

Abstract
Proteolytic processing of the amyloid-β precursor protein (APP) generates the Aβ amyloid peptide of Alzheimer's disease. The biological function of APP itself remains, however, unclear. In the current review, we study in detail the different subdomains of APP and try to assign functional significance to particular structures identified in the protein.
Keywords: Alzheimer's disease, amyloid-β precursor protein, APP, structural and functional relationships
Introduction
There are few proteins that have been studied more than amyloid-β precursor protein (APP). We have structures of several subdomains, knockout models in mice, fly and worms, detailed cell biological analysis of subcellular trafficking and post-translational modifications and a bewildering list of potential functions. Beyond discussion is the fact that ‘secretases', a series of proteolytic activities, cleave APP in different fragments (Figure 1 and reviewed in Haass, 2004), and that those fragments have very different fates. Most notoriously, β- and subsequent γ-secretase cleavage release the amyloidogenic Aβ peptides that build up the amyloid plaques in the brain of Alzheimer's disease patients (reviewed in De Strooper and Annaert, 2000; Selkoe, 2001). Some evidence implies the soluble APP ectodomain (sAPP) and the APP intracellular domain (AICD) in signaling and in a series of physiological responses (see below). While it is possible that one does not need to understand these functions to understand the role of Aβ amyloid in Alzheimer's disease, it is certainly not excluded that disturbance of normal APP function contributes to the disease process as well (see, for instance, Stokin et al, 2005). Moreover, after more than 15 years of intense research, understanding the biology of APP remains an important scientific and intellectual challenge. Finally, dysfunction of APP itself or of the protein context in which APP is operating could lead to (transient) changes in cellular APP expression and to increases in Aβ generation. For instance, brain trauma is associated with upregulation of APP expression (Murakami et al, 1998; Van den Heuvel et al, 1999; Leyssen et al, 2005) and deposition of diffuse amyloid precipitates, raising the questions whether this contributes to increased AD risk, and what the function of APP in brain trauma could be.
Figure 1.
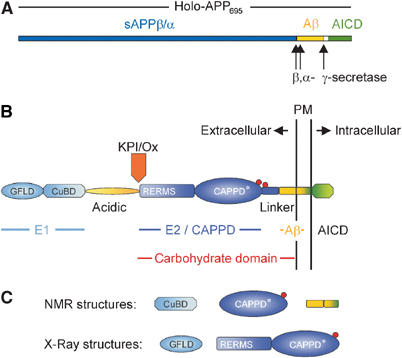
(A) Schematic representation of the holoprotein of human APP695 including the relative position of the α-, β- and γ-secretase cleavage sites. (B) Domain organization of APP: the E1 region consists of the N-terminal growth factor-like domain (GFLD) and the following copper-binding domain (CuBD). The E1 region is linked via the acidic region to the carbohydrate domain, which contains the two N-glycosylation sites of the ectodomain (red spheres). The carbohydrate domain can be subdivided into the E2 domain, also called central APP domain (CAPPD), and a linker or juxtamembrane domain. The carbohydrate domain is followed by the transmembrane and the APP intracellular domain (AICD). Aβ indicates the amyloid β-peptide sequence. The Kunitz-type protease inhibitor domain (KPI), which is present in APP751 and APP770, and the Ox2 sequence, which is present in APP770, are shown above their insertion site. (C) Known stable structures of APP.
Understanding the function of APP piece by piece
In mammalia, APP is part of a larger gene family that includes APP-like proteins 1 and 2 (APLP1 and APLP2) (De Strooper and Annaert, 2000). APP homologs are also found in Caenorhabditis elegans (Apl-1) (Daigle and Li, 1993) and Drosophila (APPL) (Martin-Morris and White, 1990).
When studying APP (or its homologs) in cell culture, many researchers have focused on specific fragments and/or subdomains of APP (Figure 1), and a plethora of functions have been attributed to each of them (summarized in Table I). Many of these studies rely on overexpression of APP and/or use high concentrations of purified protein fragments, which inherently raise the risk of nonphysiological responses. Moreover, since the turnover of APP is quite fast (∼30–90 min, Herreman et al, 2003), it is likely that APP plays a regulatory role (as opposed to a structural role) in the cell, making the study of its function inherently more difficult.
Table 1.
Various protein interactions and functions of APP
| Function | APP domain | Binding partners | References |
|---|---|---|---|
| Surface receptor | CAPPD | F-spondin | Ho and Sudhof (2004) |
| Aβ, undefined | ApoE | Strittmatter et al (1993) and reviewed in Turner et al (2003) | |
| Undefined | Aβ | Lorenzo et al (2000) | |
| Adhesion molecule | |||
| Cell/substratum | Undefined, Aβ | ECM (laminin, collagen, perlecan, etc.) | Kibbey et al (1993) and reviewed in Small et al (1999) and Turner et al (2003) |
| E1 (GFLD) | Fibulin-1 | Ohsawa et al (2001) | |
| E1, E2 (HBD 1+2) | HSPG | Reviewed in Small et al (1999) | |
| AICD | Fe65, Menaa | Sabo et al (2001) | |
| Cell/cell | CAPPD | CAPPD (in cis or trans?) | Wang and Ha (2004) |
| E1 | E1 (in cis or trans?) | Soba et al (2005) | |
| Regulator of neuronal processes | |||
| Neurite outgrowth | sAPP | Undefined | Reviewed in Mattson (1997) and Small et al (1999) |
| Aβ | Undefined | Reviewed in Mattson (1997) and Small et al (1999) | |
| Dendritic arborization | CAPPD | Undefined | Reviewed in Mattson (1997) and De Strooper and Annaert (2000) |
| AICD (C99) | Abl, profilin, JIP | Leyssen et al (2005) | |
| Synaptogenesis | sAPP | Undefined | Reviewed in Mattson (1997) and Small et al (1999) |
| Undefined, AICD | Fasciclin II, X11/Mint | Ashley et al (2005) | |
| Synaptic plasticity | sAPP | Undefined | Reviewed in Mattson (1997) and Turner et al (2003) |
| Neuronal excitability | sAPP | Undefined | Reviewed in Mattson (1997) and Turner et al (2003) |
| Axonal transport cargo receptor | AICD | JIP, kinesina | Kamal et al (2000, 2001), Taru et al (2002), Matsuda et al (2003) and Lazarov et al (2005) |
| Regulator of neuronal stem cell division | sAPP | Undefined (present on type-A and type-C cells in SVZ) | Caille et al (2004) |
| Signaling molecule | |||
| G-protein-coupled receptor protein signaling pathway | AICD | G(o) | Nishimoto et al (1993) |
| Kinase-mediated signaling cascades | AICD | Abl, Shc/Grb2, JIP, Dab | Russo et al (2002), Tarr et al (2002) and reviewed in Koo (2002) and King and Scott Turner (2004) |
| sAPP | Undefined | Reviewed in Mattson (1997) | |
| Gene transcription | AICD (C59, C57, C49) | Fe65, Fe65L, JIP, Mint/X11, Numb | Roncarati et al (2002), Scheinfeld et al (2003), Merdes et al (2004) and reviewed in Mattson (1997), Koo (2002), Turner et al (2003), Beglopoulos et al (2004) |
| Regulator of calcium homeostasis | Aβ, undefined | Undefined | Reviewed in LaFerla (2002), Abramov et al (2004) |
| AICD (C99, C57) | Undefined (via phosphoinositide) | Leissring et al (2002) | |
| Regulator of metal homeostasis | CuBD | Cu2+ | Reviewed in Maynard et al (2005) |
| Regulator of cell survival/death | |||
| Neurotrophic | sAPP | Undefined | Reviewed in Mattson (1997) |
| Neurotoxic | Aβb | Undefined, APP | Lu et al (2003) and reviewed in Mattson (1997), Walsh and Selkoe (2004) |
| AICD casp (C31) | Caspases, APP-BP1 | Chow et al (1996) and reviewed in Milligan (2000) | |
| AICD (C59, C57) | Undefined (signals through GSK3β, Tip60) | Kinoshita et al (2002a) and Kim et al (2003) | |
| |
AICD (a.a. 649–664) |
Undefined |
Bertrand et al (2001) |
| ECM=extracellular matrix; HBD=heparin-binding domain; HSPG=heparan sulfate proteoglycans; SVZ=subventricular zone. | |||
| The different suggested biological functions of APP are summarized. The proteins identified to interact with APP and relevant for the described functions are mentioned, together with further references. | |||
| aIndirect interaction. | |||
| bOligomer versus monomer? | |||
An important approach to study APP function is the identification of protein binding partners. APP can actually bind to a large number of proteins (Table I), but not all reported interactions are equally informative. For example, proteins like calreticulin (Johnson et al, 2001) or clathrin (Nordstedt et al, 1993) are reported to bind APP, but are also known to be quite generally involved in protein maturation or endocytosis, respectively. We did not include such proteins in the overview. It is interesting to note that many of the proposed functions of APP (Table I) and its derived fragments can be grouped into a series of common biological processes. For example, neurite outgrowth, dendritic arborization and synaptogenesis all require highly organized cell–cell and cell–substratum interactions, to which APP indeed appears to contribute. From the table, it also becomes clear that both the extra- and intracellular parts of APP are involved in similar biological functions like neurite outgrowth or arborization. It is tempting to speculate that holo-APP (Figure 1A) could function in these processes at both sides of the plasma membrane linking extracellular cues (e.g. ligand or substratum binding) to intracellular signaling pathways (via scaffolding proteins, Ca2+ regulation, interactions with the cytoskeleton and/or protein kinases). In this context, APP could function as a receptor-like modulatory protein in neuronal processes (see, e.g. Ashley et al, 2005).
What could be learned from APP biological models?
The use of biological models deficient for APP (or its homologs) has not yet allowed deciding definitively to what extent the listed functions in Table I are really important in the context of the whole organism. APPL null mutant flies are viable, fertile and display subtle behavioral (Luo et al, 1992) and neuronal defects including reduced synapse number (Ashley et al, 2005) and reduced synapse bouton formation (Torroja et al, 1999) at the neuromuscular junction. Interestingly, similar (synapse) defects were observed in mutant flies for fasciclin II, a cell adhesion molecule. In an elegant study by Ashley et al (2005) both genetic and biochemical evidences support the notion that APPL and fasciclin II function in a common signaling pathway, which regulates synaptic development. Overexpression of APP or APPL in Drosophila was shown to induce a series of biological effects (which could be explained in part by gene dosage effects and tissue/cell-specific expression) ranging from a blistered wing phenotype (a cell adhesion disorder) (Fossgreen et al, 1998), a Notch gain-of-function phenotype (possibly via the adaptor protein Numb) (Loewer et al, 2004), axonal transport deficits (Torroja et al, 1999; Gunawardena and Goldstein, 2001) or induction of neurite outgrowth and arborization (via Abelson tyrosine kinase and profilin) (Leyssen et al, 2005). Taken together, it is likely that neuronal APPL indeed plays an important role in neurite outgrowth, arborization and synaptogenesis. Extensive genetic and biochemical evidence from Goldstein and collaborators (Kamal et al, 2000, 2001; Gunawardena and Goldstein, 2001) in Drosophila and mouse models supports a functional interaction between the APP cytoplasmic domain and the kinesin light chain in neurons. Rapid axonal transport of APP containing transport vesicles critically depends on this interaction. Intriguingly, these vesicles also contain β- and γ-secretase and generate the pathological Aβ40 and Aβ42 peptides (Kamal et al, 2001). In the brain of mouse models and patients with Alzheimer's disease, axonal swellings containing motor proteins and vesicular elements were observed, and disturbed axonal transport resulted in increased Aβ generation (Stokin et al, 2005). Overall these data support the hypothesis that disrupted axonal transport contributes to Alzheimer's disease. However, four independent groups recently claimed that some of the salient observations of Goldstein et al could not be reproduced (Lazarov et al, 2005). While negative data are never definitive, further independent confirmation of the model proposed by Goldstein and collaborators is now needed.
A C. elegans knockout model for Apl-1 is not yet available. Worms in which Apl-1 was downregulated using double-strand RNA (dsRNA) showed a normal development with no major abnormalities (Bruni et al, 2002). Some perturbations in pharyngeal pumping were observed however, but the molecular mechanisms responsible for this phenotype remain to be elucidated.
Perhaps, the best insights into the function of APP come from the APP/APLP1/APLP2 triple knockout mice (Herms et al, 2004). Whereas APP deficiency alone results in only a subtle phenotype (Zheng et al, 1995) (likely due to a functional redundancy with APLPs; von Koch et al, 1997; Heber et al, 2000), APP/APLP1/APLP2 triple knockout mice die prematurely and display characteristic traits of a rare human neurological disorder called cobblestone (type II) lissencephaly (Herms et al, 2004). At the morphological level, these mice show a high incidence of cortical dysplasias (characterized by a fragmented basal lamina and ‘overmigration' of neurons). The molecular mechanisms responsible for this phenotype remain to be elucidated, but these observations highlight the importance of APP in neuronal cell adhesion and migration. However, it is also clear that in many other neurons, APP function is apparently not essential during embryogenesis. The notion that APP function is more important in adulthood, for example, in brain repair after traumatic stress (Leyssen et al, 2005), therefore deserves further exploration in available APP-deficient mice. Of note, a cobblestone lissencephaly phenotype (with various penetrance) is also observed in presenilin-1, β1 and α6 integrins, focal adhesion kinase, α-dystroglycan and laminin α2 knockout mice (reviewed in Lambert de Rouvroit and Goffinet, 2001; Berechid et al, 2002). Whether a direct relationship exists between the dysfunction of APP, some of these proteins and the cortical dysplasias observed in the knockout mice will need further study. The relevance for AD is also speculative. Many biological and biochemical pathways are perturbed in the AD brain, which are as diverse as defects in protein/axonal transport (Stokin et al, 2005), active neuronal cell death (apoptosis) (Cribbs et al, 2004) and oxidative stress (Aslan and Ozben, 2004), which can, in the end, influence APP metabolism and Aβ deposition. In any case, it is clear that the study of APP biology remains a major challenge in the field.
What could be learned from APP structures?
Another way to look at APP function is by trying to understand the structural basis of APP and identifying specific regions important for its function(s). These functional subdomains can hopefully be used to identify the proteins and receptors that interact with those domains. APP is a type I transmembrane protein whose single transmembrane span separates the large N-terminal extracellular domain from the short C-terminal cytoplasmic tail domain. However, the extracellular sequence of APP can be regarded as a string of several individual domains, most of them also representing independent folding units (Figures 1B and C). These individual APP units have been structurally characterized in the last years. However, in the absence of a complete crystal structure of APP, it remains to be seen to what extent the conformation of the individual subunits is conserved in the context of the whole protein.
The ectodomain of APP
The extracellular domain of APP has a complex structure and more than 70% of the amino acid (a.a.) residues participate in standard secondary structure elements (Gralle et al, 2002). A.a. residues that do not participate in any standard secondary structure cluster in two regions from a.a. residues 190–264 and approximately from a.a. residues 507–589. The first stretch is strongly negative and corresponds to the acidic domain. More than 56% of the residues are glutamate or aspartate, sometimes eight of them in a row, which makes it quite difficult to accommodate this region into an ordered, folded structure. The second stretch lies amino-terminal to the Aβ-sequence and corresponds roughly to the so-called ‘linker region' or ‘juxtamembrane region' (Dulubova et al, 2004; Wang and Ha, 2004). According to several different secondary structure prediction algorithms, this ‘linker region' is almost completely devoid of helical or extended sheet structures. These two unstructured domains also display no homology to the otherwise very similar APP-like proteins, and may therefore correspond to domains that provide specific functions to APP. However, it seems more likely that these unstructured domains represent flexible linkers that connect the individual folding units, regulating their relative distance to each other and providing mobility in three dimensions. Again, the structure of holo-APP is needed to understand the role of the acidic and the linker region in APP better.
The N-terminal head of APP (a.a. residues 23–128) has already been crystallized 6 years ago (Rossjohn et al, 1999) and was subsequently called growth factor-like domain (GFLD). The GFLD contains nine β-strands and only one α-helix (Figure 2A). The domain is cysteine rich and possesses three disulfide bridges that, together with the hydrophobic core of the structure, are well conserved across the APP family. The disulfide bridge between Cys98 and Cys105 stabilizes a β-hairpin loop, which seems to be critical for neurite outgrowth (Small et al, 1994) and MAP kinase activation (Greenberg et al, 1995) (Figure 2A, highlighted in green). Moreover, the loop consists of several basic residues and contributes largely to a positively charged surface. This surface could represent one of the APP heparin-binding sites (Figure 2B, red colored). However, the β-hairpin loop is the most mobile region of the structure and only few of the basic residues are conserved across the APP family. Immediately adjacent to this putative heparin-binding site is a hydrophobic surface patch (Figure 2B, green surface), and such patches are quite often key players in protein–protein interactions. This hydrophobic surface is conserved within the whole APP family, and could contribute to the interaction with proteoglycans by binding to their core proteins. It is also possible that it provides an APP–APP dimerization interface (see below), not unlike other growth factors that dimerize in the presence of heparin (Mohammadi et al, 2005). The possibility that this is a ligand-binding site should also be considered. Experimental data allowing one to discriminate between these possibilities are lacking until now. Moreover, since the structure of the N-terminal head domain of APP shows no similarities to any known protein, speculating on the function of this domain is quite risky. Nevertheless, since a similar disulfide-bonded hairpin loop has been identified in a couple of growth factors (reviewed in Chirgadze et al, 1998; Kadomatsu and Muramatsu, 2004) and since the (extended) N-terminal domain of APP (E1 plus the acidic region, see Figure 1B) is able to stimulate neurite outgrowth (Ohsawa et al, 1997, 1999), some authors suggest that the N-terminal head domain by itself is responsible for the growth factor-like properties of APP (see Table I). It should also be noticed that the E2 domain of APP with the RERMS sequence (see below) has been proposed to have growth-promoting properties.
Figure 2.
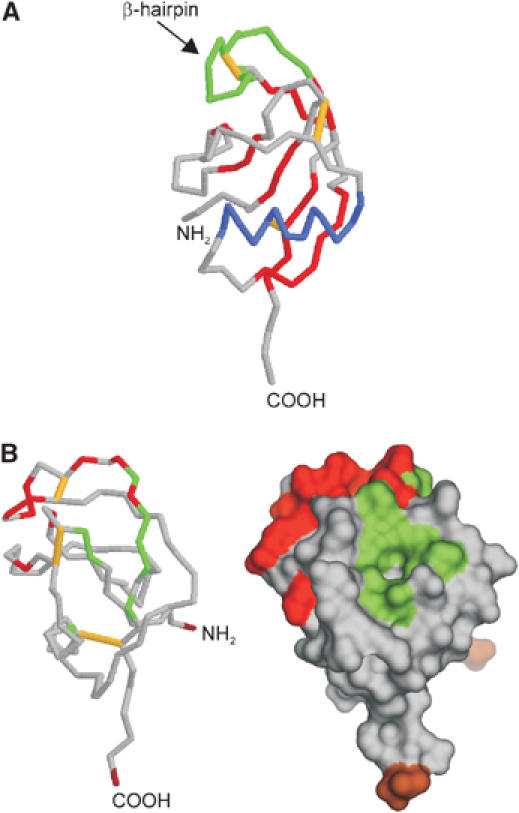
Growth factor-like domain (PDB:1MWP). (A) Backbone diagram: disulfide bridges are shown in yellow, β-sheets in red, the α-helix in blue and the β-hairpin loop in green. (B) Surface representation: the N- and C-termini are colored in brown, the hydrophobic surface patch in green and the HSPG-binging region is shown in red (note that the structure is rotated around the vertical axis by 90° compared to (A)).
The GFLD is followed by the well-studied copper-binding domain (CuBD) and both domains together constitute the E1 domain of APP (Daigle and Li, 1993). The CuBD comprises the residues 124–189 of APP and the structure consists of one α-helix packed against a triple-stranded β-sheet (Barnham et al, 2003). Three disulfide bonds and a small hydrophobic core stabilize the structure. The residues His-147, His-151, Tyr-168 and Met-170 are arranged in a tetrahedral orientation forming the copper-binding site (Figure 3). Tetrahedral coordination of Cu(II) can explain the redox chemistry associated with copper binding to APP. In general, Cu(II) ions favor a planar coordination sphere, whereas Cu(I) prefers a tetrahedral arrangement. Furthermore, histidine residues are known ligands of Cu(I) sites and oxygen ligands are more common in Cu(II) complexes (Casella and Gullotti, 1993). Thus, the unusual tyrosine residue within the copper coordination sphere in APP may facilitate binding of Cu(II), which is subsequently followed by the redox reaction. The relatively rigid tetrahedral copper-binding site would prefer Cu(I) binding and therefore facilitate the reduction of Cu(II) (Multhaup et al, 1996). Interestingly, the APP copper-binding site is surface exposed and shows strong structural similarities to copper chaperones. Although the copper chaperones have a different coordination sphere using thiol residues (CXXC motif), they also consist of α-helix packed over a triple β-sheet topology (for a review see Huffman and O'Halloran, 2001; Markossian and Kurganov, 2003). These chaperones are mainly cytosolic, whereas APP as an extracellular protein is not able to use cysteines for metal coordination. APP has therefore histidine residues to create a metal-binding motif like other membrane proteins, for example, the CopB copper ATPase from Enterococcus hirae (Cobine et al, 2002).
Figure 3.
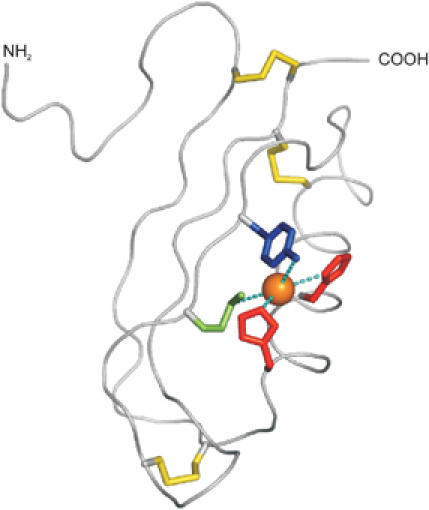
Copper-binding domain (PDB:1OWT). Backbone trace of the copper-binding domain of APP with disulphide bridges indicated in yellow. An orange sphere indicates the approximate position of a Cu(I) ion modeled to adopt a tetrahedral coordination geometry between His-147 (red), His-151 (red), Tyr-168 (blue) and Met-170 (green).
The E1 domain is connected via the unstructured and flexible acidic region to the carbohydrate domain, which can be divided into the E2 domain, also called central APP domain (CAPPD), and the linker region. The E2 domain is one of the most interesting regions within the ectodomain of APP, because it contains a couple of substructures that might provide interaction sites for binding partners. Structural analysis of the E2 domain revealed two distinct coiled-coil substructures composed of six α-helices in total (Wang and Ha, 2004). Whereas the most prominent feature of the N-terminal substructure is a two-stranded coiled coil without any twist, the C-terminal structure consists of a triple-stranded antiparallel coiled coil. The long central helix joins the two substructures (Figure 4A). Each substructure contains a hydrophobic cluster of evolutionary conserved a.a. residues and also the overall protein fold of the E2 domain is shared by other members of the APP family. The E2 domain contains the famous RERMS sequence, implicated in the growth-promoting properties of APP (Ninomiya et al, 1993; Li et al, 1997). In line with this hypothesis, the pentapeptide is surface exposed, although it is not part of a defined pocket or groove (Figure 4, highlighted in blue). Another feature of the E2 domain is the highly conserved heparan sulfate proteoglycan (HSPG)-binding site, which forms a groove on the surface (Figure 4, red colored). Compared to the HSPG-binding site of the GFLD, the elongated shape of the E2 HSPG-binding site seems to be much more suitable for tight HSPG binding, providing a large surface wrap around the helical, usually straight polysaccharide. However, binding constants of both domains have not been compared, and thus it remains speculative whether the two HSPG domains reflect a low- (GFLD) and a high-affinity (E2) binding site.
Figure 4.
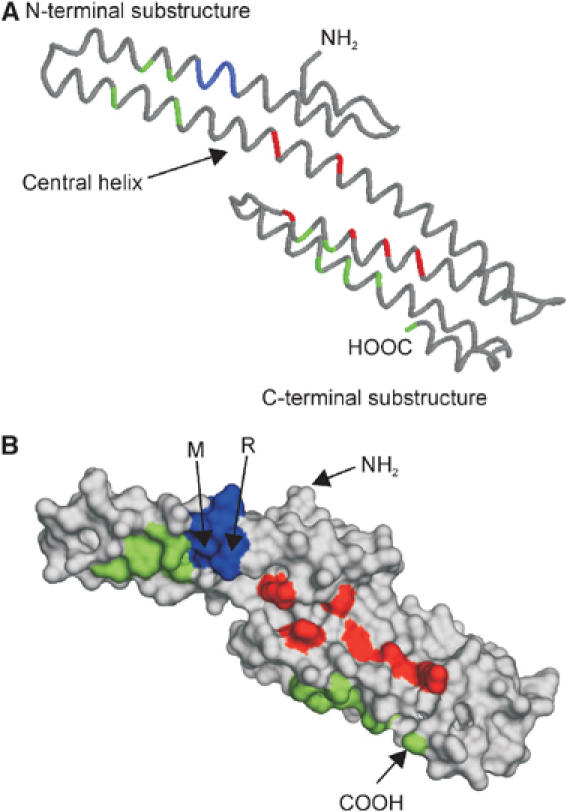
Monomeric E2 domain/central APP domain (CAPPD) (PDB:1RW6). (A) Backbone diagram: position of RERMS sequence is marked in blue, amino-acid residues forming the two hydrophobic patches are colored in green and the conserved amino-acid residues of the HSPG-binding site in red. (B) Surface representation of (A). The N- and C-termini as well as the underlined amino-acid residues of the RERMS sequence, which participate in dimerization, are labeled.
Like all solved APP structures, the folding of the E2 domain is again quite unique, displaying only some structural similarities with spectrin or α-actinin (Wang and Ha, 2004). Interestingly, like these two proteins, the E2 domain of APP is able to form an antiparallel dimer in solution. Dimerization of the E2 domain buries the hydrophobic surface cluster (Figure 4B, green colored), and also generates a better groove for the heparin-binding site, since both monomers contribute to the positively charged surface patch. However, two a.a. residues of the RERMS sequence participate directly in the dimerization (Figure 4B), and therefore the motif is no longer available for other interactions. Thus, dimerization and dissociation of APP might regulate various APP functions. One could imagine that cell-bound APP could form dimers in trans-regulating cell–cell adhesion, whereas monomeric APP could act either as a growth factor receptor when it is still bound to the cell membrane or as a growth factor once it is released by α-secretase cleavage into the external milieu. We have to mention here that the oligomerization state of the ectodomain is still under debate. Whereas some studies revealed almost exclusively dimers for the recombinant ectodomain (Scheuermann et al, 2001; Wang and Ha, 2004), others found mainly monomers, although with quite high Stokes radius, indicating that APP behaves as an elongated structure rather than a compact spherical molecule (Gralle et al, 2002; Botelho et al, 2003). Also problematic for the dimerization hypothesis is the fact that all other substructures of the ectodomain, including the CAPPD* (Dulubova et al, 2004) (Figure 1B), are monomeric in solution. Thus, it is unclear whether the secreted ectodomain of APP (sAPP) is monomeric, dimeric or adopts both conformations in solution. To make the story even more complicated, in vivo evidence indicates that cell-bound APP can dimerize after biosynthesis in the ER (cis dimerization), and disulfide-bridged dimers of APP influence Aβ generation (Scheuermann et al, 2001). Moreover, the strong homology and conserved domain organization between APP and the APP family members APLP1 and APLP2 imply that not only homo-oligomerization but also hetero-oligomerization might be possible as demonstrated recently (Soba et al, 2005). In contrast to the cis/trans-oligomerization of the E2 domain observed by Wang and colleagues (Wang and Ha, 2004), Soba and co-workers identified the E1 domain as a major interaction interface. The E1 trans-complexes promote cell adhesion and the existence of endogenous heterocomplexes in mouse brain and synaptic compartments supports a role of the APP family proteins in cell–cell interaction. In conclusion, dynamic alterations of the APP ectodomain between monomeric, homodimeric and heterodimeric status could at least partially explain some of the variety in the functions of APP (Table I).
The Aβ region
The main focus of structural studies has always been the Aβ peptide and several techniques have been employed to characterize soluble Aβ, aggregation intermediates and Aβ fibrils. Meanwhile, about 40 structures of peptides with various lengths are available, documenting the structure of soluble Aβ in a variety of conditions (for a review see: Serpell, 2000; Morgan et al, 2004). Most of the studies have been performed in membrane-mimicking environments both to avoid aggregation and as an approximation of the physiological structure of the Aβ peptide in APP. The absence of β-strand secondary structures, which are linked with insolubility and plaques formation, is striking. Aβ40/42 peptides most likely consist of two α-helices that are linked via a ‘kink' or a loop (Figure 5). In contrast to this ‘soluble' Aβ, the peptide exhibits a wide content of β-sheet in amyloid fibrils, and the conformational switch from α-helical to β-sheet structure in Aβ could be fundamental for the amyloidogenic properties of the peptide. Triggers of this conformational switch are still not fully understood, but disease-related mutants like the Dutch and Arctic mutations are located in the first α-helix (a.a. 10–24) and cause its destabilization favoring a β-sheet conformation. Indeed, the Dutch mutation was one of the earliest identified mutations that increase Aβ-fibril formation (Levy et al, 1990; Wisniewski et al, 1991).
Figure 5.

Solution structure of Aβ42 (A) in 40% trifluorethanol (PDB:1AML) and (B) in 80% hexafluoroisopropanol (PDB:1IYT). Three models out of 20 and 10 are depicted. Note that the structure of Aβ42 is less stable in 40% trifluorethanol.
Nevertheless, in vivo amyloidogenesis cannot be explained simply by the Aβ concentration and/or Aβ40/42 ratio. The process involves additional factors like metal ions, glycosaminoglycans and other extracellular matrix components. While the role of Aβ in Alzheimer's disease has received enormous attention (for reviews see Morgan et al, 2004; Walsh and Selkoe, 2004), some recent studies suggest that Aβ is more than only a ‘wear and tear' product. Aβ was proposed as a regulator of ion channel function (Ramsden et al, 2001, 2002) and as essential for neuronal health (Plant et al, 2003). Perhaps, the more striking results came from the work of Kamenetz et al (2003). They observe that Aβ is secreted from neurons in response to synaptic activity and that Aβ, in turn, downregulates synaptic transmission. This negative feedback loop could operate as a physiological homeostatic mechanism to keep the levels of neuronal activity in check. The hypothesis that Aβ has a physiological function remains nevertheless quite speculative. For instance, the Aβ sequence is the less conserved part between the human, and mouse sequence and mouse APP (when compared to human) is processed poorly by β-secretase, resulting in about three-fold lower amounts of Aβ peptide generated per mol mouse APP compared to human APP (De Strooper et al, 1995). This suggests that Aβ is not critical in the mouse brain. Further work in vivo in mice totally lacking Aβ generation (e.g. in BACE knockout mice) is needed to confirm whether Aβ has a biological function.
The intracellular domain
The cytoplasmic domain of APP (referred to as the APP intracellular domain, AID or AICD) is proteolytically released by a presenilin/γ-secretase-dependent process and comprises 49–50 a.a. residues (Gu et al, 2001; Sastre et al, 2001; Yu et al, 2001; Weidemann et al, 2002). This is 7–9 a.a.'s shorter than expected from γ-secretase cleavage (Figure 1A; and for a detailed scheme see Annaert and De Strooper, 2002). It is possible that APP is first cleaved at this ‘ɛ-site' and that the remaining membrane-bound part is then further processed to yield finally an Aβ-peptide of 40–42 residues. The recent identification of long, cell-bound Aβ species would agree with this hypothesis (Qi-Takahara et al, 2005; Zhao et al, 2005).
Most of the identified binding partners of APP interact with the intracellular domain (Table I). Of particular interest, the YENPTY motif (a.a. residues 682–687) present in the AICD is completely conserved from C. elegans to humans. This motif is important for Clathrin-mediated endocytosis and binding to numerous proteins including Fe65, JIP and X11/Mint (reviewed in De Strooper and Annaert, 2000; King and Scott Turner, 2004). At the structural level, AICD adopts no stable conformation in solution. However, several transient structures could be observed over a broad pH range including a hydrophobic cluster, a type I β-turn and a nascent helix character in the C-terminal half of the peptide (Ramelot et al, 2000). Interestingly, the structural analysis shows no tertiary contacts and might therefore describe an early state in the protein folding process. Interaction with a binding partner would induce further protein folding and stabilize the resulting structure. Thus, the structure of AICD could be different depending on the binding partner. While some structural features would be retained and stabilized in the folded (=bound) state, other structures can then be rearranged with minimal energetic costs. Indeed, this behavior was observed in the cocrystal of AICD with the PDI domain of X11 (Zhang et al, 1997). Thus, an explanation is at hand for the manifold ‘specific' interactions with different intracellular proteins (Table I). Such phenomenon is called ‘binding promiscuity' or ‘one-to-many' signaling and could be relevant for the understanding of the activity of a variety of intrinsic unstructured proteins, for example, α-synuclein or the estrogen receptor α (for a review see Uversky et al, 2005). Stability of the AICD–ligand complex can be further influenced by phosphorylation of the AICD domain. Phosphorylation of threonine 668, for instance, is crucial for Fe65 binding, but has little or no impact on the interaction with X11 (Ando et al, 2001). Thus, phosphorylation of the AICD might regulate complex formation and stability and also signaling that is possibly associated with γ-secretase cleavage of APP. As shown in Table I, AICD is believed to function in multiple signaling pathways ranging from phosphoinositide-mediated calcium signaling (Leissring et al, 2002) and apoptosis (Passer et al, 2000; Kinoshita et al, 2002a; Kim et al, 2003) to gene transcription regulation (Cao and Südhof, 2001; Cupers et al, 2001; Kimberly et al, 2001, 2005; Kinoshita et al, 2002b; Walsh et al, 2003; von Rotz et al, 2004; Pardossi-Piquard et al, 2005). However, until now little hard in vivo evidence is available supporting these claims. The putative function of AICD in nuclear signaling was proposed back in 1999 based on the analogies between Notch and APP processing (Annaert and De Strooper, 1999). In contrast to the large Notch intracellular domain, which contains several signal sequences, the AICD is extremely short and simple. Moreover, most evidence implying AICD in gene transcription regulation is based on quite strong overexpression experiments using artificial reporter paradigms (Cao and Südhof, 2001). It is not clear to what extent these experiments are relevant for natural transduction pathways. A series of endogenous candidate AICD target genes have been identified over the last years including APP, Tip60, neprilysin, GSK3β, KAI1 and others (Baek et al, 2002; Kim et al, 2003; von Rotz et al, 2004; Pardossi-Piquard et al, 2005). Interestingly, γ-secretase cleavage of APP, which releases Aβ, also generates AICD that induces expression of the Aβ degrading neprilysin, providing a negative feedback control on Aβ generation (Pardossi-Piquard et al, 2005). While impressive genetic and cell biological evidence was provided, other authors report difficulties to consistently reproduce this observation (Hass and Yankner, 2005). We would like to note that most reported gene expression alterations upon AICD overexpression are very moderate. Moreover, given its relaxed conformation, exogenously overexpressed AICD might perturb many different protein–protein interactions in nonspecific ways, making the interpretation of these experiments very difficult. Recent work suggests that APP acts at the cell membrane to recruit and/or activate signaling molecules via its intracellular domain (Cao and Sudhof, 2004; Hass and Yankner, 2005). In addition, the phosphorylation of threonine 668 in the APP cytoplasmic domain varies largely during in vitro neuronal differentiation (Kimberly et al, 2005), which also influences, as discussed above, the structure, and therefore function, of the AICD polypeptide.
Summary and perspectives
Gradually, the structure–function relationships in APP become unraveled. Cell biological studies are strongly suggestive for a regulatory role of APP in cell adhesion, which could contribute to neurite outgrowth and synaptogenesis. The in vivo data are also partially compatible with such a role. However, APP and its gene family members do not share strong structural and functional similarities with other proteins, including other cell adhesion molecules (i.e. integrins) and HSPGs, and it remains unclear what exactly APP contributes to cell–cell or cell–matrix interactions. The fact that, for instance, APP-induced neurite outgrowth depends on the intracellular domain but not on the ectodomain (Leyssen et al, 2005) suggests that intracellular interactions with the cytoskeleton and signaling contribute importantly to this adhesion function. Several proteins interact indeed with the intracellular domain of APP including kinases and adaptor proteins, and it seems likely that γ-secretase-mediated release of the AICD leads to disassembly of these complexes. This could contribute to intracellular changes in cytoskeleton organization or kinase localization, and possibly also cause indirect effects on nuclear transcription. The role of the ectodomain remains even more enigmatic: Cu2+ homeostasis is certainly one aspect of its function, but how relevant this is for the organism remains to be further explored. Also, the possible role of APP as a growth factor, either or not in combination with HSPGs, remains to be further explored. The structures that are available show indeed that APP is quite a unique protein. They provide, however, important insights to guide our experiments when making novel mutations in the protein to further explore specific aspects of APP function (e.g. omission of potential HSPG-binding or dimerization motifs). In the future, these structures will help us to isolate relevant binding partners for the ectodomain of APP. For instance, the identification of growth factor receptors binding this domain would greatly support the growth factor hypothesis for APP. We foresee in the next years an intensification of focused research on specific aspects of APP function and we hope that such approaches will finally lead to a better understanding of the role of this intriguing protein in the whole organism and in particular in the brain.
Acknowledgments
We are very grateful to Dr Lieven Buts from the Department of Molecular and Cellular Interactions, Vrije Universiteit Brussel (Belgium) for the help with the structural representations of the figures. Work in the laboratory is supported by a Freedom to Discover grant from Bristol Myers Squib, a Pioneer award from the Alzheimer's Association, the Fund for Scientific Research, Flanders; KU Leuven (GOA); European Union (APOPIS: LSHM-CT-2003-503330); and Federal Office for Scientific Affairs, Belgium (IUAP P5/19). Constanze Reinhard is supported by an EMBO postdoctoral fellowship (ALTF 244-2004). All structures are derived from the RCSB protein database (PDB) available online (http://www.rcsb.org/pdb/). Corresponding PDB reference numbers are shown in the figure legends.
References
- Abramov AY, Canevari L, Duchen MR (2004) Calcium signals induced by amyloid beta peptide and their consequences in neurons and astrocytes in culture. Biochim Biophys Acta 1742: 81–87 [DOI] [PubMed] [Google Scholar]
- Ando K, Iijima KI, Elliott JI, Kirino Y, Suzuki T (2001) Phosphorylation-dependent regulation of the interaction of amyloid precursor protein with Fe65 affects the production of beta-amyloid. J Biol Chem 276: 40353–40361 [DOI] [PubMed] [Google Scholar]
- Annaert W, De Strooper B (1999) Presenilins: molecular switches between proteolysis and signal transduction. Trends Neurosci 22: 439–443 [DOI] [PubMed] [Google Scholar]
- Annaert W, De Strooper B (2002) A cell biological perspective on Alzheimer's disease. Annu Rev Cell Dev Biol 18: 25–51 [DOI] [PubMed] [Google Scholar]
- Ashley J, Packard M, Ataman B, Budnik V (2005) Fasciclin II signals new synapse formation through amyloid precursor protein and the scaffolding protein dX11/Mint. J Neurosci 25: 5943–5955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aslan M, Ozben T (2004) Reactive oxygen and nitrogen species in Alzheimer's disease. Curr Alzheimer Res 1: 111–119 [DOI] [PubMed] [Google Scholar]
- Baek SH, Ohgi KA, Rose DW, Koo EH, Glass CK, Rosenfeld MG (2002) Exchange of N-CoR corepressor and Tip60 coactivator complexes links gene expression by NF-kappaB and beta-amyloid precursor protein. Cell 110: 55–67 [DOI] [PubMed] [Google Scholar]
- Barnham KJ, McKinstry WJ, Multhaup G, Galatis D, Morton CJ, Curtain CC, Williamson NA, White AR, Hinds MG, Norton RS, Beyreuther K, Masters CL, Parker MW, Cappai R (2003) Structure of the Alzheimer's disease amyloid precursor protein copper binding domain. A regulator of neuronal copper homeostasis. J Biol Chem 278: 17401–17407 [DOI] [PubMed] [Google Scholar]
- Beglopoulos V, Sun X, Saura CA, Lemere CA, Kim RD, Shen J (2004) Reduced beta-amyloid production and increased inflammatory responses in presenilin conditional knock-out mice. J Biol Chem 279: 46907–46914 [DOI] [PubMed] [Google Scholar]
- Berechid BE, Kitzmann M, Foltz DR, Roach AH, Seiffert D, Thompson LA, Olson RE, Bernstein A, Donoviel DB, Nye JS (2002) Identification and characterization of presenilin-independent Notch signaling. J Biol Chem 277: 8154–8165 [DOI] [PubMed] [Google Scholar]
- Bertrand E, Brouillet E, Caille I, Bouillot C, Cole GM, Prochiantz A, Allinquant B (2001) A short cytoplasmic domain of the amyloid precursor protein induces apoptosis in vitro and in vivo. Mol Cell Neurosci 18: 503–511 [DOI] [PubMed] [Google Scholar]
- Botelho MG, Gralle M, Oliveira CL, Torriani I, Ferreira ST (2003) Folding and stability of the extracellular domain of the human amyloid precursor protein. J Biol Chem 278: 34259–34267 [DOI] [PubMed] [Google Scholar]
- Bruni P, Minopoli G, Brancaccio T, Napolitano M, Faraonio R, Zambrano N, Hansen U, Russo T (2002) Fe65, a ligand of the Alzheimer's beta-amyloid precursor protein, blocks cell cycle progression by down-regulating thymidylate synthase expression. J Biol Chem 277: 35481–35488 [DOI] [PubMed] [Google Scholar]
- Caille I, Allinquant B, Dupont E, Bouillot C, Langer A, Muller U, Prochiantz A (2004) Soluble form of amyloid precursor protein regulates proliferation of progenitors in the adult subventricular zone. Development 131: 2173–2181 [DOI] [PubMed] [Google Scholar]
- Cao X, Südhof TC (2001) A transcriptionally (correction of transcriptively) active complex of APP with Fe65 and histone acetyltransferase Tip60. Science 293: 115–120 [DOI] [PubMed] [Google Scholar]
- Cao X, Sudhof TC (2004) Dissection of amyloid-beta precursor protein-dependent transcriptional transactivation. J Biol Chem 279: 24601–24611 [DOI] [PubMed] [Google Scholar]
- Casella L, Gullotti M (1993) Dioxygen activation by biomimetic dinuclear complexes. In Bioinorganic Chemistry of Copper, Karlin KD, Teyklar, Z (eds) pp 292–305. New York: Chapmann & Hall [Google Scholar]
- Chirgadze DY, Hepple J, Byrd RA, Sowdhamini R, Blundell TL, Gherardi E (1998) Insights into the structure of hepatocyte growth factor/scatter factor (HGF/SF) and implications for receptor activation. FEBS Lett 430: 126–129 [DOI] [PubMed] [Google Scholar]
- Chow N, Korenberg JR, Chen XN, Neve RL (1996) APP-BP1, a novel protein that binds to the carboxyl-terminal region of the amyloid precursor protein. J Biol Chem 271: 11339–11346 [DOI] [PubMed] [Google Scholar]
- Cobine PA, George GN, Jones CE, Wickramasinghe WA, Solioz M, Dameron CT (2002) Copper transfer from the Cu(I) chaperone, CopZ, to the repressor, Zn(II)CopY: metal coordination environments and protein interactions. Biochemistry 41: 5822–5829 [DOI] [PubMed] [Google Scholar]
- Cribbs DH, Poon WW, Rissman RA, Blurton-Jones M (2004) Caspase-mediated degeneration in Alzheimer's disease. Am J Pathol 165: 353–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cupers P, Orlans I, Craessaerts K, Annaert W, De Strooper B (2001) The amyloid precursor protein (APP)-cytoplasmic fragment generated by gamma-secretase is rapidly degraded but distributes partially in a nuclear fraction of neurones in culture. J Neurochem 78: 1168–1178 [DOI] [PubMed] [Google Scholar]
- Daigle I, Li C (1993) apl-1, a Caenorhabditis elegans gene encoding a protein related to the human beta-amyloid protein precursor. Proc Natl Acad Sci USA 90: 12045–12049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Strooper B, Annaert W (2000) Proteolytic processing and cell biological functions of the amyloid precursor protein. J Cell Sci 113 (Part 11): 1857–1870 [DOI] [PubMed] [Google Scholar]
- De Strooper B, Simons M, Multhaup G, Van Leuven F, Beyreuther K, Dotti CG (1995) Production of intracellular amyloid-containing fragments in hippocampal neurons expressing human amyloid precursor protein and protection against amyloidogenesis by subtle amino acid substitutions in the rodent sequence. EMBO J 16: 4932–4938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulubova I, Ho A, Huryeva I, Sudhof TC, Rizo J (2004) Three-dimensional structure of an independently folded extracellular domain of human amyloid-beta precursor protein. Biochemistry 43: 9583–9588 [DOI] [PubMed] [Google Scholar]
- Fossgreen A, Bruckner B, Czech C, Masters CL, Beyreuther K, Paro R (1998) Transgenic Drosophila expressing human amyloid precursor protein show gamma-secretase activity and a blistered-wing phenotype. Proc Natl Acad Sci USA 95: 13703–13708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gralle M, Botelho MM, de Oliveira CL, Torriani I, Ferreira ST (2002) Solution studies and structural model of the extracellular domain of the human amyloid precursor protein. Biophys J 83: 3513–3524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg SM, Qiu WQ, Selkoe DJ, Ben-Itzhak A, Kosik KS (1995) Amino-terminal region of the beta-amyloid precursor protein activates mitogen-activated protein kinase. Neurosci Lett 198: 52–56 [DOI] [PubMed] [Google Scholar]
- Gu Y, Misonou H, Sato T, Dohmae N, Takio K, Ihara Y (2001) Distinct intramembrane cleavage of the beta-amyloid precursor protein family resembling gamma-secretase-like cleavage of Notch. J Biol Chem 276: 35235–35238 [DOI] [PubMed] [Google Scholar]
- Gunawardena S, Goldstein LS (2001) Disruption of axonal transport and neuronal viability by amyloid precursor protein mutations in Drosophila. Neuron 32: 389–401 [DOI] [PubMed] [Google Scholar]
- Haass C (2004) Take five—BACE and the gamma-secretase quartet conduct Alzheimer's amyloid beta-peptide generation. EMBO J 23: 483–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hass MR, Yankner BA (2005) A Gamma-secretase independent mechanism of signal transduction by the amyloid precursor protein. J Biol Chem (Epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heber S, Herms J, Gajic V, Hainfellner J, Aguzzi A, Rulicke T, von Kretzschmar H, von Koch C, Sisodia S, Tremml P, Lipp HP, Wolfer DP, Muller U (2000) Mice with combined gene knock-outs reveal essential and partially redundant functions of amyloid precursor protein family members. J Neurosci 20: 7951–7963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herms J, Anliker B, Heber S, Ring S, Fuhrmann M, Kretzschmar H, Sisodia S, Muller U (2004) Cortical dysplasia resembling human type 2 lissencephaly in mice lacking all three APP family members. EMBO J 23: 4106–4115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herreman A, Van Gassen G, Bentahir M, Nyabi O, Craessaerts K, Mueller U, Annaert W, De Strooper B (2003) Gamma-secretase activity requires the presenilin-dependent trafficking of nicastrin through the Golgi apparatus but not its complex glycosylation. J Cell Sci 116: 1127–1136 [DOI] [PubMed] [Google Scholar]
- Ho A, Sudhof TC (2004) Binding of F-spondin to amyloid-beta precursor protein: a candidate amyloid-beta precursor protein ligand that modulates amyloid-beta precursor protein cleavage. Proc Natl Acad Sci USA 101: 2548–2553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huffman DL, O'Halloran TV (2001) Function, structure, and mechanism of intracellular copper trafficking proteins. Annu Rev Biochem 70: 677–701 [DOI] [PubMed] [Google Scholar]
- Johnson RJ, Xiao G, Shanmugaratnam J, Fine RE (2001) Calreticulin functions as a molecular chaperone for the beta-amyloid precursor protein. Neurobiol Aging 22: 387–395 [DOI] [PubMed] [Google Scholar]
- Kadomatsu K, Muramatsu T (2004) Midkine and pleiotrophin in neural development and cancer. Cancer Lett 204: 127–143 [DOI] [PubMed] [Google Scholar]
- Kamal A, Almenar-Queralt A, LeBlanc JF, Roberts EA, Goldstein LS (2001) Kinesin-mediated axonal transport of a membrane compartment containing beta-secretase and presenilin-1 requires APP. Nature 414: 643–648 [DOI] [PubMed] [Google Scholar]
- Kamal A, Stokin GB, Yang Z, Xia CH, Goldstein LS (2000) Axonal transport of amyloid precursor protein is mediated by direct binding to the kinesin light chain subunit of kinesin-I. Neuron 28: 449–459 [DOI] [PubMed] [Google Scholar]
- Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R (2003) APP processing and synaptic function. Neuron 37: 925–937 [DOI] [PubMed] [Google Scholar]
- Kibbey MC, Jucker M, Weeks BS, Neve RL, Van Nostrand WE, Kleinman HK (1993) Beta-amyloid precursor protein binds to the neurite-promoting IKVAV site of laminin. Proc Natl Acad Sci USA 90: 10150–10153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HS, Kim EM, Lee JP, Park CH, Kim S, Seo JH, Chang KA, Yu E, Jeong SJ, Chong YH, Suh YH (2003) C-terminal fragments of amyloid precursor protein exert neurotoxicity by inducing glycogen synthase kinase-3beta expression. FASEB J 17: 1951–1953 [DOI] [PubMed] [Google Scholar]
- Kimberly WT, Zheng JB, Guenette SY, Selkoe DJ (2001) The intracellular domain of the beta-amyloid precursor protein is stabilized by Fe65 and translocates to the nucleus in a notch-like manner. J Biol Chem 276: 40288–40292 [DOI] [PubMed] [Google Scholar]
- Kimberly WT, Zheng JB, Town T, Flavell RA, Selkoe DJ (2005) Physiological regulation of the beta-amyloid precursor protein signaling domain by c-Jun N-terminal kinase JNK3 during neuronal differentiation. J Neurosci 25: 5533–5543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King GD, Scott Turner R (2004) Adaptor protein interactions: modulators of amyloid precursor protein metabolism and Alzheimer's disease risk? Exp Neurol 185: 208–219 [DOI] [PubMed] [Google Scholar]
- Kinoshita A, Whelan CM, Berezovska O, Hyman BT (2002a) The gamma secretase-generated carboxyl-terminal domain of the amyloid precursor protein induces apoptosis via Tip60 in H4 cells. J Biol Chem 277: 28530–28536 [DOI] [PubMed] [Google Scholar]
- Kinoshita A, Whelan CM, Smith CJ, Berezovska O, Hyman BT (2002b) Direct visualization of the gamma secretase-generated carboxyl-terminal domain of the amyloid precursor protein: association with Fe65 and translocation to the nucleus. J Neurochem 82: 839–847 [DOI] [PubMed] [Google Scholar]
- Koo EH (2002) The beta-amyloid precursor protein (APP) and Alzheimer's disease: does the tail wag the dog? Traffic 3: 763–770 [DOI] [PubMed] [Google Scholar]
- LaFerla FM (2002) Calcium dyshomeostasis and intracellular signalling in Alzheimer's disease. Nat Rev Neurosci 3: 862–872 [DOI] [PubMed] [Google Scholar]
- Lambert de Rouvroit C, Goffinet AM (2001) Neuronal migration. Mech Dev 105: 47–56 [DOI] [PubMed] [Google Scholar]
- Lazarov O, Morfini GA, Lee EB, Farah MH, Szodorai A, DeBoer SR, Koliatsos VE, Kins S, Lee VM, Wong PC, Price DL, Brady ST, Sisodia SS (2005) Axonal transport, amyloid precursor protein, kinesin-1, and the processing apparatus: revisited. J Neurosci 25: 2386–2395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leissring MA, Murphy MP, Mead TR, Akbari Y, Sugarman MC, Jannatipour M, Anliker B, Muller U, Saftig P, De Strooper B, Wolfe MS, Golde TE, LaFerla FM (2002) A physiologic signaling role for the gamma-secretase-derived intracellular fragment of APP. Proc Natl Acad Sci USA 99: 4697–4702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy E, Carman MD, Fernandez-Madrid IJ, Power MD, Lieberburg I, van Duinen SG, Bots GT, Luyendijk W, Frangione B (1990) Mutation of the Alzheimer's disease amyloid gene in hereditary cerebral hemorrhage, Dutch type. Science 248: 1124–1126 [DOI] [PubMed] [Google Scholar]
- Leyssen M, Ayaz D, Hebert SS, Reeve S, De Strooper B, Hassan BA (2005) Amyloid precursor protein promotes post-developmental neurite arborization in the Drosophila brain. EMBO J 24: 2944–2955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HL, Roch JM, Sundsmo M, Otero D, Sisodia S, Thomas R, Saitoh T (1997) Defective neurite extension is caused by a mutation in amyloid beta/A4 (A beta) protein precursor found in familial Alzheimer's disease. J Neurobiol 32: 469–480 [PubMed] [Google Scholar]
- Loewer A, Soba P, Beyreuther K, Paro R, Merdes G (2004) Cell-type-specific processing of the amyloid precursor protein by presenilin during Drosophila development. EMBO Rep 5: 405–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzo A, Yuan M, Zhang Z, Paganetti PA, Sturchler-Pierrat C, Staufenbiel M, Mautino J, Vigo FS, Sommer B, Yankner BA (2000) Amyloid beta interacts with the amyloid precursor protein: a potential toxic mechanism in Alzheimer's disease. Nat Neurosci 3: 460–464 [DOI] [PubMed] [Google Scholar]
- Lu DC, Shaked GM, Masliah E, Bredesen DE, Koo EH (2003) Amyloid beta protein toxicity mediated by the formation of amyloid-beta protein precursor complexes. Ann Neurol 54: 781–789 [DOI] [PubMed] [Google Scholar]
- Luo L, Tully T, White K (1992) Human amyloid precursor protein ameliorates behavioral deficit of flies deleted for Appl gene. Neuron 9: 595–605 [DOI] [PubMed] [Google Scholar]
- Markossian KA, Kurganov BI (2003) Copper chaperones, intracellular copper trafficking proteins. Function, structure, and mechanism of action. Biochemistry (Moscow) 68: 827–837 [DOI] [PubMed] [Google Scholar]
- Martin-Morris LE, White K (1990) The Drosophila transcript encoded by the beta-amyloid protein precursor-like gene is restricted to the nervous system. Development 110: 185–195 [DOI] [PubMed] [Google Scholar]
- Matsuda S, Matsuda Y, D'Adamio L (2003) Amyloid beta protein precursor (AbetaPP), but not AbetaPP-like protein 2, is bridged to the kinesin light chain by the scaffold protein JNK-interacting protein 1. J Biol Chem 278: 38601–38606 [DOI] [PubMed] [Google Scholar]
- Mattson MP (1997) Cellular actions of beta-amyloid precursor protein and its soluble and fibrillogenic derivatives. Physiol Rev 77: 1081–1132 [DOI] [PubMed] [Google Scholar]
- Maynard CJ, Bush AI, Masters CL, Cappai R, Li QX (2005) Metals and amyloid-beta in Alzheimer's disease. Int J Exp Pathol 86: 147–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merdes G, Soba P, Loewer A, Bilic MV, Beyreuther K, Paro R (2004) Interference of human and Drosophila APP and APP-like proteins with PNS development in Drosophila. EMBO J 23: 4082–4095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan CE (2000) Caspase cleavage of APP results in a cytotoxic proteolytic peptide. Nat Med 6: 385–386 [DOI] [PubMed] [Google Scholar]
- Mohammadi M, Olsen SK, Ibrahimi OA (2005) Structural basis for fibroblast growth factor receptor activation. Cytokine Growth Factor Rev 16: 107–137 [DOI] [PubMed] [Google Scholar]
- Morgan C, Colombres M, Nunez MT, Inestrosa NC (2004) Structure and function of amyloid in Alzheimer's disease. Prog Neurobiol 74: 323–349 [DOI] [PubMed] [Google Scholar]
- Multhaup G, Schlicksupp A, Hesse L, Beher D, Ruppert T, Masters CL, Beyreuther K (1996) The amyloid precursor protein of Alzheimer's disease in the reduction of copper(II) to copper(I). Science 271: 1406–1409 [DOI] [PubMed] [Google Scholar]
- Murakami N, Yamaki T, Iwamoto Y, Sakakibara T, Kobori N, Fushiki S, Ueda S (1998) Experimental brain injury induces expression of amyloid precursor protein, which may be related to neuronal loss in the hippocampus. J Neurotrauma 15: 993–1003 [DOI] [PubMed] [Google Scholar]
- Ninomiya H, Roch JM, Sundsmo MP, Otero DA, Saitoh T (1993) Amino acid sequence RERMS represents the active domain of amyloid beta/A4 protein precursor that promotes fibroblast growth. J Cell Biol 121: 879–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimoto I, Okamoto T, Matsuura Y, Takahashi S, Okamoto T, Murayama Y, Ogata E (1993) Alzheimer amyloid protein precursor complexes with brain GTP-binding protein G(o). Nature 362: 75–79 [DOI] [PubMed] [Google Scholar]
- Nordstedt C, Caporaso GL, Thyberg J, Gandy SE, Greengard P (1993) Identification of the Alzheimer beta/A4 amyloid precursor protein in clathrin-coated vesicles purified from PC12 cells. J Biol Chem 268: 608–612 [PubMed] [Google Scholar]
- Ohsawa I, Takamura C, Kohsaka S (1997) The amino-terminal region of amyloid precursor protein is responsible for neurite outgrowth in rat neocortical explant culture. Biochem Biophys Res Commun 236: 59–65 [DOI] [PubMed] [Google Scholar]
- Ohsawa I, Takamura C, Kohsaka S (2001) Fibulin-1 binds the amino-terminal head of beta-amyloid precursor protein and modulates its physiological function. J Neurochem 76: 1411–1420 [DOI] [PubMed] [Google Scholar]
- Ohsawa I, Takamura C, Morimoto T, Ishiguro M, Kohsaka S (1999) Amino-terminal region of secreted form of amyloid precursor protein stimulates proliferation of neural stem cells. Eur J Neurosci 11: 1907–1913 [DOI] [PubMed] [Google Scholar]
- Pardossi-Piquard R, Petit A, Kawarai T, Sunyach C, Alves da Costa C, Vincent B, Ring S, D'Adamio L, Shen J, Muller U, St George Hyslop P, Checler F (2005) Presenilin-dependent transcriptional control of the Abeta-degrading enzyme neprilysin by intracellular domains of betaAPP and APLP. Neuron 46: 541–554 [DOI] [PubMed] [Google Scholar]
- Passer B, Pellegrini L, Russo C, Siegel RM, Lenardo MJ, Schettini G, Bachmann M, Tabaton M, D'Adamio L (2000) Generation of an apoptotic intracellular peptide by gamma-secretase cleavage of Alzheimer's amyloid beta protein precursor. J Alzheimers Dis 2: 289–301 [DOI] [PubMed] [Google Scholar]
- Plant LD, Boyle JP, Smith IF, Peers C, Pearson HA (2003) The production of amyloid beta peptide is a critical requirement for the viability of central neurons. J Neurosci 23: 5531–5535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi-Takahara Y, Morishima-Kawashima M, Tanimura Y, Dolios G, Hirotani N, Horikoshi Y, Kametani F, Maeda M, Saido TC, Wang R, Ihara Y (2005) Longer forms of amyloid beta protein: implications for the mechanism of intramembrane cleavage by gamma-secretase. J Neurosci 25: 436–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramelot TA, Gentile LN, Nicholson LK (2000) Transient structure of the amyloid precursor protein cytoplasmic tail indicates preordering of structure for binding to cytosolic factors. Biochemistry 39: 2714–2725 [DOI] [PubMed] [Google Scholar]
- Ramsden M, Henderson Z, Pearson HA (2002) Modulation of Ca2+ channel currents in primary cultures of rat cortical neurones by amyloid beta protein (1–40) is dependent on solubility status. Brain Res 956: 254–261 [DOI] [PubMed] [Google Scholar]
- Ramsden M, Plant LD, Webster NJ, Vaughan PF, Henderson Z, Pearson HA (2001) Differential effects of unaggregated and aggregated amyloid beta protein (1–40) on K(+) channel currents in primary cultures of rat cerebellar granule and cortical neurones. J Neurochem 79: 699–712 [DOI] [PubMed] [Google Scholar]
- Roncarati R, Sestan N, Scheinfeld MH, Berechid BE, Lopez PA, Meucci O, McGlade JC, Rakic P, D'Adamio L (2002) The gamma-secretase-generated intracellular domain of beta-amyloid precursor protein binds Numb and inhibits Notch signaling. Proc Natl Acad Sci USA 99: 7102–7107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossjohn J, Cappai R, Feil SC, Henry A, McKinstry WJ, Galatis D, Hesse L, Multhaup G, Beyreuther K, Masters CL, Parker MW (1999) Crystal structure of the N-terminal, growth factor-like domain of Alzheimer amyloid precursor protein. Nat Struct Biol 6: 327–331 [DOI] [PubMed] [Google Scholar]
- Russo C, Dolcini V, Salis S, Venezia V, Violani E, Carlo P, Zambrano N, Russo T, Schettini G (2002) Signal transduction through tyrosine-phosphorylated carboxy-terminal fragments of APP via an enhanced interaction with Shc/Grb2 adaptor proteins in reactive astrocytes of Alzheimer's disease brain. Ann NY Acad Sci 973: 323–333 [DOI] [PubMed] [Google Scholar]
- Sabo SL, Ikin AF, Buxbaum JD, Greengard P (2001) The Alzheimer amyloid precursor protein (APP) and FE65, an APP-binding protein, regulate cell movement. J Cell Biol 153: 1403–1414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sastre M, Steiner H, Fuchs K, Capell A, Multhaup G, Condron MM, Teplow DB, Haass C (2001) Presenilin-dependent gamma-secretase processing of beta-amyloid precursor protein at a site corresponding to the S3 cleavage of Notch. EMBO Rep 2: 835–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheinfeld MH, Matsuda S, D'Adamio L (2003) JNK-interacting protein-1 promotes transcription of A beta protein precursor but not A beta precursor-like proteins, mechanistically different than Fe65. Proc Natl Acad Sci USA 100: 1729–1734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheuermann S, Hambsch B, Hesse L, Stumm J, Schmidt C, Beher D, Bayer TA, Beyreuther K, Multhaup G (2001) Homodimerization of amyloid precursor protein and its implication in the amyloidogenic pathway of Alzheimer's disease. J Biol Chem 276: 33923–33929 [DOI] [PubMed] [Google Scholar]
- Selkoe DJ (2001) Alzheimer's disease: genes, proteins, and therapy. Physiol Rev 81: 741–766 [DOI] [PubMed] [Google Scholar]
- Serpell LC (2000) Alzheimer's amyloid fibrils: structure and assembly. Biochim Biophys Acta 1502: 16–30 [DOI] [PubMed] [Google Scholar]
- Small DH, Clarris HL, Williamson TG, Reed G, Key B, Mok SS, Beyreuther K, Masters CL, Nurcombe V (1999) Neurite-outgrowth regulating functions of the amyloid protein precursor of Alzheimer's disease. J Alzheimers Dis 1: 275–285 [DOI] [PubMed] [Google Scholar]
- Small DH, Nurcombe V, Reed G, Clarris H, Moir R, Beyreuther K, Masters CL (1994) A heparin-binding domain in the amyloid protein precursor of Alzheimer's disease is involved in the regulation of neurite outgrowth. J Neurosci 14: 2117–2127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soba P, Eggert S, Wagner K, Zentgraf H, Siehl K, Kreger S, Lower A, Langer A, Merdes G, Paro R, Masters CL, Muller U, Kins S, Beyreuther K (2005) Homo- and heterodimerization of APP family members promotes intercellular adhesion. EMBO J (Epub ahead of print) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokin GB, Lillo C, Falzone TL, Brusch RG, Rockenstein E, Mount SL, Raman R, Davies P, Masliah E, Williams DS, Goldstein LS (2005) Axonopathy and transport deficits early in the pathogenesis of Alzheimer's disease. Science 307: 1282–1288 [DOI] [PubMed] [Google Scholar]
- Strittmatter WJ, Weisgraber KH, Huang DY, Dong LM, Salvesen GS, Pericak-Vance M, Schmechel D, Saunders AM, Goldgaber D, Roses AD (1993) Binding of human apolipoprotein E to synthetic amyloid beta peptide: isoform-specific effects and implications for late-onset Alzheimer disease. Proc Natl Acad Sci USA 90: 8098–8102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarr PE, Roncarati R, Pelicci G, Pelicci PG, D'Adamio L (2002) Tyrosine phosphorylation of the beta-amyloid precursor protein cytoplasmic tail promotes interaction with Shc. J Biol Chem 277: 16798–16804 [DOI] [PubMed] [Google Scholar]
- Taru H, Iijima K, Hase M, Kirino Y, Yagi Y, Suzuki T (2002) Interaction of Alzheimer's beta-amyloid precursor family proteins with scaffold proteins of the JNK signaling cascade. J Biol Chem 277: 20070–20078 [DOI] [PubMed] [Google Scholar]
- Torroja L, Chu H, Kotovsky I, White K (1999) Neuronal overexpression of APPL, the Drosophila homologue of the amyloid precursor protein (APP), disrupts axonal transport. Curr Biol 9: 489–492 [DOI] [PubMed] [Google Scholar]
- Turner PR, O'Connor K, Tate WP, Abraham WC (2003) Roles of amyloid precursor protein and its fragments in regulating neural activity, plasticity and memory. Prog Neurobiol 70: 1–32 [DOI] [PubMed] [Google Scholar]
- Uversky VN, Oldfield CJ, Dunker AK (2005) Showing your ID: intrinsic disorder as an ID for recognition, regulation and cell signaling. J Mol Recogn 18: 343–384 [DOI] [PubMed] [Google Scholar]
- Van den Heuvel C, Blumbergs PC, Finnie JW, Manavis J, Jones NR, Reilly PL, Pereira RA (1999) Upregulation of amyloid precursor protein messenger RNA in response to traumatic brain injury: an ovine head impact model. Exp Neurol 159: 441–450 [DOI] [PubMed] [Google Scholar]
- von Koch CS, Zheng H, Chen H, Trumbauer M, Thinakaran G, van der Ploeg LH, Price DL, Sisodia SS (1997) Generation of APLP2 KO mice and early postnatal lethality in APLP2/APP double KO mice. Neurobiol Aging 18: 661–669 [DOI] [PubMed] [Google Scholar]
- von Rotz RC, Kohli BM, Bosset J, Meier M, Suzuki T, Nitsch RM, Konietzko U (2004) The APP intracellular domain forms nuclear multiprotein complexes and regulates the transcription of its own precursor. J Cell Sci 117: 4435–4448 [DOI] [PubMed] [Google Scholar]
- Walsh DM, Fadeeva JV, LaVoie MJ, Paliga K, Eggert S, Kimberly WT, Wasco W, Selkoe DJ (2003) Gamma-secretase cleavage and binding to FE65 regulate the nuclear translocation of the intracellular C-terminal domain (ICD) of the APP family of proteins. Biochemistry 42: 6664–6673 [DOI] [PubMed] [Google Scholar]
- Walsh DM, Selkoe DJ (2004) Deciphering the molecular basis of memory failure in Alzheimer's disease. Neuron 44: 181–193 [DOI] [PubMed] [Google Scholar]
- Wang Y, Ha Y (2004) The X-ray structure of an antiparallel dimer of the human amyloid precursor protein E2 domain. Mol Cell 15: 343–353 [DOI] [PubMed] [Google Scholar]
- Weidemann A, Eggert S, Reinhard FB, Vogel M, Paliga K, Baier G, Masters CL, Beyreuther K, Evin G (2002) A novel epsilon-cleavage within the transmembrane domain of the Alzheimer amyloid precursor protein demonstrates homology with Notch processing. Biochemistry 41: 2825–2835 [DOI] [PubMed] [Google Scholar]
- Wisniewski T, Ghiso J, Frangione B (1991) Peptides homologous to the amyloid protein of Alzheimer's disease containing a glutamine for glutamic acid substitution have accelerated amyloid fibril formation. Biochem Biophys Res Commun 179: 1247–1254 [DOI] [PubMed] [Google Scholar]
- Yu C, Kim SH, Ikeuchi T, Xu H, Gasparini L, Wang R, Sisodia SS (2001) Characterization of a presenilin-mediated amyloid precursor protein carboxyl-terminal fragment gamma. Evidence for distinct mechanisms involved in gamma-secretase processing of the APP and Notch1 transmembrane domains. J Biol Chem 276: 43756–43760 [DOI] [PubMed] [Google Scholar]
- Zhang Z, Lee CH, Mandiyan V, Borg JP, Margolis B, Schlessinger J, Kuriyan J (1997) Sequence-specific recognition of the internalization motif of the Alzheimer's amyloid precursor protein by the X11 PTB domain. EMBO J 16: 6141–6150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao G, Cui MZ, Mao G, Dong Y, Tan J, Sun L, Xu X (2005) Gamma -cleavage is dependent on zeta -cleavage during the proteolytic processing of amyloid precursor protein within its transmembrane domain. J Biol Chem (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- Zheng H, Jiang M, Trumbauer ME, Sirinathsinghji DJ, Hopkins R, Smith DW, Heavens RP, Dawson GR, Boyce S, Conner MW, Stevens KA, Slunt HH, Sisoda SS, Chen HY, Van der Ploeg LH (1995) Beta-Amyloid precursor protein-deficient mice show reactive gliosis and decreased locomotor activity. Cell 81: 525–531 [DOI] [PubMed] [Google Scholar]