

Abstract
We have created P1 artificial chromosome transgenic mice expressing the human mitochondrial superoxide dismutase 2 (SOD2) and thus generated mice with a physiologically controlled augmentation of SOD2 expression leading to increased SOD2 enzyme activities and lowered superoxide levels. In the transgenic mice, effects on mitochondrial function such as enhanced oxidative capacity and greater resistance against inducers of mitochondrial permeability were observed. Superoxide in the mitochondrial matrix has been proposed to activate uncoupling proteins (UCPs), thus providing a feedback mechanism that will lower respiratory chain superoxide production by increasing a proton leak across the inner mitochondrial membrane. However, UCP1 and UCP3 activities and mitochondrial ATP production rates were not altered in isolated mitochondria from SOD2 transgenic mice, despite lowered superoxide levels. Globally, the transgenic mice displayed normal resting metabolic rates, indicating an absence of effect on any UCP activities, and normal oxygen consumption responses after norepinephrine injection. These results strongly suggest that endogenously generated matrix superoxide does not regulate UCP activity and in vivo energy expenditure.
Keywords: energy expenditure, mitochondria, superoxide, transgenic mice, uncoupling protein
Introduction
Manganese superoxide dismutase 2 (SOD2) is a critical mitochondrial antioxidant defense against superoxide produced by respiration. Homozygous SOD2 knockout mice develop severe symptoms in heart (Li et al, 1995) and brain (Lebovitz et al, 1996; Melov et al, 1998) leading to early postnatal death. Heterozygous SOD2 knockout mice exhibit numerous alterations in mitochondrial function such as a reduction in complex I and II activities, lowered aconitase activity (Melov et al, 1999), increased susceptibility for induction of permeability transition (Kokoszka et al, 2001), ultrastructural abnormalities (mitochondrial swelling) and enhanced lipid peroxidation (Strassburger et al, 2005).
Correspondingly, SOD2 overexpression protects mitochondrial respiratory function and blocks apoptosis induction during heart ischemia–reperfusion injury (Suzuki et al, 2002), and attenuates mitochondrial reactive oxygen species (ROS) generation, intracellular lipid peroxidation and cell death (Motoori et al, 2001). However, the antioxidative effect of SOD is strongly dependent on its expression level. Excessive SOD overexpression may significantly alter the levels of related ROS and cause enhanced lipid peroxidation and hypersensitivity to ROS (Kowald and Klipp, 2004). Careful regulation of SOD expression is thus critical in order for cells and tissues to benefit from its antioxidative effect. Introduction of large genomic sequences into the mouse genome by P1 artificial chromosomes (PAC) confers physiological control of gene expression due to the presence of both positive and negative regulatory sequence elements. Here, we used this transgenic approach to overexpress SOD2, particularly to study the regulation of uncoupling protein (UCP) activities by superoxide.
The UCPs belong to the large family of mitochondrial inner membrane carriers. The first identified UCP isoform, UCP1, is specifically expressed in brown adipose tissue (BAT) and confers a regulated proton leak across the inner mitochondrial membrane (Cannon and Nedergaard, 2004). Two other UCP isoforms, UCP2 and UCP3, were identified based on sequence homology to UCP1 (Ricquier and Bouillaud, 2000), but their physiological functions remain elusive (Nedergaard and Cannon, 2003; Brand and Esteves, 2005). UCP function has been implicated in the control of ROS homeostasis. Macrophages from UCP2 knockout mice have increased ROS levels (Arsenijevic et al, 2000). UCP3 knockout mice display signs of increased oxidative damage in muscle mitochondria, indicative of increased ROS production (Brand et al, 2002).
In both mice and rats, generation of superoxide by xanthine plus xanthine oxidase increases the proton conductance in mitochondria expressing UCP3 (skeletal muscle), as well as in mitochondria expressing UCP1 (BAT) and UCP2 (kidney, pancreatic β-cells, spleen) (Echtay et al, 2002b), suggesting that superoxide activates all UCP isoforms in vitro. This effect is inhibited by addition of SOD, by GDP or by molecules that bind fatty acids. Activation of the UCPs by superoxide has been indicated to occur from the matrix side (Echtay et al, 2002a; Talbot et al, 2004). Mitochondrial uncoupling is expected to result in a more oxidized state of the electron transport chain, thereby preventing superoxide generation (Korshunov et al, 1997). Superoxide-mediated activation of UCPs has therefore been proposed to serve as a negative feedback mechanism to limit further superoxide production (Echtay et al, 2002b). Superoxide-mediated activation of UCPs may also be expected to have a significant regulatory effect on energy metabolism, due to the widespread expression of UCP2 and UCP3.
However, the hypothesis that superoxide activates UCPs is not universally accepted (Couplan et al, 2002). Many of the studies described above were performed in isolated mitochondria exposed to exogenous superoxide, and only one study has indicated that superoxide-stimulated UCP-mediated uncoupling might also occur in vivo (Krauss et al, 2003).
In the genetically modified mouse strain used here having a regulated increase of SOD2 enzyme activity, superoxide levels were reduced and effects on mitochondrial function were observed. However, UCP1 and UCP3 activities were not altered in isolated mitochondria in SOD2-overexpressing mice, which also showed normal resting metabolic rates (indicating no global effects on any UCP activities). The results strongly suggest that endogenously generated superoxide does not regulate UCP activity and energy expenditure in vivo.
Results and discussion
Creation and characterization of mice with increased SOD2 enzyme activity
We created transgenic mice with an increased SOD2 enzyme activity by introducing PAC clones with the human SOD2 gene into the mouse. The chosen transgenic strategy has several advantages. The large size of the PAC clones will make it likely that all sequence elements needed for physiological regulation of gene expression are present and will minimize positional effects caused by random integration of the transgene (Heintz, 2000). The use of specific probes for human SOD2 makes it easy to establish that the transgene is present and expressed in the transgenic mice (Ekstrand et al, 2004) and the overexpression will be within a physiologically relevant interval (Ekstrand et al, 2004).
We screened a human genomic PAC library for the presence of the SOD2 gene and identified 16 positive clones. The clones were ordered in a contig by using Southern blotting with end probes (generated by vectorette PCR) and by size determination with pulsed-field gel electrophoresis as described previously (Ekstrand et al, 2004). Three PAC clones with overlapping 5′ and 3′ sequences (Figure 1A) named PAC662, PAC817 and PAC737 were chosen for pronuclear injections of embryos of the inbred FVB/N mouse strain. We performed Southern blots with a human SOD2 cDNA probe and found integration of the human SOD2 gene into the mouse genome without signs of any rearrangements (Figure 1B). We obtained germline transmission from nine of 12 founder animals (Figure 1B). Two independent lines, PAC662D1 (from the founder D1 of clone PAC662) and PAC737D2 (from the founder D2 of clone PAC737), were characterized further (Figure 1B). The transgenes of these two lines were constantly transmitted at less than the expected Mendelian ratio of 50% (PAC662D1 34% and PAC737D2 38%; P<0.001, χ2 test for uneven genotype distribution). Increased expression of SOD2 thus had some effects on fertility, consistent with a previous report (Raineri et al, 2001). Western blot analyses with a polyclonal antibody against human SOD2 demonstrated expression of the human SOD2 protein mainly in kidney and muscle in the PAC737D2 line (Figure 1C) and a widespread tissue distribution in the PAC662D1 line (Figure 1C and D). In the PAC737D2 line, SOD enzyme activity was increased in isolated mitochondria from skeletal muscle only (Figure 1E), but in the PAC662D1 line, increased activities were found in all tissues examined (Figure 1E). The observed SOD enzyme activity pattern (Figure 1E) was thus consistent with the observed human SOD2 protein expression pattern (Figure 1C). Further studies were focused on the PAC662D1 line.
Figure 1.
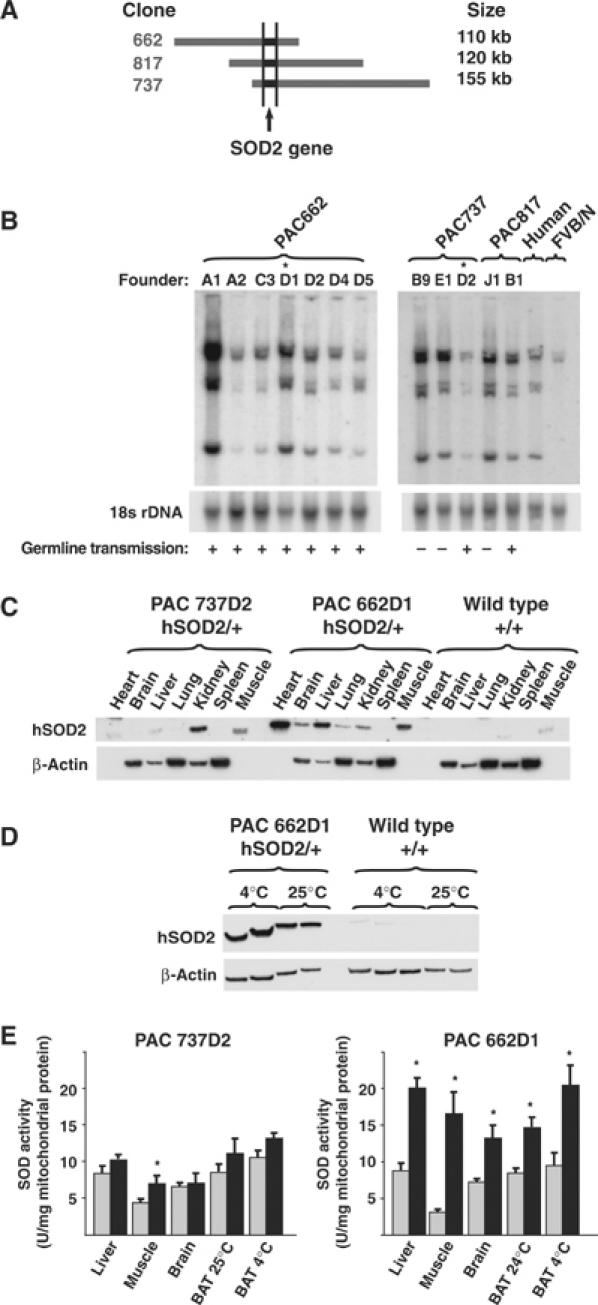
Creation of SOD2-overexpressing mice. (A) Contig of the PAC clones used to generate transgenic mice. (B) Southern blot analysis of tail DNA from transgenic founder animals probed with human SOD2 cDNA. The blot was rehybridized with an 18S rDNA probe to assess loading. Human DNA and FVB/N mouse DNA were used as controls. Symbols are used to indicate the founders with germline transmission of the SOD2 transgene (+) and the lines studied further (*). (C) Western blot analysis of levels of human SOD2 protein in different tissues. β-Actin was used for information on loading. β-Actin is not present in heart and muscle. (D) Western blot analysis of levels of human SOD2 protein (hSOD2) in BAT of wild-type or SOD2-overexpressing mice of the PAC662D1 line acclimated to 25 and 4°C. β-Actin was used as a loading control. (E) SOD enzyme activity in mitochondria from different tissues of wild-type (gray bars) and SOD2-overexpressing mice (black bars) (n=3–5).
As seen in Supplementary Figure 1, no evidence was found that indicated any influence of SOD overexpression on mitochondrial biogenesis or gene expression.
SOD2 overexpression lowers superoxide release rates
Net superoxide release rates were assessed directly in isolated skeletal muscle and brown fat mitochondria and in mouse embryonic fibroblasts (MEF) by fluorescence with the dye dihydroethidium (DHE) (Benov et al, 1998). The conversion of DHE to ethidium is superoxide-induced, and the fluorescence emitted by the ethidium formed was followed. A chemical validation of this method was first performed. Generation of superoxide by the xanthine plus xanthine oxidase system in the presence of DHE resulted in a significant increase in ethidium-emitted fluorescence (Figure 2A, upper panel). This was blocked by further addition of recombinant SOD (Figure 2A, upper panel). The method was then biologically validated. Wild-type mitochondria incubated with succinate and rotenone showed a linear increase in fluorescence (Figure 2A, lower panel). Antimycin A was added as a positive control to enhance mitochondrial superoxide generation. This resulted in a much more pronounced increase in fluorescence that was inhibited by further addition of recombinant SOD (Figure 2A, lower panel). This assay thus detected mitochondrial superoxide release. Skeletal muscle mitochondria were therefore incubated under conditions of reverse electron flow, that is, in the presence of succinate without rotenone, to promote superoxide generation (Figure 2B). The resulting endogenous superoxide release rate was of the same order of magnitude as that generated exogenously (Figure 2A, upper panel). PAC662D1 mitochondria displayed a lower increase in ethidium-emitted fluorescence compared with wild-type mitochondria (Figure 2B and C), thus demonstrating reduced superoxide release rates under reverse electron flow conditions compared with wild-type mitochondria. A similar reduction in superoxide release rate was observed in brown fat mitochondria under all conditions tested (Figure 2D).
Figure 2.
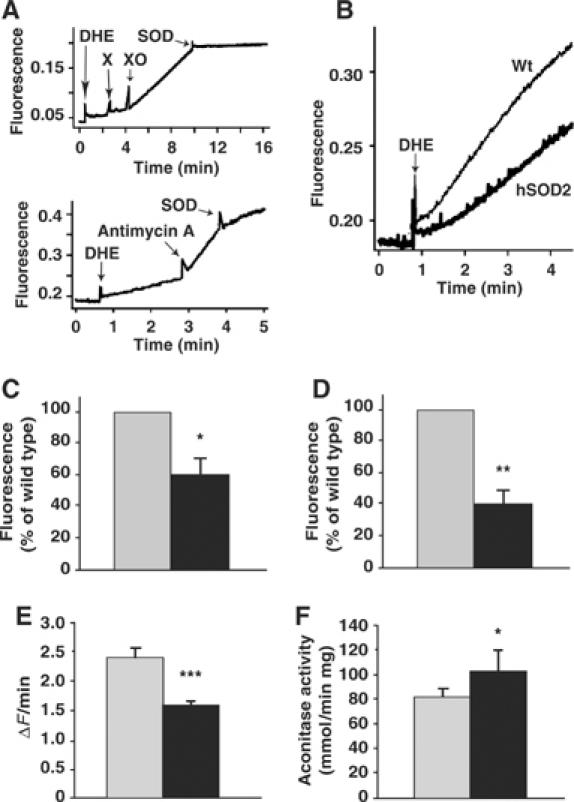
Superoxide measurements. (A) Validation. Upper panel: Chemical validation. Sequential addition of 63.5 μM DHE, 375 μM xanthine (X), 5 μg/ml xanthine oxidase (XO) and 68 U/ml recombinant SOD was performed and fluorescence emission was followed. Lower panel: Biological validation. Mitochondria, 0.5 mg, from skeletal muscle were incubated with 5 mM succinate, 2 μg/ml oligomycin and 3.3 μg/ml rotenone followed by addition of 63.5 μM DHE. Further additions of 0.8 μg/ml antimycin A and of 24 U/ml recombinant SOD were made. (B) Mitochondria, 0.5 mg, from skeletal muscle (as in B) were incubated under conditions of reverse electron flow using 5 mM succinate and 2 μg/ml oligomycin followed by addition of 63.5 μM DHE. (C) Quantification of the change in fluorescence signal intensity over time under conditions of reverse electron flow as exemplified in (B). The rate of superoxide release was calculated as the change in fluorescence intensity during the linear response after addition of DHE. The y-axis denotes the rate of superoxide release in PAC662D1 mitochondria (black bar) relative to wild-type mitochondria (gray bar) and is given in percentage. The values represent the means±s.e.m. (n=3 independent mitochondrial preparations, each analyzed in triplicate). (D) The rate of superoxide release from brown fat mitochondria under conditions similar to those in (B) but with 5 mM glycerol-3-phosphate as substrate and 3.3 μg/ml rotenone. Other conditions (e.g. presence/absence of GDP or rotenone) yielded similarly decreased superoxide release rates in the SOD2-overexpressing mitochondria (not shown) (n=3 independent mitochondrial preparations, each analyzed in triplicate). (E) Superoxide measurements of PAC662D1 (black bar) and wild-type (gray bar) MEF. MEF were incubated in Dulbecco's modified Eagle's medium (DMEM) containing 10 μM DHE. The change in fluorescence signal intensity of each cell was monitored online by confocal microscopy. For statistical analyses, data from six separate experiments were pooled. The values represent the means±s.e.m. (hSOD2, n=198; wild type, n=201). (F) Aconitase enzyme activity measurements in PAC662D1 (black bar) and wild-type skeletal muscle mitochondria (gray bar). The values represent the means±s.e.m. (n=5).
We then investigated whether SOD2 overexpression altered superoxide levels in vivo. Superoxide levels were determined in MEF isolated from PAC662D1 mice and from wild-type littermate controls. MEF were incubated in the presence of DHE and monitored online by confocal microscopy (Krauss et al, 2003). MEF of PAC662D1 mice showed a reduced increase in fluorescence signal intensity over time compared with wild-type MEF (Figure 2E). As a positive control, MEF were incubated with 10 μM antimycin A, which led to a 150% increase in ethidium-emitted fluorescence (not shown). These results thus demonstrate lowered endogenous superoxide release rates in SOD2-overexpressing MEF. Aconitase enzyme activities are readily and specifically impaired by superoxide (Gardner et al, 1995). Higher aconitase enzyme activities were found in PAC662D1 mitochondria compared with wild-type mitochondria (Figure 2F). We conclude that SOD2 overexpression reduced net superoxide release rates in isolated mitochondria and in MEF.
SOD2 overexpression increases resistance to inducers of mitochondrial permeability
Oxidative stress results in damage of cellular membranes (Davies, 1995). Mitochondria isolated from heterozygous SOD2 knockout mice are more prone to undergo permeability transition (Kokoszka et al, 2001). In order to investigate the functional significance of the SOD2 overexpression, liver and muscle mitochondria were isolated from wild-type and PAC662D1 mice. They showed no spontaneous swelling when incubated for 30 min at 37°C (Figure 3A and B). High-amplitude swelling was induced by addition of Ca2+ (Figure 3A) or oleate (Figure 3B), and somewhat less swelling was observed in PAC662D1 mitochondria in comparison with wild-type mitochondria. The response to alamethicin, which induces maximal mitochondrial swelling, was greater in PAC662D1 mitochondria, showing that prior to the addition of alamethicin, these mitochondria were significantly less swollen than wild-type mitochondria (Figure 3C). We thus conclude that SOD2 overexpression confers enhanced resistance to induction of mitochondrial permeability. These data are in line with reports of enhanced ROS production and cardiolipin peroxidation in liver mitochondria undergoing Ca2+- or fatty acid-induced permeability transition (Catisti and Vercesi, 1999; Petrosillo et al, 2004). The increased resistance to induction of mitochondrial permeability conferred by SOD2 overexpression probably results from lowered generation of hydroxyl (OH•) radicals by the Fenton reaction leading to lowered lipid peroxidation of the inner mitochondrial membrane (Brand et al, 2004).
Figure 3.
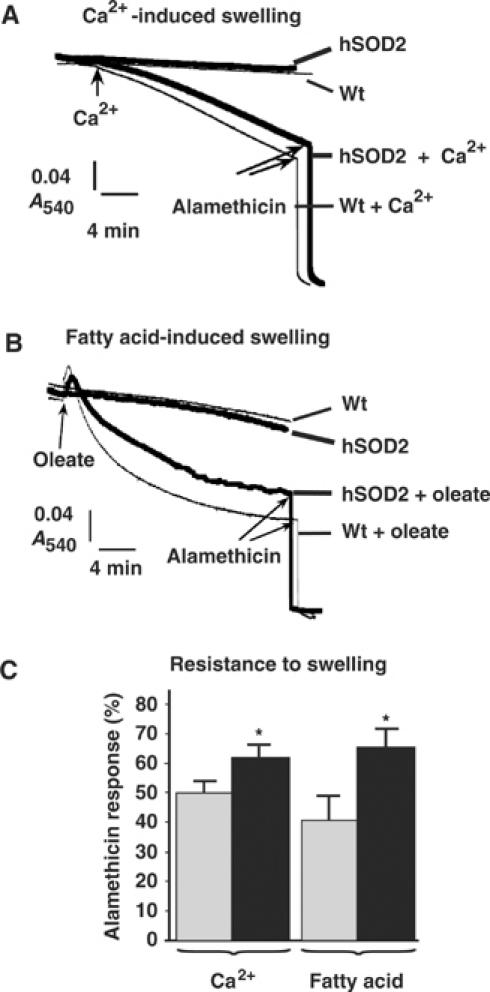
Induction of mitochondrial high-amplitude swelling. (A) Ca2+-induced swelling of liver mitochondria isolated from wild-type (Wt, thin lines) or SOD2-overexpressing (hSOD2, thick lines) mice. The substrate used was 5 mM each of glutamate and malate. Arrows indicate the time points for addition of 30 μM Ca2+ and 10 μg/ml alamethicin. (B) Oleate-induced swelling of skeletal muscle mitochondria from wild-type (Wt, thin lines) or SOD2-overexpressing mice (hSOD2, thick lines). The substrate used was 5 mM each of pyruvate and malate. Arrows indicate the time points for addition of 180 μM oleate and 10 μg/ml alamethicin. (C) Quantification of the amplitude of swelling 20 min after the addition of Ca2+ or oleate. Values are indicated in percent of the maximum response (defined as the absorbance difference between the starting value and the value after alamethicin addition) in mitochondria from wild-type (gray bars) or SOD2-overexpressing (black bars) mice (n=3 independent mitochondrial preparations, each analyzed in duplicate).
SOD2 overexpression does not diminish basal proton leak but enhances mitochondrial oxidative capacity
If matrix superoxide is a significant activator of UCPs, mitochondria overexpressing SOD2 would be expected to show a markedly lower basal respiration because of reduced proton leak. To examine this, mitochondria were isolated from tissues with different expression of UCPs, that is, liver, brain, skeletal muscle and BAT, and respiratory chain function was measured (Figure 4 and Tables I and and II). In liver mitochondria, which lack all three UCPs, no difference in basal (i.e. state 4, after oligomycin addition) respiration was observed (Table I). Also in brain mitochondria (that have been reported to express UCP2) and in skeletal muscle mitochondria (that express UCP3), SOD2 overexpression failed to influence state 4 respiration (Table I). In brown fat mitochondria, which express UCP1, the equivalent respiration (after GDP addition) was also unchanged (Table II). As an alternative indicator for the basal proton leak, the mitochondrial membrane potential (MMP) was also measured but was found to be unaltered (Table I). Thus, these data indicate that, in the basal state, mitochondrial superoxide does not have an observable activating effect on proton conductance mediated by UCPs (or by any other mitochondrial proteins).
Figure 4.
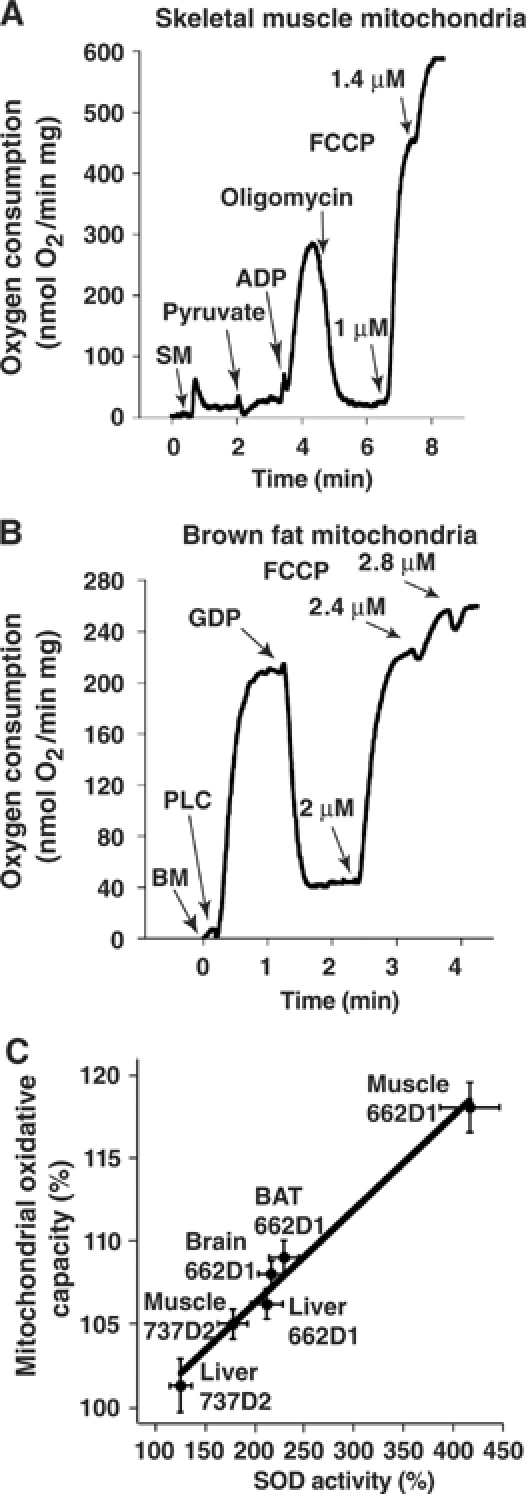
Mitochondrial respiratory measurements. (A) Measurement of oxygen consumption in skeletal muscle mitochondria (SM). FCCP was added successively to reach a final concentration of 1.4 μM. (B) Measurement of oxygen consumption in brown fat mitochondria (BM). Palmitoyl-L-carnitine (PLC), GDP and FCCP (to a final concentration of 2.8 μM) were added as indicated. (C) Correlation between mitochondrial oxidative capacity and mitochondrial SOD enzyme activity. The respiratory measurements were performed as described in (A, B) and the SOD enzyme activities as in Figure 1E. The relative increase in mitochondrial oxidative capacity after FCCP addition (mutant/wild type, %, calculated from the parallel preparations each day) was plotted against the relative increase in mitochondrial SOD enzyme activity (mutant/wild type, %). The results are from 2–5 independent experiments.
Table 1.
Parameters of oxidative phosphorylation and MMP in different tissues
| State 3 | State 4 | State 3/state 4 | Unc. (FCCP) | Unc./state 4 | MMP | |
|---|---|---|---|---|---|---|
| Liver | ||||||
| Wild type (n=3) | 118±5 | 21.8±0.7 | 5.5±0.4 | 173±2 | 7.9±0.1 | |
| hSOD2/+ (n=3) | 119±3 | 21.1±0.4 | 5.7±0.1 | 182±2* | 8.6±0.2 | |
| Brain | ||||||
| Wild type (n=3) | 87±9 | 10.6±0.9 | 8.9±0.9 | 170±10 | 18.6±1.6 | |
| hSOD2/+ (n=2) | 99±10 | 9.8±1.6 | 10.9±1.4 | 189±4* | 19.1±1.9 | |
| Skeletal muscle | ||||||
| Wild type (n=5) | 249±8 | 15.0±1.4 | 16.0±1.2 | 505±10 | 33.6±2.9 | 179±6 |
| hSOD2/+ (n=4) | 252±7 | 14.5±1.2 | 17.6±1.3 | 577±4* | 39.5±3.1 | 180±10 |
| Values are from experiments as illustrated in Figure 4A and are expressed as nmol O2/min mg protein. Substrates for brain and muscle are 5 mM pyruvate and for liver 5 mM glutamate, all with 3 mM malate. State 3 respiration is after 450 μM ADP and state 4 after oligomycin (2 μg/ml). Uncoupled state is that after FCCP (1.4 μM) and is denoted by Unc. MMP refers to the mitochondrial membrane potential in state 4 and is expressed in mV. | ||||||
Table 2.
Respiration measurements in brown fat mitochondria
| Substrate | GDP | UCP1 dependent (substrate−GDP) | Uncoupled state (FCCP) | |
|---|---|---|---|---|
| PLC+malate | ||||
| Wild type (n=4) | 209±8 | 45.0±1.4 | 164±9 | 260±3 |
| hSOD2/+ (n=4) | 211±7 | 40.5±2.2 | 170±8 | 277±4* |
| Pyruvate+malate | ||||
| Wild type (n=4) | 160±15 | 42.2±2.9 | 118±5 | 231±8 |
| hSOD2/+ (n=5) | 168±11 | 40.3±2.6 | 128±9 | 251±10* |
| Values are from experiments as illustrated in Figure 4B and are expressed as nmol O2/min 1 mg protein. PLC (palmitoyl carnitine) 50 μM, pyruvate 5 mM, malate 3 mM. | ||||
ADP-stimulated state 3 respiration in liver, brain and muscle was unaffected by the SOD2 overexpression (Table I). However, in all tissues, effects of SOD2 overexpression were observed on maximal respiratory chain capacity (after FCCP addition; Tables I and II). We plotted the relative increase in mitochondrial oxidative capacity caused by SOD2 overexpression against the relative increase in SOD enzyme activity and obtained a linear correlation (Figure 4C). This increase in mitochondrial oxidative capacity of SOD2-overexpressing mitochondria is probably explained by the protection of superoxide-sensitive iron–sulfur clusters in enzymes of the Krebs cycle such as aconitase (Gardner et al, 1995) and subunits of the electron transport chain (Zhang et al, 1990). The higher aconitase activity of PAC662D1 skeletal muscle mitochondria (Figure 2F) supports this view.
Unchanged UCP1 activity in SOD2-overexpressing BAT mitochondria
The basal proton leak in BAT mitochondria was not affected by SOD2 overexpression. It is well accepted that respiration inhibitable by GDP in BAT mitochondria is mediated by UCP1 (see Figure 4B). The UCP1-mediated respiration was identical in wild-type and PAC662D1 BAT mitochondria (Table II). This result indicates that maximal UCP1 activity is independent of SOD2 activity and hence of matrix superoxide levels. It has been suggested that the superoxide activation of UCPs is fatty acid dependent (Echtay et al, 2002b). UCP1 was therefore activated by fatty acids as described previously (Shabalina et al, 2004) and it was examined whether the sensitivity to fatty acids was influenced by SOD2 overexpression (Figure 5A). SOD2 overexpression did not influence sensitivity to fatty acids (Km of wild type=24±5 nM, n=5 and of PAC662D1=26±8 nM, n=5), indicating that the fatty acid effect is not influenced by superoxide (Figure 5B). We also investigated whether SOD2 overexpression affected the GDP-mediated inhibition of UCP1 (Figure 5C and D). Also here, no influence of SOD2 overexpression could be observed (Km of wild type=162±25 μM, n=3 and of PAC662D1=168±28 μM, n=3).
Figure 5.
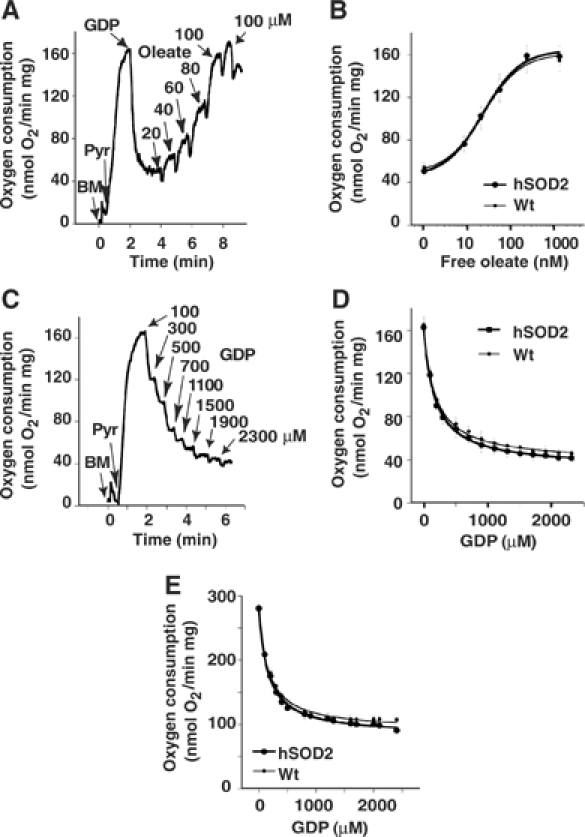
Regulation of UCP1 activity in brown fat mitochondria. (A) Oleate and (C) GDP titration of oxygen consumption in wild-type brown fat mitochondria (BM) using pyruvate (Pyr) as a substrate. Different amounts of oleate (20–120 μM) and GDP (100–2300 μM) were added at time points indicated by arrows. In (A), GDP was used at a final concentration of 1 mM. (B) oleate- and (D) GDP–response curves of oxygen consumption in BM of wild-type and SOD2-overexpressing mice, compiled from five independent experiments (means±s.e.m.) as shown in (A) and (C), respectively. The x-axis in (B) refers to the free oleate concentration. (E) GDP–response curve of oxygen consumption compiled from five independent experiments (means±s.e.m.) as shown in (C) but using glycerol-3-phosphate instead of pyruvate as substrate.
These measurements were then repeated with glycerol 3-phosphate, which gives rise to superoxide (hydrogen peroxide) in these mitochondria (Drahota et al, 2002), as in other mitochondria (Miwa and Brand, 2005). Similar oxygen consumption rates were found in SOD2-overexpressing and control mitochondria (Figure 5E), and GDP titration experiments showed no significant difference between wild-type (Km=169±30 μM, n=5) and PAC662D1 (Km=146±28 μM, n=5) BAT mitochondria (Figure 5E). Thus, even when superoxide generation was augmented, no effects of SOD2 overexpression on UCP1 activity or on GDP sensitivity were observed. (The levels of UCP1 protein were similar in BAT from wild-type and PAC662D1 mice (Supplementary Figure 1D).)
Unchanged UCP3 activity in SOD2-overexpressing muscle mitochondria
As also superoxide activation of UCP3 has been suggested to be fatty acid dependent (Echtay et al, 2002b), palmitate titration curves were performed with wild-type and PAC662D1 skeletal muscle mitochondria (Figure 6A and B) and no significant difference was found between them (Km of wild type=2.6±0.6 μM, n=4 and of PAC662D1=2.5±0.8 μM, n=4). In skeletal muscle mitochondria, the Km of palmitate was not influenced by the presence of GDP (Figure 6A and B), indicating that skeletal muscle mitochondria, in contrast to BAT mitochondria (Shabalina et al, 2004), do not display competitive interaction between GDP and fatty acids. The maximal response to fatty acid was slightly decreased by GDP treatment (Figure 6A and B), to the same extent in wild-type and PAC662D1 mitochondria (ΔGDP of wild type=21±4 nmol O2, n=4 and of PAC662D=25±5 nmol O2, n=4). Skeletal muscle mitochondria did not exhibit innate GDP-responsive uncoupling (not shown), in accordance with a previous report (Cadenas et al, 2002). However, it has been reported that conditions giving rise to reverse electron flow will generate high levels of superoxide, and that GDP can inhibit basal proton leak under such conditions (Talbot et al, 2004). Reverse electron flow was induced by using succinate in the absence of rotenone. This leads to a significantly lower rate of superoxide production in PAC662D1 skeletal muscle mitochondria than in wild type (Figure 2B and C). A small GDP-mediated inhibitory effect on basal respiration (Figure 6C) could be observed under these conditions. However, the degree of inhibition was the same in wild-type and PAC662D1 skeletal muscle mitochondria (ΔGDP effect=4.5±2.5 nmol O2 (n=2) versus 3.9±1.9 nmol O2 (n=2)). The relation between MMP and oxygen consumption was also measured under conditions of reverse electron flow (basal proton leak kinetics) (Figure 6D) by successive inhibition of respiration with malonate. However, the leak current characteristics were the same in PAC662D1 mitochondria as in wild type (Figure 6D), despite the lowered superoxide levels under reverse electron flow conditions (Figure 2C) in PAC662D1 mitochondria. These results thus demonstrate that increased SOD2 expression influences neither the basal proton leak nor the effects of fatty acids or GDP on skeletal muscle mitochondria and, provided that these effects are UCP3 mediated, demonstrate that UCP3 activity is not influenced by SOD2 overexpression.
Figure 6.
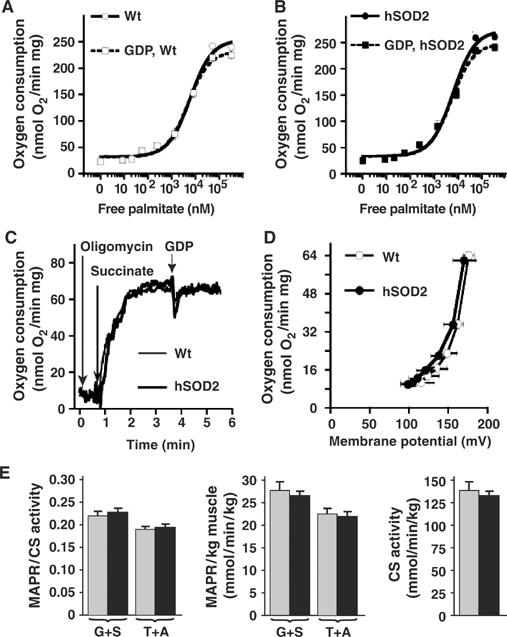
Effects of SOD2 overexpression on skeletal muscle mitochondria. (A, B) Palmitate–oxygen consumption response curves in skeletal muscle mitochondria of wild-type (A) and SOD2-overexpressing (B) mice in the presence (dashed line) or absence (solid line) of 1 mM GDP. The curves in (A, B) are means (±s.e.m.) from four independent experiments. The x-axes in (A, B) refer to the free palmitate concentration. (C) GDP-induced inhibition of oxygen consumption in skeletal muscle mitochondria of wild-type (thin lines) and SOD2-overexpressing (thick lines) mice under conditions inducing reverse electron flow. A 1 mM portion of GDP was added as indicated. (D) Basal proton leak kinetic measurements in skeletal muscle mitochondria of SOD2-overexpressing (thick lines, filled symbols) and wild-type (empty symbols, thin lines) mice. The MMP and oxygen consumption of 0.4 mg skeletal muscle mitochondria were measured in parallel in the presence of 5 mM succinate (without rotenone), 2 μg/ml oligomycin and 5 μM safranin O and were titrated by sequential additions of up to 0.33 mM malonate. The membrane potential was calculated as described in Materials and methods. The values represent the means±s.e.m. of three separate mitochondrial preparations, each analyzed in triplicate. (E) MAPR and citrate synthase (CS) activities (Wredenberg et al, 2002) in skeletal muscle of wild-type (gray bars) and SOD2-overexpressing (black bars) mice. MAPR were measured by using either glutamate plus succinate (G+S) or TMPD plus ascorbate (T+A) as substrates and are given per unit of CS activity or per kg skeletal muscle (n=4–6).
It has previously been indicated that loss of UCP3 activity through gene ablation leads to increased ATP production in skeletal muscle (Cline et al, 2001). Mitochondrial ATP production rates (MAPR) were determined in skeletal muscle of wild-type and PAC662D1 mice and no differences were found (Figure 6E). These results support the conclusion that UCP3 activity is not regulated by matrix superoxide, because SOD2 overexpression would be expected to decrease the activation of UCP3 and thereby increase MAPR.
SOD2-overexpressing mice do not display alterations in energy metabolism
The experiments presented above do not indicate significant effects of SOD2 overexpression on proton leak or on UCP activities. However, these experiments were performed in isolated systems, and potentially important regulatory factors may have been absent. We therefore also investigated whether signs of a regulatory role for superoxide could be observed in the intact animal. No difference in body weight was found when wild-type and PAC662D1 mice of both sexes were compared (Figure 7A). Mice were also acclimated to 25 or 4°C for 4 weeks after which the resting metabolic rates were determined at thermoneutrality. No differences were found between wild-type and PAC662D1 mice (Figure 7B). These results show that SOD2 overexpression does not affect energy expenditure, that is, the innate UCP activation summated for all tissues was apparently not altered. To estimate the importance of SOD2 for UCP1 activity in vivo, we induced nonshivering thermogenesis by injecting norepinephrine (NE) into the mice and measured oxygen consumption (Figure 7C). The increase in oxygen consumption in response to NE was similar in wild-type and PAC662D1 mice (Figure 7C). These results demonstrate that SOD2 overexpression does not affect UCP1 activity in vivo. In addition, there was no indication that a putative decrease in UCP1 activity was compensated for by augmented recruitment of the tissue (Supplementary Table I).
Figure 7.
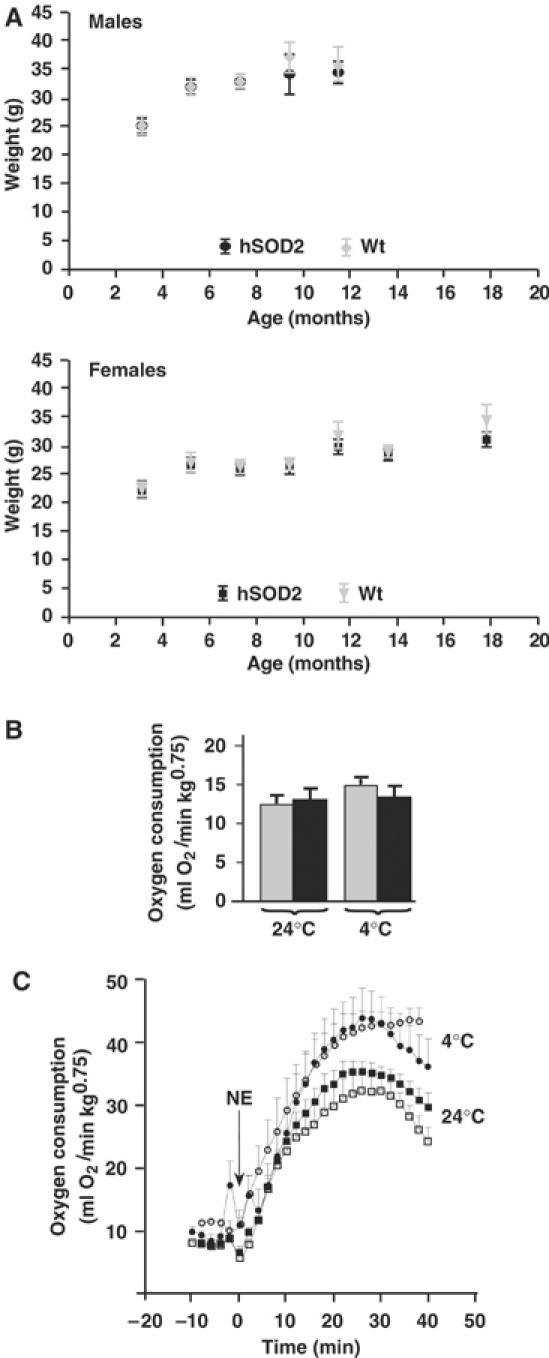
Body weights and metabolic rate measurements. (A) Body weights of males and females followed for up to 18 months of age. (B) Resting metabolic rate measurements of wild-type (gray bars) and SOD2-overexpressing (black bars) mice, 2 months old, measured for 3 h in a metabolic chamber at 30°C (Golozoubova et al, 2004) following acclimation to 25 or 4°C for 4 weeks. The values are from 4–6 wild-type and SOD2-overexpressing mice. (C) Respiratory response to NE of wild-type mice acclimated to 25°C (open squares) or to 4°C (open circles) or of SOD2-overexpressing mice acclimated to 25°C (filled squares) or to 4°C (filled circles). Oxygen consumption was measured at 33°C in pentobarbital-anesthetized mice prior to and after injection of NE (1 mg/kg body weight) (Golozoubova et al, 2004). The values are from 4–6 wild-type and SOD2-overexpressing mice.
Conclusions
Careful regulation of SOD2 expression is critical in order to avoid deleterious effects associated with excessive SOD2 overexpression (Kowald and Klipp, 2004). To obtain transgenic mice with a regulated increase of SOD2 enzyme activities, we introduced PAC clones from a human genomic library containing the SOD2 gene into the mouse genome. The SOD2-overexpressing mice displayed increased SOD2 activities in several tissues and reduced superoxide release rates. This in turn led to improved mitochondrial parameters such as enhanced resistance against inducers of mitochondrial permeability and enhanced oxidative capacity. These mice thus provide a valuable tool to investigate the role of superoxide in bioenergetics and aging.
In vitro studies of isolated mitochondria have indicated that superoxide activates UCPs from the matrix side and thereby increases the proton conductance (Echtay et al, 2002a; Talbot et al, 2004). This effect of exogenously generated superoxide was counteracted by added SOD enzyme (Echtay et al, 2002b) or by mitochondrially targeted antioxidant chemicals (Echtay et al, 2002a). Based on such effects of in vitro-generated superoxide, it has been proposed that endogenous superoxide would have a physiologically relevant effect on UCP activity. The level of endogenously generated superoxide would be expected to lead to a decrease in the activity of UCPs, which should result in measurable metabolic changes. However, we found no differences in UCP1 and UCP3 activities between wild-type and SOD2-overexpressing mice. Also, in a global approach, that of basal metabolic rate in the intact animal, we found no evidence for effects of SOD2 overexpression, that is, no significant part of basal metabolism appears to derive from superoxide-regulated UCP activities.
Materials and methods
Creation of PAC transgenic mice
A total of 16 clones carrying the SOD2 gene were isolated from a human genomic PAC library, RPCI (Roswell Park Cancer Institute), and ordered into a contig by Southern blotting with probes derived from the end fragments of the inserts and by size determination of the inserts by pulsed-field gel electrophoresis (Ekstrand et al, 2004). Transgenic lines were generated from two clones, PAC662 and PAC737, by injection of CsCl-purified PAC DNA into pronuclear-stage embryos of the FVB/N strain (Figure 1A) (Ekstrand et al, 2004). Founders were identified by PCR genotyping (see below) and Southern blot analysis of tail DNA using the human SOD2 cDNA as a probe (Figure 1B). Searches with the Celera (Applera Corporation©) and NCBI databases revealed no additional genes in the 3′-flanking region of the human SOD2 gene but two genes on its 5′ flanking region, encoding the Wilms tumor 1-associated protein (WTAP) and acyl-CoA:cholesterol acyltransferase 2 (ACAT2).
Mating and genotyping of transgenic animals
SOD2-overexpressing mice were maintained as inbred stocks by crossing heterozygous hSOD2/+ mice to wild-type FVB/N mice. Transgenic animals were identified by PCR genotyping of tail DNA using primers to specifically amplify exon 1 (GCTCCCGGCGCTTTCTTAAGGC and CCCTTCCTTCTCACCCGCACACT, annealing temperature 63°C) and exon 5 (TTCTAACAGGCCTTATTCCAC and AGAAATGTCCCAATCAGATTC, annealing temperature 51°C) of the human SOD2 gene.
Western blotting
Proteins were extracted as described previously (Ekstrand et al, 2004; Lindgren et al, 2004). Primary antibodies were diluted as follows: anti-human SOD2 (Biosite) 1:2000 and anti-β-actin (Sigma) 1:2000. Secondary horseradish peroxidase-conjugated antibodies were used at a dilution of 1:2000.
Animals
Age-matched animals were kept single-caged in climate-controlled rooms at 25°C on a 12–12-h light–dark cycle. They were fed a pelleted standard diet (R70, Lactamin) ad libitum and had free access to water. Experiments were approved by the Animal Ethics Committees of the north or south Stockholm regions.
Isolation of mitochondria
Mitochondria were isolated in parallel from SOD2-overexpressing and wild-type mice kept at 25 or 4°C for 4–5 weeks by differential centrifugation. Brain and liver mitochondria were isolated in 10 mM K+-Tes (pH 7.2), 250 mM sucrose, 1 mM EDTA and 0.2% fatty acid-free BSA, and skeletal muscle mitochondria were isolated in 20 mM K+-Tes (pH 7.2), 100 mM sucrose, 50 mM KCl, 1 mM EDTA and 0.2% fatty acid-free BSA following treatment of skeletal muscle homogenate with 1 mg/g nagarse. BAT mitochondria were isolated according to Cannon and Nedergaard (2001) and diluted in 20 mM K+-Tes (pH 7.2), 100 mM KCl and 0.2% fatty acid-free bovine serum.
Oxygen consumption measurements
Oxygen consumption rates were monitored with a Clark-type oxygen electrode in a sealed chamber at 37°C, as described previously (Shabalina et al, 2004). Skeletal muscle (0.25 mg protein/ml), liver (0.5 mg protein/ml) and brain (0.5 mg protein/ml) mitochondria were incubated in a medium consisting of 125 mM sucrose, 20 mM K+-Tes (pH 7.2), 30 mM KCl, 2 mM MgCl2, 1 mM EDTA, 4 mM KPi and 0.1% fatty acid-free BSA. BAT mitochondria (0.35 mg protein/ml) were incubated in the same medium without KCl.
In BAT mitochondria, respiration was initiated by adding the substrate and inhibited by adding 2 mM GDP. UCP1-dependent respiration was calculated as the difference between substrate-stimulated and GDP-inhibited oxygen consumption rates. Maximal oxygen consumption rates (uncoupled state) were obtained by addition of FCCP (2.8 μM).
The free concentration of oleate and palmitate was calculated using previously described equations (Richieri et al, 1993) for the binding of fatty acid to bovine serum albumin at 37°C: [Free oleate] (nM)=6.5n−0.19+0.13 exp(1.54n) and [Free palmitate] (nM)=4.4n−0.03+0.23 exp(1.16n), where n is the molar ratio of fatty acid to albumin. Concentration–response curve data were analyzed with the general fit option of the KaleidaGraph application for Macintosh for adherence to simple Michaelis–Menten kinetics, V(x)=basal+ΔVmax(x/(Km+x)) (for fatty acid concentration–response curve) and V(x)=Vmax−ΔVmax(x/(Km+x)) (for GDP concentration–response curve).
Measurement of mitochondrial membrane potential
MMP measurements were performed in skeletal muscle mitochondria using the dye safranin O (5 μM) (Akerman and Wikstrom, 1976). Conditions and media were otherwise exactly the same as those for oxygen consumption measurements. The changes in absorbance of safranin O at 511–533 nm were followed at 37°C and used to calculate the membrane potential (mV) by the Nernst equation. Calibration curves were made for each mitochondrial preparation (Nedergaard, 1983). To determine the basal proton leak, MMP and oxygen consumption measurements were performed in parallel using the same media as described above. MMP and state 4 respiration were titrated by sequential additions of up to 0.33 mM malonate. Proton leak kinetics measurements were performed within 10 min.
Monitoring of mitochondrial high-amplitude swelling
Mitochondrial swelling was monitored as absorbance change at 540 nm upon addition of 30 μM Ca2+ or 180 μM oleate in the medium and experimental conditions described for the oxygen consumption measurements.
Measurements of superoxide and SOD activity
Total SOD activity was measured in isolated mitochondria from liver, muscle, brain and BAT by the adrenaline method (Misra and Fridovich, 1972). One unit of SOD was defined as the amount of enzyme reducing the rate of auto-oxidation of adrenaline by 50%. In control experiments with total heart homogenate, SOD activity in the absence and presence (20 min) of 4 mM KCN (to inhibit SOD1) (Salin et al, 1978) was determined in wild-type and SOD2-overexpressing mice. We found that the increased SOD activity in the overexpressing mice resulted entirely from an increase in SOD2 activity.
Superoxide was measured using superoxide-induced conversion of DHE (Molecular Probes) to ethidium (Benov et al, 1998) at 37°C using an excitation wavelength of 495 nm and collecting the emission via a cutoff filter at 580 nm.
MEF were isolated from SOD2-overexpressing embryos and littermate control embryos at embryonic day E13.5. MEF were maintained in DMEM (Gibco) containing 22.7 mM glucose, 10% fetal bovine serum and 1% penicillin–streptomycin and plated exactly three times before superoxide measurements. Superoxide measurements were performed sequentially in subconfluent mutant and control MEF cultures. Superoxide production was measured in intact cells by ethidium imaging and analysis as described previously (Krauss et al, 2003). Owing to low inter-experimental variation of the mean slope (<25%), MEF data from separate experiments were pooled.
Aconitase measurements were performed on isolated skeletal muscle mitochondria following the procedure described by Gardner et al (1995).
RNA isolation, Northern blots and quantitative real-time PCR analyses
Animals were killed by cervical dislocation. Tissues were rapidly harvested, frozen in liquid nitrogen and stored at −80°C. RNA was extracted using Trizol® (Invitrogen). MtRNA was measured by Northern blot analysis using a [α-32P]dCTP random-labeled COXI DNA probe as described previously (Ekstrand et al, 2004). PGC1α mRNA and PPARγ mRNA were determined by quantitative real-time PCR using SYBR Green PCR Master Mix (Applied Biosystems) and primers to specifically amplify PGC1α mRNA (GGTGTAGCGACCAATCGGAA and GGCAATCCGTCTTCATCCA), PPARγ mRNA (GGTGTAGCGACCAATCGGAA and GGCAATCCGTCTTCATCCA) and β-actin mRNA (CATGTACCCAGGCATTGCTGAC and GAGCCACCGATCCACACAGAG).
Statistics
All data are expressed as means±s.e.m. Statistical analyses were performed using paired or unpaired, two-tailed Student's t-test. Significant differences between wild-type and transgenic mice are indicated as *P<0.05, **P<0.01 and ***P<0.001.
Supplementary Material
Supplementary Figure 1
Supplementary Table I
Supplementary Data
Acknowledgments
NGL is supported by the Swedish Research Council, the Torsten and Ragnar Söderbergs Foundation, the Swedish Heart and Lung Foundation, the Swedish Foundation for Strategic Research (Functional Genomics and INGVAR) and Funds of Karolinska Institute. BC and JN are supported by the European Union (DLARFID), the Swedish Research Council and the Swedish Cancer Society.
References
- Akerman KE, Wikstrom MK (1976) Safranine as a probe of the mitochondrial membrane potential. FEBS Lett 68: 191–197 [DOI] [PubMed] [Google Scholar]
- Arsenijevic D, Onuma H, Pecqueur C, Raimbault S, Manning BS, Miroux B, Couplan E, Alves-Guerra MC, Goubern M, Surwit R, Bouillaud F, Richard D, Collins S, Ricquier D (2000) Disruption of the uncoupling protein-2 gene in mice reveals a role in immunity and reactive oxygen species production. Nat Genet 26: 435–439 [DOI] [PubMed] [Google Scholar]
- Benov L, Sztejnberg L, Fridovich I (1998) Critical evaluation of the use of hydroethidine as a measure of superoxide anion radical. Free Radic Biol Med 25: 826–831 [DOI] [PubMed] [Google Scholar]
- Brand MD, Buckingham JA, Esteves TC, Green K, Lambert AJ, Miwa S, Murphy MP, Pakay JL, Talbot DA, Echtay KS (2004) Mitochondrial superoxide and aging: uncoupling-protein activity and superoxide production. Biochem Soc Symp 71: 203–213 [DOI] [PubMed] [Google Scholar]
- Brand MD, Esteves TC (2005) Physiological functions of the mitochondrial uncoupling proteins UCP2 and UCP3. Cell Metab 2: 85–93 [DOI] [PubMed] [Google Scholar]
- Brand MD, Pamplona R, Portero-Otin M, Requena JR, Roebuck SJ, Buckingham JA, Clapham JC, Cadenas S (2002) Oxidative damage and phospholipid fatty acyl composition in skeletal muscle mitochondria from mice underexpressing or overexpressing uncoupling protein 3. Biochem J 368: 597–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadenas S, Echtay KS, Harper JA, Jekabsons MB, Buckingham JA, Grau E, Abuin A, Chapman H, Clapham JC, Brand MD (2002) The basal proton conductance of skeletal muscle mitochondria from transgenic mice overexpressing or lacking uncoupling protein-3. J Biol Chem 277: 2773–2778 [DOI] [PubMed] [Google Scholar]
- Cannon B, Nedergaard J (2001) Respiratory and thermogenic capacities of cells and mitochondria from brown and white adipose tissue. Methods Mol Biol 155: 295–303 [DOI] [PubMed] [Google Scholar]
- Cannon B, Nedergaard J (2004) Brown adipose tissue: function and physiological significance. Physiol Rev 84: 277–359 [DOI] [PubMed] [Google Scholar]
- Catisti R, Vercesi AE (1999) The participation of pyridine nucleotides redox state and reactive oxygen in the fatty acid-induced permeability transition in rat liver mitochondria. FEBS Lett 464: 97–101 [DOI] [PubMed] [Google Scholar]
- Cline GW, Vidal-Puig AJ, Dufour S, Cadman KS, Lowell BB, Shulman GI (2001) In vivo effects of uncoupling protein-3 gene disruption on mitochondrial energy metabolism. J Biol Chem 276: 20240–20244 [DOI] [PubMed] [Google Scholar]
- Couplan E, del Mar Gonzalez-Barroso M, Alves-Guerra MC, Ricquier D, Goubern M, Bouillaud F (2002) No evidence for a basal, retinoic, or superoxide-induced uncoupling activity of the uncoupling protein 2 present in spleen or lung mitochondria. J Biol Chem 277: 26268–26275 [DOI] [PubMed] [Google Scholar]
- Davies KJ (1995) Oxidative stress: the paradox of aerobic life. Biochem Soc Symp 61: 1–31 [DOI] [PubMed] [Google Scholar]
- Drahota Z, Chowdhury SK, Floryk D, Mracek T, Wilhelm J, Rauchova H, Lenaz G, Houstek J (2002) Glycerophosphate-dependent hydrogen peroxide production by brown adipose tissue mitochondria and its activation by ferricyanide. J Bioenerg Biomembr 34: 105–113 [DOI] [PubMed] [Google Scholar]
- Echtay KS, Murphy MP, Smith RA, Talbot DA, Brand MD (2002a) Superoxide activates mitochondrial uncoupling protein 2 from the matrix side. Studies using targeted antioxidants. J Biol Chem 277: 47129–47135 [DOI] [PubMed] [Google Scholar]
- Echtay KS, Roussel D, St-Pierre J, Jekabsons MB, Cadenas S, Stuart JA, Harper JA, Roebuck SJ, Morrison A, Pickering S, Clapham JC, Brand MD (2002b) Superoxide activates mitochondrial uncoupling proteins. Nature 415: 96–99 [DOI] [PubMed] [Google Scholar]
- Ekstrand MI, Falkenberg M, Rantanen A, Park CB, Gaspari M, Hultenby K, Rustin P, Gustafsson CM, Larsson NG (2004) Mitochondrial transcription factor A regulates mtDNA copy number in mammals. Hum Mol Genet 13: 935–944 [DOI] [PubMed] [Google Scholar]
- Gardner PR, Raineri I, Epstein LB, White CW (1995) Superoxide radical and iron modulate aconitase activity in mammalian cells. J Biol Chem 270: 13399–13405 [DOI] [PubMed] [Google Scholar]
- Golozoubova V, Gullberg H, Matthias A, Cannon B, Vennstrom B, Nedergaard J (2004) Depressed thermogenesis but competent brown adipose tissue recruitment in mice devoid of all hormone-binding thyroid hormone receptors. Mol Endocrinol 18: 384–401 [DOI] [PubMed] [Google Scholar]
- Heintz N (2000) Analysis of mammalian central nervous system gene expression and function using bacterial artificial chromosome-mediated transgenesis. Hum Mol Genet 9: 937–943 [DOI] [PubMed] [Google Scholar]
- Kokoszka JE, Coskun P, Esposito LA, Wallace DC (2001) Increased mitochondrial oxidative stress in the Sod2 (+/−) mouse results in the age-related decline of mitochondrial function culminating in increased apoptosis. Proc Natl Acad Sci USA 98: 2278–2283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korshunov SS, Skulachev VP, Starkov AA (1997) High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett 416: 15–18 [DOI] [PubMed] [Google Scholar]
- Kowald A, Klipp E (2004) Alternative pathways might mediate toxicity of high concentrations of superoxide dismutase. Ann NY Acad Sci 1019: 370–374 [DOI] [PubMed] [Google Scholar]
- Krauss S, Zhang CY, Scorrano L, Dalgaard LT, St-Pierre J, Grey ST, Lowell BB (2003) Superoxide-mediated activation of uncoupling protein 2 causes pancreatic beta cell dysfunction. J Clin Invest 112: 1831–1842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebovitz RM, Zhang H, Vogel H, Cartwright J Jr, Dionne L, Lu N, Huang S, Matzuk MM (1996) Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice. Proc Natl Acad Sci USA 93: 9782–9787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Huang TT, Carlson EJ, Melov S, Ursell PC, Olson JL, Noble LJ, Yoshimura MP, Berger C, Chan PH, Wallace DC, Epstein CJ (1995) Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat Genet 11: 376–381 [DOI] [PubMed] [Google Scholar]
- Lindgren EM, Nielsen R, Petrovic N, Jacobsson A, Mandrup S, Cannon B, Nedergaard J (2004) Noradrenaline represses PPAR (peroxisome-proliferator-activated receptor) gamma2 gene expression in brown adipocytes: intracellular signalling and effects on PPARgamma2 and PPARgamma1 protein levels. Biochem J 382: 597–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melov S, Coskun P, Patel M, Tuinstra R, Cottrell B, Jun AS, Zastawny TH, Dizdaroglu M, Goodman SI, Huang TT, Miziorko H, Epstein CJ, Wallace DC (1999) Mitochondrial disease in superoxide dismutase 2 mutant mice. Proc Natl Acad Sci USA 96: 846–851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melov S, Schneider JA, Day BJ, Hinerfeld D, Coskun P, Mirra SS, Crapo JD, Wallace DC (1998) A novel neurological phenotype in mice lacking mitochondrial manganese superoxide dismutase. Nat Genet 18: 159–163 [DOI] [PubMed] [Google Scholar]
- Misra H, Fridovich I (1972) The role of superoxide anion in the autoxidation of epinephrine and a simple assay for superoxide dismutase. J Biol Chem 247: 3170–3175 [PubMed] [Google Scholar]
- Miwa S, Brand MD (2005) The topology of superoxide production by complex III and glycerol 3-phosphate dehydrogenase in Drosophila mitochondria. Biochim Biophys Acta 1709: 214–219 [DOI] [PubMed] [Google Scholar]
- Motoori S, Majima HJ, Ebara M, Kato H, Hirai F, Kakinuma S, Yamaguchi C, Ozawa T, Nagano T, Tsujii H, Saisho H (2001) Overexpression of mitochondrial manganese superoxide dismutase protects against radiation-induced cell death in the human hepatocellular carcinoma cell line HLE. Cancer Res 61: 5382–5388 [PubMed] [Google Scholar]
- Nedergaard J (1983) The relationship between extramitochondrial Ca2+ concentration, respiratory rate, and membrane potential in mitochondria from brown adipose tissue of the rat. Eur J Biochem 133: 185–191 [DOI] [PubMed] [Google Scholar]
- Nedergaard J, Cannon B (2003) The ‘novel' ‘uncoupling' proteins UCP2 and UCP3: what do they really do? Pros and cons for suggested functions. Exp Physiol 88: 65–84 [DOI] [PubMed] [Google Scholar]
- Petrosillo G, Ruggiero FM, Pistolese M, Paradies G (2004) Ca2+-induced reactive oxygen species production promotes cytochrome c release from rat liver mitochondria via mitochondrial permeability transition (MPT)-dependent and MPT-independent mechanisms: role of cardiolipin. J Biol Chem 279: 53103–53108 [DOI] [PubMed] [Google Scholar]
- Raineri I, Carlson EJ, Gacayan R, Carra S, Oberley TD, Huang TT, Epstein CJ (2001) Strain-dependent high-level expression of a transgene for manganese superoxide dismutase is associated with growth retardation and decreased fertility. Free Radic Biol Med 31: 1018–1030 [DOI] [PubMed] [Google Scholar]
- Richieri GV, Anel A, Kleinfeld AM (1993) Interactions of long-chain fatty acids and albumin: determination of free fatty acid levels using the fluorescent probe ADIFAB. Biochemistry 32: 7574–7580 [DOI] [PubMed] [Google Scholar]
- Ricquier D, Bouillaud F (2000) The uncoupling protein homologues: UCP1, UCP2, UCP3, StUCP and AtUCP. Biochem J 345: 161–179 [PMC free article] [PubMed] [Google Scholar]
- Salin MD, Day EDJ, Crapo JD (1978) Isolation and characterization of a manganese-containing superoxide dismutase from rat liver. Arch Biochem Biophys 187: 223–228 [DOI] [PubMed] [Google Scholar]
- Shabalina I, Jacobsson A, Cannon B, Nedergaard J (2004) Native UCP1 displays simple competitive kinetics between the regulators purine nucleotides and fatty acids. J Biol Chem 279: 38236–38248 [DOI] [PubMed] [Google Scholar]
- Strassburger M, Bloch W, Sulyok S, Schuller J, Keist AF, Schmidt A, Wenk J, Peters T, Wlaschek M, Krieg T, Hafner M, Kumin A, Werner S, Muller W, Scharffetter-Kochanek K (2005) Heterozygous deficiency of manganese superoxide dismutase results in severe lipid peroxidation and spontaneous apoptosis in murine myocardium in vivo. Free Radic Biol Med 38: 1458–1470 [DOI] [PubMed] [Google Scholar]
- Suzuki K, Murtuza B, Sammut IA, Latif N, Jayakumar J, Smolenski RT, Kaneda Y, Sawa Y, Matsuda H, Yacoub MH (2002) Heat shock protein 72 enhances manganese superoxide dismutase activity during myocardial ischemia–reperfusion injury, associated with mitochondrial protection and apoptosis reduction. Circulation 106: I270–I276 [PubMed] [Google Scholar]
- Talbot DA, Lambert AJ, Brand MD (2004) Production of endogenous matrix superoxide from mitochondrial complex I leads to activation of uncoupling protein 3. FEBS Lett 556: 111–115 [DOI] [PubMed] [Google Scholar]
- Wredenberg A, Wibom R, Wilhelmsson H, Graff C, Wiener HH, Burden SJ, Oldfors A, Westerblad H, Larsson NG (2002) Increased mitochondrial mass in mitochondrial myopathy mice. Proc Natl Acad Sci USA 99: 15066–15071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Marcillat O, Giulivi C, Ernster L, Davies KJ (1990) The oxidative inactivation of mitochondrial electron transport chain components and ATPase. J Biol Chem 265: 16330–16336 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Table I
Supplementary Data