

Abstract
Poly(A)-specific ribonuclease (PARN) is a processive, poly(A)-specific 3′ exoribonuclease. The crystal structure of C-terminal truncated human PARN determined in two states (free and RNA-bound forms) reveals that PARNn is folded into two domains, an R3H domain and a nuclease domain similar to those of Pop2p and ɛ186. The high similarity of the active site structures of PARNn and ɛ186 suggests that they may have a similar catalytic mechanism. PARNn forms a tight homodimer, with the R3H domain of one subunit partially enclosing the active site of the other subunit and poly(A) bound in a deep cavity of its nuclease domain in a sequence-nonspecific manner. The R3H domain and, possibly, the cap-binding domain are involved in poly(A) binding but these domains alone do not appear to contribute to poly(A) specificity. Mutations disrupting dimerization abolish both the enzymatic and RNA-binding activities, suggesting that the PARN dimer is a structural and functional unit. The cap-binding domain may act in concert with the R3H domain to amplify the processivity of PARN.
Keywords: deadenylation, mRNA decay, PARN, processivity, X-ray crystallography
Introduction
Modulation of mRNA stability has been shown to play important roles in the regulation of gene expression, quality control of mRNA biogenesis (Maquat and Carmichael, 2001) and antiviral defenses (van Hoof and Parker, 1999; Dykxhoorn et al, 2003). Studies performed primarily in yeast and human have identified two general mRNA decay pathways (Meyer et al, 2004; Parker and Song, 2004). The first step in general mRNA turnover in eukaryotes is deadenylation. In yeast, the predominant deadenylase complex contains two main nucleases, Ccr4p and Pop2p (Tucker et al, 2001). Ccr4p is a member of the ExoIII family of nucleases (Dlakic, 2000), whereas Pop2 has a fold similar to deoxyribonucleases (DNases) of the DnaQ family (Thore et al, 2003). The complex containing Ccr4p and Pop2p is conserved in all eukaryotes and has been shown to be involved in mRNA deadenylation in Drosophila (Temme et al, 2004) and in human cells (Chang et al, 2004).
In mammalian cells, an additional enzyme responsible for deadenylase activity is the poly(A)-specific exonuclease referred to as poly(A)-specific ribonuclease (PARN) (Astrom et al, 1992; Korner and Wahle, 1997). The important properties of PARN are its high specificity for single-stranded poly(A) and a requirement for a 3′ hydroxyl group for activity (Astrom et al, 1992; Korner and Wahle, 1997). Sequence analysis shows that PARN also belongs to the ribonuclease (RNase) D superfamily (Moser et al, 1997) and has a novel conserved R3H domain, which may function as a single-stranded nucleic-acid-binding domain to facilitate binding of PARN to polyadenylated mRNA (Grishin, 1998). PARN is conserved in many eukaryotes but notably absent from Saccharomyces cerevisiae and Drosophila melanogaster, suggesting that this enzyme is not required by all eukaryotes (Parker and Song, 2004).
PARN is a major deadenylase in mammalian cell extracts and its activity is inhibited by the poly(A)-binding protein (Pab1p) under physiological salt conditions (Korner and Wahle, 1997). In Xenopus laevis, PARN is required for an evolutionarily conserved mechanism (default deadenylation) to silence the translation of maternal mRNAs during oocyte maturation (Korner et al, 1998; Copeland and Wormington, 2001). PARN in Arabidopsis thaliana has been shown to be an essential enzyme during early development (Chiba et al, 2004) and to be responsible for the deadenylation of a subset of mRNAs (Reverdatto et al, 2004). Moreover, PARN has been reported to be required for the rapid deadenylation of mRNAs containing AU-rich element (ARE), promoted by the ARE-binding protein tristetraprolin, and to affect the process of nonsense-mediated mRNA decay (Lai et al, 2003; Lejeune et al, 2003). Mutagenesis and biochemical studies show that PARN is a divalent metal ion-dependent, oligomeric and poly(A)-specific exonuclease producing 5′ AMP (Korner and Wahle, 1997; Martinez et al, 2000, 2001; Ren et al, 2002, 2004). The shortest substrate sufficient for PARN activity appeared to be adenosine di- or trinucleotide depending on the presence of divalent metal ions (Ren et al, 2004). PARN is a cap-binding protein, and the enzyme's interaction with the 5′ cap not only stimulates the deadenylation activity but also enhances the processivity of the deadenylation reaction (Dehlin et al, 2000; Gao et al, 2000; Martinez et al, 2001).
The RNase D family belongs to the DEDD superfamily composed of RNases as well as DNases defined by four conserved acidic residues, three aspartic acids (D) and one glutamic acid (E), distributed among three separate sequence motifs (ExoI–III) (Moser et al, 1997; Zuo and Deutscher, 2001). These four conserved amino acids form the active site and are involved in binding of the two-metal ions, which are crucial for catalysis (Steitz et al, 1994). The DEDD superfamily can be divided into two subgroups, DEDDy and DEDDh, which are distinguished according to whether a fifth conserved residue is tyrosine (Y) or histidine (H). Based on the crystal structures of the exonuclease domain of DNA polymerase I (Pol I) (Beese and Steitz, 1991) and the ɛ-subunit of DNA polymerase III (ɛ186) (Hamdan et al, 2002), these Tyr and His residues are postulated to play an equivalent role in activating a water molecule or a hydroxide ion during phosphodiester bond cleavage. Recently, three crystal structures of RNases in the DEDD superfamily have been determined, including Pop2, a DEDD-related exoribonuclease (Thore et al, 2003), ISG20, an interferon-induced antiviral exoribonuclease (Horio et al, 2004), and the Escherichia coli RNase D (Zuo et al, 2005).
To gain insight into the poly(A) recognition and the catalytic mechanism of PARN, we have determined the crystal structure of the C-terminal truncated PARN (PARNn) in both ligand-free and poly(A)-bound forms. These results reveal that PARNn is folded into two domains, the nuclease domain and R3H domain, with the nuclease domain highly resembling that of Pop2p. PARN forms a homodimer, with each subunit binding one RNA oligonucleotide. Mutagenesis combined with structural data suggests that the dimeric form of PARN is essential for its poly(A)-specific activity. The catalytic mechanism of PARN and how cap binding increases the processivity of PARN are discussed.
Results and discussion
Structure determination
The C-terminal truncated human PARN (residues 1–537), which contains the nuclease domain, the R3H domain and the putative cap-binding domain, was initially expressed and purified. The purified PARN(1–537) is fully active in poly(A) cleavage (see below) and capable of binding to m7GTP Sepharose (M Wu and H Song, unpublished results). However, the protein underwent substantial proteolysis and was not amenable for crystallization. Therefore, another C-terminal truncated human PARN (residues 1–430), which consists of just the nuclease domain and the R3H domain, but does not bind the cap structure, designated as PARNn, was expressed in E. coli and purified to homogeneity. The purified protein is catalytically active for cleavage of a 15-nucleotide-long noncapped poly(A) molecule albeit with lower activity (see below). Crystals of PARNn co-crystallized with a 10-nucleotide poly(A) RNA (designated as PARNn–RNA) in the presence of 5 mM EDTA belong to space group P212121 with two protein–RNA complexes in the asymmetric unit (AU). The addition of 5 mM EDTA in the crystallization buffer is to annihilate the enzymatic activity of PARNn. The structure of the PARNn–RNA complex was determined using the MAD method at a resolution of 2.6 Å. Of the 10 nucleotides present in the crystals (Supplementary Figure 1), only the last three adenosine nucleotides (A8–10) are visible in the electron density map. For both PARNn–RNA complexes in the AU, the last nucleotide A10 has well-defined electron density, whereas the adenine bases of A8 and A9 are less well defined (Figure 1A). Four regions in the PARNn–RNA complex are not visible in the electron density map and are assumed to be disordered, namely residues 41–45, 144–256, 370–374 and 424–430 for molecule A and 40–49, 144–260, 370–374 and 424–430 for molecule B. Crystals of native PARNn were obtained in the presence of 5 mM MgCl2. The structure of native PARNn was solved by the molecular replacement method using the nuclease domain in the PARNn–RNA complex as the search model. Several regions of native PARNn are disordered, namely residues 37–45, 145–169 and 370–374 for molecule A, residues 40–46, 148–172, 370–374 and 428–430 for molecule B, residues 36–47, 148–169 and 370–374 for molecule C and residues 39–45, 144–169 and 370–374 for molecule D. Statistics of structure determination and refinement are summarized in Table I (see Materials and methods).
Figure 1.
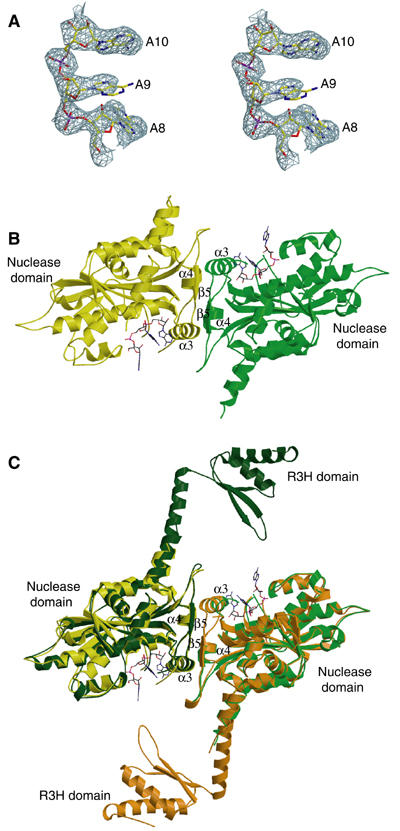
Structures of PARNn in free and RNA-bound forms. (A) Stereo diagram of 2.6 Å simulated annealing (SA) omit map contoured at 2σ covering the bound poly(A) in the PARNn–RNA complex. The last three nucleotides are shown in stick model. (B) A ribbon diagram of the PARNn–RNA complex. The two molecules are shown in yellow and green, respectively. Nucleotides are shown in stick model. α3, α4 and β5 are labeled in (B, C). (C) Superimposition of the PARNn–RNA complex with native PARNn. The color coding for the PARNn–RNA complex is as in (B). The two molecules (chain A and chain B) of native PARNn are highlighted with dark green and orange, respectively. Nucleotides are shown in stick model.
Table 1.
Data Collection and refinement statistics
| Crystals | The PARNn–RNA complex |
Native PARNn | ||
|---|---|---|---|---|
| λ1 (peak) | λ2 (edge) | λ3 (remote) | ||
| Wavelength (Å) | 0.9791 | 0.9793 | 0.9762 | |
| Data collection | ||||
| Cell dimension | ||||
| a/b/c (Å) | 92.79/92.40/159.64 | 205.54/123.02/82.84 | ||
| α/β/γ (deg) | 90.0/90.0/90.0 | 90.0/112.6/90.0 | ||
| Space group | P212121 | C2 | ||
| Resolution (Å) | 2.6 | 2.8 | 2.7 | 2.6 |
| Unique reflections (N) | 36 375 | 34 991 | 35 991 | 58 519 |
| Completeness (%) | 95.7 (98.0) | 92.1 (95.5) | 93.2 (96.4) | 99.5 (99.5) |
| Redundancy | 5.0 | 4.5 | 5.3 | 3.5 |
| Rmerge a (%) | 7.8 (35.4) | 8.4 (49.5) | 8.4 (41.5) | 7.9 (43.7) |
| I/σ | 7.6 (2.0) | 7.3 (2.1) | 7.0 (1.9) | 6.2 (2.0) |
| Number of Se sites | 15 | |||
| Figure of merit | ||||
| Before density modification | 0.31 | |||
| After density modification | 0.85 | |||
| Refinement | ||||
| Resolution range (Å) | 20–2.6 | 20–2.6 | ||
| Total atoms | 5116 | 13 192 | ||
| Rwork b (%) | 21.8 (29.0) | 21.9 (31.0) | ||
| Rfree c (%) | 23.5 (32.0) | 25.3 (35.0) | ||
| R.m.s.d. | ||||
| Bond distance (Å) | 0.012 | 0.013 | ||
| Bond angle (deg) | 1.29 | 1.33 | ||
| Averaged B-values (Å2) | ||||
| Protein molecules | 52.4 | 55.7 | ||
| RNA molecules | 74.0 | |||
| Ramchandran plot | ||||
| Most favored region | 90.2% | 89.1% | ||
| Number of outliers | 1 | 4 | ||
| Values in parentheses indicate the specific values in the highest resolution shell. | ||||
| aRmerge=∑∣Ij−〈I〉∣/∑Ij, where Ij is the intensity of an individual reflection and 〈I〉 is the average intensity of that reflection. | ||||
| bRwork=∑∣∣Fo∣−∣Fc∣∣/∑∣Fc∣, where Fo denotes the observed structure factor amplitude and Fc denotes the structure factor amplitude calculated from the model. | ||||
| cRfree is as for Rwork but calculated with randomly chosen reflections omitted from the refinement. In PARNn–RNA and PARNn, 5 and 3% of reflections were used, respectively. | ||||
Overall structure description
As shown in Figure 1B, PARNn forms a homodimer in the PARNn–RNA complex, with each subunit binding to three adenosine nucleotides. A subunit of PARNn has an α/β structure with a large central eight-stranded β-sheet flanked by 12 α-helices. The fifth β-strand (β5) and the corresponding strand from the other subunit form a small antiparallel β-sheet. This β-sheet combined with helices α3 and α4 from both subunits forms the dimer interface. The structures of two subunits are highly similar with pairwise Cα root-mean-square deviation (r.m.s.d.) of 0.35 Å. The trinucleotide is bound to a large cavity that is located on the opposite sides of the dimer (Figure 1B). The R3H domain, which is supposed to be located between helices α5 and α8, is disordered.
In the structure of PARNn in the absence of oligo(A) (Figure 1C and Supplementary Figure 2), the polypeptide chain of PARNn is folded into two domains, the nuclease domain (residues 1–133 and 269–430) and the R3H domain (residues 175–245), which is disordered in the PARNn–RNA complex. The overall shape of the structure is reminiscent of a sea horse (Figure 2C). The long helix α8, which is partly disordered in the PARNn–RNA complex, links the R3H domain and the main nuclease domain. No interdomain contacts exist between the nuclease domain and the R3H domain. There are two homodimers in the AU, with the nuclease domain mediating the dimerization as observed in the PARNn–RNA complex. No substantial differences are observed among the structures of four PARNn molecules in the AU (mean pairwise Cα r.m.s.d. of 0.5 Å). Therefore, only a homodimer consisting of molecules A and B is used for the subsequent analysis. Moreover, the overall structures of all the nuclease domains in both the native PARNn and the PARNn–RNA complex are similar to one another with mean pairwise Cα r.m.s.d. of ∼0.61 Å (Figure 1C), with the exception that PARNn in the native state has two additional antiparallel β-strands formed by residues 423–430.
Figure 2.
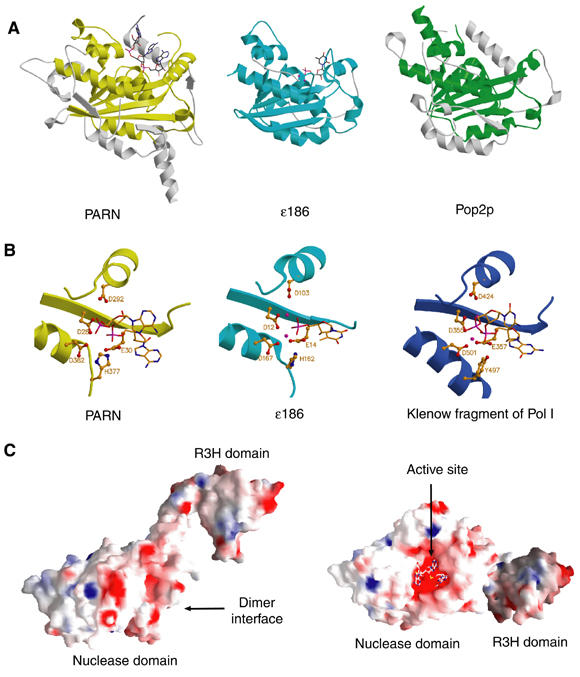
Comparison of PARNn with other members of the DEDD family. (A) Structural comparison of the nuclease domain of PARNn with those of ɛ186 and Pop2p. The DEDD core domains are colored yellow, cyan and green for PARN, ɛ186 and Pop2p, respectively, with the rest of the molecules colored in pale gray. Bound nucleotides are shown in stick model. (B) Structures of the active sites of PARNn, ɛ186 of Pol III and the klenow fragment of Pol I. Bound nucleotides are shown in stick model, catalytic residues in ball-and-stick model and metal ions in CPK model colored with magenta. (C) Solvent-accessible and electrostatic potential of PARNn colored from blue (basic) to red (acidic). For simplicity, only one subunit is shown. Left panel: the side view of the electrostatic potential surface. Right panel: the top view of the surface rotated about 90° around y axis relative to the view in the left panel.
Similarity to other DEDD exonucleases
Strong similarity between the nuclease domain of PARN and other 3′–5′ exonucleases was identified using the DALI server. These structural homologs of the nuclease domain of PARN include the nuclease domain of Pop2p (Z-score: 21.1; PDB entry: 1UOC) (Thore et al, 2003), ɛ186 (Z-score: 13.8; PDB entry: 1J53) (Hamdan et al, 2002), the nuclease domain of E. coli exonuclease I (ExoI) (Z-score: 11.2; PDB entry: 1FXX) (Breyer and Matthews, 2000) and ISG20 (Z-score: 9.9; PDB entry: 1WLJ) (Horio et al, 2004). All of these exonucleases except Pop2p belong to the DEDDh subfamily, suggesting that PARN is a member of the DEDDh family.
Superposition of the structure of the nuclease domain of PARNn either in ligand-free form or in poly(A)-bound form with that of ɛ186 showed that the core structures of these nuclease domains, which are composed of a five-stranded β-sheet surrounded by seven α-helices, are very similar with pairwise Cα r.m.s.d. of less than 1.5 Å (Figure 2A). Despite the fact that PARN and Pop2p only share 17% of sequence identity and Pop2p contains two noncanonical residues in its active site, the nuclease domain of PARNn superimposes strikingly well with that of Pop2p with an r.m.s.d. of 1.4 Å (Figure 2A).
In spite of these similarities, some notable structural differences exist between the nuclease domains of PARN, ɛ186 and Pop2p. For example, two loops linking α12 and α13, and α13 and α14, respectively, show large positional shifts. The C-terminus of PARNn is much longer than that of ɛ186, and displays a large structural deviation compared to that of Pop2p. Another distinguishing feature of PARN is that its nuclease domain contains the R3H subdomain, whereas ɛ186 or Pop2p does not contain.
Probably, the most striking difference is that the nuclease domain of PARN is a homodimer, whereas the isolated nuclease domain in both ɛ186 and Pop2p is a monomer.
Active site and catalytic mechanism
Based on site-directed mutagenesis and comparison with a number of 3′–5′ exonucleases of the DEDD superfamily (Figure 3A), four conserved residues, Asp28, Glu30, Asp292 and Asp382, that are essential for the catalytic activity of PARN and are required for the binding of divalent metal ions to PARN have been identified (Ren et al, 2002). Based on these observations, it was proposed that PARN utilizes the two-metal ion mechanism for its catalysis, as suggested for the exonuclease domain of Pol I (Beese and Steitz, 1991), although the detailed catalytic mechanism remains elusive.
Figure 3.
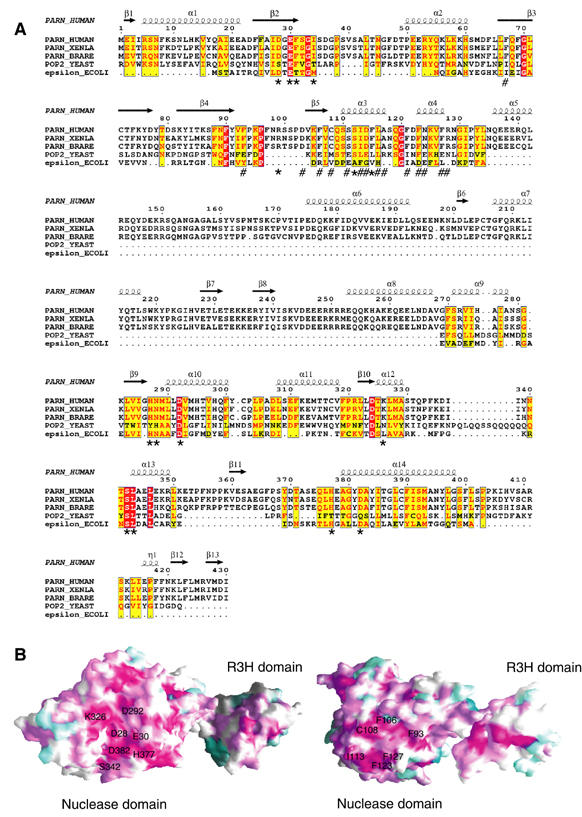
Sequence alignment and conservation mapping. (A) Structure-based sequence alignment of the nuclease domains of PARN proteins from human, Xenopus and Zebra fish with yeast Pop2p and E. coli ɛ186 of Pol III. The invariant residues are colored in red. Conserved residues are colored in yellow. The residues involved in interactions with RNA are marked with ‘*' and those involved in PARN dimerization are marked with ‘#'. (B) Molecular surface of PARNn showing regions of high to low sequence conservation shared by the PARN proteins, corresponding to a color ramp from violet and cyan, respectively. Left panel: a conserved RNA-binding cavity. Right panel: a conserved region involved in dimer formation of PARN. The conserved residues in two interfaces are labeled.
In both the ligand-free and poly(A)-bound forms of PARNn, Asp28, Glu30, Asp292 and Asp382 form a negatively charged cavity (Figure 2C). The trinucleotide is bound in this deep cavity in the PARNn–RNA complex (Figure 2C; see below). Upon poly(A) binding, no significant conformational changes occur for Asp28, Asp292 and Asp382, whereas a small positional shift is observed for Glu30. These observations are consistent with the finding that these four residues are not required for stabilization of the PARN–RNA substrate complex (Ren et al, 2002). No Mg2+ ion is found in the active site of the native PARNn even though 5 mM MgCl2 was included in the crystallization buffer and there is enough space to accommodate the metal ions in the active site. Structural comparison showed that residues Asp28, Glu30, Asp292 and Asp382 of the PARNn–RNA complex can be aligned spatially with the active site residues Asp12, Glu14, Asp103 and Asp167 in the ɛ186–TMP complex, with only small differences in the positions of side-chain groups (Figure 2B). The missing metal ions could possibly occupy positions similar to those of the two Mn2+ ions in the ɛ186–TMP complex. His162 of ɛ186, which was proposed to play a similar role to that of Tyr497 in the exonuclease domain of Pol I (Figure 2B) (Hamdan et al, 2002), corresponds spatially to His377 of PARN, confirming that PARN belongs to the DEDDh subfamily. Consistent with a role of His377 in catalysis, substitution of His377 by Ala, in the PARN(1–537) context, inhibits the PARN activity (Figure 4A), suggesting that His377 is essential for the catalytic activity of PARN.
Figure 4.
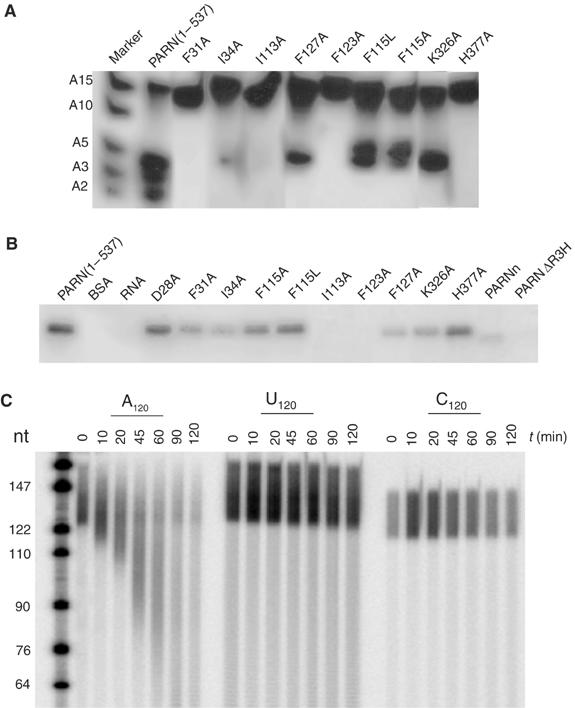
Effects of mutations on the poly(A)-binding and cleavage activities of PARN. (A) Deadenylation activity assay of the PARN mutants. The mixture of labeled A15, A10, A5, A3 and A2 is used as RNA ladder. PARN (1–537) is used as a control. A 0.1 pmol portion of mutants was incubated with labeled A15 for 10 min at room temperature and analyzed on denaturing polyacrylamide gel. (B) Photochemical crosslinking assay of the PARN mutants. BSA (lane BSA) and labeled 15-mer poly(A) (lane RNA) without protein were used as negative control. A 100 pmol portion of mutants was incubated with labeled A15 and exposed to UV for 30 min. (C) Poly(A) specificity of PARNn. Activity assays were carried out with 5′-end-labeled, size-fractionated RNAs (poly(A), poly(C) and poly(U), as indicated). RNAs were used at approximately 1–2 pmol (as mononucleotides) per time point, and PARNn was used at 0.45 pmol per time point. Reactions were stopped after the times indicated, and RNA was analyzed on a 10% polyacrylamide/urea gel.
Although no metal ion is present in the active sites of our PARN structures, mutagenesis and biochemical studies have suggested that residues Asp28, Glu30, Asp292 and Asp382 are involved in coordinating metal ions (Ren et al, 2002, 2004). The high similarity of the active-site structures between PARN and ɛ186 strongly suggests that the five conserved residues in these two enzymes play the same catalytic roles in the respective exonuclease reactions, and therefore the catalytic mechanisms for these two enzymes are probably identical.
PARN is a homodimer
Previously, based on gel filtration and protein/protein crosslinking, the N-terminal 54 kDa fragment of PARN with 3′ exonuclease activity has been proposed to be an oligomeric structure, most likely consisting of three subunits (Martinez et al, 2000). Against this finding, our crystal structures showed that in both ligand-free and poly(A)-bound forms, PARNn forms a homodimer in the crystal lattice. In support of our observations, analytical gel filtration chromatography using purified PARN(1–537) showed that PARN(1–537) is a homodimer in solution as well (Supplementary Figure 3). The association state of full-length PARN in solution was analyzed by analytical ultracentrifugation. A sedimentation velocity experiment showed that the protein was a homogeneous preparation with an apparent sedimentation coefficient of 4.77 S. During the lengthy sedimentation equilibrium run, however, about 20% of the protein appeared as an oligomeric species of higher molecular mass at the bottom of the cell, probably due to the high local protein concentration. The remaining protein in solution could be described by an apparent molecular mass of 143.8±4.2 kDa. This result was independent of whether the complete data set was analyzed with a fit including a separate mass term for the aggregated species or a processed data set in which the part of the data dominated by the aggregates were deleted (Figure 5A). As the molecular mass of His-tagged monomeric PARN is 74.45 kDa, the protein exists predominantly as a dimer in solution.
Figure 5.
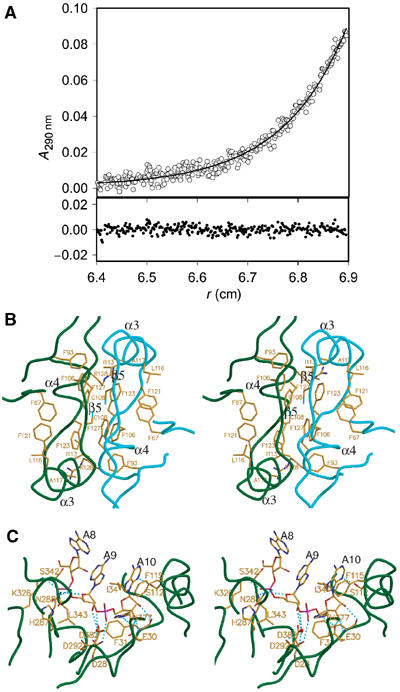
PARN dimer and PARN-poly(A) interfaces. (A) Association state of PARN. Native molecular mass determination of PARN was performed by sedimentation equilibrium measurements at 8000 r.p.m. and 10°C. The experimental data (o) could be fitted (−) to a protein of molecular mass 143.8±4.2 kDa. For clarity, the part of the data dominated by aggregates (radial position 6.9–7.1 cm) is not shown. The upper panel contains the experimental data and the fit and the lower panel shows the deviation of the fit from the data. (B) Stereo diagram of the dimer interface of PARNn. The two molecules of PARNn are colored with dark green and cyan, respectively. Residues involved in dimerization are shown in stick model. (C) Stereo diagram of the RNA-binding site of PARNn. PARN is colored with dark green. Hydrogen bonds are shown with cyan lines, nucleotides in stick model and residues involved in poly(A) recognition in stick model.
Consistent with the observations that PARN is in a dimeric state in solution, our structures show that the nuclease domain of PARN is involved in the dimer formation, with strand β5 and helices α3 and α4 contributing mainly to the dimer interface (Figure 5B). The buried accessible surface area of the dimer interface is 2261 Å2. Extensive hydrophobic interactions and hydrogen-bond networks are involved in dimerization. Briefly, helices α3 and α4 of one protomer pack against helices α4 and α3 from the other protomer, respectively. The strand β5 of one protomer forms an antiparallel β-sheet with its counterpart from the other protomer. The side chains of conserved residues Phe93, Cys108, Phe106, Ile113, Phe123 and Phe127 (Figures 3 and 5B and 5B) from one protomer make extensive hydrophobic interactions with the same set of residues from the other protomer. Strikingly, Phe123, an invariant residue in PARN across species (Figure 3A), is located in the center of the hydrophobic core formed by the side chains of residues Phe67, Ile113, Leu116, Ala117, Phe121 and Phe127 from the same molecule, and the side chain of Phe127 and the methylene group of Arg128 from the other molecule (Figure 5B). Additional contacts are contributed by hydrogen bonds, including the hydrogen-bond network formed between the two antiparallel β5 strands in the dimer interface.
The extensive dimer interface and involvement of some highly conserved residues in PARN suggest that the homodimer of PARN may function as a structural unit for its enzymatic activity. Consistent with this view, substitution of Ile113 or Phe123 by Ala in PARN(1–537) inactivated the enzyme, whereas mutation of Phe127 to Ala substantially reduced the enzyme activity (Figure 4A). Quantitative assays of the F123A mutation in the context of the full-length protein showed at least a 150-fold reduction in activity. In contrast, the F127A mutation had little effect on the activity of the full-length protein, suggesting that perhaps regions outside PARNn stabilize the dimeric structure (Supplementary Table I). Moreover, mutants I113A and F123A showed little, if any, affinity for the poly(A) substrate in the PARN(1–537) context, whereas F127A showed reduced binding to the poly(A) substrate (Figure 4B). Analytic gel filtration analysis of PARN(1–537) mutants indicated that F127A exists mostly in a monomeric state, whereas mutations of Ile113 and Phe123 to Ala completely convert PARN from a dimeric to a monomeric state (Supplementary Figure 3). One likely explanation for these observations is that mutations disrupting the dimer formation may also destabilize the architecture of the RNA-binding cavity, thus affecting the PARN activity, as the dimer interface and the active site in PARNn are located in a back-to-back manner, with helix α3 involved in both dimerization and formation of the RNA-binding cavity (Figure 1B; see below).
Binding of poly(A)
As the structures of two protomers are highly similar in the PARNn–RNA complex, for simplicity, only the interactions between molecule A and the bound trinucleotide are described here. The trinucleotide, which consists of the last three nucleotides A8, A9 and A10, is bound to the deep cavity formed by the central portion of strand β2, surrounded by helices α3, α10, α12, α13, the N-terminal segment of α14, the loop connecting α12 and α13, the extended segment linking strand β2 and the loop between β4 and β5 from the other molecule. The last two nucleotides A9 and A10 bind to the cavity in a similar fashion to the dinucleotide observed in the structure of the exonuclease domain of Pol I in complex with a normal single-stranded DNA substrate (Figure 2B) (Brautigam et al, 1999).
As shown in Figure 5C, recognition of the bound trinucleotide by the enzyme is mediated by a combination of hydrogen bonds, hydrophobic interactions and van der Waals (VDW) contacts. Specifically, the adenine base of A10 stacks against the aromatic ring of Phe115, whereas the N3 atom of the adenine base is hydrogen-bonded to the hydroxyl group of Ser112. The amino nitrogen and carbonyl oxygen of Phe31 are hydrogen-bonded to the ribose O3′ and O2′ of A10, respectively, whereas its side chain makes VDW contacts with the ribose moiety and the phosphate group of A10. The ribose O3′ of A10 is hydrogen-bonded to the OE1 group of Glu30, which in turn is hydrogen-bonded to the NE2 atom of His377. The side chain of His377 interacts with the phosphate group of A10 via VDW contacts, whereas the OD2 atom of Asp28 is hydrogen-bonded to the O2P atom in the phosphate group of A10. Compared to A10, there are much fewer contacts between the adenine base of A9 and the enzyme. The side chain of Ile34 stacks against the adenine base of A9. The NZ group of Lys326 and the main-chain NH group of Leu343 make hydrogen bonds with the O1P and O2P atoms of A9, respectively, whereas the ND2 atom of Asn288 is hydrogen-bonded to the ribose sugar O4′ atom. Moreover, the side chain of Asp292 makes two water-mediated hydrogen bonds with the ribose O2′ and O3′ atoms of A9. Additional contacts are made between the side chain of His287 and the phosphate group of A9 by VDW interactions. The third adenosine nucleotide A8 counted from the 3′ end is exposed to the solvent, with its ribose ring making VDW contacts with the side chains of Ser342 and Lys326.
To probe the role of the residues involved in multiple contacts with poly(A) biochemically, we constructed several mutations in the RNA-binding pocket of PARN(1–537). Mutations of Phe31 or Ile34 to Ala caused severe defects in both the poly(A) cleavage and binding activities, underscoring their importance in poly(A) recognition, whereas substitution of Lys326 by Ala substantially reduced the poly(A) cleavage and binding activities (Figure 4A and B). Phe115 stacks against the adenine base of the 3′-end nucleotide (A10). Mutation of this residue to either Ala or Leu reduced both the nuclease and poly(A)-binding activities of PARN, with F115A mutant exhibiting more severe defects in RNA binding (Figure 4A and B). The point mutations were also introduced into the full-length protein and assayed in a quantitative manner by the release of acid-soluble mononuocleotides. The mutations F31A, I34A, F115A and K326A all behaved similarly with KM values increased two to eightfold and kcat reduced 10- to 50-fold (Supplementary Table I). These data support a role of F31, I34, F115 and K326 in binding the poly(A) substrate.
Previously, it has been demonstrated that PARN prefers to cleave a poly(A) substrate with a free 3′-OH group (Korner and Wahle, 1997; Martinez et al, 2000). Consistent with this, our structure shows that Glu30 specifically interacts with the 3′ hydroxyl group of the ribose of A10 (Figure 5C). In analogy with the role of Glu14 in ɛ186 (Hamdan et al, 2002), Glu30 may be involved in activating the attacking water molecule during the catalytic reaction of PARN. Therefore, Glu30 may fulfill two roles, one as the catalytic residue and the other conferring the specificity for recognition of the poly(A) tail. One of the unresolved issues about the functions of PARN is how PARN distinguishes poly(A) from poly(U), poly(C) and poly(G). Biochemical studies have demonstrated that PARN degrades poly(A) efficiently and poly(U) moderately under certain conditions, but not poly(C) and poly(G) (Korner and Wahle, 1997; Martinez et al, 2000). Surprisingly, there are no specific hydrogen-bonding interactions between the adenine bases and the protein, and the backbone phosphates and ribose moieties actually make more contacts with the protein than the adenine bases (Figure 5C). Therefore, how PARN achieves the high specificity for poly(A) recognition remains elusive.
R3H domain
The R3H domain is a conserved sequence motif, identified in over 100 proteins from a diverse range of organisms. The most prominent feature of the R3H domain is the presence of an invariant arginine residue and a highly conserved histidine residue that are separated by three residues (Grishin, 1998). The three-dimensional structures of the R3H domain from both human Sμbp-2 (Liepinsh et al, 2003) and mouse PARN (PDB entry: 1UG8) determined by NMR spectroscopy showed that the R3H domain consisting of a three-stranded antiparallel β-sheet and two α-helices is a structural homolog to the C-terminal domain of the translational initiation factor IF3 (Grishin, 1998; Liepinsh et al, 2003).
In the structure of native PARNn, the R3H domain from one molecule is located at the top of the poly(A)-binding site from the other molecule (Figure 1C and Supplementary Figure 2), serving as a ‘cap' to enclose the poly(A)-binding cavity. Such a unique structural arrangement of the R3H domain in the native PARNn is not caused by the crystal packing, as the R3H domain of PARNn co-crystallized with 10 adenosine nucleotides in a different space group (P21) is arranged in the same way albeit with less well-defined electron density (M Wu and H Song, unpublished results).
Superposition of the R3H domain of PARNn with those of Sμbp-2 (PDB code: 1MSZ) and mouse PARN gives an r.m.s.d. of 2.3 and 1.1 Å for 26 and 53 equivalent Cα atoms, respectively, indicating that the R3H domain of PARNn is structurally similar to that of Sμbp-2 and highly resembles that of mouse PARN (Figure 6A). Despite these similarities, some notable differences exist between these three R3H domains. For example, the R3H domains in both PARNn and mouse PARN lack the canonical arginine and histidine residues, which correspond to Arg755 and His759 in Sμbp-2, respectively (Liepinsh et al, 2003). The structural differences between the R3H domains in PARNn and mouse PARN are mainly confined to the N- and C-termini of the domain and the loop connecting strands β7 and β8.
Figure 6.
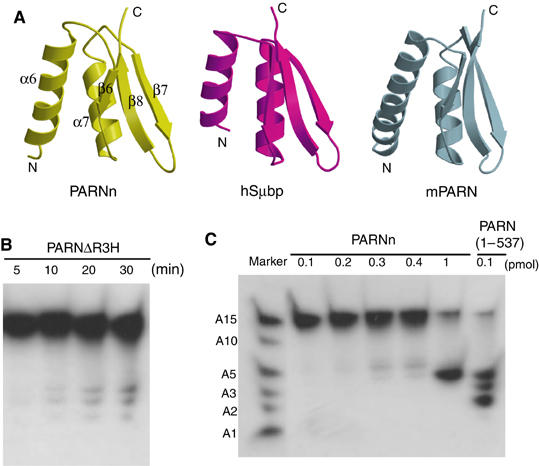
Structure of the R3H domain and its role in enzymatic activity of PARN. (A) Structural comparison of the R3H domain of PARNn with that of human Sμbp-2 and mouse PARNn. (B) Deadenylation activity assays of PARNΔR3H. A 100 pmol portion of PARNΔR3H was used for the deadenylation assay. Aliquots were taken at the times indicated. Products were analyzed on denaturing polyacrylamide gel and exposed to hyperfilm for 4 h. (C) Deadenylation activity assays of PARNn and PARN(1–537). The mixture of labeled A15, A10, A5, A3, A2 and A1 is used as RNA ladder. PARNn (0.1, 0.2, 0.3, 0.4 and 1 pmol) and PARN(1–537) (0.1 pmol) were incubated with labeled A15 for 10 min. The resulting products were analyzed on denaturing polyacrylamide gel and exposed to hyperfilm for 2 h.
The R3H domain is believed to be involved in nucleic acid binding (Grishin, 1998); however, electrostatic potential mapping on the surface of PARNn failed to reveal a positively charged patch that is large enough for RNA binding (Figure 2C). Given the close proximity of the R3H domain with the poly(A)-binding pocket (Figure 1C and Supplementary Figure 2), the R3H may be involved in the catalytic reaction of PARN by increasing the affinity and/or specificity of PARN for binding to poly(A) or by coordinating with the cap-binding domain to amplify the processivity of PARN. The possibility of the R3H domain contributing to poly(A) specificity is suggested by the observation that the oligonucleotide in the active site exposes its bases to the outside, facing the R3H domain (Figures 1C and 5C).
To examine the functional role of R3H, we created the deletion mutant PARNΔR3H, in which residues 134–268 containing the R3H domain and the neighboring α-helices (α5 and α8) was deleted from PARN(1–537), and tested its ability to bind and cleave poly(A) RNA substrate. As shown in Figure 6B, removal of the R3H domain dramatically reduced the cleavage activity of PARN, as 1000-fold more mutant than wild-type protein was required to observe the deadenylation activity (compared to Figure 6C, last lane). Interestingly, deletion of the R3H domain prevents it from binding to the poly(A) substrate as well (Figure 4B). A similar deletion mutant of the full-length protein in which residues 134–269 were removed caused poor solubility and a complete loss of activity. However, a smaller deletion removing just the R3H domain (residues 175–245) led to an increase in the KM value of about 15-fold, with no reduction of kcat, strongly suggesting that the R3H domain contributes to poly(A) binding (Supplementary Table I). However, preliminary assays did not reveal any loss of substrate specificity. Thus, the R3H domain does not appear to make a major contribution to the preference of PARN for poly(A). This raises an intriguing possibility that the cap-binding domain may contribute to the poly(A) specificity of PARN. To reveal the role of the cap-binding domain in conferring the poly(A) specificity, the activity of PARNn, which lacks the putative cap-binding domain to digest the poly(A), poly(U) and poly(C), was examined. However, the results showed that PARNn is specific for poly(A) (Figure 4C).
Insight into processivity
Processivity is important for many enzymes such as DNA polymerases and nucleases (Breyer and Matthews, 2001). For λ-exonuclease (Kovall and Matthews, 1997), processivity is achieved through a multisubunit toroidal structure that completely encloses the substrate. DNA polymerases achieve their processivity through the action of the external processivity factors, the sliding clamps and the globular accessory factor (Breyer and Matthews, 2001). The sliding clamps are protein rings that encircle the DNA, thereby keeping the DNA polymerase attached to the DNA template (Baker and Bell, 1998). For T7 DNA polymerase, which is not very processive in its native state, processivity is achieved by formation of a stable complex in which the accessory factor thioredoxin associates with an extension at the tip of the polymerase thumb domain. This association with thioredoxin is thought to stabilize the extension in the polymerase, which in turn is predicted to contact the duplex DNA, thereby increasing the polymerase processivity (Doublie et al, 1998).
Biochemical studies have shown that the processivity of PARN with uncapped RNA substrates is low (Korner and Wahle, 1997; Martinez et al, 2001). In contrast, capped RNA substrates are degraded in a processive manner (Dehlin et al, 2000; Martinez et al, 2000, 2001). When provided in trans, the cap structure stimulates deadenylation at low concentrations and inhibits deadenylation at high concentrations (Martinez et al, 2001). However, the molecular basis for these observations remains unclear. Our structures show that PARN forms a homodimer, with the R3H domain from one protomer forming a lid over the RNA-binding cavity of the other protomer, thereby partially enclosing the active site (Figure 1C and Supplementary Figure 2). As an auxiliary processivity factor is absent for PARN, and a small degree of processivity has been detected even in the absence of a cap structure (Martinez et al, 2001), the R3H domain may contribute to the enzyme's processivity by playing a role similar to that of thioredoxin for T7 DNA polymerase. Consistent with this hypothesis, deletion of the R3H domain substantially reduced the RNA affinity of PARN (Figure 4B and Supplement Table I).
Although PARNn lacks the cap-binding domain, the structures suggest that the cap-binding domain is most likely distinct from the active site (Figure 1C), consistent with the observation that cap analog is a noncompetitive inhibitor for PARN (Martinez et al, 2001). The importance of the cap-binding domain is underscored by the observations that PARN lacking the cap-binding domain (PARNn) showed reduced activity, whereas PARN(1–537) containing the cap-binding domain displayed robust activity (Figure 6C and Supplement Table I). Moreover, the cap-binding domain may act in concert with the R3H domain to increase the processivity of PARN, although the underlying mechanism remains elusive.
Conclusion
The structures of PARNn in two distinct functional states presented here reveal for the first time how the nuclease binds the poly(A) tail. Structures combined with mutational and biochemical data strongly suggest that the dimeric form of PARN is a structural and functional unit for poly(A) tail binding and cleavage. The R3H domain and, possibly, the cap-binding domain are involved in poly(A) binding, but these domains alone do not appear to contribute to poly(A) specificity. Although we failed to observe the presence of metal ions in the active site in the ligand-free form of PARNn, given the high similarity of the active site geometry in PARNn to that of ɛ186, these two enzymes may share a common two-metal ion-dependent catalytic mechanism. Our work also provides the starting point for further crystallographic, biochemical and genetic studies of PARN and its role in mRNA deadenylation and translation initiation.
Materials and methods
Protein expression and purification
The C-terminal truncated human PARN (residues 1–430; PARNn) was cloned into the pGEX-6p-1 (Amersham) vector and expressed as a glutathione-S-transferase fusion protein in E. coli. PARNn was purified using glutathione-Sepharose 4B, MonoQ and Superdex 200 gel filtration columns (Amersham). The protein was concentrated to 10 mg/ml for crystallization. The SeMet-substituted PARNn was expressed in a minimal medium containing 20 mg/l SeMet (Neidhardt et al, 1974), and purified in the same way as that used for the native protein except that the dithiothreitol (DTT) concentration was 10 mM.
The cDNA encoding the truncated PARN including the putative cap-binding domain (residue 1–537) was cloned into pET28a (Novagen). All mutant proteins were created using the QuickChange mutagenesis kit (Stratagene) and purified using TALON affinity resin (BD Biosciences).
The plasmid pGMMCS 645295 (Korner et al, 1998) was used for the production of full-length PARN and also to generate point mutations in the full-length sequence by the same procedure as above. These proteins were expressed in E. coli and purified on a Ni2+-NTA column (Qiagen), followed by chromatography on a MonoQ FPLC column (Amersham), essentially as described before (Korner et al, 1998).
Crystallization, data collection and structure determination
Crystals of native PARNn were initially obtained at 20°C using the hanging-drop method, and further improved by macroseeding from a condition consisting of 13–14% PEG3350, 200 mM ammonium tartrate and 5 mM MgCl2. Crystals of the PARNn–RNA complex were grown from a condition containing 15–16% PEG3350, 200 mM ammonium tartrate, 5 mM EDTA, 5 mg/ml PARNn and 500 μM 10-mer oligo(A) purchased from Dharmacon Research Inc. All the crystals were cryoprotected by inclusion of 20% PEG400 in the precipitant solution and flash-frozen in liquid nitrogen.
Diffraction data were collected on ID-29 at ESRF (Grenoble, France) and processed with MOSFLM and CCP4 (CCP4, 1994). Crystals of native PARNn belong to space group C2 with four molecules in the AU, whereas those of the PARNn–RNA complex belong to space group P212121 with two molecules in the AU. The structure of the PARNn–RNA complex was solved with MAD method. The selenium sites were located with SnB (Miller et al, 1994) and refined with SHARP (De la Fortelle and Bricogne, 1997). About 80% of the final model was automatically built with ARP/wARP (Perrakis et al, 1999). The rest of the model was built manually with O (Jones et al, 1991). Crystallographic refinement was carried out with CNS (Brunger et al, 1998) and REFMAC5 (Murshudov et al, 1997). The structure of native PARNn was solved using AMoRe (Navaza and Saludjian, 1997), with the nuclease domain of the PARNn–RNA complex as a search model. The model was rebuilt manually using O and refined by CNS and REFMAC5. All data statistics of these two structures are shown in Table I.
Deadenylation assays
The 2-, 3-, 5-, 10- and 15-mer oligo(A) (A2, A3, A5, A10 and A15) were purchased from Dharmacon Research Inc. The short substrates were deprotected according to the instructions from the manufacturer. Approximately 100 pmol of RNAs described above were labeled with [γ-32P]ATP (3000 Ci/mmol; Amersham Biosciences) by T4 polynucleotide kinase (Invitrogen) in 50 μl reactions at 37°C for 1 h. A microspin G-20 column (Amersham) was used to separate the labeled RNAs from extra [γ-32P]ATP. The labeled A15 was used for deadenylation assay. Deadenylation reactions were started by the addition of the purified proteins to the reaction buffer (30 mM Tris, pH 8.0, 200 mM NaCl, 2 mM DTT, 2 mM MgCl2) and incubated at room temperature for 10 min. After the reaction was stopped with EDTA, the products were separated with 25% polyacrylamide (19:1 acrylamide/bisacrylamide)/7 M urea gels and exposed to Hyperfilm (Amersham). The details of deadenylation assays for full-length PARN and its mutants are described in Supplementary data.
Photochemical crosslinking assay
The labeled 15-mer oligo(A) was used to test the poly(A)-binding activity of PARN mutants. The PARN mutants were added to the reaction buffer (30 mM Tris, pH 8.0, 200 mM NaCl, 2 mM DTT) containing 10 mM EDTA mixed with labeled oligo(A). The crosslinking reaction was carried out for 0.5 h at room temperature using UV Stratalinker 2400 (Stratagene). The reaction products were analyzed with 10% SDS–polyacrylamide (29:1 acrylamide/bisacrylamide) gels. The gels were visualized using Hyperfilm (Amersham) exposed overnight.
Analytical ultracentrifugation
Sedimentation velocity and sedimentation equilibrium were measured in an analytical ultracentrifuge Optima XL-A (Beckman Instruments, Palo Alto, CA). An An50 Ti rotor and double sector cells were used at 30 000 and 8000 r.p.m. for velocity and equilibrium analysis, respectively. The protein at a concentration of 0.35 mg/ml was dissolved in 50 mM Tris, pH 7.9, 100 mM KCl, 1 mM DTT, 0.002% NP-40 and 10% (w/w) glycerol. Centrifugation was performed at 10°C. To avoid changes in absorption during data collection due to oxidation of DTT, the protein was monitored at 290 nm. The data were analyzed using the software provided by Beckman Instruments or the program Sedeq developed by Allan Minton (http://www.rasmb.bbri.org/rasmb/windows/sedeq-minton/). All equilibrium measurements were corrected for water density at 20°C.
Supplementary Material
Supplementary Table 1
Supplementary materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Acknowledgments
We thank the beamline scientists at ID29 (ESRF, France) for assistance and access to synchrotron radiation facilities. We are grateful to Eva Dehlin for supplying the PARN preparation used in the ultracentrifugation experiment and to Uwe Kühn for reagents. This work is financially supported by the Agency for Science, Technology and Research (A*Star) in Singapore (HS), and by the Deutsche Forschungsgemeinschaft and Fonds der Chemischen Industrie (EW).
Coordinates: The coordinates and structure-factor amplitudes for PARNn and the PARNn–RNA complex have been deposited in the Protein Data Bank with accession codes 2A1S and 2A1R, respectively.
References
- Astrom J, Astrom A, Virtanen A (1992) Properties of a HeLa cell 3′ exonuclease specific for degrading poly(A) tails of mammalian mRNA. J Biol Chem 267: 18154–18159 [PubMed] [Google Scholar]
- Baker TA, Bell SP (1998) Polymerases and the replisome: machines within machines. Cell 92: 295–305 [DOI] [PubMed] [Google Scholar]
- Beese LS, Steitz TA (1991) Structural basis for the 3′–5′ exonuclease activity of Escherichia coli DNA polymerase I: a two metal ion mechanism. EMBO J 10: 25–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brautigam CA, Sun S, Piccirilli JA, Steitz TA (1999) Structures of normal single-stranded DNA and deoxyribo-3′-S-phosphorothiolates bound to the 3′–5′ exonucleolytic active site of DNA polymerase I from Escherichia coli. Biochemistry 38: 696–704 [DOI] [PubMed] [Google Scholar]
- Breyer WA, Matthews BW (2000) Structure of Escherichia coli exonuclease I suggests how processivity is achieved. Nat Struct Biol 7: 1125–1128 [DOI] [PubMed] [Google Scholar]
- Breyer WA, Matthews BW (2001) A structural basis for processivity. Protein Sci 10: 1699–1711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D 54: 905–921 [DOI] [PubMed] [Google Scholar]
- CCP4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr D 50: 760–763 [DOI] [PubMed] [Google Scholar]
- Chang TC, Yamashita A, Chen CY, Yamashita Y, Zhu W, Durdan S, Kahvejian A, Sonenberg N, Shyu AB (2004) UNR, a new partner of poly(A)-binding protein, plays a key role in translationally coupled mRNA turnover mediated by the c-fos major coding-region determinant. Genes Dev 18: 2010–2023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba Y, Johnson MA, Lidder P, Vogel JT, van Erp H, Green PJ (2004) AtPARN is an essential poly(A) ribonuclease in Arabidopsis. Gene 328: 95–102 [DOI] [PubMed] [Google Scholar]
- Copeland PR, Wormington M (2001) The mechanism and regulation of deadenylation: identification and characterization of Xenopus PARN. RNA 7: 875–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De la Fortelle E, Bricogne G (1997) Maximum-likelihood heavy-atom parameter refinement for multiple isomorphous replacement and multiwavelength anomalous diffraction method. Methods Enzymol 276: 472–494 [DOI] [PubMed] [Google Scholar]
- Dehlin E, Wormington M, Korner CG, Wahle E (2000) Cap-dependent deadenylation of mRNA. EMBO J 19: 1079–1086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dlakic M (2000) Functionally unrelated signalling proteins contain a fold similar to Mg2+-dependent endonucleases. Trends Biochem Sci 25: 272–273 [DOI] [PubMed] [Google Scholar]
- Doublie S, Tabor S, Long AM, Richardson CC, Ellenberger T (1998) Crystal structure of a bacteriophage T7 DNA replication complex at 2.2 Å resolution. Nature 391: 251–258 [DOI] [PubMed] [Google Scholar]
- Dykxhoorn DM, Novina CD, Sharp PA (2003) Killing the messenger: short RNAs that silence gene expression. Nat Rev Mol Cell Biol 4: 457–467 [DOI] [PubMed] [Google Scholar]
- Gao M, Fritz DT, Ford LP, Wilusz J (2000) Interaction between a poly(A)-specific ribonuclease and the 5′ cap influences mRNA deadenylation rates in vitro. Mol Cell 5: 479–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grishin NV (1998) The R3H motif: a domain that binds single-stranded nucleic acids. Trends Biochem Sci 23: 329–330 [DOI] [PubMed] [Google Scholar]
- Hamdan S, Carr PD, Brown SE, Ollis DL, Dixon NE (2002) Structural basis for proofreading during replication of the Escherichia coli chromosome. Structure (Camb) 10: 535–546 [DOI] [PubMed] [Google Scholar]
- Horio T, Murai M, Inoue T, Hamasaki T, Tanaka T, Ohgi T (2004) Crystal structure of human ISG20, an interferon-induced antiviral ribonuclease. FEBS Lett 577: 111–116 [DOI] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard M (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A 47 (Part 2): 110–119 [DOI] [PubMed] [Google Scholar]
- Korner CG, Wahle E (1997) Poly(A) tail shortening by a mammalian poly(A)-specific 3′-exoribonuclease. J Biol Chem 272: 10448–10456 [DOI] [PubMed] [Google Scholar]
- Korner CG, Wormington M, Muckenthaler M, Schneider S, Dehlin E, Wahle E (1998) The deadenylating nuclease (DAN) is involved in poly(A) tail removal during the meiotic maturation of Xenopus oocytes. EMBO J 17: 5427–5437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovall R, Matthews BW (1997) Toroidal structure of lambda-exonuclease. Science 277: 1824–1827 [DOI] [PubMed] [Google Scholar]
- Lai WS, Kennington EA, Blackshear PJ (2003) Tristetraprolin and its family members can promote the cell-free deadenylation of AU-rich element-containing mRNAs by poly(A) ribonuclease. Mol Cell Biol 23: 3798–3812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lejeune F, Li X, Maquat LE (2003) Nonsense-mediated mRNA decay in mammalian cells involves decapping, deadenylating, and exonucleolytic activities. Mol Cell 12: 675–687 [DOI] [PubMed] [Google Scholar]
- Liepinsh E, Leonchiks A, Sharipo A, Guignard L, Otting G (2003) Solution structure of the R3H domain from human Smubp-2. J Mol Biol 326: 217–223 [DOI] [PubMed] [Google Scholar]
- Maquat LE, Carmichael GG (2001) Quality control of mRNA function. Cell 104: 173–176 [DOI] [PubMed] [Google Scholar]
- Martinez J, Ren YG, Nilsson P, Ehrenberg M, Virtanen A (2001) The mRNA cap structure stimulates rate of poly(A) removal and amplifies processivity of degradation. J Biol Chem 276: 27923–27929 [DOI] [PubMed] [Google Scholar]
- Martinez J, Ren YG, Thuresson AC, Hellman U, Astrom J, Virtanen A (2000) A 54-kDa fragment of the Poly(A)-specific ribonuclease is an oligomeric, processive, and cap-interacting Poly(A)-specific 3′ exonuclease. J Biol Chem 275: 24222–24230 [DOI] [PubMed] [Google Scholar]
- Meyer S, Temme C, Wahle E (2004) Messenger RNA turnover in eukaryotes: pathways and enzymes. Crit Rev Biochem Mol Biol 39: 197–216 [DOI] [PubMed] [Google Scholar]
- Miller R, Gallo SM, Khalak HG, Weeks CM (1994) SnB: crystal structure determination via shake-and-bake. J Appl Crystallogr 27: 613–621 [Google Scholar]
- Moser MJ, Holley WR, Chatterjee A, Mian IS (1997) The proofreading domain of Escherichia coli DNA polymerase I and other DNA and/or RNA exonuclease domains. Nucleic Acids Res 25: 5110–5118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D 53: 240–255 [DOI] [PubMed] [Google Scholar]
- Navaza J, Saludjian P (1997) Amore: an automated molecular replacement program package. Methods Enzymol 276: 581–594 [DOI] [PubMed] [Google Scholar]
- Neidhardt F, Bloch PL, Smith DF (1974) Culture medium for enterobacteria. J Bacteriol 119: 736–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker R, Song H (2004) The enzymes and control of eukaryotic mRNA turnover. Nat Struct Mol Biol 11: 121–127 [DOI] [PubMed] [Google Scholar]
- Perrakis A, Morris R, Lamzin VS (1999) Automated protein model building combined with iterative structure refinement. Nat Struct Biol 6: 458–463 [DOI] [PubMed] [Google Scholar]
- Ren YG, Kirsebom LA, Virtanen A (2004) Coordination of divalent metal ions in the active site of poly(A)-specific ribonuclease. J Biol Chem 279: 48702–48706 [DOI] [PubMed] [Google Scholar]
- Ren YG, Martinez J, Virtanen A (2002) Identification of the active site of poly(A)-specific ribonuclease by site-directed mutagenesis and Fe(2+)-mediated cleavage. J Biol Chem 277: 5982–5987 [DOI] [PubMed] [Google Scholar]
- Reverdatto SV, Dutko JA, Chekanova JA, Hamilton DA, Belostotsky DA (2004) mRNA deadenylation by PARN is essential for embryogenesis in higher plants. RNA 10: 1200–1214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steitz TA, Smerdon SJ, Jager J, Joyce CM (1994) A unified polymerase mechanism for nonhomologous DNA and RNA polymerases. Science 266: 2022–2025 [DOI] [PubMed] [Google Scholar]
- Temme C, Zaessinger S, Meyer S, Simonelig M, Wahle E (2004) A complex containing the CCR4 and CAF1 proteins is involved in mRNA deadenylation in Drosophila. EMBO J 23: 2862–2871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thore S, Mauxion F, Seraphin B, Suck D (2003) X-ray structure and activity of the yeast Pop2 protein: a nuclease subunit of the mRNA deadenylase complex. EMBO Rep 4: 1150–1155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker M, Valencia-Sanchez MA, Staples RR, Chen J, Denis CL, Parker R (2001) The transcription factor associated Ccr4 and Caf1 proteins are components of the major cytoplasmic mRNA deadenylase in Saccharomyces cerevisiae. Cell 104: 377–386 [DOI] [PubMed] [Google Scholar]
- van Hoof A, Parker R (1999) The exosome: a proteasome for RNA? Cell 99: 347–350 [DOI] [PubMed] [Google Scholar]
- Zuo Y, Deutscher MP (2001) Exoribonuclease superfamilies: structural analysis and phylogenetic distribution. Nucleic Acids Res 29: 1017–1026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo Y, Wang Y, Malhotra A (2005) Crystal structure of Escherichia coli RNase D, an exoribonuclease involved in structured RNA processing. Structure (Camb) 13: 973–984 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1
Supplementary materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3