

Abstract
Objective:
To evaluate the effect of impending HIPAA regulations on Applications for Exemptions from Institutional Review Board (IRB) approval.
Summary Background Data:
HIPAA was implemented to reduce potential for misuse of personal information and restricts access to medical records by insurers, employers, and clinical researchers. We hypothesized that HIPAA regulations adversely impact medical records research.
Methods:
The UW–Madison Human Subjects Committee database was accessed to evaluate success and delays in processing Applications for Exemption between September 1999 and March 2003. The number of protocols submitted, number of required revisions, and number considered nonexempt (requiring full IRB review) were determined.
Results:
Prior to 2000, applications for medical records research were rare (11 applications in 1999–2000). In anticipation of the implementation of HIPAA regulations, a new application process was instituted in 2001. During that year, 92 of 103 were approved by an expedited process with few requiring full IRB approval. In 2002 to 2003, submissions increased to 199 and approval without revision dropped to 59% (P < 0.0001) as the number requiring revision (25%) and full IRB approval (16%) increased significantly (P < 0.0001 and P < 0.05, respectively). Of the 31 requiring full IRB approval, 7 were pursued while 24 (77%) were abandoned.
Conclusion:
HIPAA appears to inhibit medical record and database research. Ethical considerations in healthcare research are paramount, but current HIPAA implementation strategies increase workload for HSC and researchers, and increase the dropout rate for proposed studies when investigators are unable or unwilling to meet the regulatory requirements. It is unclear whether or to what degree the new requirements add to protection of privacy. Studies designed to investigate the costs and effects on quantity and/or quality of research should be prospectively implemented.
New HIPAA regulations complicate the process involving permission to do medical records research. Significant delays in approval due to more stringent IRB expectations/requirements and an increase in abandoned requests likely due to additional criteria for approval have been noted in the immediate pre-HIPAA era. These regulations may have significant impact upon subsequent medical records research.
Research involving medical records has played a critical role in medical progress. It is also an important and efficient component in introducing students, residents, and fellows in training to research methods. Formulation of a clinical postulate, creation of a data collection sheet, identification of appropriate study patients, review and rereview of patient charts, data analysis, abstract development and submission, and eventual publication provide excellent training for young investigators. Like all clinical research, medical records studies are time-consuming, particularly when they rely on paper records, and present challenges in time management for trainees and faculty who are increasingly obligated to provide clinical services. Recent changes in the regulation of medicine and medical records do not appear to be making this task easier.1–8
As our field rushes into a highly electronic world with readily transferable data, the risks and benefits of research must be balanced against risks to patient privacy and confidentiality. Misuse of sensitive clinical information could lead to discrimination in insurance or employment, or embarrassment to individual patients or patient populations. The “Common Rule” has been a central component in the protection of human subjects in research, including issues of privacy and confidentiality.1,2 It represents regulations adopted by multiple federal agencies that govern the protection of human subjects in research. Although the National Committee on Vital Health Statistics found no evidence of a medical or health research threat to privacy and confidentiality in a 1997 report, Congress, as part of the Health Insurance Portability and Accountability Act (HIPAA), directed the Commissioner of the U.S. Department of Health and Human Services (DHHS) to write regulations providing protection against inappropriate use of protected health information (PHI) if Congress failed to do so by 2000.1 Although these HIPAA guidelines were not created to address research per se, the guidelines apply to records and data sets that contain PHI used in clinical research. In academic institutions, the onus of assuring privacy and confidentiality in research falls upon Institutional Review Boards (IRBs), which have responded by creating policies and procedures to comply with HIPAA guidelines. With substantial penalties possible for noncompliance, it is not surprising that the guidelines are interpreted conservatively to protect the institution. It is less clear whether these regulations and policies serve the best interests of patients when analysis of clinical databases and medical records might lead to new insights into prevention or treatment of disease, but are discouraged or abandoned because of regulatory obstacles.
Enormous efforts have been invested by attorneys, investigators, IRB members, and others to prepare for HIPAA implementation, which began April 14, 2003. New or expanded IRBs and staffs became necessary as requirements for documentation increased dramatically. This paper examines the extent to which this new regulatory system affects one aspect of clinical research, the Application for Exemption. It focuses primarily on a most basic element of clinical outcome evaluation, the chart review, which remains an important research and teaching tool. Although subtle differences exist between HIPAA and the “Common Rule” in defining protocols exempt from full IRB evaluation, the central issue is whether identifiable data are incorporated into the database. This paper examines experiences of clinical investigators submitting Applications for Exemption at the University of Wisconsin Hospitals and Clinics.
METHODS
After obtaining approval from the University of Wisconsin (UW) Health Sciences IRB (also known as the Human Subjects Committee or HSC), all records of submissions to the IRB for chart review requesting evaluation for exempt status between September 1999 and March 2003 were obtained for review. These records were evaluated for the number of revisions required per protocol, time from protocol submission to approval, and proposals ultimately not approved for exemption. These rejected protocols were cross-referenced against submissions for full Human Subjects Committee (HSC) review. Data were retrospectively reviewed and a database created for the purpose of this study.
Chart reviews submitted for exemption were divided into 3 time periods. Prior to 1999, no specific application process was in place for research involving retrospective review of medical record data. From September 1999 to December 2000 (period I), some investigators submitted written documentation to the UW HSC that met the criteria for exemption. These applications were handled using an expedited review process. In January 2001, the UW HSC implemented a standardized Application for Exemption form to comply with federal regulations protecting human subjects research and period II encompasses January to December 2001. Period III includes requests submitted between January 2002 and March 2003, immediately prior to implementation of HIPAA regulations. During this period, the IRB aggressively organized efforts to assure compliance with full HIPAA implementation effective April 2003. Results were evaluated for statistical significance using both the Fisher exact test and Student’s t test using Sigma Plot (v 8.0)/ Sigma Stat (v. 3.0) Software (Aspire Software International, Leesburg, VA).
RESULTS
Between September 1999 and March 2003, the UW IRB received 303 Applications for Exemption. Of the 303 applications, 170 were for chart or database analysis, 68 for evaluating existing tissue samples, and the remaining 65 were surveys, educational studies/observations, etc. In period I, the 11 protocols submitted were reviewed using an expedited process, kept on file, and not reviewed by the full IRB. Beginning January 2001, the IRB initiated a more formal process for reviewing these studies. The number of Applications for Exemption increased from 103 in period II (12 months) to 189 applications in the final period (15 months).
In period II, 92 of 103 (89%) proposals received approval without revision in 13 ± 18 days (Fig. 1). First-pass approval dropped significantly to 59% (119 of 199, P < 0.001) in period III in 12 ± 23 days. Requested revisions typically consisted of removal of database identifiers, including name, hospital identification number, birth dates, surgical procedure with date, etc, which could allow identification of individuals. Of the 11 charts requiring revision in period II, 4 (36%) were revised and successfully approved an average of 75 ± 64 days following the initial submission (Fig. 2). Seven failed exempt status, required formal IRB submission, but eventually obtained final approval.

FIGURE 1. Percent of applications and time (number of days) for approval for Internal Review Board exemption obtaining “first pass” approval.
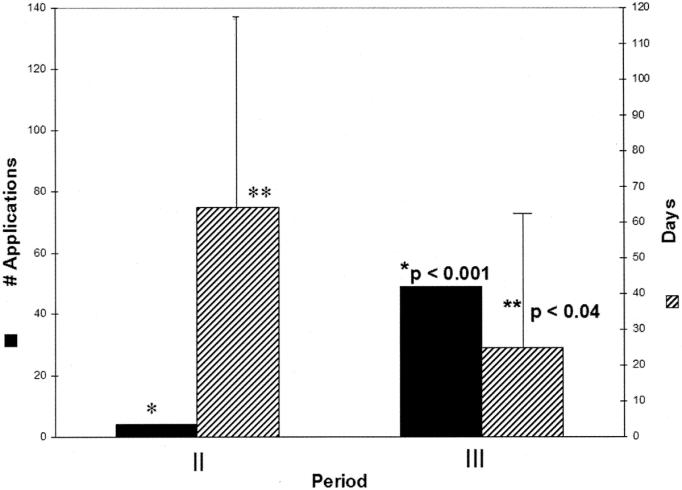
FIGURE 2. Number of applications and time for approval for Internal Review Board exemption obtaining “second pass” approval.
In period III, the numbers of proposals requiring revision increased to 41% (80 of 199 P < 0.001 compared with period II) and more than half of these proposals (49 of 80, 61%) were successfully revised and approved 29 ± 35 days following initial submission (Fig. 2; P < 0.04 vs. period II). This decrease in delay occurred with administrative response by the UW IRB to the increased workload. Charts requiring full IRB review increased significantly between periods II and III from 7% (7 of 103) to 16% (31 of 199) (P < 0.05). Of the 31 proposals requiring full IRB review in period III, 24 (77%) were abandoned by the clinical investigators while 7 (23%) were pursued to successful approval. Almost all of these abandoned proposals were chart reviews.
DISCUSSION
The electronic age permits researchers to retain and use large data sets and perform complex analyses that would have been unthinkable 40 years ago. This technology can provide significant benefit by increasing understanding of the variables that affect health and disease, and the effectiveness of therapeutics to a greater extent than ever before. The technology also increases the opportunity for misuse, which could jeopardize the well-being of patients. Medical information should be private, and its sensitivity warrants scrutiny over its distribution and use. Because of the potential for serious harm through discrimination, loss of insurance, unemployability, or stigmatization, HIPAA regulations provide federal protection for this health information. Unfortunately, a negative effect of this legislation is demonstrated in this study.
Study of medical records and databases provide the opportunity to advance knowledge and understanding about factors that affect health and disease. There are major challenges in designing, implementing, analyzing, and completing such studies. The HIPAA regulations create new impediments, which, although not insurmountable, appear to have had a detrimental effect in our institution. The IRBs obtained resources to meet the new responsibilities, but impediments and delays were observed in the approval process. Our data show that despite these delays the motivated researcher who invests the time and effort to meet the new requirements can accomplish the work. When, however, these efforts to obtain an exemption fail and full IRB review (and the work associated with it) is mandated, the work was abandoned in most cases.
We cannot measure the effects of research not completed as a result of the HIPAA regulations. Since it is unknown how many chart review requests result in completed studies and there are often multiyear delays between completion and publication, we did not expect any observable or meaningful effect on clinically derived publications over this 3-year period. We also cannot measure the degree to which privacy was improved for the patient. Presumably, the HIPAA regulations increased awareness among investigators of the requirements of the Common Rule regarding medical records research and improved understanding of the complicated notion of “identifiability” that reduces the possibility of unwarranted breaches of privacy or confidentiality. For studies that required full IRB review, the separate consent form adds to the costs of research, and it is unclear whether this has had any beneficial effect on privacy.
While ethical considerations in health care and health care research are paramount, the current HIPAA implementation strategies have dramatically increased the workload and responsibility of IRBs, which must now more closely scrutinize the investigator’s proposal.1–8 The new HIPAA rules require specific authorization from a patient to use their protected health information for nonclinical purposes such as research. Although HIPAA guidelines do allow an IRB to grant waivers of authorization if certain criteria are met, the guidelines are not self-implementing, and each institution must define how to apply them. IRBs and investigators are evolving in formulating a shared understanding of the new rules and how they work in specific situations.
The cost to the institution is high. At our institution, panels of attorneys and clinicians struggled to understand the new HIPAA guidelines and define appropriate mechanisms to implement them. HIPAA-specific computerized education courses were developed and individuals with access to, or responsibility for, PHI took intensive, time-consuming training in the new guidelines. The new requirements dramatically increased the number of applications for exemption. Delays in processing these applications occurred early, as our IRB struggled with the increased demands. Subsequently, the IRB expanded and new employees, investigators, and IRB staff were schooled in the intricacies of the “Common Rule” and HIPAA so they could understand and appropriately respond to the requirements. The percentage of Applications for Exemption, which passed a first review, dropped between periods II and III as the institution prepared for the impending HIPAA compliance date of April 2003. While one might suggest that the increase in applications from 103 (period II) to 199 (period III) might have produced “poorer” quality applications and a higher failure rate, it is unlikely that investigators would deliberately sabotage their own requests to increase their chance of failure. Most likely, the increase in number is partially due to the longer period of study (12 vs. 15 months), and the higher failure rate results from poorer understanding of the new guidelines for approval, which were HIPAA-induced. Fortunately, as IRBs received more resources and personnel and costs increased, the initial delays, which had exceeded an average of 2 months initially (through IRB issues and part through clinician delays), dropped and delays and the backlog of proposals decreased significantly. Some protocols could not meet the criteria to be considered exempt by the IRBs and required resubmission to full Human Subjects Committee review. This entailed considerably greater effort from the investigator to meet the more stringent review and, as our data showed, most were abandoned.
To our knowledge, this is the first empiric study that investigates the impact of HIPAA guidelines on clinical outcomes research. In an environment of increasing scrutiny, decreasing time and resources, and increasing clinical demands, these regulations pose another major challenge in the effort to improve the health and welfare of patients. We did not determine the reasons for abandonment of proposals by investigators for practical reasons. This would have required another IRB proposal as well as development of a time-consuming consent process. Some investigators in several private conversations expressed certain frustration with the delays and roadblocks caused by the new guidelines. This question is better addressed prospectively.
We hope this work will stimulate prospective studies designed to investigate the costs involved and measure changes in quantity or quality of this clinical research, which must be balanced with concerns for patient privacy. While patient privacy and confidentiality are paramount and scrutiny is required to protect against misuse of this information, the costs of this scrutiny must not be ignored. If investigations are abandoned due to new impediments, how many important insights will be lost?
Discussions
Dr. Anthony A. Meyer (Chapel Hill, North Carolina): I want to thank the authors for the opportunity to review the manuscript and for their presentation. I appreciate the opportunity to comment on this paper, which is like an early warning of an iceberg. It briefly identifies a relatively small but important problem that is only part of a much larger entity with potentially great threat to clinical research, education, and practice.
HIPAA limits the continuity of existing prospectively gathered data sets and places real impediments on retrospective reviews as described in this study, and threatens the effective methods of teaching and patient care. I have 3 questions and a comment.
First, what were the reasons described by the investigators for abandoning the 29 proposals?
Second, have you been able to measure a change in clinically derived publications from your institution over the same time period?
Third, do you have any measures of the cost of this process at the University of Wisconsin, including the cost to establish the process, the cost of the personnel involved to process the applications, the cost of additional personnel to monitor compliance, and the hidden opportunity costs of lost revenue or lost academic activities of clinicians involved in these endeavors?
I believe that the cost-benefit analyses of HIPAA at the academic medical centers are significantly negative and its impact on society is negative as well.
I agree with the concerns for improperly used protective health information and feel that much less onerous restrictions could have addressed the issues with equal efficacy. In general, I feel privacy of medical information is an important concern that has been carried much too far by the present HIPAA legislation.
Dr. Joseph B. Cofer (Chattanooga, Tennessee): In the manuscript, Dr. Kudsk and his co-authors have examined the outcome of submissions of research protocols to their institutional review boards for projects that requested an application for exemption from full IRB review. They studied 292 applications for exemption for projects involving chart reviews or examination of preexisting tissue samples among others. They essentially compared 2 time periods.
The first was period II, which is in essence a historical control group, which was calendar year 2001. During this time period, they examined 103 proposals to the IRB for exemption from full IRB review. Eighty-nine percent of these were approved on first pass, in 13 days on average. Of those not approved, 36% were successfully revised in 75 days. Seven of these failed, but all eventually received IRB approval.
The second group, in essence the study group, period III, comprised January 2002 to March 2003. In this group, there were 189 proposals for exemption, almost double those in the first group. Fifty-nine percent were approved in 12 days. Of those not approved, 75% were revised to approval. Thirty-one needed full IRB approval. And of these, 23% were approved but 77% were inexplicably abandoned.
So between these 2 periods, they demonstrated a significant decrease in proposals for full IRB exemption approved on first pass, a significant decrease in time required to successfully revise a proposal for eventual approval. They demonstrated a significant increase in projects eventually requiring full IRB review and a dramatic increase in projects abandoned by the investigators. All this in a setting where the absolute number of projects almost doubled in the second period. I have a comment and then 4 questions.
Although Dr. Kudsk and his colleagues have nicely demonstrated the differences between the 2 groups as previously described, comparing the most recent period with the set of internal historic controls. And while the conclusion that these differences are due to the new HIPAA regulations may be intuitive, I see nothing in the manuscript that clearly proves this seminal conclusion.
Some questions for the authors: First, how do you know these differences were due to the new HIPAA regulations? In other words, you had almost twice as many submissions in the second period; maybe some were not as good. You had the same IRB functioning over both periods, and I assume they were using the same regulations. Maybe the IRB was doing a better job during the second period.
Second, much like Dr. Meyer’s. Why were these abandoned projects abandoned? In other words, did you analyze these? Did you look at whether it was from a small set of the same investigators who wrote bad proposals? Interestingly, none in the control group was abandoned and all were passed. So why the sudden change in the second time period?
Third, how did you cut the time successfully revised from 75 days in the first period to 29 days in the second period? That is dramatic improvement. Did you have a better IRB the second time? Or were they better proposals?
Finally, a comment and then a final question. HIPAA is here to stay, along with such other unfunded mandates as the 80-hour workweek, and we could go on and on. Although these regulations will clearly increase the paperwork to do the time-honored chart review, even in your own institution the absolute number, not the percent, of proposals receiving first pass actually increased from 91 to 119 in the 2 time periods. This was comparing 12 to 14 months. So as we get better with these new rules and better learn how to play the game, won’t we work through this, too? So my final and controversial question: Are these regulations really bad for patients or just bad for academic physicians? And which is more important?
Dr. Richard E. Goldstein (Louisville, Kentucky): I wanted to just proceed a little bit along the lines of what Dr. Cofer just talked about in the sense that I think that these HIPAA regulations are here to stay. And they may be modulated to some extent down the line, but I don’t think that we should look to the federal government to make it easier for us.
Our goal in our Division of Surgical Surgery in Louisville is to try to develop protocols so that we can follow many of our patients on a prospective basis, and our ultimate goal is that virtually every patient that we see will be signed up early from protocol to allow those patients to be followed. And I just wanted to get your comment on what you think of that as a potential way around this.
Dr. William C. Lineaweaver (Jackson, Mississippi): I have 3 questions. One, do you have any data on the specific flaws or reasons why these proposals were rejected by the IRB?
Second, does your IRB do any education now for your grant people to make it easier to understand the regulations and to submit passable applications? And have you done any analysis at all of the ultimately rejected and abandoned proposals? Is there any chance that these regulations are snagging bad research or poorly written proposals?
Dr. Basil A. Pruitt, Jr. (San Antonio, Texas): We need more information to evaluate your findings. There were 3 periods of study, but they were each of different duration. If we annualize those periods, tell us what the effect of that is. In the years before approval was required, how many clinical studies were in force? That number would permit us to determine whether HIPAA really had a significant effect on total research activity. Then thirdly, since in many instances you are going to have to de-identify records, can you tell us what the cost is per record to be de-identified?
Dr. Kenneth A. Kudsk (Madison, Wisconsin): I will leave the reasons for the abandonment to the very end.
Dr. Meyer asked whether there was any difference in the publications that have come out. It is very hard to track how many publications came from UW that were related to chart reviews. I really don’t have a handle on that. Certainly some of those that are approved never get published.
There are tremendous costs from HIPAA. A panel of attorneys together with a panel of clinicians spent countless hours trying to figure out how the university would deal with these compliance issues.
An educational tool was created that took me 70 minutes to complete on a computer. That had to be completed by anyone who used protected health information within the university. Not just the medical center, but athletics, the college of nutritional sciences, etc. So this is a huge time sink. If everybody spent 70 minutes, you can imagine the number of man-hours that were invested.
Dr. Cofer, I appreciate your comments. This whole process was HIPAA driven. The IRB changed tremendously between period II and III. There was a large increase in staff and another IRB, the Minimal Risk IRB that I was recruited into. It has been a significant time sink for me. And I would advise that a surgeon be put on these committees. It will give a surgical perspective and will give someone within your department to help an individuals get through the maze. It took the committee approximately 2.5 months to figure out how to interpret HIPAA rules and it is just now becoming more streamlined.
We are trying to educate individuals on what they need to do to get a second pass. Almost all problems relate to the data collection sheet, which we require to have no identifiers on it. The process of de-identification, even with information like county and an occupation, has to be considered by the IRB. It makes it very difficult. Your data collection sheet better be correct the first time you review the charts, because there is no way to figure out a specific chart to add more information.
We are getting better education to the people, but does it help the patients? I am not sure, but it certainly has a negative impact upon the time commitment of the academic researchers.
In regard to Dr. Goldstein’s suggestion about signing individuals early, it has been proposed that consent be obtained from the patients when they are admitted to the hospital. I think there is some reluctance to allow institutions to have a blanket approval for the use of protected health information from everyone that enters the hospital. It was Johns Hopkins, which tried to get that through, but it was denied. I don’t think they have been successful so far.
Dr. Lineaweaver asked about the data flaws. It is not what you collect, it is the fact there is a link, even obscure links, allowing someone to find out what patient that data came from. Our new submission process will be put on the Internet, and hopefully there will be clues of the sort of appropriate answers to ease approval. We will try to make a checklist so that as people work their way through the list they get educated to what the right and wrong answers are.
I think there were 3 periods, as Dr. Pruitt pointed out, but I have no idea what was happening prior to HIPAA.
The question about abandonment is an excellent example of what has happened with HIPAA. We wanted to interview those 31 people and find out why they abandoned the studies. It would have required going to the IRB submission of an informed consent (or at least an information sheet), a list of what the questions were that we were going to ask, and then final approval up front. When we would look at the gain and compared it to the cost, we abandoned the project.
Footnotes
Reprints: Kenneth A. Kudsk, MD, University of Wisconsin Hospital and Clinics, Department of Surgery, H4/736 Clinical Science Center, 600 Highland Avenue, Madison, WI 53792-7375. E-mail: Kudsk@surgery.wisc.edu.
REFERENCES
- 1.Kulynych J, Korn D. The new HIPAA (Health Insurance Portability and Accountability Act of 1996) Medical Privacy Rule: help or hindrance for clinical research? Circulation. 2003;108:912–914. [DOI] [PubMed] [Google Scholar]
- 2.Kulynych J, Korn D. The effect of the New Federal Medical–Privacy Rule on Research. N Engl J Med. 2002;346:201–204. [DOI] [PubMed] [Google Scholar]
- 3.Kilbridge P. The cost of HIPAA compliance. N Engl J Med. 2003;348:1423–1424. [DOI] [PubMed] [Google Scholar]
- 4.Califf R, Muhlbaier L. Health Insurance Portability and Accountability Act (HIPAA): must there be a trade-off between privacy and quality of health care or can we advance both? Circulation. 2003;108:915–918. [DOI] [PubMed] [Google Scholar]
- 5.Willerson J, Kereiakes D. Clinical research and future improvement in clinical care: the Health Insurance Portability and Accountability Act (HIPAA) and future difficulties but optimism for the way forward. Circulation. 2003;108:919–920. [DOI] [PubMed] [Google Scholar]
- 6.Durham M. How research will adapt to HIPAA: a view from within the healthcare delivery system. Am J Law Med. 2002;28:491–502. [PubMed] [Google Scholar]
- 7.Dimond K. The impact of privacy regulations on clinical research. J Biolaw Bus. 2002. 2002;5:50–53. [PubMed] [Google Scholar]
- 8.Melton LJ. The threat to medical-records research. N Engl J Med. 1997;337:1466–1470. [DOI] [PubMed] [Google Scholar]