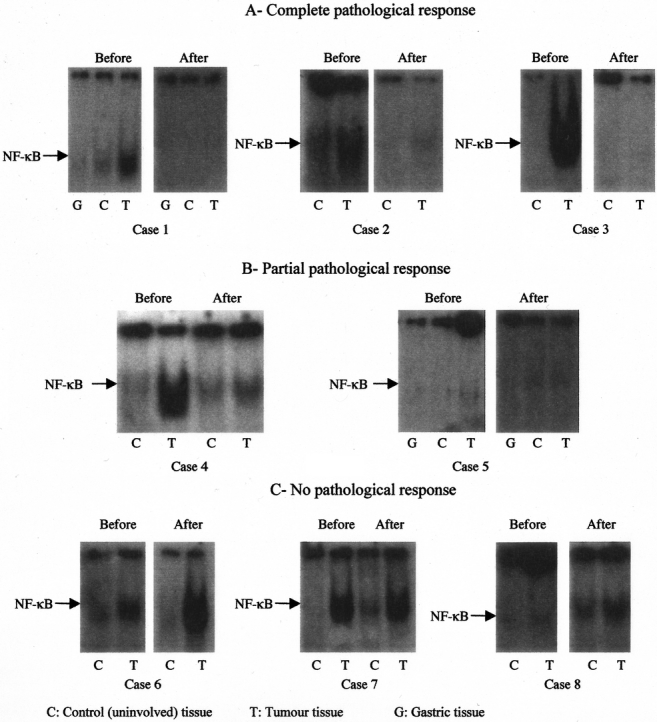
FIGURE 4. Correlation of NF-κB levels to response to neoadjuvant therapy in EMSA assays. Nuclear extracts before and after neoadjuvant therapy from the same patients were used to detect NF-κB levels. A, Complete pathologic response was observed in 3 of 8 specimens. B, Partial inhibition of NF-κB was observed in 2 of 8 specimens. C, No difference in NF-κB levels was seen in 3 of 8 specimens before and after neoadjuvant therapy. A representative gel is shown.
Survival
The cumulative survival with regard to pretreatment NF-κB status as determined by immunocytochemistry is depicted in Figure 5. The median survival of those who were positive for NF-κB (n = 59) was 7 months (95% CI; 4 to 12 months), compared with 12 months (95% CI; 5–19 months) in the NF-κB negative group (n = 38). The absence of NF-κB is associated with significantly (P < 0.05) improved survival in esophageal adenocarcinoma. By multivariate analysis NF-κB is not at this point an independent predictor of survival (Hazards ratio (95%CI) 1.65 (0.8–3.2) P = 0.07. This compares with lymph node status, 3.08 (1.7–8.6) P < 0.0001) as a predictor of survival.
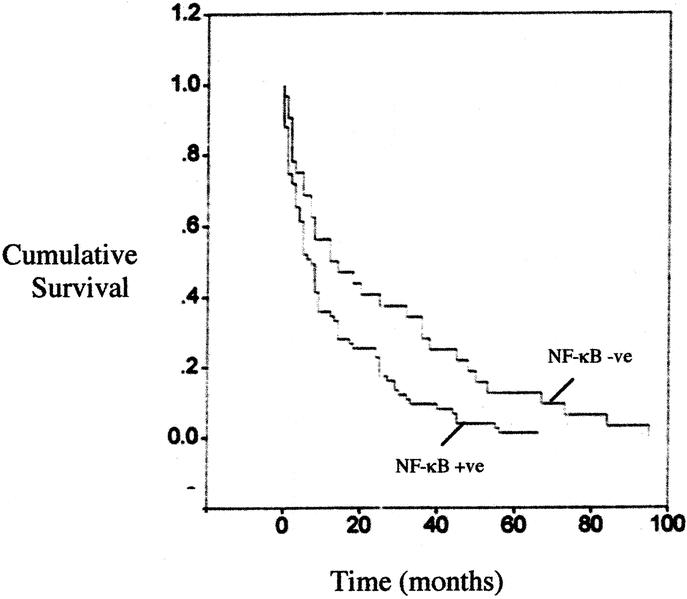
FIGURE 5. The cumulative survival with regard to NF-κB status. The median survivals of those were positive for NF-κB group (n = 59) was 7 months (95% CI, P < 0.05), compared with 12 months (95% CI; P = 0.04) in the NF-κB-negative group (n = 38).
Activation of NF-κB in Barrett's Esophagus
To investigate the expression of NF-κB in biopsy specimens from patients with Barrett's esophagus and Barrett's adjacent to adenocarcinoma, nuclear extracts were prepared and probed with the labeled 32P-NF-κB oligonucleotide. The results showed increased levels of activated NF-κB in 6/15 (40%) of Barrett's tissues compared with uninvolved tissues (Fig. 6A). Moreover, Barrett's tissues adjacent to esophageal adenocarcinoma also showed activated NF-κB and this expression was greater in tumor tissues from the same patients compared with Barrett's tissues (Fig. 6B).
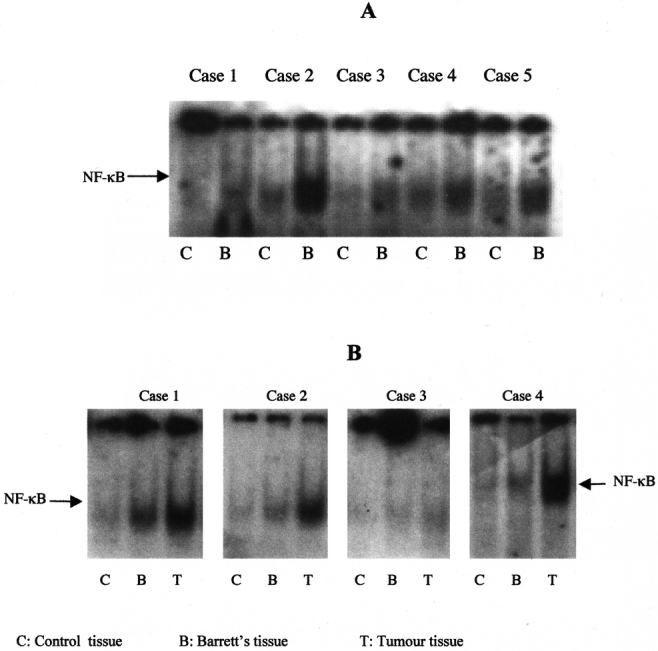
FIGURE 6. Presence of activated NF-κB transcription factor in biopsy specimens from Barrett's esophagus patients. Nuclear extracts were isolated from Barrett's tissues (A) or Barrett's tissues adjacent to esophageal adenocarcinomas (B) and uninvolved tissues and analyzed for NF-κB binding activity by EMSA. Each experiment was performed 3 times and a representative gel is shown.
Esophageal Cell Lines
Effect of DCA and Low pH on NF-κB Expression in Esophageal Cancer Cells
Barrett's metaplasia develops in response to chronic acid and bile reflux. We aimed to examine the effects of the bile acid deoxycholic acid, a strong tumor promoter, and low pH on NF-κB expression in esophageal cancer cells. Treatment of OE33 cells with DCA caused NF-κB activation in a dose-dependent manner. Figure 7A shows increased NF-κB DNA-binding activity at amounts of 300 μm DCA at 2 hours (Fig. 7A). No further induction of NF-κB was observed beyond this dose, which are toxic to the cells as evaluated by cell viability. Time-course experiments showed increased levels of NF-κB as the exposure time of DCA was increased from 30 minutes to 8 hours (data not shown).
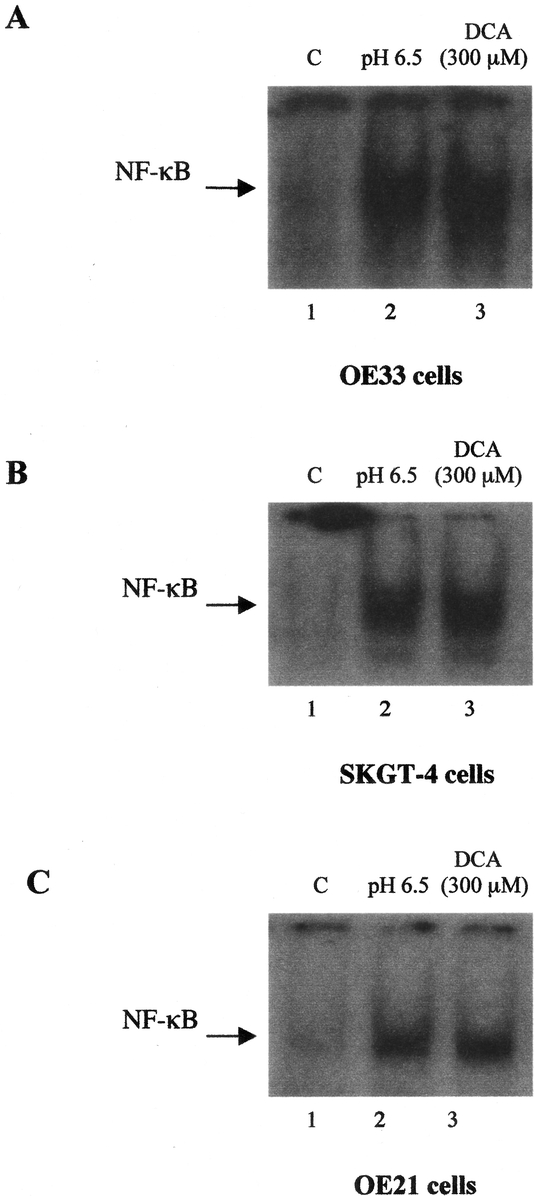
FIGURE 7. Effect of deoxycholic acid and low pH on NF-κB DNA-binding activity in esophageal cell lines. OE33 (A), OE21 (B), or SKGT-4 (C) cells were treated with DCA (300 μm) for 2 hours or exposed to pH 6.5 for 1 hour. NF-κB binding activity was assayed by EMSA on nuclear extracts. Each experiment was repeated 3 times with similar results and a representative gel is shown.
To examine whether exposure of esophageal cancer cells to low pH induces the expression of NF-κB, OE33 cells were incubated in media of different low pH values for 1 hour. Nuclear extracts from OE33 cells in the resting state revealed no activation of NF-κB and only minimal levels of active NF-κB but the exposure of OE33 cells to low pH resulted in induction of NF-κB activity, and this DNA-binding activity was increased as pH was reduced from pH 7.4 (pH of the culture medium) to pH 4 by adding 0.1 M HCl. Incubation of OE33 cells at pH 6.5 resulted in enhanced NF-κB DNA-binding activity compared with that seen at pH 7.4 of the resting OE33 cells (Fig. 7A). However, the incubation of OE33 cells below pH 4 resulted in very marked reduction in cell viability as assessed by fluorescence staining.
Moreover, nuclear extracts from control OE21, esophageal squamous carcinoma cells and SKGT-4, esophageal adenocarcinoma cells cultured at neutral pH or in the absence of DCA exhibited no DNA binding activity to the NF-κB site containing oligonucleotide. When OE21 or SKGT-4 cells treated with DCA (300 μm) or incubated at pH 6.5, NF-κB DNA-binding activity was increased (Fig. 7B and C).
DISCUSSION
This study demonstrates that NF-κB is activated in esophageal adenocarcinoma. The overexpression of NF-κB in tumor tissues was also evidenced by higher expression of RelA and low expression of IκB-α compared with nontumor tissues as demonstrated in Western blot analysis. The lower IκB-α levels in tumor tissues are associated with higher NF-κB activation. The NF-κB dimer is sequestered in the cytoplasm of most cells through interaction with the inhibitory IκB protein that prevents NF-κB from entering the nucleus.10,11 Dejardin et al24 have shown a high constitutive NF-κB activity and low IκB-α expression in a number of adenocarcinoma cells. The activation of NF-κB in tumor tissues could be possibly due to a direct consequence of the degradation of IκB-α or the presence of a mutant form of IκB-α. The dysregulation of IκB-α protein levels may be due to genetic mutations that occur from the consequences of the malignant state rather than from the induction of malignant state.14 Immunohistochemical studies have also demonstrated that 61% of resected tumors displayed NF-κB immunoreactivity. Thirty-one percent of the NF-κB negative tumors were Stage I or IIa compared with 12% of the NF-κB positive tumors, whereas 87.5% NF-κB positive tumors were Stage IIb or III compared with 69.5% of the NF-κB negative tumors.
NF-κB comprises a family of inducible transcription factors that serve as important regulators of the host immune and inflammatory response.11,12 The NF-κB pathway is a key mediator of many genes involved in proliferation and apoptosis. It is thought that TNF-α and IL-1 released in response to chronic gastro-esophageal reflux disease (GORD) may contribute to by activation of the NF-κB pathway.25 Cytokines that are stimulated by NF-κB, such as IL-1 and TNF-α, can also directly activate the NF-κB pathway and amplify the inflammatory response and increase the duration of chronic inflammation. NF-κB also activates expression of several antiapoptotic genes including the cellular inhibitors of apoptosis (c-1AP1, c-1AP2 and 1XAP), the TNF receptor-associated factors (TRAF1 and TRAF2), the Bcl-2 homologue A1/Bfl-1, and 1EX-IL and p53.25,26 Moreover, overexpression of NF-κB or its activation by IL-1 protects against TNF induced apoptosis, possibly by induction of antiapoptotic genes.27
The exact association of the Barrett's phenotype of SIM with esophageal adenocarcinoma is unclear, in particular whether reflux-induced SIM is an essential step in the development of all cases of adenocarcinoma. In this study, NF-κB activation was evident in patients with Barrett's esophagus, and a sequential progression in NF-κB activation from metaplasia to adenocarcinoma was evident in patients with adenocarcinoma and associated SIM. In patients with true Barrett's adenocarcinoma, therefore, this data may be consistent with a multistep process during which the activation of NF-κB is progressive from normal epithelium through metaplasia, dysplasia to adenocarcinoma.
Chronic gastroesophageal reflux of bile acids and acid are the major factors linked to the development of Barrett's metaplasia. Recent epidemiological studies also associate chronic reflux independent of Barrett's esophagus with risk of esophageal adenocarcinoma.28 Our results demonstrated that the bile acid DCA activates NF-κB in esophageal cancer cells. The activation of NF-κB is also markedly increased when the esophageal cancer cells are exposed to conditions of low pH. Bile acids, principally DCA, are known endogenous promoters of gastrointestinal cancer and enhance cell transformation.29 Conjugated bile acids exacerbate esophageal mucosal injury either alone or in combination with acid, both in vitro and in experimental models.30 Acid directly affects cell proliferation and differentiation of Barrett's epithelium and acid damage to the esophagus is followed by re-epithelialization of the esophagus with columnar epithelium of esophageal origin. Fitzgerald et al31 have suggested that both cultured intestinal cells and Barrett's epithelial cells may be induced to change their phenotype on culture in conditions of low pH.
Multimodal therapy is increasingly used in the management of esophageal adenocarcinoma. In this study, neoadjuvant therapy decreased NF-κB protein levels and the absence of NF-κB activity significantly correlated with response to chemoradiotherapy. Twenty-two percent of the 58 patients who received chemoradiotherapy showed a complete pathologic response and the NF-κB activity was down-regulated in those patients. The median survival (12 months vs. 7 months) among this group of patients was significantly higher compared with the NF-κB positive group. Several cellular mechanisms that determine sensitivity of tumor cells to the cytotoxic agents and affect the ability of a cancer cell to undergo apoptosis have been elucidated. One of these mechanisms is inducible chemoresistance, a process whereby exposure of tumor cells to cancer chemotherapy leads to their resistance to apoptosis. It has been suggested that the activation of NF-κB inhibits the apoptotic response in multiple types of tumor cells. Wang and colleagues32 have shown that inhibition of NF-κB enhances antitumor therapy through increased apoptosis. Also, the activation of NF-κB induces chemoresistance to the effects of chemotherapy agents and radiation. Constitutive NF-κB activation has been shown to inhibit apoptosis and to promote cell proliferation in breast cancer.11 The inhibition of NF-κB could trigger apoptosis in cancer cells or sensitize them to cytotoxic drug-induced cell death.
The present study demonstrated that esophageal adenocarcinoma tumor tissues contain high levels of activated NF-κB and neoadjuvant therapy results in the inhibition of constitutive NF-κB activation. Moreover, the absence of NF-κB in esophageal cancer correlates with the early-stage of the disease and with the responsiveness to neoadjuvant therapy. Our results also demonstrated a role for the bile acid DCA and low pH in activating NF-κB DNA-binding activity. Therefore, correction of reflux and exposure to bile acids might protect against further epithelial injury and mucosal inflammation. NF-κB activation may have an important role in the development of esophageal adenocarcinoma. Moreover, given the pleiotropic effects (Fig. 8) of NF-κB and its central pro-inflammatory and antiapoptotic roles, this pattern may explain the association of NF-κB activation with later stage of disease and resistance to neoadjuvant therapy. NF-κB may be become a new target for the study of Barrett's esophagus and management of adenocarcinoma of the esophagus, and NF-κB inhibitors may be used as a novel approach in the prevention and management of this increasingly prevalent tumor.
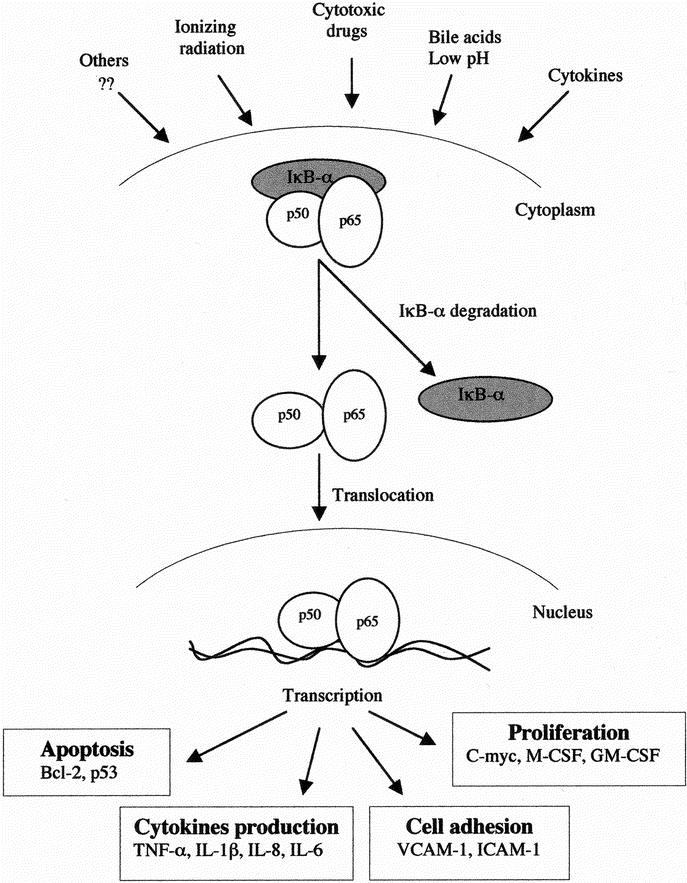
FIGURE 8. Potential model illustrates the activation of NF-κB transcription factor and induction of NF-κB-dependent genes in esophageal adenocarcinoma. Stimuli such as bile acids, low pH, cytokines, radiation and cytotoxic drugs induce NF-κB, which subsequently regulates many genes involved in cell proliferation, cytokine production, cell adhesion and apoptosis.
Footnotes
Supported in part by the Baggot Street Research Fund.
Presented in part at the Society of Academic and Research Surgeons, London, UK, January 2001.
Reprints: Professor John V. Reynolds, Department of Clinical Surgery and Dublin Molecular Medicine Centre, Trinity Centre for Health Sciences, St. James's Hospital, Dublin 8, Ireland. E-mail: reynoljv@tcd.ie.
REFERENCES
- 1.Pera M, Cameron AJ, Trastek VF, et al. Increasing incidence of adenocarcinoma of the esophagus and esophagogastric junction. Gastroenterology. 1993;104:510–513. [DOI] [PubMed] [Google Scholar]
- 2.Blot WJ, Devesa SS, Kneller RW, et al. Rising incidence of adenocarcinoma of the esophagus and gastric cardia. Gastroenterology. 1993;104:510. [PubMed] [Google Scholar]
- 3.DeMeester SR, DeMeester TR. Columnar mucosa and intestinal metaplasia of the esophagus: fifty years of controversy. Ann Surg. 2000;231:303–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hameetman W, Tytgat GNJ, Houthoff HJ, et al. Barrett's esophagus: development of dysplasia and adenocarcinoma. Gastronneterolgy. 1989;96:1249–1256. [DOI] [PubMed] [Google Scholar]
- 5.Altorki NA, Giraldi L, Skinner DB. En bloc esophagectomy improves survival for Stage III esophageal cancer. J Thorac Cardiovasc Surg. 1997;114:948–956. [DOI] [PubMed] [Google Scholar]
- 6.Lerut T, Coosemans W, De Lyn P, et al. Reflections on three-field lymphadenectomy in carcinoma of the esophagus and gastro-esophageal junction. Hepato-Gastroenterology. 1999;46:717–725. [PubMed] [Google Scholar]
- 7.Menke-Pluymers MBE, Schoute NW, Mulder AH, et al. Outcome of surgical treatment of adenocarcinoma in Barrett's oesophagus. Gut. 1992;33:1454–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Geh JI, Crellin Am, Glynne-Jones R. Preoperative (neoadjuvant) chemoradiotherapy in esophageal cancer. Br J Surg. 2001;88:338–348. [DOI] [PubMed] [Google Scholar]
- 9.Kastan MB. Molecular determinants of sensitivity to antitumor agents. Biochim Biophys Acta. 1999;1424:R37–42. [DOI] [PubMed] [Google Scholar]
- 10.Baldwin AS Jr. The NF-κB and IκB proteins: new discoveries and insights. Ann Rev Immunol 1996;14:649–683. [DOI] [PubMed] [Google Scholar]
- 11.Baeuerle PA, Henkel T. Function and activation of NFκB in the immune system. Annu Rev Immunol. 1994;12:141–179. [DOI] [PubMed] [Google Scholar]
- 12.Barnes PJ, Karin M. Nuclear factor-κB, a pivotal transcription factor in chronic inflammatory diseases. N Engl J Med. 1997;336:1066–1077. [DOI] [PubMed] [Google Scholar]
- 13.Sovak MA, Bellas RE, Kim DW, et al. Aberrant nuclear factor-κB/Rel expression and the pathogenesis of breast cancer. J Clin Invest. 1997;100:2952–2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tai D-I, Tsai S-L, Chang Y-H, et al. Constitutive activation of nuclear factor B in hepatocellular carcinoma. Cancer. 2000;89:2274–2281. [PubMed] [Google Scholar]
- 15.Beg AA, Baltimore D. An essential role for NF-κB in preventing TNF-α-induced cell death. Science. 1996;274:782–784. [DOI] [PubMed] [Google Scholar]
- 16.Wang C-Y, Mayo MW, Baldwin AS. TNF-α and cancer therapy-induced apoptosis: potentiation by inhibition of NF-κB. Science. 1996;274:784–787. [DOI] [PubMed] [Google Scholar]
- 17.Aldulaimi D, Jankowski J. Barrett's oesophagus: an overview of the molecular biology. Dis Esoph. 1999;12:177–180. [DOI] [PubMed] [Google Scholar]
- 18.Walsh TN, Noonan N, Hollywood D, et al. A comparison of multimodel therapy and surgery for esophageal adenocarcinoma. New Engl J Med. 1996;335:462–467. [DOI] [PubMed] [Google Scholar]
- 19.Beahrs OH, Henson DE, Hutter RVP, Myers MH, eds. Oesophagus. Manual for Staging of Cancer. 3rd ed. Philadelphia; J. P. Lippincott; 1988:63–67. [Google Scholar]
- 20.Madrigal L, Lynch S, Feighery C, et al. Flow cytometric analysis of surface major histocompatibility complex class II expression on human epithelial cells prepared from small intestinal biopsies. J Immunol Methods. 1993;158:207–214. [DOI] [PubMed] [Google Scholar]
- 21.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;297:680–685. [DOI] [PubMed] [Google Scholar]
- 22.Osborn L, Kunkel S, Nabel GJ. Tumor necrosis factor alpha and interleukin-1 stimulate the human immunodeficiency virus enhancer by activation of the nuclear factor kappa B. Pro Natl Acad Sci USA. 1989;86:2336–2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1979;72:248–254. [DOI] [PubMed] [Google Scholar]
- 24.Dejardin E, Deregowski V, Chapelier M, et al. Regulation of NF-kappaB activity by I kappaB-related proteins in adenocarcinoma cells. Oncogene. 1999;18:2567–2577. [DOI] [PubMed] [Google Scholar]
- 25.Yamamoto Y, Gaynor RB. Therapeutic potential of inhibition of the NF-κB pathway in the treatment of inflammation and cancer. J Clin Invest. 2001;107:135–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bours V, Bentires-Alj M, Hellin A-C, et al. Nuclear factor-B, cancer and apoptosis. Biochem Pharm. 2000;60:1085–1090. [DOI] [PubMed] [Google Scholar]
- 27.Liu Z, Hsu H, Goeddel DV, et al. Dissection of TNF receptor 1 effector functions: JNK activation prevents cell death. Cell. 1996;87:565–576. [DOI] [PubMed] [Google Scholar]
- 28.Lagergren J, Bergstrom LA, Nyren O. Symptomatic gastroesophageal reflux as a risk factor for esophageal adenocarcinoma. N Engl J Med. 1999;340:825–831. [DOI] [PubMed] [Google Scholar]
- 29.Debruyne PR, Bruyneel EA, Li X, et al. The role of bile acids in carcinogenesis. Mutat Res. 2001;480–481:359-69. [DOI] [PubMed] [Google Scholar]
- 30.Cohen BI, Raicht RF. Effects of bile acids on colon carcinogenesis in rats treated with carcinogens. Cancer Res. 1981;41:3759–3760. [PubMed] [Google Scholar]
- 31.Fitzgerald RC, Omary MB, Triadafilopoulos G. Dynamic effects of acid on Barrett's esophagus. An ex vivo proliferation and differentiation model. J Clin Invest. 1996;98:2120–2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang C-Y, Cusack JC Jr, Liu R, et al. Control of inducible chemoresistance: increased apoptosis by inhibition of NF-κB. Nat Med. 1999;5:412–417. [DOI] [PubMed] [Google Scholar]