

Abstract
Amyloid diseases like Alzheimer's disease and familial amyloidosis of Finnish type (FAF) stem from endoproteolytic cleavage of a precursor protein to generate amyloidogenic peptides that accumulate as amyloid deposits in a tissue-specific manner. FAF patients deposit both 8 and 5 kDa peptides derived from mutant (D187Y/N) plasma gelsolin in the extracellular matrix (ECM). The first of two aberrant sequential proteolytic events is executed by furin to yield a 68 kDa (C68) secreted fragment. We now identify the metalloprotease MT1-matrix metalloprotease (MMP), an integral membrane protein active in the ECM, as a protease that processes C68 to the amyloidogenic peptides. We further demonstrate that ECM components are capable of accelerating gelsolin amyloidogenesis. Proteolysis by MT1-MMP-like proteases proximal to the unique chemical environment of the ECM offers an explanation for the tissue-specific deposition observed in FAF and provides critical insight into new therapeutic strategies.
Keywords: amyloid, gelsolin, glycosaminoglycan, matrix metalloprotease, MT1-MMP
Introduction
Proteins involved in human amyloidogenesis have distinct native structures and functions, yet they are believed to form a common cross β-sheet conformation in the misfolded/misassembled state that leads to the characteristic fibrillar structures of amyloid deposits. The process of amyoidogenesis is genetically and biochemically linked to pathology, irrespective of the type of amyloidogenic protein. Amyloidoses emerge from three classes of proteins, including native folds that undergo amyloidogenic conformational changes, proteins that are normally endoproteolytically cleaved to an amyloidogenic fragment, and proteins where a mutation alters the fold, leading to aberrant endoproteolytic release of an amyloidogenic fragment(s). Plasma gelsolin, a protein of the latter class, when aggregated leads to familial amyloidosis of Finnish type (FAF), which is one of more than 30 different human diseases (Carrell and Gooptu, 1998; Dobson, 2003) in which amyloid is deposited in the extracellular matrix (ECM) of tissues. While Alzheimer's disease (AD) is the result of amyloidogenesis in the brain, FAF is characterized by extensive skin, arterial, neurologic, and ophthalmologic amyloid deposition (Kiuru, 1998). Understanding the basis for tissue-specific amyloid deposition and pathology remains a formidable challenge. Tissue-specific expression differences, influencing the concentration and thus the rate of aggregation, are likely to affect the tissue selectivity of amyloidoses (Sekijima et al, 2005). However, there are clearly additional mechanistic factors that contribute to the highly selective deposition of 8 and 5 kDa gelsolin fragments in the ECM.
Unlike many amyloid diseases, there is no sporadic disease associated with wild-type gelsolin deposition. In contrast, inheritance of either the D187N or D187Y mutations leads to FAF with unknown penetrance. Two forms of gelsolin are generated by alternative splicing (Kwiatkowski et al, 1986; Yin, 1987; Burtnick et al, 1997; Sun et al, 1999), intracellular 81 kDa and secreted 83 kDa versions, and, while both splice variants contain the mutation, only the secreted form (plasma gelsolin) is associated with amyloidosis (Kangas et al, 1996). Plasma gelsolin variants (D187N/Y) are aberrantly processed by at least two successive proteolytic events to generate the amyloidogenic peptides. The protease (α-gelsolinase) responsible for the initial intracellular cleavage of plasma gelsolin to generate the 68 kDa fragment was identified as furin (Chen et al, 2001), a member of the proprotein convertase family that cycles between the endocytic and exocytic pathways (Molloy et al, 1999). D187N/Y mutations abolish Ca2+ binding in domain 2, destabilizing and rendering the domain accessible to aberrant proteolysis by furin as it transits through the Golgi. However, the identity of β-gelsolinase(s), whose activity results in formation of the 8 and 5 kDa fragments found in amyloid deposits (Ghiso et al, 1990; Haltia et al, 1990; Levy et al, 1990; Maury and Baumann, 1990; Maury et al, 1990; Paunio et al, 1994; Kangas et al, 1999), is unknown, as is the location of proteolysis. Moreover, the physiological basis for the ECM-localized deposition of FAF-associated amyloid fibrils remains unclear.
Herein we show that members of the matrix metalloprotease (MMP) family with MT1-MMP-like activity have a β gelsolinase activity that generates the 8 and 5 kDa amyloidogenic peptides from the 68 kDa furin-derived gelsolin fragment. While several MMPs generate the 8 kDa amyloidogenic fragment in vitro, a fibroblast cell line from a mouse model lacking MT1-MMP and HT1080 cells transfected with siRNA directed to MT1-MMP demonstrates that MT1-MMP activity can be solely responsible for the 8 kDa fragment produced in a cellular context. Given that MT1-MMP specifically and MMPs in general are located proximal to the ECM, the site of amyloid deposition in FAF, we investigated whether the chemical composition of the ECM hastened gelsolin amyloidogenesis. We demonstrate that GAGs, a major component of the ECM, accelerate gelsolin amyloidogenesis in vitro. We suggest that the unique enzymological and chemical environment of the ECM region may contribute to selective gelsolin amyloid deposition, emphasizing the role of the ECM in FAF, and likely other amyloidoses.
Results
HT1080 cells have β-gelsolinase activity
The fragments found in FAF amyloid deposits result from sequential endoproteolysis of D187N/Y plasma gelsolin (Figure 1A). Initial processing of full-length (83 kDa) variant plasma gelsolin occurs during secretion, at R172–A173, by the intracellular protease furin in the Golgi (Chen et al, 2001). This generates a secreted 68 kDa carboxy (C)-terminal fragment (C68) that contains the amyloidogenic region at its N-terminus. C68 is the substrate for a second protease, β-gelsolinase, whose activity yields the major 8 kDa (70–71 residue) and minor 5 kDa (approximately 53 residue) amyloidogenic fragments. An extensive search for the β-gelsolinase(s) within the exocytic or endocytic pathways, or in blood plasma, did not yield candidate proteases (unpublished results).
Figure 1.
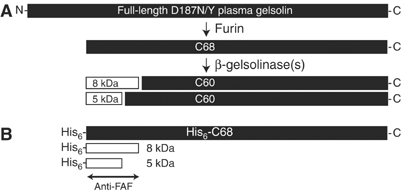
Processing of plasma gelsolin to the 5 and 8 kDa amyloidogenic peptides. (A) Schematic representation of the full-length FAF variant plasma gelsolin (83 kDa) and the cleavage products detected in FAF patients. The first cleavage event, carried out by furin in the TGN, generates a 68 kDa product (C68). C68 is further processed by one or more β-gelsolinases to form the 8 and 5 kDa fragments found in FAF amyloid deposits, as well as a 60 kDa C-terminal fragment (C60). (B) Schematic representation of the recombinant 68 kDa protein generated with a His6 tag at the N-terminus (His6-C68) that is used as a substrate to detect β-gelsolinase activity. In addition, the 8 and 5 kDa products of His6-C68 cleavage by β-gelsolinase are shown. Detection on immunoblots was carried out using either an antibody raised against the major 8 kDa product (anti-FAF; indicated) and/or a probe to the His6 tag.
Although the sequences of the β-gelsolinase cleavage sites do not provide substantial insight into the identity of this protease(s), a hint regarding its identity came from the observation that FAF amyloid deposits are localized in the ECM of a variety of tissues (Kiuru, 1998). A number of proteases function in the formation and remodeling of the ECM. These proteases were investigated as potential β-gelsolinases. A recombinant 68 kDa plasma gelsolin construct corresponding to the furin cleavage product was expressed with a His6 tag at the N-terminus (His6-C68) to facilitate the identification of β-gelsolinase (Figure 1B). The presence of the His6 tag could be detected using a His-specific antibody. In addition, an antibody that recognizes an epitope in both the 8 and 5 kDa fragments was generated (anti-FAF; Figure 1B).
To provide initial insight into the identity of β-gelsolinase, HT1080 cells, a human fibrosarcoma-derived cell line, were utilized as a known source of numerous ECM proteases. Incubation of His6-C68 with medium recovered from HT1080 cells yielded no cleavage into the fragments of interest even when APMA, an MMP activator, was present (data not shown), suggesting that proteases secreted by HT1080 cells do not have β-gelsolinase activity. In contrast, incubation of His6-C68 with the HT1080 whole-cell lysate resulted in the generation of multiple fragments, including peptides having the approximate molecular weight expected for both the major (8 kDa) and minor (5 kDa) amyloidogenic peptides (Figures 1B and 2A). The observed band at approximately 8 kDa runs slightly higher than does the band for a recombinant 8 kDa FAF fragment, due to the presence of the N-terminal His6 tag in the cleaved fragment (Figure 2A). A lower band is observed at approximately 5 kDa (Figure 2A). These bands were reactive to the anti-FAF antibody (Figure 2A) and the antibody against the His6 tag (Figure 2Aii), indicating that the peptides are derived from the N-terminus of His6-C68, as expected for β-gelsolinase cleavage products (Figure 1B).
Figure 2.
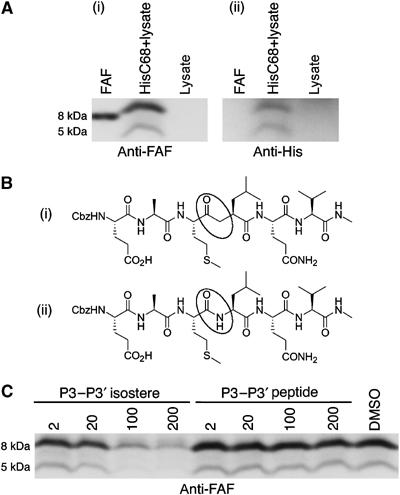
Cleavage of His6-C68 by HT1080 cell lysate. (A) HT1080 lysate and His6-C68 were incubated either alone or together at 37°C for 17 h. The proteolytic products were detected with (i) anti-FAF and (ii) a His probe. The purified recombinant 8 kDa peptide (FAF), used as a standard, runs slightly faster on the gel than the HT1080-derived fragment because it lacks the His6 tag on the N-terminus. (B) P3–P3′ isostere (i) and control peptide (ii) designed from residues E241–V246 of gelsolin to probe the specificity of the β-gelsolinase activity in HT1080 lysate. The scissile amide moiety in the peptide (ii) and the noncleavable equivalent in the isostere (i) are circled. (C) Analysis of the specificity of cleavage in HT1080 lysate. HT1080 lysate was incubated with either P3–P3′ isostere or P3–P3′ peptide, at the concentrations indicated (μM), for 15 min (37°C) prior to addition of His6-C68. The sample labeled DMSO is a control incubated with neither the P3–P3′ isostere nor the P3–P3′ peptide, but with the solvent used. The blots were probed using anti-FAF.
Proteolysis by the HT1080 lysate is specific for the β-gelsolinase cleavage site
The major 8 kDa fragment of variant gelsolin deposited in FAF patients is thought to result from β-gelsolinase cleavage of C68 at either the A242–M243 or M243–L244 amide bonds, yielding an unknown distribution of 70- and 71-residue peptides that have been isolated from ex vivo samples (Maury, 1991).
To determine whether the protease activity in HT1080 lysate cleaves His6-C68 in the region defined by the A242–M243–L244 sequence to form the 8 kDa fragment observed in FAF deposits, a peptidomimetic inhibitor was designed (supplementary data). This compound mimics the six-residue peptide sequence matching the E241–A–M–L–Q–V246 β-gelsolinase site in gelsolin, with the exception that the amide bond between M243 and L244 is replaced by a proteolytically uncleavable keto-ethylene dipeptide isostere (P3–P3′ isostere; Figure 2Bi). Dose-dependent inhibition of the protease activity found in the HT1080 lysate by this isostere would demonstrate that the protease binds and cleaves at or near this amino-acid sequence in gelsolin. The corresponding peptide containing an amide bond at M243–L244 was used as a control (P3–P3′ peptide; Figure 2Bii). The isostere or peptide was incubated with HT1080 lysate for 15 min (37°C) before the addition of the His6-C68 substrate. The production of the 8 and 5 kDa fragments exhibited concentration-dependent inhibition by the P3–P3′ isostere, but not the P3–P3′ peptide (Figure 2C), indicating that the active protease in HT1080 lysate inhibited by the P3–P3′ isostere was specific for cleavage at or near the site implicated by sequencing studies on the 8 kDa ex-vivo amyloid fragment (Maury, 1991). The appearance of the minor 5 kDa fragment also decreased with increasing P3–P3′ isostere concentration, suggesting that the two cleavages are carried out either by the same enzyme or by two closely related proteases whose activities are effected by the same inhibitor. Alternatively, generation of the 5 kDa fragment may require initial generation of the 8 kDa fragment.
Cleavage in HT1080 lysate is carried out by MMPs
The properties of the HT1080 protease were investigated using a panel of protease inhibitors. Inhibitors to serine, cysteine, and aspartic proteases had little effect on C68 processing. However, EDTA inhibited the production of both the 8 and 5 kDa fragments, suggesting the involvement of a metalloprotease (Figure 3A).
Figure 3.
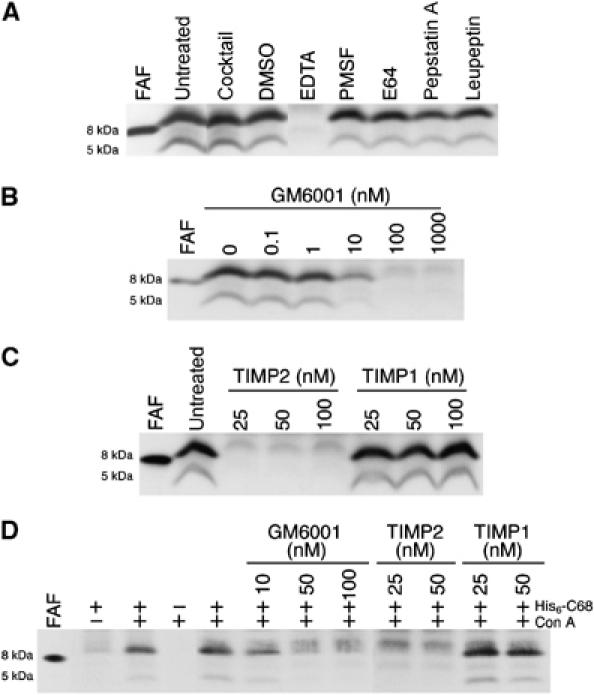
Inhibition of the HT1080-derived β-gelsolinase. (A) HT1080 lysates were incubated (37°C) with or without the protease inhibitors (EDTA (15 mM), PMSF (1 mM), E64 (100 μM), pepstatin A (100 μM), leupeptin (100 μM)), indicated for 15 min prior to addition of His6-C68. The protease inhibitor cocktail contains AEBSF (1.4 mM), aprotinin (0.8 μM) leupeptin (21 μM), bestatin (36 μM), pepstatin A (15 μM), and E64 (14 μM). (B) GM6001 was added to HT1080 lysate at the concentrations indicated 15 min before addition of HisC68. (C) HT1080 lysate was preincubated for 2 h (37°C) with or without TIMP1 or 2 at the concentrations indicated prior to the addition of His6-C68. (D) His6-C68 was added to intact HT1080 cells in serum-free DMEM in the presence or absence of Con A (50 μg/ml). GM6001, TIMP1, or 2 were added 1 h prior to the addition of His6-C68 at the concentrations indicated. After 20 h at 37°C, the media was removed and analyzed for the presence of His6-C68 cleavage products. All blots were probed with the anti-FAF antibody and have the recombinant 8 kDa fragment without the His6 tag for reference.
An important family of metalloproteases that are highly expressed in tumor cells, such as HT1080 cells, are the MMPs. The potential involvement of MMPs in the HT1080-mediated His6-C68 cleavage was assessed using the general MMP inhibitor GM6001 (Galardy et al, 1994a, 1994b). Concentrations of GM6001 as low as 10 nM inhibited the production of both the 8 and 5 kDa fragments from His6-C68 in HT1080 lysate, implicating MMPs as potential β-gelsolinases (Figure 3B).
MMPs are not the only metalloproteases in the ECM. A related family of metalloproteases, the ADAM (A Disintegrin And Metalloprotease domain) family, may also be inhibited by hydroxamate inhibitors such as GM6001 (Yong et al, 2001). To differentiate between MMP and ADAM activity in the lysate of HT1080 cells, tissue inhibitors of metalloproteases (TIMPs) were utilized. TIMPs are endogenous metalloprotease inhibitors that function to regulate MMP activity in vivo (Gomez et al, 1997). TIMPs inhibit all MMPs tested so far, with the exception that TIMP1 fails to inhibit transmembrane MMPs (the MT-MMPs, MT1-, MT2-, MT3-, and MT5-MMP) (Will et al, 1996; Butler et al, 1997; Matsumoto et al, 1997; Llano et al, 1999; Shimada et al, 1999; Lee et al, 2003). ADAM family members are generally insensitive to TIMPs, although some examples of TIMP1 and 3 inhibition have been reported (Amour et al, 2000; Baker et al, 2002). Incubation of TIMP2 with HT1080 lysate prior to addition of His6-C68 resulted in the inhibition of production of both the 8 and 5 kDa gelsolin fragments at all concentrations tested (Figure 3C). Inhibition of β-gelsolinase activity by TIMP2 indicates that the activity resides in an MMP family member and not in an ADAM family member. In contrast, no inhibition was observed in the case of TIMP1 (Figure 3C). The lack of inhibition of cleavage of His6-C68 by TIMP1 implicates an MT-MMP, and more specifically MT1-MMP, an MT-MMP member known to be expressed in HT1080 cells (Strongin et al, 1995; Lohi et al, 1996), as the primary β-gelsolinase in HT1080 lysate.
Since MT1-MMP is an integral membrane protein with catalytic activity in the extracellular environment, cleavage of C68 by this protease should not require cell lysis. MT1-MMP expression and cell-surface levels have been shown to increase when cells are treated with concanavalin A (Con A) (Lohi et al, 1996; Jiang et al, 2001). His6-C68 was incubated with intact HT1080 cells in the presence or absence of Con A (Figure 3D). In the absence of Con A, production of the 8 kDa fragment is detectable, albeit at very low levels (Figure 3D). Treatment of the cells with Con A prior to His6-C68 addition dramatically increases the level of the 8 kDa fragment produced with the 5 kDa fragment becoming detectable, and this production displays sensitivity to the inhibitors GM6001 and TIMP2, but not to TIMP1, consistent with the activity of MT1-MMP (Figure 3D).
MT1-MMP is a β-gelsolinase
Incubation of the purified recombinant catalytic domain of MT1-MMP with His6-C68 directly demonstrates the ability of MT1-MMP to cleave this substrate to produce 8 and 5 kDa peptides (Figure 4Ai). The specificity of recombinant MT1-MMP for the β-gelsolinase cleavage site in His6-C68 was assayed in the absence or presence of the P3–P3′ isostere. Incubation of the isostere with MT1-MMP inhibited its ability to cleave His6-C68 in a concentration-dependent fashion, while the control peptide had little effect (Figure 4B), demonstrating a similar specificity to the protease present in HT1080 lysate for the β-gelsolinase cleavage site. Although β-gelsolinase generates an 8 kDa amyloidogenic fragment that has either an Ala (70-residue) or Met (71-residue) at its C-terminus, the proportions of these peptides in ex vivo amyloid have not been determined quantitatively (Maury, 1991). Inhibition by the P3–P3′ isostere indicates that MT1-MMP may be specific for the M243–L244 cleavage site in C68. However, cleavage at the alternative A242–M243 site cannot be ruled out from these experiments, as the isostere could potentially function as a competitive inhibitor for the adjacent cleavage site.
Figure 4.
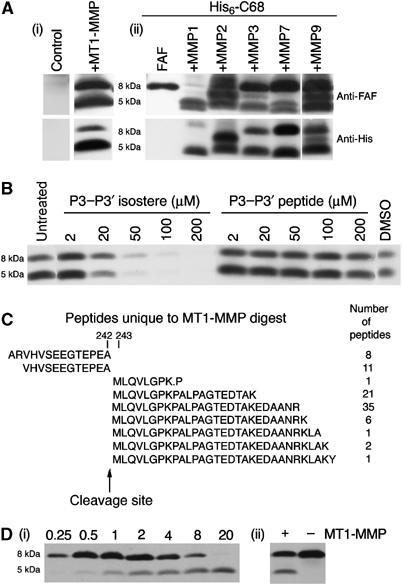
MT1-MMP cleavage of 68 kDa C-terminal gelsolin fragment. (A) His6-C68 was incubated at 37°C for 17 h either alone, with control, or with the purified recombinant MMPs indicated. The blots were probed with either anti-FAF (top) or His-probe (bottom). The recombinant 8 kDa fragment (FAF) is shown for reference. (B) MT1-MMP was preincubated for 2 h with either the P3–P3′ isostere or the P3–P3′ peptide at the concentrations indicated. His6-C68 was added and the incubation continued for 2 h at 37°C. The lane labeled untreated was incubated with buffer only and the lane labeled DMSO was incubated with DMSO only. The blot was incubated with the antibody probe to the His6 tag. (C) MudPIT analyses of MT1-MMP cleavage products of 68 kDa PG performed as described in Materials and methods. The cleavage site of MT1-MMP (between A and M) is indicated by the arrow. (D) (i) Recombinant MT1-MMP was incubated with His6-C68 for the time periods indicated above each lane (in h). The blot was incubated with the anti-FAF antibody. (ii) The recombinant 8 kDa fragment was incubated in the presence or absence of recombinant MT1-MMP for 4 h. The blot was probed with the anti-FAF antibody.
To determine the precise site of cleavage, we analyzed the gelsolin cleavage products generated by the purified soluble proteolytic domain of MT1-MMP using mass spectrometry. Following incubation of the His6-C68 substrate in the presence or absence of MT1-MMP, tryptic digests of the resulting peptide(s) were prepared and the distribution of the fragments was determined by multi-dimensional protein identification technology (MudPIT) (Wolters et al, 2001; Washburn et al, 2002). MudPIT revealed a striking recovery of cleavage products implicating proteolysis between A242 and M243 (Figure 4C), affording the 70-residue amyloidogenic peptide identified in patients (Maury, 1991). No evidence was found for cleavage between M243 and L244 to yield the 71-residue peptide terminating with M243 also found in patients (Maury, 1991). From the 70-residue peptide, we recovered 86 independent mass spectra of tryptic peptides terminating with A242 or beginning with the M243 residue in the MT1-MMP-treated tryptic digest compared to two spectra in the untreated, control sample. This cleavage pattern is consistent with the known preference that MT1-MMP has for the Pro residue being in the P3 position (–P240–E–A–M–L–Q–V246–) (Zucker et al, 2003). Although the 5 kDa fragment from FAF patients has not been carefully characterized to determine the site of cleavage, incubation of purified MT1-MMP for increasing time with His6-C68 shows that sequential cleavage events by this protease may result in generation of a 5 kDa fragment from the 8 kDa fragment (Figure 4Di). Indeed, incubation of the recombinant 8 kDa fragment with MT1-MMP results in the generation of a 5 kDa fragment (Figure 4Dii), providing a potential explanation for the source of this proteolytic fragment.
Collectively, these data strongly implicate MT1-MMP activity in generating the 8 and 5 kDa fragments observed in the media of HT1080 cells incubated with His6-C68. However, it remains possible that other β-gelsolinases could contribute to the generation of the fragments observed in tissues. MMPs are a large family of metalloproteases and, due to similarities in their catalytic activity, it is likely that they have overlapping substrate specificities. Using a panel of purified recombinant MMPs with a range of enzyme concentrations in each case to identify early- and late-cleavage products (data not shown), we determined that, in addition to MT1-MMP, soluble MMPs 3, 7, and 9, but not other MMPs such as MMP2, have an MT1-MMP-like activity that could cleave the substrate to produce fragments of approximately 8 and 5 kDa, which are immunoreactive for both the FAF and His6 epitopes (Figure 4Aii). These data indicate that multiple MMP family members may contain β-gelsolinase activity and contribute to the generation of the amyloidogenic peptides in FAF patients.
Cells lacking MT1-MMP fail to generate the 8 kDa FAF fragment
Given the possibility that MMPs besides MT1-MMP could potentially contribute to FAF, we analyzed two distinct cellular systems where MT1-MMP is absent, or greatly reduced, to look at the importance of this family member in regard to β-gelsolinase activity in these cells.
The first cell system examined utilized HT1080 cells, where expression levels of MT1-MMP were manipulated with siRNA. HT1080 cells transfected with siRNA directed to MT1-MMP show complete loss of the 57 kDa active MT1-MMP enzyme (Figure 5A, upper panel) compared to cells exposed to control scrambled RNA. When these cells were analyzed for their ability to cleave the His6-C68 substrate to the 8 kDa fragment, it was found that the cells lacking MT1-MMP were unable to generate the 8 kDa fragment (Figure 5A, lower panel).
Figure 5.
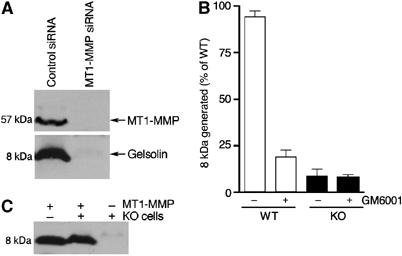
Cells lacking MT1-MMP do not contain β-gelsolinase activity. (A) HT1080 cells were transfected with either control-scrambled RNA or siRNA directed to MT1-MMP. Cell lysates were blotted and probed with an antibody raised against MT1-MMP, shown in the upper panel. Cell lysates were incubated with His6-C68 at 37°C for 17 h and the blots probed with the anti-FAF antibody, lower panel. Each assay was executed in triplicate and representative data are shown. (B) Cell lysates (at 4 mg/ml) were generated from either wild type (WT, white columns) or MT1-MMP knockout fibroblasts (KO, black columns) and incubated with His6-C68 at 37°C in the presence or absence of GM6001 (200 μM) for 20 h. Blots were probed using the anti-FAF antibody and densitometry utilized to quantify levels of 8 kDa produced. Each condition is an average of five replicates from two independent experiments. Data are normalized to the highest level of 8 kDa generated by WT lysate in the absence of the inhibitor. Error bars indicate the standard error of the mean. (C) His6-C68 was incubated either alone or with lysate from knockout fibroblasts, in the presence or absence of recombinant MT1-MMP for 4 h at 37°C. The blots were probed with the anti-FAF antibody.
The second system analyzed involved the generation of fibroblast cell lines from both wild-type and MT1-MMP knockout mouse embryos (Holmbeck et al, 1999). If MT1-MMP is the predominant or sole source of β-gelsolinase in these cells, it is expected that only the wild-type and not the MT1-MMP knockout cells would be capable of generating the 8 kDa peptide from His6-C68. Cells prepared from both the wild-type and heterozygous mouse embryos (data not shown), but not two cell lines derived from the two knockout mouse embryos, had β-gelsolinase activity. Whereas the lysate of wild-type cells incubated with His6-C68 produced an 8 kDa fragment with a protease that is sensitive to the MMP inhibitor GM6001 (Figure 5B), the lysate from the MT1-MMP knockout cells is unable to generate significant levels of the 8 kDa fragment (Figure 5B, dark bars). Transient overexpression of MT1-MMP in the knockout fibroblasts failed to recover processing of His6-C68 to 8 kDa—further analyses revealed that the knockout cell line fails to process MT1-MMP from the inactive 60 kDa precursor to the active 57 kDa (data not shown); presumably, the rapid overexpression overwhelms the processing machinery (Lohi et al, 1996). Consistent with this interpretation, addition of recombinant MT1-MMP to the knockout cell lysate enables the recovery of the activity that generates the 8 kDa fragment, indicating that it is the lack of MT1-MMP that is responsible for loss of processing and not the loss or gain of some other factor (Figure 5C). These results provide additional data in an in vivo cellular context, suggesting that MT1-MMP can be a critical β-gelsolinase in FAF.
The ECM environment contributes to FAF by hastening amyloidogenesis
That MT1-MMP generates amyloidogenic gelsolin peptides proximal to the ECM raises the possibility that the chemical environment of the ECM could contribute to selective gelsolin deposition. We have previously shown that amyloidogenesis of the recombinant 8 kDa (71 residue) gelsolin fragment (A173–M243) is optimal at low pH, leading to speculation that disease-associated fibril formation may occur in an acidic environment (Ratnaswamy et al, 1999). However, the identification of the extracellular MT1-MMP as a β-gelsolinase now suggests that gelsolin is cleaved and forms fibrils in the neutral pH environment of the ECM. To address whether these peptides could form fibrils at neutral pH, we re-examined the pH dependence of fibril formation in vitro using the recombinant 70- and 71-residue FAF 8 kDa fragments, corresponding to those found in FAF patients. Amyloidogenicity was monitored utilizing thioflavin T (TfT), an environmentally sensitive fluorophore that fluoresces upon binding to a cross β-sheet structure. Whereas amyloid production was not detected at pH 7.0 in nonagitated solutions on a laboratory timescale (Ratnaswamy et al, 1999), when agitation is used to increase the rate of amyloidogenesis (Cohlberg et al, 2002) both the 70- and 71-residue fragments formed TfT-positive aggregates over the pH range of 4–7. Transmission electron microscopy (TEM) confirmed a well-defined fibrillar morphology for all conditions (Figure 6Aii, inset).
Figure 6.
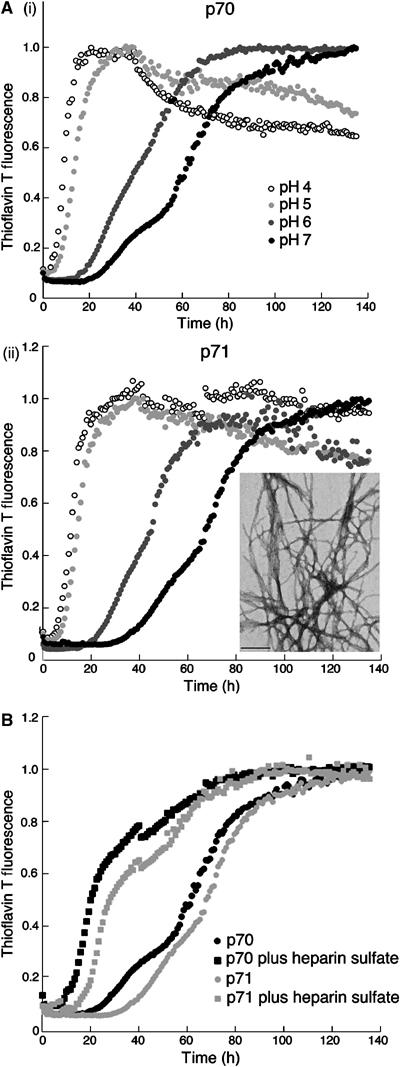
Effects of pH and ECM components on FAF fibril formation. (A) pH dependence of agitated 70-residue (i) and 71-residue (ii) fibril formation monitored by TfT fluorescence. (Aii, inset) EM image of FAF fibrils (70-residue peptide) formed at pH 7 in an agitation assay. The photograph is taken at × 52 000 magnification and is a representative of at least three experiments. Scale bar equals 200 nm. (B) Fibril formation in an agitation assay with 20 μM 70-residue (black points) and 71-residue (gray points) peptides at pH 7 in either the presence (squares) or absence (circles) of 0.1 mg/ml heparin monitored by TfT fluorescence intensity. Heparin alone did not affect the fluorescence of TfT (data not shown). The fibril formation assays were repeated on at least 5–6 independent occasions and the representative data are shown. TfT fluorescence units were measured every 10 min and plotted against time. The data were then normalized to the TfT fluorescence value at maximal fibril formation to account for the pH dependence of TfT fluorescence intensity.
Given that FAF-associated amyloid is predominantly deposited in the ECM (Kiuru, 1998), the role of ECM components in the formation of fibrils was examined. Several ECM components, such as heparan sulfate proteoglycans (HSPGs) and GAGs, including heparan sulfate and heparin, are known to be associated with amyloid fibrils in many amyloidoses (Sipe and Cohen, 2000). To determine whether ECM components may contribute to FAF amyloidogenesis, the rate of fibril formation by the 70- and 71-residue amyloidogenic (8 kDa) fragments was evaluated in response to the addition of heparin. Heparin was found to accelerate the formation of TfT-positive aggregates at pH 4–7 (Figure 6B; pH 7 data shown). TEM analysis of agitated samples in the presence of heparin revealed well-defined fibrillar structures for both fragments (data not shown). These results suggest that GAGs accelerate amyloidogenesis at neutral pH, consistent with previous reports that ECM components stimulate other amyloidogenic polypeptides to form the cross β-sheet structure, rationalizing their presence in the ECM (McLaurin et al, 1999; Cohlberg et al, 2002; Ancsin, 2003).
Discussion
MMPs have an established pathological role in diseases such as cancer and arthritis (Knauper and Murphy, 1998; Folgueras et al, 2004). We now provide strong evidence that MMPs, specifically MT1-MMP, can fulfill the specific function of β-gelsolinase, cleaving the secreted variant C68 to generate the 8 and 5 kDa amyloidogenic fragments found in FAF patients. While in vitro studies show that several MMPs can generate the 8 and 5 kDa amyloidogenic fragments, a fibroblast cell line from a mouse model lacking the membrane-localized MMP, MT1-MMP, and HT1080 cells transfected with siRNA directed to MT1-MMP demonstrates that MT1-MMP activity can be responsible for generation of the 8 kDa amyloidogenic fragment. We also reveal that GAGs, a major component of the ECM, accelerate gelsolin amyloidogenesis in vitro. Given the close juxtaposition of the cell membrane and the ECM, a unique chemical and enzymological environment may exist that significantly contributes to deposition of gelsolin amyloid and onset of FAF. A more quantitative examination of the composition of patient deposits in future may reveal further ECM-associated proteolytic events. Circumstantial evidence also exists to suggest that MMPs have a role in fibril formation in AA amyloidosis (Stix et al, 2001) and in the most prevalent amyloid disease, AD (Leake et al, 2000; Chong et al, 2001; Jung et al, 2003; Lorenzl et al, 2003; Saarela et al, 2004). The precise mechanism by which MMPs effect AD pathology is unknown, though it has been proposed that their action may facilitate deposition through re-modeling of the ECM (Saarela et al, 2004).
A potentially important factor influencing the location of FAF deposits is the local concentration of C68. C68 is highly abundant in the serum of patients with FAF (Maury and Rossi, 1993; Maury et al, 1997). If transcytosed by the endothelial cells composing the arterial wall to the extravascular space, C68 would be expected to encounter MT1-MMP activity in the area adjacent to the ECM. In this scenario, serum C68 could contribute significantly to the extensive peripheral systemic amyloid deposition associated with FAF pathology. A second possibility is that C68 produced proximal to the site of deposition would have direct access to the local ECM. In this view, the circulating plasma pool may play no role or a reduced role in disease. Though plasma gelsolin is highly expressed by muscle cells, many cell types also synthesize and secrete the protein (Kwiatkowski et al, 1988; Paunio et al, 1997). Thus, secretion of the C68 fragment into the adjacent ECM by these cells may directly account for the extent and rate of tissue-selective gelsolin amyloid deposition. These possibilities are not mutually exclusive. It remains possible that the extent of tissue-specific systemic deposition reflects the activity of one or both pools.
While C68 levels are likely to play a role in FAF, the distribution, concentration, and level of activity of MT1-MMP-like proteases that have related cleavage specificities are also expected to influence the extent of and site of gelsolin amyloid deposition. MT1-MMP-like proteases are expressed in a variety of adult tissues, with each MMP having distinct expression patterns and levels (Knauper and Murphy, 1998; Pei, 1999). The arterial wall, a site where FAF patients exhibit substantial deposition, is composed of smooth muscle cells. Smooth muscle cells have been shown to express a high level of both gelsolin (Kwiatkowski et al, 1988) and MT1-MMP (Su et al, 2004). Thus, there is a strong correlation between this major site of deposition and high levels of both substrate and the MT1-MMP enzyme. The potential contribution of other MT1-MMP-like proteases remains to be explored.
Our in vitro data suggest that amyloid deposition is also likely to be dependent on components of the ECM in a given tissue. GAGs are known to lower the critical concentration required for misassembly, accelerating the cross β-sheet formation of other amyloidogenic peptides (Snow and Wight, 1989). The colocalization of the GAG accelerators and the MT1-MMP cleavage enzyme provides an explanation for the proclivity of selective gelsolin amyloid deposition in the ECM.
The discovery that furin- and MT1-MMP-like proteases have activities that are potentially responsible for the FAF pathological cascade in humans now makes it possible to target FAF using protease inhibitors. Given the central importance of furin in protein-trafficking pathways (Molloy et al, 1999), it seems unlikely that we can inhibit the formation of C68 with furin inhibitors without encountering unwanted side effects. In contrast, numerous potent MMP inhibitors, some of which are quite selective, could prove useful for inhibiting the processing of C68 into the amyloidogenic peptides. A second possibility, revealed by our observation that ECM components accelerate plasma gelsolin amyloidogenesis, is to antagonize the acceleration of gelsolin amyloidogeneis by ECM mimetics. Fibrillex™, a compound recently approved by the FDA to ameliorate SAA amyloidosis, functions by antagonizing the interaction between the ECM components and fibrils, and could be useful for treating FAF.
The identification of furin (α-gelsolinase) (Chen et al, 2001) and now MT1-MMP-like proteases (β-gelsolinase), in combination with the influence of the ECM on the rate of gelsolin amyloidogenesis, provides important clues for understanding of FAF pathology in humans. The link between the site of origin of precursor substrates and conditions supporting generation and deposition of amyloid remains an elusive feature of many amyloid diseases. The current studies provide important insights into a new general mechanism by which processing at multiple sites and the local environment strongly influence the onset and accumulation of amyloid.
Materials and methods
Materials
MMPs and GM6001 were purchased from Oncogene Research products (Boston, MA). General protease inhibitors, Con A and heparin sulfate, were purchased from Sigma-Aldrich (St Louis, MO), and TIMP1 and 2 and the polyclonal rabbit anti-MT1-MMP antibody (#AB815) from Chemicon Inc. (Temecula, CA).
Design and construction of His6-C68
Primers CGTGTGCATATGGCCACCGAGGTACCTGTGTCC and GGCCAGCTTGCGGTTGGCCGC were used for PCR. Digestion of pET11d with NdeI/BamHI, plasma gelsolin pCMV3 with PstI/BamHI, and the PCR product with NdeI/PstI followed by a three-part ligation created His6-C68 in pET11d. Expression was carried out in BL21 cells and His6-C68 purified on Ni-NTA agarose (Qiagen, Valencia, CA).
Preparation of recombinant FAF fragment and antibodies
The 8 kDa p70 and p71 FAF peptides were recombinantly expressed and purified as described (Ratnaswamy et al, 1999). The polyclonal antibodies against GST-8 kDa gelsolin fragment (Ratnaswamy et al, 1999) were raised in rabbits. Anti-His probe was obtained from Pierce Biotechnology, Inc. (Rockford, IL).
Preparation of gelsolin peptides and peptide isosteres
See Supplementary data.
HT1080 assays
See Supplementary data.
siRNA
See Supplementary data.
Generation of mouse fibroblast cell lines
See Supplementary data.
Fibril formation assay
See Supplementary data.
Mass spectrometry
See Supplementary data.
Supplementary Material
Supplemental Materials and Methods
Acknowledgments
This work was supported by grants from the National Institutes of Health to JWK and WEB (AG18917), as well by the Skaggs Institute for Chemical Biology, and the Lita Annenberg Hazen Foundation. We thank Songpon Deechongkit for assistance with TEM and Peiqing Sun for aid in generating the mouse fibroblast cell lines.
References
- Amour A, Knight CG, Webster A, Slocombe PM, Stephens PE, Knauper V, Docherty AJ, Murphy G (2000) The in vitro activity of ADAM-10 is inhibited by TIMP-1 and TIMP-3. FEBS Lett 473: 275–279 [DOI] [PubMed] [Google Scholar]
- Ancsin JB (2003) Amyloidogenesis: historical and modern observations point to heparan sulfate proteoglycans as a major culprit. Amyloid 10: 67–79 [DOI] [PubMed] [Google Scholar]
- Baker AH, Edwards DR, Murphy G (2002) Metalloproteinase inhibitors: biological actions and therapeutic opportunities. J Cell Sci 115: 3719–3727 [DOI] [PubMed] [Google Scholar]
- Burtnick LD, Koepf EK, Grimes J, Jones EY, Stuart DI, McLaughlin PJ, Robinson RC (1997) The crystal structure of plasma gelsolin: implications for actin severing, capping, and nucleation. Cell 90: 661–670 [DOI] [PubMed] [Google Scholar]
- Butler GS, Will H, Atkinson SJ, Murphy G (1997) Membrane-type-2 matrix metalloproteinase can initiate the processing of progelatinase A and is regulated by the tissue inhibitors of metalloproteinases. Eur J Biochem 244: 653–657 [DOI] [PubMed] [Google Scholar]
- Carrell RW, Gooptu B (1998) Conformational changes and disease—serpins, prions and Alzheimer's. Curr Opin Struct Biol 8: 799–809 [DOI] [PubMed] [Google Scholar]
- Chen CD, Huff ME, Matteson J, Page L, Phillips R, Kelly JW, Balch WE (2001) Furin initiates gelsolin familial amyloidosis in the Golgi through a defect in Ca(2+) stabilization. EMBO J 20: 6277–6287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong YH, Sung JH, Shin SA, Chung JH, Suh YH (2001) Effects of the beta-amyloid and carboxyl-terminal fragment of Alzheimer's amyloid precursor protein on the production of the tumor necrosis factor-alpha and matrix metalloproteinase-9 by human monocytic THP-1. J Biol Chem 276: 23511–23517 [DOI] [PubMed] [Google Scholar]
- Cohlberg JA, Li J, Uversky VN, Fink AL (2002) Heparin and other glycosaminoglycans stimulate the formation of amyloid fibrils from alpha-synuclein in vitro. Biochemistry 41: 1502–1511 [DOI] [PubMed] [Google Scholar]
- Dobson CM (2003) Protein folding and disease: a view from the first Horizon Symposium. Nat Rev Drug Discov 2: 154–160 [DOI] [PubMed] [Google Scholar]
- Folgueras AR, Pendas AM, Sanchez LM, Lopez-Otin C (2004) Matrix metalloproteinases in cancer: from new functions to improved inhibition strategies. Int J Dev Biol 48: 411–424 [DOI] [PubMed] [Google Scholar]
- Galardy RE, Cassabonne ME, Giese C, Gilbert JH, Lapierre F, Lopez H, Schaefer ME, Stack R, Sullivan M, Summers B, Tressler R, Tyrrell D, Wee J, Allen SD, Castellot JJ, Barletta JP, Schultz GS, Fernandez LA, Fisher S, Cui T-Y, Foellmer HG, Grobelny D, Holleran WM (1994a) Low molecular weight inhibitors in corneal ulceration. Ann NY Acad Sci 732: 315–323 [DOI] [PubMed] [Google Scholar]
- Galardy RE, Grobelny D, Foellmer HG, Fernandez LA (1994b) Inhibition of angiogenesis by the matrix metalloprotease inhibitor N-[2R-2-(hydroxamidocarbonymethyl)-4-methylpentanoyl)]-L-tryptophan methylamide. Cancer Res 54: 4715–4718 [PubMed] [Google Scholar]
- Ghiso J, Haltia M, Prelli F, Novello J, Frangione B (1990) Gelsolin variant (Asn-187) in familial amyloidosis, Finnish type. Biochem J 272: 827–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez DE, Alonso DF, Yoshiji H, Thorgeirsson UP (1997) Tissue inhibitors of metalloproteinases: structure, regulation and biological functions. Eur J Cell Biol 74: 111–122 [PubMed] [Google Scholar]
- Haltia M, Ghiso J, Prelli F, Gallo G, Kiuru S, Somer H, Palo J, Frangione B (1990) Amyloid in familial amyloidosis, Finnish type, is antigenically and structurally related to gelsolin. Am J Pathol 136: 1223–1228 [PMC free article] [PubMed] [Google Scholar]
- Holmbeck K, Bianco P, Caterina J, Yamada S, Kromer M, Kuznetsov SA, Mankani M, Robey PG, Poole AR, Pidoux I, Ward JM, Birkedal-Hansen H (1999) MT1-MMP-deficient mice develop dwarfism, osteopenia, arthritis, and connective tissue disease due to inadequate collagen turnover. Cell 99: 81–92 [DOI] [PubMed] [Google Scholar]
- Jiang A, Lehti K, Wang X, Weiss SJ, Keski-Oja J, Pei D (2001) Regulation of membrane-type matrix metalloproteinase 1 activity by dynamin-mediated endocytosis. Proc Natl Acad Sci USA 98: 13693–13698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung SS, Zhang W, Van Nostrand WE (2003) Pathogenic A beta induces the expression and activation of matrix metalloproteinase-2 in human cerebrovascular smooth muscle cells. J Neurochem 85: 1208–1215 [DOI] [PubMed] [Google Scholar]
- Kangas H, Paunio T, Kalkkinen N, Jalanko A, Peltonen L (1996) In vitro expression analysis shows that the secretory form of gelsolin is the sole source of amyloid in gelsolin-related amyloidosis. Hum Mol Genet 5: 1237–1243 [DOI] [PubMed] [Google Scholar]
- Kangas H, Ulmanen I, Paunio T, Kwiatkowski DJ, Lehtovirta M, Jalanko A, Peltonen L (1999) Functional consequences of amyloidosis mutation for gelsolin polypeptide—analysis of gelsolin–actin interaction and gelsolin processing in gelsolin knock-out fibroblasts. FEBS Lett 454: 233–239 [DOI] [PubMed] [Google Scholar]
- Kiuru S (1998) Gelsolin-related familial amyloidosis, Finnish type (FAF), and its variants found worldwide. Amyloid 5: 55–66 [DOI] [PubMed] [Google Scholar]
- Knauper V, Murphy G (1998) Matrix Metalloproteinases. New York: Academic Press [Google Scholar]
- Kwiatkowski DJ, Mehl R, Izumo S, Nadal-Ginard B, Yin HL (1988) Muscle is the major source of plasma gelsolin. J Biol Chem 263: 8239–8243 [PubMed] [Google Scholar]
- Kwiatkowski DJ, Stossel TP, Orkin SH, Mole JE, Colten HR, Yin HL (1986) Plasma and cytoplasmic gelsolins are encoded by a single gene and contain a duplicated actin-binding domain. Nature 323: 455–458 [DOI] [PubMed] [Google Scholar]
- Leake A, Morris CM, Whateley J (2000) Brain matrix metalloproteinase 1 levels are elevated in Alzheimer's disease. Neurosci Lett 291: 201–203 [DOI] [PubMed] [Google Scholar]
- Lee MH, Rapti M, Murphy G (2003) Unveiling the surface epitopes that render tissue inhibitor of metalloproteinase-1 inactive against membrane type 1-matrix metalloproteinase. J Biol Chem 278: 40224–40230 [DOI] [PubMed] [Google Scholar]
- Levy E, Haltia M, Fernandez-Madrid I, Koivunen O, Ghiso J, Prelli F, Frangione B (1990) Mutation in gelsolin gene in Finnish hereditary amyloidosis. J Exp Med 172: 1865–1867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llano E, Pendas AM, Freije JP, Nakano A, Knauper V, Murphy G, Lopez-Otin C (1999) Identification and characterization of human MT5-MMP, a new membrane-bound activator of progelatinase a overexpressed in brain tumors. Cancer Res 59: 2570–2576 [PubMed] [Google Scholar]
- Lohi J, Lehti K, Westermarck J, Kahari VM, Keski-Oja J (1996) Regulation of membrane-type matrix metalloproteinase-1 expression by growth factors and phorbol 12-myristate 13-acetate. Eur J Biochem 239: 239–247 [DOI] [PubMed] [Google Scholar]
- Lorenzl S, Albers DS, Relkin N, Ngyuen T, Hilgenberg SL, Chirichigno J, Cudkowicz ME, Beal MF (2003) Increased plasma levels of matrix metalloproteinase-9 in patients with Alzheimer's disease. Neurochem Int 43: 191–196 [DOI] [PubMed] [Google Scholar]
- Matsumoto S, Katoh M, Saito S, Watanabe T, Masuho Y (1997) Identification of soluble type of membrane-type matrix metalloproteinase-3 formed by alternatively spliced mRNA. Biochim Biophys Acta 1354: 159–170 [DOI] [PubMed] [Google Scholar]
- Maury CP (1991) Gelsolin-related amyloidosis. Identification of the amyloid protein in Finnish hereditary amyloidosis as a fragment of variant gelsolin. J Clin Invest 87: 1195–1199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maury CP, Alli K, Baumann M (1990) Finnish hereditary amyloidosis. Amino acid sequence homology between the amyloid fibril protein and human plasma gelsoline. FEBS Lett 260: 85–87 [DOI] [PubMed] [Google Scholar]
- Maury CP, Baumann M (1990) Isolation and characterization of cardiac amyloid in familial amyloid polyneuropathy type IV (Finnish): relation of the amyloid protein to variant gelsolin. Biochim Biophys Acta 1096: 84–86 [DOI] [PubMed] [Google Scholar]
- Maury CP, Rossi H (1993) Demonstration of a circulating 65K gelsolin variant specific for familial amyloidosis, Finnish type. Biochem Biophys Res Commun 191: 41–44 [DOI] [PubMed] [Google Scholar]
- Maury CP, Sletten K, Totty N, Kangas H, Liljestrom M (1997) Identification of the circulating amyloid precursor and other gelsolin metabolites in patients with G654A mutation in the gelsolin gene (Finnish familial amyloidosis): pathogenetic and diagnostic implications. Lab Invest 77: 299–304 [PubMed] [Google Scholar]
- McLaurin J, Franklin T, Kuhns WJ, Fraser PE (1999) A sulfated proteoglycan aggregation factor mediates amyloid-beta peptide fibril formation and neurotoxicity. Amyloid 6: 233–243 [DOI] [PubMed] [Google Scholar]
- Molloy SS, Anderson ED, Jean F, Thomas G (1999) Bi-cycling the furin pathway: from TGN localization to pathogen activation and embryogenesis. Trends Cell Biol 9: 28–35 [DOI] [PubMed] [Google Scholar]
- Paunio T, Kangas H, Kalkkinen N, Haltia M, Palo J, Peltonen L (1994) Toward understanding the pathogenic mechanisms in gelsolin-related amyloidosis: in vitro expression reveals an abnormal gelsolin fragment. Hum Mol Genet 3: 2223–2229 [DOI] [PubMed] [Google Scholar]
- Paunio T, Kangas H, Kiuru S, Palo J, Peltonen L, Syvanen AC (1997) Tissue distribution and levels of gelsolin mRNA in normal individuals and patients with gelsolin-related amyloidosis. FEBS Lett 406: 49–55 [DOI] [PubMed] [Google Scholar]
- Pei D (1999) Identification and characterization of the fifth membrane-type matrix metalloproteinase MT5-MMP. J Biol Chem 274: 8925–8932 [DOI] [PubMed] [Google Scholar]
- Ratnaswamy G, Koepf E, Bekele H, Yin H, Kelly JW (1999) The amyloidogenicity of gelsolin is controlled by proteolysis and pH. Chem Biol 6: 293–304 [DOI] [PubMed] [Google Scholar]
- Saarela MS, Lehtimaki T, Rinne JO, Hervonen A, Jylha M, Roytta M, Ahonen JP, Mattila KM (2004) Interaction between matrix metalloproteinase 3 and the epsilon4 allele of apolipoprotein E increases the risk of Alzheimer's disease in Finns. Neurosci Lett 367: 336–339 [DOI] [PubMed] [Google Scholar]
- Sekijima Y, Wiseman RL, Matteson J, Hammarstrom P, Miller SR, Sawkar AR, Balch WE, Kelly JW (2005) The biological and chemical basis for tissue-selective amyloid disease. Cell 121: 73–85 [DOI] [PubMed] [Google Scholar]
- Shimada T, Nakamura H, Ohuchi E, Fujii Y, Murakami Y, Sato H, Seiki M, Okada Y (1999) Characterization of a truncated recombinant form of human membrane type 3 matrix metalloproteinase. Eur J Biochem 262: 907–914 [DOI] [PubMed] [Google Scholar]
- Sipe JD, Cohen AS (2000) Review: history of the amyloid fibril. J Struct Biol 130: 88–98 [DOI] [PubMed] [Google Scholar]
- Snow AD, Wight TN (1989) Proteoglycans in the pathogenesis of Alzheimer's disease and other amyloidoses. Neurobiol Aging 10: 481–497 [DOI] [PubMed] [Google Scholar]
- Stix B, Kahne T, Sletten K, Raynes J, Roessner A, Rocken C (2001) Proteolysis of AA amyloid fibril proteins by matrix metalloproteinases-1, -2, and -3. Am J Pathol 159: 561–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strongin AY, Collier I, Bannikov G, Marmer BL, Grant GA, Goldberg GI (1995) Mechanism of cell surface activation of 72-kDa type IV collagenase. Isolation of the activated form of the membrane metalloprotease. J Biol Chem 270: 5331–5338 [DOI] [PubMed] [Google Scholar]
- Su AI, Wiltshire T, Batalov S, Lapp H, Ching KA, Block D, Zhang J, Soden R, Hayakawa M, Kreiman G, Cooke MP, Walker JR, Hogenesch JB (2004) A gene atlas of the mouse and human protein-encoding transcriptomes. Proc Natl Acad Sci USA 101: 6062–6067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun HQ, Yamamoto M, Mejillano M, Yin HL (1999) Gelsolin, a multifunctional actin regulatory protein. J Biol Chem 274: 33179–33182 [DOI] [PubMed] [Google Scholar]
- Washburn MP, Ulaszek R, Deciu C, Schieltz DM, Yates JR III (2002) Analysis of quantitative proteomic data generated via multidimensional protein identification technology. Anal Chem 74: 1650–1657 [DOI] [PubMed] [Google Scholar]
- Will H, Atkinson SJ, Butler GS, Smith B, Murphy G (1996) The soluble catalytic domain of membrane type 1 matrix metalloproteinase cleaves the propeptide of progelatinase A and initiates autoproteolytic activation. Regulation by TIMP-2 and TIMP-3. J Biol Chem 271: 17119–17123 [DOI] [PubMed] [Google Scholar]
- Wolters DA, Washburn MP, Yates JR III (2001) An automated multidimensional protein identification technology for shotgun proteomics. Anal Chem 73: 5683–5690 [DOI] [PubMed] [Google Scholar]
- Yin HL (1987) Gelsolin: calcium- and polyphosphoinositide-regulated actin-modulating protein. BioEssays 7: 176–179 [DOI] [PubMed] [Google Scholar]
- Yong VW, Power C, Forsyth P, Edwards DR (2001) Metalloproteinases in biology and pathology of the nervous system. Nat Rev Neurosci 2: 502–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker S, Pei D, Cao J, Lopez-Otin C (2003) Membrane type-matrix metalloproteinases (MT-MMP). Curr Top Dev Biol 54: 1–74 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Materials and Methods