

Abstract
Herpes simplex virus (HSV) entry into cells requires binding of the envelope glycoprotein D (gD) to one of several cell surface receptors. The 50 C-terminal residues of the gD ectodomain are essential for virus entry, but not for receptor binding. We have determined the structure of an unliganded gD molecule that includes these C-terminal residues. The structure reveals that the C-terminus is anchored near the N-terminal region and masks receptor-binding sites. Locking the C-terminus in the position observed in the crystals by an intramolecular disulfide bond abolished receptor binding and virus entry, demonstrating that this region of gD moves upon receptor binding. Similarly, a point mutant that would destabilize the C-terminus structure was nonfunctional for entry, despite increased affinity for receptors. We propose that a controlled displacement of the gD C-terminus upon receptor binding is an essential feature of HSV entry, ensuring the timely activation of membrane fusion.
Keywords: glycoprotein D, herpes simplex virus, receptor binding, viral entry
Introduction
Herpes simplex virus type 1 and 2 (HSV-1 and -2) are widespread human pathogens. HSV infects mainly epithelial tissues and then spreads to neurons of the peripheral nervous system, where it establishes latency. Recurrent HSV-1 infection typically causes oral lesions, whereas HSV-2 causes genital lesions (Whitley, 2001).
Similar to other enveloped viruses, HSV entry into target cells requires fusion between viral and cell membranes. Of the 11 or more proteins present on the viral envelope, five (gC, gB, gD, gH, and gL) are involved in entry (Spear and Longnecker, 2003). Initially, gB and gC interact with cell surface heparan sulfate (HS) proteoglycans, allowing virus-cell attachment, then glycoprotein D (gD) binds to a cell surface receptor (Campadelli-Fiume et al, 2000; Spear et al, 2000). The latter event is followed by membrane fusion mediated by gB and the heterodimer gH/gL (Spear and Longnecker, 2003).
Binding of gD to a functional cellular receptor such as herpes virus entry mediator (HVEM), a TNF receptor family member, or nectin-1, a member of the immunoglobulin superfamily, is essential for HSV cell entry (for a recent review on HSV gD receptors, see Spear, 2004). Moreover, the soluble gD ectodomain gD306 (residues 1–306) is able to mediate entry of an HSV virion lacking gD on its envelope (gD-null virus), demonstrating that the gD receptor interaction also participates in an entry step that differs from virus–cell attachment (Cocchi et al, 2004). In the gamma-herpes virus Epstein–Barr virus (EBV), the structurally unrelated envelope glycoprotein gp42 plays a functional role similar to gD by binding to MHC class II on B cells and triggering virus entry in a process that also requires gH/gL and gB (Li et al, 1997; Haan et al, 2000; Mullen et al, 2002). Thus, herpes viruses have evolved an entry mechanism that relies on a receptor-binding protein to activate the membrane fusion process, which in turn involves other glycoproteins.
We previously solved the structures of a C-terminally truncated form of the gD ectodomain, gD285 (residues 1–285; the full ectodomain encompasses residue 1–316), alone and bound to HVEM (Carfi et al, 2001). gD consists of a V-like Ig domain with N- and C-terminal extensions. The N-terminus of gD285 is flexible and extended and folds into a hairpin structure that contains all of the HVEM-binding residues when gD is bound to HVEM (Carfi et al, 2001; Connolly et al, 2002). The final 26 amino acids of the C-terminus (from residue 260 to 285), although present in the crystals, were disordered and therefore not visible in either structure.
The C-terminus of the gD ectodomain (residues 260–316) plays an important functional role in HSV entry. Virions carrying forms of gD with insertions/deletions between residues 275 and 300, or a chimera containing amino acids 1–260 of gD linked to two or four domains of CD8, are impaired in cell entry (Chiang et al, 1994; Milne et al, 2003; Zhou et al, 2003). Moreover, soluble gD306 or gD285, but not gD260, can mediate entry of a gD-null virus, confirming the functional importance of the C-terminal region (Cocchi et al, 2004). Interestingly, the defects of these mutants are not caused by the lack of receptor binding, as all of them bind HVEM and nectin-1. In addition, truncated forms of gD lacking part of the C-terminus (e.g. gD285, gD275, and gD260) bound each receptor with greater affinity than gD306 (Whitbeck et al, 1997; Rux et al, 1998; Willis et al, 1998b; Krummenacher et al, 1999). The link between enhanced receptor affinity and defects in entry has been so far elusive.
Here we report the structures of two gD mutant proteins that include the C-terminal residues not resolved by the previous structural studies. The structures and the accompanying biochemical and functional evidence demonstrate that we have trapped gD in its pre-receptor-binding conformation, and suggest a mechanism whereby receptor binding initiates HSV entry.
Results
Structure determination
Unlike the gD285 protein, the longer gD306 failed to produce crystals, perhaps due to the flexibility of its longer C-terminus (Figure 1A) (Carfi et al, 2002). Although soluble gD285 and gD306 ectodomains are monomeric, previous crosslinking experiments suggest that virion envelope gD is dimeric (Handler et al, 1996). Therefore, we speculated that dimerization, possibly mediated by the high local concentration of gD on the viral membrane or by the transmembrane (TM) region, could stabilize the conformation of the C-terminus. We reasoned that the C-terminus could also be stabilized by the addition of an extra cysteine at the end of the gD ectodomain (gD306307C), which would favor gD dimerization through the formation of an intermolecular disulfide bond. The gD306307C molecule was purified from the supernatant of insect cells infected with a recombinant baculovirus. Approximately half of the protein formed disulfide-linked dimers that were separated from the monomers by size exclusion chromatography (data not shown). The dimeric gD306307C was recognized by all the tested conformation-dependent monoclonal antibodies (Mabs) and also bound to HVEM and nectin-1 (JC Whitbeck, unpublished results). Importantly, the dimeric gD306307C crystallized, although the first 22 amino acids were cleaved during the crystallization process, presumably by trace amounts of a copurified protease. The structure of the resulting dimeric gD(23–306)307C was determined by molecular replacement and refined at 2.1 Å resolution (Table I and Materials and methods).
Figure 1.
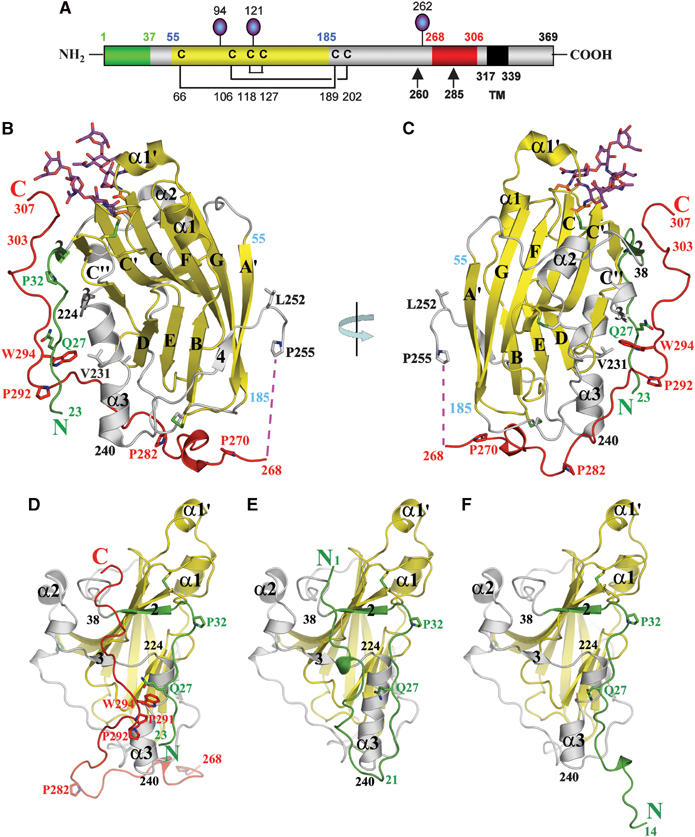
gD(23–306)307C structure. (A) Schematic representation of HSV-1 gD. Disulfide bonds are shown as black lines and N-linked oligosaccharides as lollipops. Colors of the N-terminal region, forming the HVEM-binding hairpin in the gD285–HVEM complex, the Ig-like core, and the C-terminal region past residue 255 are represented in green, yellow, and red, respectively. Positions of important amino acids and domain boundaries are numbered according to the mature form of gD. TM: transmembrane region. (B) Ribbon representation of the gD(23–306)307C subunit. The secondary structure elements are labeled as in Carfi et al (2001). The color code used is same as in (A). (C) As in (B) after 180° rotation as indicated. (D) Front view of gD(23–306)307C structure. The model is rotated 90° clockwise with respect to panel B to show the front side of gD, where the N- and C-terminal regions interact. (E) Front view of gD285 from the gD285–HVEM complex, with HVEM removed for clarity. (F) Front view of gD285 in the unbound state.
Table 1.
X-ray data collection and refinement statistics
| gD(23–306)307C | gD316A37C–V302C | |
|---|---|---|
| Data collection | ||
| Beam line | APS BM14 | ESRF ID-14 |
| Wavelength (Å) | 1.000 | 0.931 |
| Space group | P212121 | P212121 |
| Cell dimensions (Å) | a=74.25 | a=67.63 |
| b=106.19 | b=79.10 | |
| c=130.31 | c=123.56 | |
| α=β=γ=90° | α=β=γ=90° | |
| Resolution range (Å)a | 50.0–2.1 (2.18–2.10) | 50.0–2.5 (2.59–2.50) |
| Number of reflections | ||
| Total | 247 116 | 172 344 |
| Unique | 55 770 | 23 602 |
| Redundancy | 4.3 (3.9) | 7.3 (7.1) |
| Completeness (%) | 92.46 (78.12) | 99.61 (98.22) |
| Rmerge | 5.8 (42.0) | 8.1 (34.4) |
| Refinement | ||
| Resolution range (Å) | 30.0–2.1 | 30.0–2.5 |
| Reflections | 54 886 | 23 271 |
| Number of molecules per AU | 2 | 2 |
| Rcryst (%) | 19.89 | 20.72 |
| Rfree (%)b | 24.68 | 25.63 |
| Overall average B-factor (Å2) | 40.52 | 44.47 |
| R.m.s.d. bond lengths (Å) | 0.009 | 0.012 |
| R.m.s.d. bond angles (deg) | 1.317 | 1.462 |
| Ramachandran plot regions | ||
| Allowed | 89.6% | 88.6% |
| Additionally allowed | 9.2% | 9.8% |
| Generally allowed | 1.2% | 1.2% |
| Disallowed |
0.0% |
0.4% |
| aValues in parenthesis correspond to the highest resolution shell. | ||
| bThe R-free factor (Rfree) was calculated with 5.1% of the data omitted from refinement for both data collections. Rmerge=∑(∣(I−〈I〉)∣)/∑(I). | ||
Overall structure of the subunits of the gD(23–306)307C dimer
The overall structure of the subunits of the gD(23–306)307C dimer (Figure 1B–D) is very similar to that of the gD285 monomer when bound to HVEM (Figure 1E) or when in the unbound form (Figure 1F) (Carfi et al, 2001; Connolly et al, 2005). The secondary structure elements and loops of the Ig-like core (in yellow in Figure 1A) are almost identical in the three structures and overlap with r.m.s. deviations between 0.38 and 0.42 Å for residues 32–223. However, the N- and C-terminal extensions differ in the three structures. The first 22 amino acids that are missing in gD(23–306)307C (Figure 1D), form a hairpin when gD contacts HVEM (Figure 1E), and are extended in gD285 alone (Figure 1F). The C-terminal residues 224–240 are completely α-helical in the gD–HVEM complex, but the same α-helix is kinked at residues 231–232 and shifted by 2–3 Å in its C-terminal portion in both gD(23–306)307C and gD285 structures (Figure 1D–F). The kink is presumably caused by the lack of interaction with the N-terminal hairpin that is formed only when gD is in complex with HVEM. Of most importance, the gD(23–306)307C structure revealed the positions of a large number of C-terminal residues, past Glu259 (in red in Figure 1B and C), none of which were previously defined.
The structure of the gD C-terminus
In gD(23–306)307C, all of the C-terminal 50 amino acids are ordered and well defined in the electron density map (Supplementary Figure S1) except residues 256 to 267 for which no electron density is present (Figure 1B and C). From residue 270 to 282, the polypeptide chain first contacts residues at the bottom of the Ig-like core and subsequently turns upward in the direction of the N-terminal region. As a result, the last 16 C-terminal amino acids run roughly parallel to residues 23–32 and presumably point toward the viral membrane (Figure 1B–D). Residues from the 286–306 region contact residues 23–38 from the N-terminal region as well as residues 220–240 near or within the α3 α-helix (Figure 1D). A prominent feature of the C-terminal extension is the insertion of Pro291 and the side chain of Trp294 into a groove on the gD surface (Figure 1D). These two residues contact the main chain atoms of residues 23–27 and the side chains of Leu25 and Gln27 from the N-terminal region, as well as Phe223, Asn227, Thr230, Val231, and Tyr234 within the α3 α-helix and finally Ile290 from the C-terminus (Figure 2A and B). The relative positions of the N- and C-termini are further constrained by hydrogen bonds between the side chain of Gln27 and the main chain nitrogen and oxygen atoms of His295, as well as between the carbonyl oxygen of Leu25 and the side chain nitrogens of Asn293 and Trp294 (Figure 2A).
Figure 2.
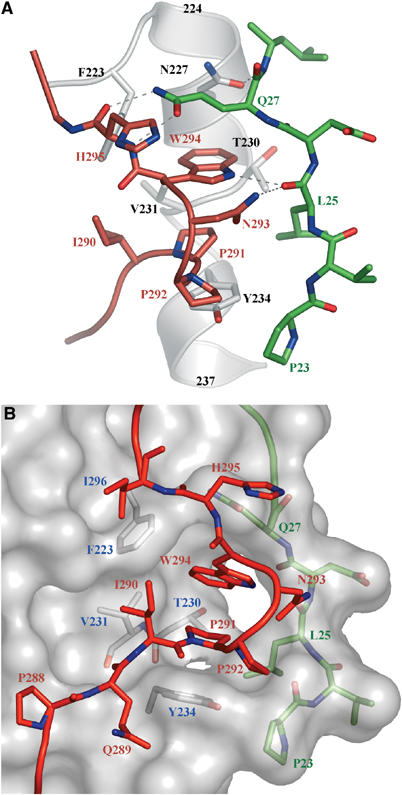
Interactions of the C-terminal region in the gD(23–306)307C structure. (A) Interactions of the 290–295 region (in red), with residues 23–27 (in green) and the α3 α-helix (in white). Dashed lines represent hydrogen bonds. (B) Surface representation of gD core (in gray) with residues involved in the formation of the Trp294- and Pro291-binding crevice shown underneath in white. The side chains of Pro291, Trp294, and other C-terminal residues are shown in red as ball-and-stick representation, whereas residues from the N-terminal region are shown in green under the surface.
The gD dimer and the C-terminus
In the crystal structure of gD(23–306)307C, a disulfide bond links the two subunits together to form a butterfly-shaped dimer (Figure 3A–C). An ion that we interpreted to be Zn2+ from the crystallization solution was trapped at the dimer interface by the tetra coordination of His39 and Asp215 of both subunits (light blue sphere in Figure 3A). A total of 1710 Å2 of solvent-accessible surface is buried in the dimer interface. Of this, the two C-termini contribute nearly 900 Å2 of the buried area. It is likely that the formation of this large dimer interface stabilized the conformation of the C-terminus and allowed us to trap it in the pre-receptor-binding conformation (see below).
Figure 3.
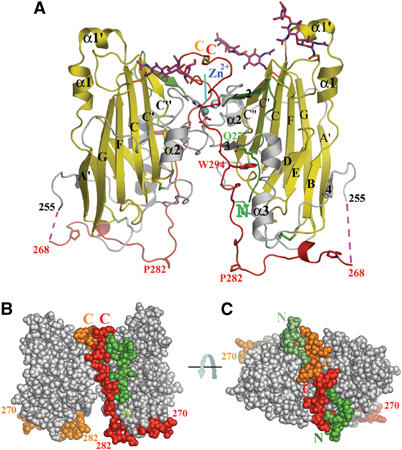
The disulfide-linked dimer of gD(23–306)307C. (A) Ribbon representation of the dimer. A Zn2+ ion, trapped at the dimer interface, and the disulfide bonds are shown. The red dotted lines represent a disordered part of the C-terminal region. N- and C-termini and important amino acids are indicated. (B) Sphere representation of the dimer. The C-termini (268–307) of the two subunits are in red (subunit A, on the right) and orange (subunit B), and the N-terminal residues (23–28) in dark green (A) and pale green (B). The view is as in A. (C) As in panel B after a 90° rotation around an axis parallel to the plane.
The gD C-terminus and receptor binding
A comparison of structures shows that the last 18 C-terminal residues of gD(23–306)307C occupy the same space as the first 16 N-terminal amino acids of gD285 in the gD285–HVEM complex (in red and green, respectively, in Figure 4A). Thus, formation of the N-terminal HVEM-binding hairpin requires the displacement of the gD C-terminus (see also Figure 1C–E). The atomic details of the gD/nectin-1 interaction are presently unknown, but mutagenesis studies (Whitbeck et al, 1999; Yoon et al, 2003; Jogger et al, 2004; Manoj et al, 2004; Connolly et al, 2005) showed that the first 32 amino acids of gD do not contribute to nectin-1 binding and their deletion does not affect HSV entry into nectin-1-expressing cells. Moreover, these studies pointed to a number of gD residues involved in the interaction with nectin-1. In gD(23–306)307C, three of these residues, Tyr38, Arg222, and Phe223, are buried under the C-terminal residues 290–303 (Figure 4A, and see also Figure 2B). This arrangement suggests that binding to nectin-1 would also require a displacement of the C-terminus. Therefore, gD binding to two unrelated receptors would require a displacement of the C-terminus and would cause similar conformational changes.
Figure 4.
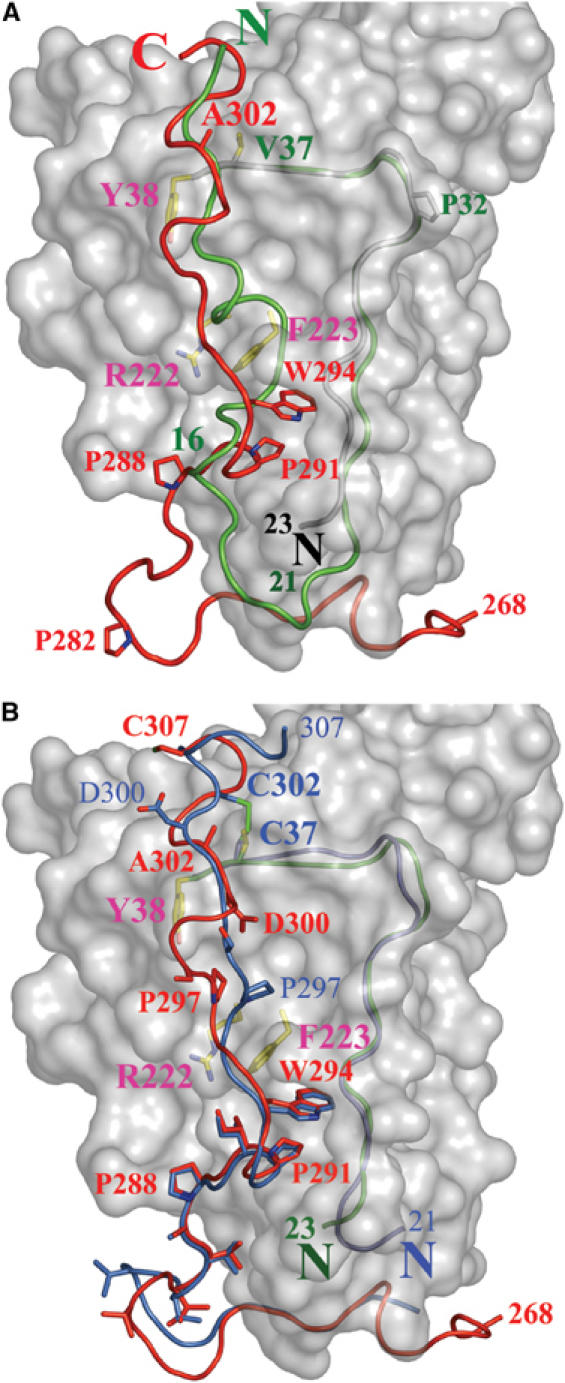
Superimposition of gD(23–306)307C and gD285 from the gD285–HVEM complex structure. (A) The gD(23–306)307C core is represented as a white surface. The N-terminal residues of gD285 (1–16, in green) from the gD285–HVEM complex occupy the same space as residues from the C-terminus of gD(23–306)307C (289–307, in red). The side chains of Tyr38, Phe223, and Arg222, three residues involved in nectin-1 binding and buried under C-terminal residues in gD(23–306)307C, are shown. Val37 and Ala302, two residues predicted to form a disulfide if mutated in cysteines, are also shown. (B) The C-terminus of gD(23–306)307C (in red) and of gD316V37C–A302C (in blue) occupy identical positions in the 285–296 region, but adopt different conformations in the 297–307 region. The disulfide bond between Cys302 and Cys37, the side chains of several C-terminal residues as well as those of Tyr38, Phe223, and Arg222 are shown. The N-terminal residues of gD(23–306)307C and gD316V37C–A302C are colored in green and blue, respectively.
To validate the position of the C-terminus and to discern how it modulates receptor binding, we used the structure of gD(23–306)307C to engineer an intramolecular disulfide bond in gD that should lock the C-terminus in the position observed in the gD(23–306)307C structure (Hazes and Dijkstra, 1988). The presence of several prolines in the gD C-terminus and the suboptimal relative orientations of the side chains of the N- and C-terminal residues limited the choice of residues suitable to form disulfide bonds when mutated to cysteines. Ultimately, we chose Val37 and Ala302, whose Cα are 5.0 Å apart in the gD(23–306)307C structure (Figure 4A). Both residues were simultaneously mutated to cysteines in a soluble form of gD encompassing its entire ectodomain (gD316V37C–A302C). Wild-type (wt) gD306 and gD316 have similar antigenic properties and bind to HVEM and nectin-1 with the same affinities (data not shown). The mutant protein was expressed in insect cells, purified, and formation of the correct intramolecular disulfide bonds was verified by mass spectrometry. gD316V37C–A302C was crystallized and its structure determined at 2.5 Å resolution (see Materials and methods and Table I). The first 20 amino acids and the last 10 amino acids of the C-terminus had poorly defined electron density, suggesting flexibility of these two regions.
The structure of gD316V37C–A302C was very similar to that of gD(23–306)307C, with an r.m.s. deviation of 0.34 Å for 215 Cα positions (residues 38–253). In particular, the 256–267 region was disordered in both structures, the C-terminal α3 α-helix was kinked and shifted compared to gD285-HVEM, and residues 285–296 occupied equivalent positions, with Pro291 and Trp294 side chains filling the same space in both structures (Figure 4B). Only residues 297–306 of gD316V37C–A302C assumed a conformation different from that seen in the gD(23–306)307C structure (Figure 4B). This was probably due to a repositioning of the region proximal to Cys302, whose position in the gD(23–306)307C structure was not optimal to form a disulfide bond with Cys37 (see above). Although this conformational difference is likely caused by the constraints imposed by the introduction of disulfide bonds, it highlights the conformational flexibility of the gD C-terminus past residue Pro297. Nevertheless, the C-terminus of gD316V37C–A302C still buries two residues, Phe223 and Arg222, both of which are proposed to be involved in nectin-1 binding (Whitbeck et al, 1999; Manoj et al, 2004). Interestingly, gD316V37C–A302C did not form dimers similar to those observed in the gD(23–306)307C structure, suggesting that the presence of the entire C-terminus is not sufficient to generate stable gD dimers. Moreover, the N-terminal 22 amino acids in the gD316V37C–A302C structure, although present, are disordered, suggesting that in the unliganded gD these residues are conformationally flexible even in the presence of the C-terminal amino acids.
The conformation of gD316V37C–A302C was validated by probing with conformation-dependent Mabs in native Western blots (Figure 5A). Under nondenaturing and nonreducing conditions, gD316V37C–A302C migrated faster compared to other soluble gDs (Figure 5A). This is consistent with gD316V37C–A302C adopting a more compact configuration when the C-terminus is locked. Furthermore, gD316V37C–A302C was recognized by the conformation-dependent Mab AP7. This is critical because the AP7 epitope is known to require correct folding of residues at both the N- and C-termini of gD (Chiang et al, 1994). In fact, AP7 binds both virion-associated gD (Minson et al, 1986) and soluble wt-gD306 (Chiang et al, 1994; Nicola et al, 1997), but does not bind to the N-terminal mutants Leu25Pro (Minson et al, 1986) or Gln27Pro (Rid-1) (Dean et al, 1994; Nicola et al, 1997), nor does it bind to gD285 (Rux et al, 1998) or denatured wt-gD306. Therefore, AP7 binding to gD316V37C–A302C provides strong evidence that the C-terminus is correctly folded in this molecule and that the ‘folded back' structure of the C-terminus observed in both gD(23–306)307C and gD316V37C–A302C is also present in virion gD.
Figure 5.
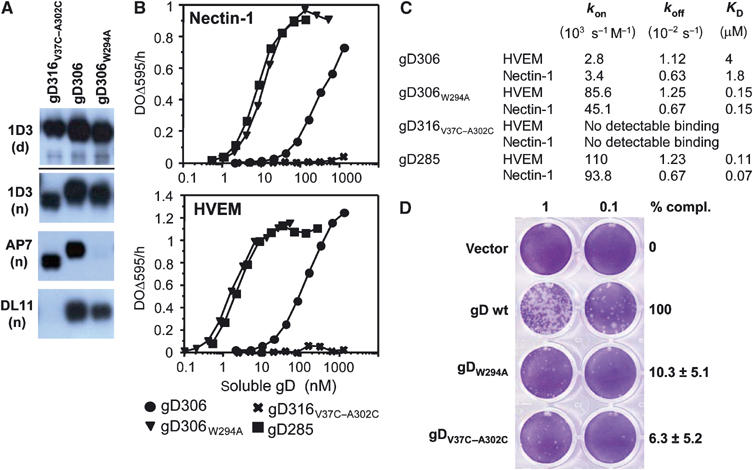
Biochemical and functional characterization of gD mutants. (A) Western blots of purified gD ectodomains were probed by various Mabs. Mab 1D3 detects a linear epitope (aa 11–19) under both denaturing/reducing (d) and native conditions (n) electrophoresis. Mabs AP7 and DL11 detect conformation-dependent epitopes under native conditions of electrophoresis. (B) Receptor binding as detected by ELISA. Various concentrations of the indicated forms of purified gD ectodomains were added to immobilized receptor ectodomains (i.e. nectin or HVEM). The bound gD was detected with anti-gD polyclonal serum R7. (C) Kinetic constants (kon and koff) and affinity constant (KD) for gD–HVEM and gD–nectin-1 complexes were measured by surface plasmon resonance and fitted to a 1:1 Langmuir interaction model (see Materials and methods). (D) Complementation of HSV FgDβ with gD mutants and titration of complemented virus on VD60 cells. VD60 cells were stained with crystal violet to visualize plaques. Complemented virus present in the culture medium of transfected–infected cells was undiluted or diluted 10-fold. Titers of complemented viruses are represented as percentage of wt on the right. These numbers are an average of four experiments, including the one presented on the left. Error bars represent ±1 s.d. Vector indicates the use of pcDNA3.1 for transfection as a negative control.
In spite of having a native-like conformation as assessed by Mab binding, soluble gD316V37C–A302C did not bind either HVEM or nectin-1 (Figure 5B and C). The lack of receptors binding of gD316V37C–A302C is consistent with our hypothesis that the gD C-terminus has to be displaced for either receptor to bind. The lack of HVEM binding is explained by our structural analysis showing that when the C-terminus is in the folded back position residues 289–306 occupy the same space as the first 16 amino acids of the N-terminal HVEM-binding hairpin (Figure 4A). On the other hand, the lack of nectin binding is consistent with the observation that the C-terminal gD residues 289–303 bury residues involved in nectin binding (Figure 4B). It should be noted that the C-terminus of gD does not interact with receptors and that the point mutation Val37Ala did not affect nectin binding (Connolly et al, 2005), suggesting that the locked position of the C-terminal residues rather than the cysteine mutations prevents the interaction of gD316V37C–A302C with nectin. A similar mechanism may explain why gD316V37C–A302C was not recognized by the potent virus-neutralizing antibody DL11 (Figure 5A). The binding of Mab DL11 to gD on the virus envelope or as a soluble protein completely blocks interaction of the glycoprotein with nectin-1 and HVEM (Nicola et al, 1998; Whitbeck et al, 1999; Connolly et al, 2005).
The contribution of Trp294 to the structure of the gD C-terminus
In the structures of gD(23–306)307C and gD316V37C–A302C, the Trp294 side chain points towards a groove on the gD surface and contacts residues at both the N- and C-termini. This suggests that Trp294 plays an important role in anchoring the C-terminus in proximity of the N-terminal region. To test this hypothesis and to further validate the position of the C-terminus observed in the two structures, we mutated Trp294 to Ala. The gD306W294A mutant was expressed in insect cells and the protein was purified and characterized for its ability to bind Mabs and receptors (Figure 5A and B). The mutant protein bound to nectin-1 and HVEM 10–50 times better than did gD306, which is similar to what was observed for gD285 (Rux et al, 1998; Willis et al, 1998b; Krummenacher et al, 1999) (Figure 5B and C). Similar to gD285, the difference in affinity between the point mutant gD306W294A and gD306 was mainly due to an increase in the rate of complex formation (kon), as determined by surface plasmon resonance experiments (Figure 5C). Likely, the Trp294Ala mutation destabilizes the structure of the C-terminus, which is normally constrained by the large Trp side chain. Thus, this mutation would favor both exposure of the nectin-1-binding site and formation of the N-terminal HVEM-binding hairpin, thereby accounting for the increase in the rate of complex formation (kon) without affecting the rate of dissociation (koff) (Figure 5C). Importantly, the mutant gD306W294A was recognized on a Western blot under native conditions by all the conformation-dependent Mabs tested except for AP7 (Figure 5A and data not shown). Since this Mab bound to gD316V37C–A302C, where the Trp294 indole side chain is buried, we infer that Mab AP7 does not interact directly with this amino-acid side chain, and that the position of Trp294 is critical for formation of the AP7 epitope. Therefore, the lack of AP7 binding to gD306W294A results from disruption of its conformational epitope, plausibly caused by an unfolding of the C-terminus. Moreover, the fact that AP7 bound to gD316V37C–A302C but not gD306W294A demonstrates that the region in proximity of Trp294 as well as the region containing Gln27 and Leu25, two residues critical for AP7 binding, are correctly folded in gD316V37C–A302C. The conformations of both regions are revealed by the crystal structure of gD316V37C–A302C, and, importantly, are the same as in the gD(23–306)307C structure (Figure 4B). Thus, the receptor and antibody binding data for gD306W294A along with those for gD316V37C–A302C support the crystallographic model and suggest that Trp294 plays a crucial role in holding the C-terminal residues (aa 282–300) in proximity of N-terminal residues (aa 23–30), in the pre-receptor-binding conformation of gD.
The role of the C-terminus of gD in HSV cell entry
The ability of full-length gD-carrying mutations Trp294Ala or Val37Cys/Ala302Cys to complement a gD-null virus for entry was compared to that of wt-gD. Complemented viruses were titered on VD60 cells (Figure 5D). Both gD mutants were impaired in promoting HSV entry and complemented gD-null virus entry at levels of about 10% of wt-gD. This phenotype has been previously observed in the case of gD(∇290–299), where the indicated residues were substituted by a linker insertion (Chiang et al, 1994; Milne et al, 2003). Interestingly, despite a more flexible C-terminus and an increased affinity for receptors, gDW294A has reduced activity in a viral entry assay. Furthermore, the reduced infectivity of the Val37Cys/Ala302Cys double mutant is consistent with the inability of the corresponding soluble gD to bind HVEM and nectin-1. As noted above, the C-terminus of gD, past amino acid 260, does not participate in a direct interaction with either receptor, and the Val37Ala point mutation did not affect nectin binding (Connolly et al, 2005), suggesting that the defect in complementation is not due to a lack of receptor binding caused by the single cysteine mutations, but rather that the intramolecular disulfide bond between these cysteines formed also when this mutant protein was expressed on the viral envelope.
Discussion
The structures reported here include all of the C-terminal residues of the gD ectodomain, specifically those involved in regulating receptor binding, and provides a structural framework for understanding the mechanism of receptor-induced activation of gD during HSV cell entry. The structure of gD(23–306)307C was obtained by introducing a cysteine at the C-terminus of gD306 that allowed formation of dimeric molecule. In the gD dimer, the C-termini of each subunit interact with residues derived from the other subunit and contribute to the formation of the dimer interface. It is important to note that, although the surface buried in the dimer interface is in the range observed for protein–protein interactions (Lo Conte et al, 1999), the data do not allow us to ascertain if the covalent gD dimer observed in the crystals represents the gD dimer on the viral membrane. Of significance, however, this structure along with the biochemical and functional data in this and previous reports clearly show that we have trapped the C-terminus of the gD ectodomain in its pre-receptor-binding conformation.
In the structure of gD(23–306)307C, the C-terminus wraps around the gD-core and amino acids 289–306 impede the formation of the HVEM-binding hairpin and simultaneously bury residues involved in nectin-1 binding (Manoj et al, 2004; Connolly et al, 2005). The structural data are consistent with a recent report showing that C-terminal peptides (aa 260–285 or 285–306) can Co-IP the core of gD (aa 1–260), but not the full-length gD ectodomain (aa 1–306) or gD260–receptor complexes (Fusco et al, 2005). In addition, our finding also explains previous results showing that gD285, lacking those C-terminal residues, binds both receptors 100 times faster than gD306 (Rux et al, 1998; Willis et al, 1998b; Krummenacher et al, 1999). We observed that the point mutant gD306W294A exhibited a similar increase in kon and in receptor affinity. Interestingly, another point mutant gD306Q27P bound nectin-1 10-fold better than wt-gD306 (Willis et al, 1998b; Krummenacher et al, 1999). In the gD(23–306)307C structure, the Trp294 side chain protrudes into a crevice on the gD surface and the side chain of Gln27 stacks on top of the tryptophan indole ring (Figure 2). Thus, the properties of the Trp294Ala and Gln27Pro mutants are in agreement with the structure and emphasize the key role of Trp294 in keeping the C- and N-termini together.
To further discern the structure and function of the gD C-terminus, we introduced two cysteines into gD to lock the C-terminus in the same position observed in the gD(23–306)307C structure through an intramolecular disulfide bond. The structure of gD316V37C–A302C was similar to that of each subunit of gD(23–306)307C with Pro291 and the Trp294 side chain protruding into the same crevice in both structures. The gD316V37C–A302C molecule was recognized by the Mab AP7, as was gD306 and virion gD (Minson et al, 1986; Nicola et al, 1998), indirectly confirming the biological relevance of the structural model and the proximity of the N- and C-termini of gD in the virion envelope. Importantly, this ‘locked' gD was unable to bind to nectin-1 or HVEM, suggesting that the gD C-terminus must move for receptor to bind. When the Val37Cys/Ala302Cys mutations were introduced into full-length gD, the mutant protein failed to complement the infectivity of a gD-null virus. This suggests that the full-length form of this mutant in the virion envelope contains the engineered disulfide bond and shows that an ‘unlocked' and flexible C-terminus is required for gD function.
Although only residues 1–260 of HSV gD are sufficient for interaction with receptor, the region encompassing residues 260–285 of HSV is also required in an HSV/pseudorabies virus (PrV) gD hybrid for this molecule to promote cell fusion in collaboration with HSV gB and gH/gL (Zago et al, 2004). Indeed, soluble gD285 and gD306, but not gD260, rescued, albeit with low efficiency, the entry of a gD-null virus when added after virus attachment to the cell surface (Cocchi et al, 2004). This report showed that the region past amino acid 285 is not required for soluble gD to facilitate entry of a gD-null HSV after this virus is stably attached to cells, and identified the region 260–285, referred to as the profusion domain (PFD), as important in a post-receptor-binding step of the entry process. However, when a form of gD with an insertion/deletion in the 290–299 region (i.e. gD(∇290–299)) replaced full-length gD on the viral envelope, the resulting virus was impaired in cell entry (Chiang et al, 1994; Milne et al, 2003). Similarly, when we mutated Trp294, which we have shown to play a central role in positioning the gD C-terminus, to Ala in the virion gD, the resulting virus was defective in cell entry. Trp294 is outside the PFD, indicating that the entry defect of the Trp294Ala mutant is directly linked to the destabilization of the conformation of the gD C-terminus and not due to a post-receptor-binding role of this amino acid. Therefore, the biochemical and functional data demonstrate that prior to receptor binding this region assumes the folded back structure, as observed in gD(23–306)307C and gD316V37C–A302C. In addition, these data suggest that the gD C-terminus must be displaced concomitant with receptor binding since the ‘pretriggered' gDW294A and gD(∇290–299), when incorporated into the viral envelope, are impaired in function. Importantly, Pro291 and Trp294 are conserved in HSV-2 gD and a PxxW motif (where x is any amino acid; Supplementary Figure S2) is also present at the C-terminus of both bovine herpes virus (BHV)-1 and PrV gDs, suggesting a conserved role for these two residues and for the C-terminus in gD-mediated alphaherpesvirus entry.
We propose a model for activation of gD and initiation of membrane fusion based on data in this paper as well as recent functional data (Cocchi et al, 2004; Zago et al, 2004; Fusco et al, 2005) (Figure 6). We suggest that HSV has a ‘switch-on' mechanism that allows activation only when the virion is apposed to the target cell surface. We postulate that gD on the viral envelope is in an autoinhibited conformation, in which the folded-back C-terminus of the ectodomain is stabilized by contacts that include Trp294. Masking critical receptor sites might also prevent efficient neutralization by antibodies, as described for HIV-1 gp120 (Wyatt et al, 1998). We postulate that the flexible C-terminus of gD is in a conformational equilibrium between a closed state and a partially opened state, with the side chain of Trp294 in and out of the groove, respectively. Nectin-1 and HVEM bind and trap gD in the open form, stably exposing the C-terminal 20 residues of the ectodomain by displacing the gD-core. How many residues N-terminal to the 289–306 segment are displaced by receptor binding could be determined by introducing additional disulfide bonds, based on the structure presented here, to lock different C-terminal segments to the gD core. Were the entire 268–306 segment to peel away from the core, the latter could be translocated away from its TM anchor by more than 100 Å. Artificial destabilization of the gD C-terminus does not lead to a constitutively active form of gD, arguing that its displacement must be concomitant with receptor binding. This mechanism would prevent premature activation of the fusion apparatus by provoking the conformational change in gD only when the virion is productively attached to a target cell.
Figure 6.
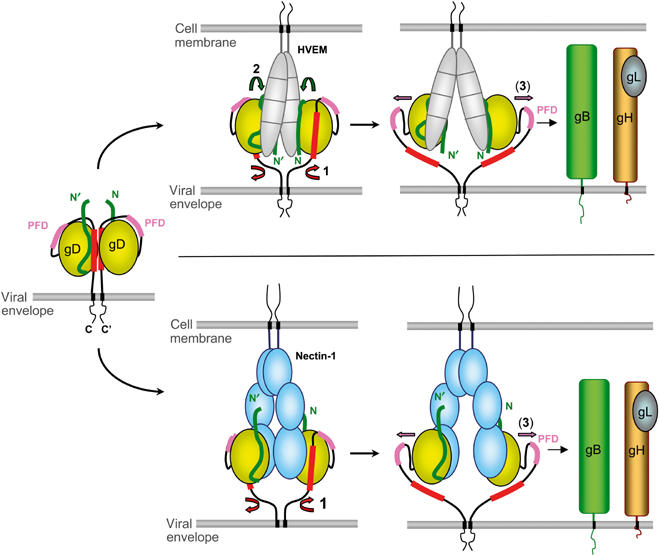
Proposed mechanism for receptor-mediated activation of HSV gD. Envelope gD is shown, as a putative dimer, in its unbound state as well as during interaction with HVEM (top) or nectin-1 (bottom). Conformational changes are chronologically indicated by numbered arrows: (1) displacement of the C-terminus, (2) folding of the gD N-terminus in the case of HVEM binding, and (3) exposure of the PFD. The N-terminus of gD is shown in green, the C-terminus (290–299) is colored red, and the PFD (260–285) is pink.
How exactly gD triggers fusion after being activated by receptor binding remains to be elucidated. After receptor binding, the ‘activated' gD core might interact with another glycoprotein such as gB and/or gH/gL and induce a conformational change in one of them. This interaction could involve the conversion of a flexible 256–268 region, not defined in our structure, to a more ordered structure upon interaction with other proteins. Alternatively, gD could play a role in stabilizing (i.e. decreasing the energy barrier) a fusion intermediate of gB and/or gH/gL, the formation of which would have been previously triggered by other factors such as a specific cellular receptor or local physiological conditions.
In summary, we have presented evidence that the C-terminus of the gD ectodomain is a flexible structural element that interferes with receptor binding. Our data suggest that in the presence of a receptor this region is displaced, possibly causing a movement of the gD core. We postulate that Trp294 plays a key role in ensuring that this conformational change happens only in the presence of a functional gD receptor and that triggering occurs only in proximity to the cell membrane. Our structure provides a glimpse of the initial conformational changes that are likely to occur when receptors bind to the virus, and provide a mechanism for receptor-mediated gD activation during HSV entry.
Materials and methods
Protein expression and purification
Purifications of soluble HVEM and nectin-1 have been described previously (Whitbeck et al, 1997; Krummenacher et al, 1998). Construction of gD306307C was performed as described for gD306 (Sisk et al, 1994; Willis et al, 1998a), but with a cysteine codon added in the downstream primer used for PCR amplification of gD sequences. Mutations Trp294Ala in plasmid pDL473 and Val37Cys/Ala302Cys in pDL485 were engineered with Quickchange mutagenesis kit (Stratagene) using pSC390 as template (Connolly et al, 2003). Subcloning in pVtBac vector and recombination into baculovirus followed previously described protocols (Sisk et al, 1994; Willis et al, 1998a).
Purifications of soluble forms of gD used in functional and biochemical assays were performed as described previously (Nicola et al, 1997; Carfi et al, 2001). Complete disulfide formation between the two newly introduced cysteines in gD316V37C–A302C occurred spontaneously during protein expression, as verified by the lack of reaction with iodoacetamide, migration on nonreducing SDS–PAGE gels, and limited proteolysis coupled to mass spectrometry. No disulfide-linked gD316V37C–A302C dimers were detected in gel filtration experiments.
Crystallization, structure determination, and refinement
The proteins under study were crystallized by the hanging drop vapor diffusion method. N-terminal sequencing of the gD306307C used in crystallization experiments revealed cleavage of the first 22 amino acids. Crystals of gD(23–306)307C dimer were obtained at pH 6.0 in 100 mM sodium cacodylate buffer, 100 mM NaCl, 100 μM ZnAc2, and 20% PEG 4K. A 2.1 Å resolution data set was collected at APS BM14 beam line on a Q4-ADSC CCD detector. Crystals belong to the orthorhombic P212121 space group and contain one dimer in the asymmetric unit (Table I). Crystals of gD316V37C–A302C were obtained at pH 4.6 in 100 mM sodium acetate buffer, 10 mM ZnCl2, and 24% PEG 6K. A 2.5 Å data set was collected at the ESRF ID14-3H beam line on a MAR CCD detector. Crystals belong to the orthorhombic P212121 space group with two molecules in the asymmetric unit (Table I). The two structures were solved independently by Molecular Replacement with the programs AMoRe and Molrep, respectively (CCP4, 1994), using the structure of gD285 as a search model. Several cycles of model building with O (Jones et al, 1991) followed by map improvement with Arp/wArp in Molrep mode (Perrakis et al, 1999) resulted in the complete models. Both structures were refined with Refmac (CCP4, 1994) using a maximum likelihood target. Individual B-factors refinement and bulk solvent corrections were applied throughout (Table I). NCS restraints were used only during the initial stages of the refinement.
ELISA, Western blot, and biosensor analysis
ELISA was performed as described previously to measure binding of soluble gD on immobilized HVEM or nectin-1 (Whitbeck et al, 1997; Krummenacher et al, 1998). Western blots on purified glycoproteins were performed under reducing/denaturing conditions using standard procedure. Low SDS ‘native' gel electrophoresis was performed without reducing agents to test conformation-dependent Abs (Cohen et al, 1986). Biosensor experiments were conducted on a BIAcore X instrument as described previously (Willis et al, 1998b; Krummenacher et al, 1999). Briefly, about 2000 RU of HVEM(200t) or of nectin-1(346t) were immobilized on a CM5 chip. Soluble gD diluted in HBS-EP (10 mM HEPES, 150 mM NaCl, 3 mM EDTA, 0.005% polysorbate 20) was flowed across the surface at 25°C at 50 μl/min. Sensorgrams were corrected for nonspecific binding and refractive index change by subtracting control sensorgrams from a receptor-free surface from the receptor surface sensorgrams. Data were analyzed with BIAevaluation software, version 3.0, using a 1:1 Langmuir interaction as global-fitting model for at least four concentrations of gD. Using this model, the χ2 value (the standard statistical measure of the closeness of fit) was below 2.5, indicating a good fit (Rux et al, 1998). Effect of mass transport was tested by increasing the flow rate up to 100 μl/min without affecting binding of the high-affinity gD285t to the immobilized receptors. Integration of mass transport parameters in the fitting model did not improve the quality of the fit, nor did it significantly affect the values of the kinetic constants. The rates of association (kon) and dissociation (koff) were used to calculate the affinity (KD=koff/kon).
Complementation assay
Plasmids pSC390 (wt gD), pDL473 (gDW294A), and pDL485 (gDV37C-A302C) or pcDNA3.1 as a negative control were used to transfect Vero cells (0.6 × 106 cells/well). The next day, cells were infected with HSV-1 FgDβ (Ligas and Johnson, 1988) at 106 pfu per well. After 2 h at 37°C, extracellular virus was inactivated with a 5-min acid wash (sodium citrate buffer, pH 3.0). At 24 h postinfection, plates were subjected to three freeze–thaw cycles. The virus-containing supernatant was cleared from cellular debris by centrifugation. Complemented virus was titered in parallel on Vero cells and on complementing VD60 cells. VD60 cells are Vero cells expressing wt-gD upon HSV infection, allowing plaque formation necessary for titration of complemented gD-null viruses (Ligas and Johnson, 1988). After 3 days, cells were fixed with formaldehyde and stained with crystal violet. Plaques were rarely observed on Vero cells; this background number was subtracted from plaque count on VD60 cells to determine the actual titer of complemented virus. Complementation with empty vector pcDNA3.1 did not yield infectious virus. Percent complementation was determined as percentage of titer of virus complemented with mutated gD compared to wt-gD.
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 1 Legend
Supplementary Figure 2 Legend
Acknowledgments
We thank Chiara Bruckmann, Huan Lou, and Haiyung Gong for help during protein purification and crystallization, as well as Melissa Sanchez, Dan Landsburg, and Wangfang Hou for help with baculovirus and plasmid constructions, and Tina Cairns, Richard Milne, Ann Rux, Sharon Willis, and Micah Luftig for advice and discussion. We are grateful to Dr David Johnson for VD60 cells, Dr Patricia Spear for KOSgDβ virus, CHO-K1 and CHO-HVEM12 cells, and Dr Tony Minson for the AP7 antibody. We would like also to thank Professor Stephen Harrison for critically reading the manuscript and Manuela Emile for help with artwork. This investigation was supported by Public Health Service grants NS-36731 from the National Institute of Neurological Disease and Stroke (RJE and GHC), AI-18289 and AI-056045 from the National Institute of Allergy and Infectious Diseases (RJE and GHC), and by a grant from the Ministero dell'Istruzione, dell'Università e della Ricerca (MIUR). The atomic coordinates have been deposited in the PDB (codes 2C3A and 2C36).
References
- Campadelli-Fiume G, Cocchi F, Menotti L, Lopez M (2000) The novel receptors that mediate the entry of herpes simplex viruses and animal alphaherpesviruses into cells. Rev Med Virol 10: 305–319 [DOI] [PubMed] [Google Scholar]
- Carfi A, Gong H, Lou H, Willis SH, Cohen GH, Eisenberg RJ, Wiley DC (2002) Crystallization and preliminary diffraction studies of the ectodomain of the envelope glycoprotein D from herpes simplex virus 1 alone and in complex with the ectodomain of the human receptor HveA. Acta Crystallogr D 58: 836–838 [DOI] [PubMed] [Google Scholar]
- Carfi A, Willis SH, Whitbeck JC, Krummenacher C, Cohen GH, Eisenberg RJ, Wiley DC (2001) Herpes simplex virus glycoprotein D bound to the human receptor HveA. Mol Cell 8: 169–179 [DOI] [PubMed] [Google Scholar]
- CCP4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr D 50: 760–763 [DOI] [PubMed] [Google Scholar]
- Chiang H-Y, Cohen GH, Eisenberg RJ (1994) Identification of functional regions of herpes simplex virus glycoprotein gD by using linker-insertion mutagenesis. J Virol 68: 2529–2543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocchi F, Fusco D, Menotti L, Gianni T, Eisenberg RJ, Cohen GH, Campadelli-Fiume G (2004) The soluble ectodomain of herpes simplex virus gD contains a membrane-proximal pro-fusion domain and suffices to mediate virus entry. Proc Natl Acad Sci USA 101: 7445–7450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen GH, Isola VJ, Kuhns J, Berman PW, Eisenberg RJ (1986) Localization of discontinuous epitopes of herpes simplex virus glycoprotein D: use of a nondenaturing (‘native' gel) system of polyacrylamide gel electrophoresis coupled with Western blotting. J Virol 60: 157–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly SA, Landsburg DJ, Carfi A, Whitbeck JC, Zuo Y, Wiley DC, Cohen GH, Eisenberg RJ (2005) Potential nectin-1 binding site on herpes simplex virus glycoprotein D. J Virol 79: 1282–1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly SA, Landsburg DJ, Carfi A, Wiley DC, Cohen GH, Eisenberg RJ (2003) Structure-based mutagenesis of herpes simplex virus glycoprotein D defines three critical regions at the gD/HveA interface. J Virol 77: 8127–8140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly SA, Landsburg DJ, Carfi A, Wiley DC, Eisenberg RJ, Cohen GH (2002) Structure-based analysis of the herpes simplex virus glycoprotein D binding site present on herpesvirus entry mediator HveA (HVEM). J Virol 76: 10894–10904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean HJ, Terhune SS, Shieh M, Susmarski N, Spear PG (1994) Single amino acid substitutions in gD of herpes simplex virus 1 confer resistance to gD-mediated interference and cause cell-type-dependent alterations in infectivity. Virology 199: 67–80 [DOI] [PubMed] [Google Scholar]
- Fusco D, Forghieri C, Campadelli-Fiume G (2005) The pro-fusion domain of herpes simplex virus glycoprotein D (gD) interacts with the gD N terminus and is displaced by soluble forms of viral receptors. Proc Natl Acad Sci USA 102: 9323–9328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haan KM, Kwok WW, Longnecker R, Speck P (2000) Epstein–Barr virus entry utilizing HLA-DP or HLA-DQ as a coreceptor. J Virol 74: 2451–2454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handler CG, Cohen GH, Eisenberg RJ (1996) Crosslinking of glycoprotein oligomers during herpes simplex virus type 1 entry. J Virol 70: 6076–6082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazes B, Dijkstra BW (1988) Model building of disulfide bonds in proteins with known three-dimensional structure. Prot Eng 2: 119–125 [DOI] [PubMed] [Google Scholar]
- Jogger CR, Montgomery RI, Spear PG (2004) Effects of linker-insertion mutations in herpes simplex virus 1 gD on glycoprotein-induced fusion with cells expressing HVEM or nectin-1. Virology 318: 318–326 [DOI] [PubMed] [Google Scholar]
- Jones TA, Zou J-Y, Cowan SW, Kjeldgaard M (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A 47 (Part 2): 110–119 [DOI] [PubMed] [Google Scholar]
- Krummenacher C, Nicola AV, Whitbeck JC, Lou H, Hou W, Lambris JD, Geraghty RJ, Spear PG, Cohen GH, Eisenberg RJ (1998) Herpes simplex virus glycoprotein D can bind to poliovirus receptor-related protein 1 or herpesvirus entry mediator, two structurally unrelated mediators of virus entry. J Virol 72: 7064–7074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krummenacher C, Rux AH, Whitbeck JC, Ponce de Leon M, Lou H, Baribaud I, Hou W, Zou C, Geraghty RJ, Spear PG, Eisenberg RJ, Cohen GH (1999) The first immunoglobulin-like domain of HveC is sufficient to bind herpes simplex virus gD with full affinity while the third domain is involved in oligomerization of HveC. J Virol 73: 8127–8137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Spriggs MK, Kovats S, Turk SM, Comeau MR, Nepom B, Hutt-Fletcher LM (1997) Epstein–Barr virus uses HLA class II as a cofactor for infection of B lymphocytes. J Virol 71: 4657–4662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ligas MW, Johnson DC (1988) A herpes simplex virus mutant in which glycoprotein D sequences are replaced by β-galactosidase sequences binds to but is unable to penetrate into cells. J Virol 62: 1486–1494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Conte L, Chothia C, Janin J (1999) The atomic structure of protein–protein recognition sites. J Mol Biol 285: 2177–2198 [DOI] [PubMed] [Google Scholar]
- Manoj S, Jogger CR, Myscofski D, Yoon M, Spear PG (2004) Mutations in herpes simplex virus glycoprotein D that prevent cell entry via nectins and alter cell tropism. Proc Natl Acad Sci USA 101: 12414–12421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne RSB, Hanna SL, Rux AH, Willis SH, Cohen GH, Eisenberg RJ (2003) Function of herpes simplex virus type 1 gD mutants with different receptor-binding affinities in virus entry and fusion. J Virol 77: 8962–8972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minson AC, Hodgman TC, Digard P, Hancock DC, Bell SE, Buckmaster EA (1986) An analysis of the biological properties of monoclonal antibodies against glycoprotein D of herpes simplex virus and identification of amino acid substitutions that confer resistance to neutralization. J Gen Virol 67: 1001–1013 [DOI] [PubMed] [Google Scholar]
- Mullen MM, Haan KM, Longnecker R, Jardetzky TS (2002) Structure of the Epstein–Barr virus gp42 protein bound to the MHC class II receptor HLA-DR1. Mol Cell 9: 375–385 [DOI] [PubMed] [Google Scholar]
- Nicola AV, Peng C, Lou H, Cohen GH, Eisenberg RJ (1997) Antigenic structure of soluble herpes simplex virus glycoprotein D correlates with inhibition of HSV infection. J Virol 71: 2940–2946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicola AV, Ponce de Leon M, Xu R, Hou W, Whitbeck JC, Krummenacher C, Montgomery RI, Spear PG, Eisenberg RJ, Cohen GH (1998) Monoclonal antibodies to distinct sites on the herpes simplex virus (HSV) glycoprotein D block HSV binding to HVEM. J Virol 72: 3595–3601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrakis A, Morris R, Lamzin VS (1999) Automated protein model building combined with iterative structure refinement. Nat Struct Biol 6: 458–463 [DOI] [PubMed] [Google Scholar]
- Rux AH, Willis SH, Nicola AV, Hou W, Peng C, Lou H, Cohen GH, Eisenberg RJ (1998) Functional region IV of glycoprotein D from herpes simplex virus modulates glycoprotein binding to the herpes virus entry mediator. J Virol 72: 7091–7098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sisk WP, Bradley JD, Leipold RJ, Stoltzfus AM, Ponce de Leon M, Hilf M, Peng C, Cohen GH, Eisenberg RJ (1994) High-level expression and purification of secreted forms of herpes simplex virus type 1 glycoprotein gD synthesized by baculovirus-infected insect cells. J Virol 68: 766–775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spear PG (2004) Herpes simplex virus: receptors and ligands for cell entry. Cell Microbiol 6: 401–410 [DOI] [PubMed] [Google Scholar]
- Spear PG, Eisenberg RJ, Cohen GH (2000) Three classes of cell surface receptors for alphaherpesvirus entry. Virology 275: 1–8 [DOI] [PubMed] [Google Scholar]
- Spear PG, Longnecker R (2003) Herpesvirus entry: an update. J Virol Methods 77: 10179–10185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitbeck JC, Muggeridge MI, Rux A, Hou W, Krummenacher C, Lou H, van Geelen A, Eisenberg RJ, Cohen GH (1999) The major neutralizing antigenic site on herpes simplex virus glycoprotein D overlaps a receptor-binding domain. J Virol 73: 9879–9890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitbeck JC, Peng C, Lou H, Xu R, Willis SH, Ponce de Leon M, Peng T, Nicola AV, Montgomery RI, Warner MS, Soulika AM, Spruce LA, Moore WT, Lambris JD, Spear PG, Cohen GH, Eisenberg RJ (1997) Glycoprotein D of herpes simplex virus (HSV) binds directly to HVEM, a member of the TNFR superfamily and a mediator of HSV entry. J Virol 71: 6083–6093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitley RJ (2001) Herpes simplex viruses. In Fields Virology, Knipe DM and Howley PM (eds), pp 2461–2509. Philadelphia: Lippincott, Williams and Wilkins [Google Scholar]
- Willis SH, Peng C, Ponce de Leon M, Nicola AV, Rux AH, Cohen GH, Eisenberg RJ (1998a) Expression and purification of secreted forms of HSV glycoproteins from baculovirus-infected insect cells. In Methods in Molecular Medicine, Brown SM, MacLean AR (eds), Vol. 10: Herpes Simplex Virus Protocols, pp 131–156. Totowa, NJ: Humana Press Inc [DOI] [PubMed] [Google Scholar]
- Willis SH, Rux AH, Peng C, Whitbeck JC, Nicola AV, Lou H, Hou W, Salvador L, Cohen GH, Eisenberg RJ (1998b) Examination of the kinetics of herpes simplex virus glycoprotein D binding to the herpesvirus entry mediator, using surface plasmon resonance. J Virol 72: 5937–5947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt R, Kwong PD, Desjardins E, Sweet RW, Robinson J, Hedrickson WA, Sodroski JG (1998) The antigenic structure of the HIV gp120 envelope glycoprotein. Nature 393: 705–711 [DOI] [PubMed] [Google Scholar]
- Yoon M, Zago A, Shukla D, Spear PG (2003) Mutations in the N termini of herpes simplex virus type 1 and 2 gDs alter functional interactions with the entry/fusion receptors HVEM, nectin-2, and 3-O-sulfated heparan sulfate but not with nectin-1. J Virol 77: 9221–9231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zago A, Jogger CR, Spear PG (2004) Use of herpes simplex virus and pseudorabies virus chimeric glycoprotein D molecules to identify regions critical for membrane fusion. Proc Natl Acad Sci USA 101: 17498–17503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou G, Avitabile E, Campadelli-Fiume G, Roizman B (2003) The domains of glycoprotein D required to block apoptosis induced by herpes simplex virus 1 are largely distinct from those involved in cell–cell fusion and binding to nectin1. J Virol 77: 3759–3767 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 1 Legend
Supplementary Figure 2 Legend