

Abstract
ATP-sensitive potassium (KATP) channels conduct potassium ions across cell membranes and thereby couple cellular energy metabolism to membrane electrical activity. Here, we report the heterologous expression and purification of a functionally active KATP channel complex composed of pore-forming Kir6.2 and regulatory SUR1 subunits, and determination of its structure at 18 Å resolution by single-particle electron microscopy. The purified channel shows ATP-ase activity similar to that of ATP-binding cassette proteins related to SUR1, and supports Rb+ fluxes when reconstituted into liposomes. It has a compact structure, with four SUR1 subunits embracing a central Kir6.2 tetramer in both transmembrane and cytosolic domains. A cleft between adjacent SUR1s provides a route by which ATP may access its binding site on Kir6.2. The nucleotide-binding domains of adjacent SUR1 appear to interact, and form a large docking platform for cytosolic proteins. The structure, in combination with molecular modelling, suggests how SUR1 interacts with Kir6.2.
Keywords: CryoEM, diabetes, KATP channel, Kir6.2, sulphonylurea receptor
Introduction
ATP-sensitive potassium (KATP) channels couple changes in cell metabolism to electrical activity of the plasma membrane. Crucially, they control insulin secretion from pancreatic β-cells, protect against cardiac stress and brain seizures, mediate ischemic preconditioning in the heart and brain, and set the tone of vascular smooth muscle (Seino and Miki, 2003; Ashcroft, 2005; Kane et al, 2005). KATP channels are of major medical importance because they serve as the target for the sulphonylurea drugs, which are used routinely to treat type II diabetes (Gribble and Reimann, 2003). Furthermore, mutations in KATP channel genes, in man or mouse, result in a range of diseases including diabetes mellitus, hyperinsulinism, epilepsy, cardiomyopathy, and Prinzmetal angina (Seino and Miki, 2003; Ashcroft, 2005; Kane et al, 2005).
Structurally, KATP channels are large hetero-octameric complexes of four pore-forming (Kir6.x) and four regulatory sulphonylurea receptor (SURx) subunits (Clement et al, 1997). Both Kir6.x and SUR subunits participate in the metabolic regulation of channel activity by nucleotides, with binding of ATP to Kir6.2 closing the channel, and binding of Mg-nucleotides (MgATP, MgADP) to SUR stimulating channel opening (Tucker et al, 1997). Consequently, metabolic inhibition leads to KATP channel opening, cessation of electrical activity, and suppression of cellular responses such as insulin secretion and muscle contraction, whereas enhanced cellular metabolism promotes KATP channel closure and stimulates insulin secretion and contraction (Seino and Miki, 2003; Ashcroft, 2005; Kane et al, 2005). Sulphonylureas, such as glibenclamide and tolbutamide, bypass cell metabolism and promote insulin secretion by binding directly to SUR and inhibiting KATP channel activity (Gribble and Reimann, 2003). Conversely, K-channel openers bind to SUR to enhance channel opening (Ashcroft and Gribble, 2000). While in most tissues Kir6.2 serves as the channel pore, SUR appears in several isoforms: SUR1 in pancreatic β-cells and neurones, SUR2A in cardiac and skeletal muscle, and SUR2B in vascular smooth muscle (Seino and Miki, 2003). This variability accounts for differences in KATP channel sensitivity to nucleotides and drugs.
Kir6.2 is a member of the inwardly rectifying K+ channel family and functions as a tetrameric channel pore permitting transmembrane flux of K+ ions (Seino and Miki, 2003). Each Kir6.2 subunit is physically coupled to a much larger SUR subunit (Clement et al. 1997). SUR belongs to the ABCC subfamily of ATP-binding cassette (ABC) transporter proteins, which includes the cystic fibrosis transmembrane conductance regulator (CFTR) and the multidrug resistance-related proteins (MRP) (Higgins and Linton, 2004). Like MRP, SUR has 17 transmembrane helices arranged in groups of 5, 6, and 6 (transmembrane domains (TMDs) 0, 1, and 2; Figure 1A). The cytosolic loop between TMDs 0 and 1 contributes to the sulphonylurea-binding site (Mikhailov et al, 2001) and interacts both physically and functionally with the N-terminus of Kir6.2 to modulate opening and closing of the pore (Babenko and Bryan, 2002; Chan et al, 2003). The large cytosolic domains following TMDs 1 and 2 contain the nucleotide-binding domains (NBDs) 1 and 2, respectively, which cooperate in nucleotide binding and hydrolysis (Higgins and Linton, 2004; Vergani et al, 2005). Most ABC proteins use the energy of ATP hydrolysis to transport a range of substrates across biological membranes. However, SUR1 is unique in that it serves as an ion channel regulator and has no known transport function: instead, Mg-nucleotide interaction with the NBDs of SUR1 modulates the gating of Kir6.2 and thereby contributes to metabolic sensing by the KATP channel (Tucker et al, 1997).
Figure 1.
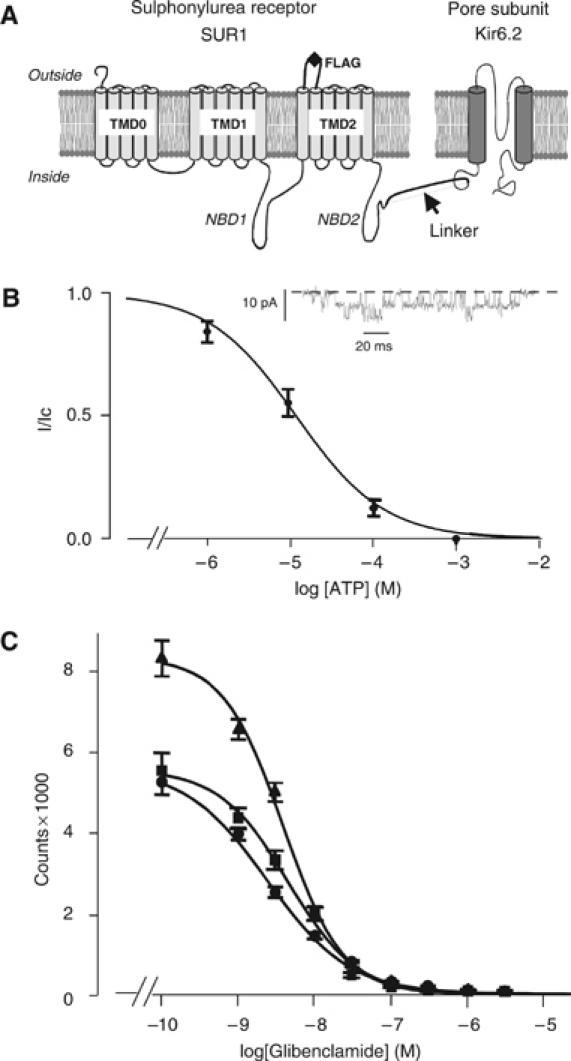
Construction and analysis of an SUR1F–Kir6.2 fusion protein. (A) Schematic of the membrane topology of SUR1 and Kir6.2 showing the location of the FLAG tag used for purification and the linker between SUR1 and Kir6.2. (B) Relationship between [ATP] and KATP current (I), expressed relative to the current in the absence of nucleotide (Ic) for SUR1F–Kir6.2 channels (n=8). The curve is the best fit of the Hill equation to the data with an IC50 11 μM and h=0.8. Inset: Single KATP channel currents at −60 mV in the absence of nucleotide. (C) Displacement of binding of [3H]glibenclamide (5 nM) to Sf9 cells expressing SUR1 (▴, n=3), SUR1–Kir6.2 (i.e. without a FLAG tag; ▪, n=3), or SUR1F–Kir6.2 (•, n=3) by unlabelled glibenclamide. The curves are fit to equation (2) and Km values are given in the text. Data points are shown as mean±s.e.m.
Despite a wealth of functional data, little is known about the three-dimensional organization of the KATP channel complex, and neither the Kir6.2 structure nor that of SUR, has yet been determined. The 3-D structures of a bacterial Kir homologue (Kuo et al, 2003) and of the cytosolic domains of two related eukaryotic Kir channels (Nishida and MacKinnon, 2002; Pegan et al, 2005) have been solved, enabling construction of a molecular model of Kir6.2 and identification of the ATP-binding site (Antcliff et al, 2005). The crystal structures of the isolated NBDs of many bacterial and eukaryotic ABC proteins have also been determined (Chen et al, 2003; Higgins and Linton, 2004; Lewis et al, 2004): all of these share the same overall fold, which allows molecular models of the NBDs of SUR to be constructed (Campbell et al, 2003; Yamada et al, 2004). The complete structure, including the TMDs, is known for only two ABC proteins, the bacterial transporters BtuCD (Locher et al, 2002) and MsbA (Reyes and Chang, 2005): no high-resolution structure for an entire mammalian ABC protein has yet been published, although the X-ray structure of NBD1 of CFTR has been solved (Lewis et al, 2004) and low-resolution structures of CFTR (Rosenberg et al, 2004) and P-glycoprotein (P-gp) (Rosenberg et al, 2003) are available. These data facilitate construction of molecular models of TMDs 1 and 2 of SUR1. However, the structure of TMD0 and its location within the KATP channel complex are unknown. Likewise, the way in which Kir6.2 and SUR associate to form the hetero-octameric KATP channel complex is unclear.
Electron microscopy of cryonegatively stained specimens, combined with single-particle analysis, provides a means of studying the structure of large-membrane protein complexes (Beis et al, 2004; Samso et al, 2005). We employed this technique to investigate the 3-D structure of a functionally active KATP channel complex. The resolution of the internal structure we obtained is sufficient to allow molecular models of Kir6.2 and SUR1 to be positioned within the complex. This reveals how the eight constituent proteins are arranged within the KATP channel, and provides a better understanding of how SUR may regulate the gating of Kir6.2.
Results
Purification of a functionally active KATP channel complex
Only octameric KATP channel complexes are correctly trafficked to the plasma membrane (Zerangue et al, 1999). However, partially assembled complexes may be present in intracellular membranes, and it is also possible that Kir6.2 and SUR subunits may dissociate following solubilization. To facilitate purification of fully assembled KATP channel complexes with the correct stoichiometry, we therefore generated a fusion protein consisting of SUR1 linked via its C-terminus to the N-terminus of Kir6.2 (Mikhailov et al, 1998). An extracellular FLAG tag was inserted between TMDs 12 and 13 to facilitate affinity purification (Figure 1A).
Expression of this construct (SUR1F–Kir6.2) in Xenopus oocytes produced functional KATP channels with conductance, kinetics, and ATP sensitivity similar to those of wild-type channels (Figure 1B). Half-maximal inhibition (IC50) was produced by 11.4±1.5 μM (n=8) ATP, which is not significantly different from that found for channels produced by coinjection of separate SUR1 and Kir6.2 mRNAs (10.8±0.2 μM, n=6). The single-channel conductance at −60 mV was 80±5 pF (n=8) for SUR1F–Kir6.2 channels, compared with 81±6 pF (n=6; NS) for wild-type channels. Furthermore, insect (Sf9) cells expressing SUR1F–Kir6.2 bound the sulphonylurea glibenclamide with affinity similar to cells expressing wild-type SUR1 or a SUR1–Kir6.2 fusion protein lacking the FLAG tag (Figure 1C). The Ki was 4.3±1.1 nM (n=3) for SUR1, 3.8±1.7 nM (n=3) for SUR1–Kir6.2, and 2.5±1.3 nM (n=3) for SUR1F–Kir6.2.
Insect cells infected with a recombinant baculovirus bearing the KATP channel fusion construct robustly express SUR1F–Kir6.2 (Figure 2A, lane 2). Following detergent solubilization, the protein was purified to homogeneity using an anti-FLAG M2 affinity gel. On SDS gels, the monomer was of molecular mass expected for the tandem SUR1F–Kir6.2 fusion construct (∼200 kDa) (Figure 2A, lane 3). On nondenaturing gels, the recombinant protein migrated as a single band with the molecular mass expected for a tetramer (∼900 kDa, Figure 2B).
Figure 2.
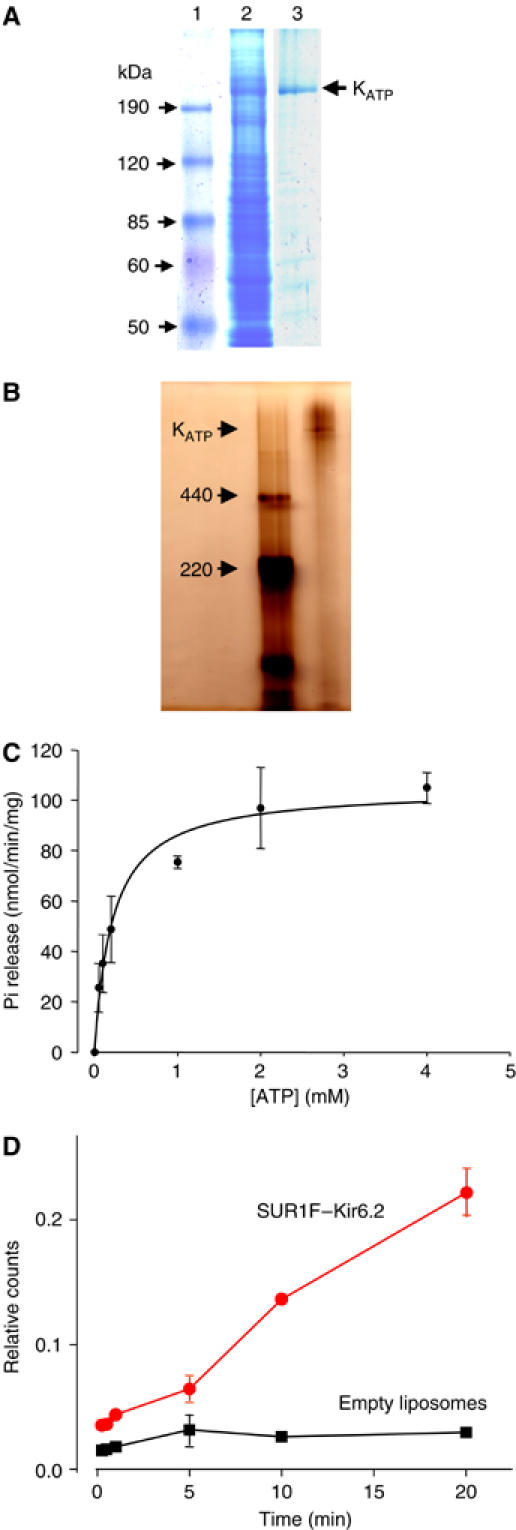
Protein purification and functional analysis. (A) Coomassie-stained SDS–PAGE (8%) gels of molecular weight markers (lane 1), solubilized Sf9 cells expressing SUR1F–Kir6.2 (lane 2) and purified SUR1F–Kir6.2 (lane 3). The purity of the protein was ∼80% as assessed using Gel Pro Analyser following digital scanning of the gel. The molecular masses calculated from the migration of molecular standards are indicated. (B) Native PFO-PAGE (5%) gel of molecular mass markers (left lane) and purified SUR1F–Kir6.2 (right lane). The high molecular mass complex (KATP, arrowed) has a molecular mass of ∼900 kDa when estimated by extrapolation from a linear plot (not shown) of log molecular mass versus migration. (C) ATPase activity of SUR1F–Kir6.2 (n=3). The line is the best fit of the Michaelis–Menten equation to the mean data and has a Vmax of 110 nmol Pi/min/mg protein and a Ki of 0.4 mM. Data points are shown as mean±s.e.m of three separate protein preparations. (D) Time course of 86Rb+ uptake into liposomes reconstituted with 2–3 μg SUR1F–Kir6.2/mg lipid ( ) or into control liposomes containing no protein (▪). Data shown are the mean±s.e.m. of three separate experiments (one protein preparation; similar results were obtained with a second protein preparation).
) or into control liposomes containing no protein (▪). Data shown are the mean±s.e.m. of three separate experiments (one protein preparation; similar results were obtained with a second protein preparation).
The enzymatic activity of the KATP channel complex was retained, as the purified protein showed ATPase activity. Maximal ATPase activity at 37°C was 110±7.6 nmol Pi/min/mg protein, with a Km of 0.4±0.2 mM (mean±s.e.m., three separate protein preparations) (Figure 2C). No ATPase activity was detected when the same protein purification procedure was applied to uninfected Sf9 cells, or cells infected with empty virus, indicating that the ATPase activity is due to the SUR1F–Kir6.2 protein. It is likely that the ATPase activity of SUR1F–Kir6.2 we observe is mediated by the NBDs of SUR1, since functional studies have shown that isolated NBDs of SUR show ATPase activity (Zingman et al, 2001; Masia et al, 2005). While the ATPase activity of SUR has not been reported previously, the value we obtained is similar to that of purified CFTR and MRP1: the hydrolytic activity of CFTR was 60 nmol Pi/min/mg, with a Km of 1.5 mM (Rosenberg et al, 2004), and that of MRP ranged from 10 (Mao et al, 1999) to 470 nmol Pi/min/mg protein (Chang et al, 1998), with a Km of 0.1–3 mM (Chang et al, 1998; Mao et al, 1999).
Importantly, the SUR1F–Kir6.2 purified protein also mediated 86Rb+ uptake when reconstituted into liposomes, demonstrating that the protein is functionally active and acts as a K+ channel (Figure 2D). No activity was found for empty liposomes.
Electron microscopy
Electron microscopy of purified and negatively stained SUR1F–Kir6.2 complexes showed ∼18 nm square-shaped particles in cross-section, which were labelled with a polyclonal rabbit antiserum raised against NBD1 of SUR1 (Mikhailov and Ashcroft, 2000) and anti-rabbit 5 nm gold-labelled antiserum (Figure 3A). This indicates that the 18 nm particles correspond to KATP channel complexes. The collective size of the primary and secondary antibodies and the gold particles probably precludes the binding of more than one gold particle-conjugated secondary antibody per KATP channel complex.
Figure 3.

Electron microscopy of the KATP channel. (A) Single SUR1F–Kir6.2 particles unlabelled (left) or labelled with 5-nm-diameter immunogold (right). Particles were negatively stained with uranyl acetate and examined at room temperature. (B) Area of an electron micrograph recorded under cryo conditions, showing trehalose-molybdate-embedded SUR1F–Kir6.2 particles (enclosed by squares). Scale bar (white), 50 nm. Inset: Fourier transform of the micrograph, with the distinctive rings representing crossover points in the contrast transfer function. The edge of the transform corresponds to 1/6.7 Å. (C) Typical projection classes with square-shaped classes displaying roughly four-fold symmetry (‘top/bottom views', upper row), hexagonal-shaped classes (‘partial side views', centre rows), and rectangular-shaped classes with some bilateral symmetry (‘side views', lowest rows). Each box is 270 Å across. (D) Preliminary 3D models calculated from the classes in panel C. Side views (left column) and top views (right column) for C4 symmetry (top row), C2 symmetry (middle row), or no symmetry (C1, bottom row) are shown. Each box is 270 Å across. (E) Fourier shell correlation versus resolution for the final 3D models generated with C1 symmetry (diamond symbols and solid line), C2 symmetry (dashed line), and C4 symmetry (solid line). The line for C4 symmetry passes below a correlation of 0.5 at a resolution of ∼18 Å.
Cryoelectron microscopy of unlabelled samples, in combination with single-particle averaging, was used to determine the 3-D structure of the KATP channel. Since the lipid PIP2 is required for KATP channel activity (Fan and Makielski, 1997) and we did not include PIP2 in the solutions used for protein purification or structure determination, we believe that the KATP channel will be in the closed state. Figure 3B and C shows a typical field of particles (B) and projection classes (C) used for analysis. Some classes had a square shape and apparent four-fold rotational symmetry, whereas others had a rectangular outline and bilateral symmetry. These features were also observed in preliminary 3-D models using C1, C2, or C4 symmetry (Figure 3D). After refinement, the structure generated with C4 symmetry had the best resolution (18 Å) (Figure 3E). As functional data indicate that the channel exists as a tetramer of heterodimers (Clement et al, 1997), C4 symmetry was selected for further analysis. The surface was rendered with a density level corresponding to 2σ above the mean, as this gave a volume corresponding to the expected molecular mass of the complex plus detergent (∼1 MDa).
Structural details of the KATP channel complex
The KATP (SUR1F–Kir6.2) channel complex is a compact structure with maximal dimensions of 18 nm in cross-section and 13 nm in height (Figure 4). There are few invaginations in the surface structure, apart from narrow crevices in the upper half that lie between four peripheral domains. Thus, contacts between the four SUR1–Kir6.2 subunits must be very intimate, especially in the bottom half of the complex. This is consistent with previous studies showing that both the cytosolic and the transmembrane regions of Kir6.2 and SUR1 interact physically and functionally (Giblin et al, 1999; Mikhailov et al, 2000; Schwappach et al, 2000; Babenko and Bryan, 2002; Chan et al, 2003; Bryan et al, 2004; Rainbow et al, 2004).
Figure 4.
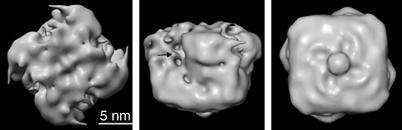
Surface representation of the KATP channel. Top (left), side (middle), and bottom (right) views of the KATP channel complex after refinement of the structure with C4 symmetry, rendered at 2σ. Arrow indicates crevice in the upper part of the structure. The identity of the ‘button' feature in the right panel is not known.
Details of the internal structure of the complex are revealed by a series of slices through the density map (Figure 5Ai–vi). In the top half of the structure, a central region of density is surrounded by four other dense domains: the dimensions of these densities are consistent with a central Kir6.2 tetramer surrounded by four SUR1 subunits. The single density seen in Figure 5Aiii may correspond to the helix bundle crossing region of Kir6.2. The lower part of the structure (Figure 5Avi) reveals four bilobed sets of densities, with positions and dimensions appropriate to those of the NBDs of SUR1. An orthogonal slice (Figure 5B and C) through the centre of the complex reveals a central V-shaped density (Figure 5B) into which the TMDs of a molecular model of Kir6.2 (Antcliff et al, 2005) can be easily placed (Figure 5C). In particular, an excellent fit was found between the angle of the V-shaped density and the tepee-like arrangement of the TMDs of Kir6.2 (Figure 5C). A combination of automated and interactive methods was used to position the Kir6.2 model. Importantly, the ‘foldhunter' algorithm within the image analysis software package EMAN (Ludtke et al, 1999) located the Kir6.2 model within the main central span of density: this enabled the nonsubjective orientation of the complex, something that is notoriously difficult for most single-particle structures. It also provides an indication of where the membrane boundaries are located, because the amphipathic slide helix of Kir6.2 is predicted to lie along the cytosolic surface of the membrane (Kuo et al, 2003). The ease with which Kir6.2 can be positioned within the EM density also provides an independent means of verifying the validity of the structure.
Figure 5.
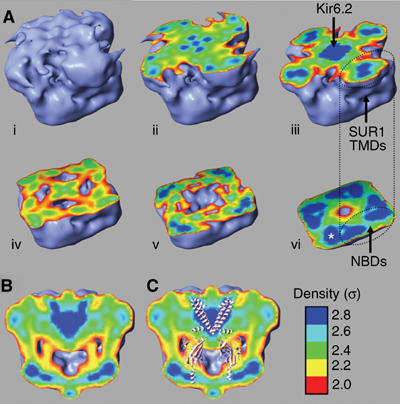
Slices through the 3D structure. The outer surface is rendered at 2σ (light blue). Colours in the interior indicate increasing density: σ=2 (red), 2.2 (yellow), 2.4 (green), 2.6 (cyan), and 2.8 (dark blue). (A) Sequential slices (i–vi) from the presumed extracellular surface. The white asterisk (vi) indicates the bilobed densities presumed to be the NBDs. The position of TMDs 1 and 2 assumed to correspond to the NBDs is indicated. (B, C) Slice perpendicular to the presumed plane of the membrane without (B) and with (C) a molecular model of Kir6.2 positioned in the density. For clarity, only the TMDs of two Kir6.2 subunits, and the cytosolic domains of the two other subunits, are shown.
After inserting the model of Kir6.2 within the density (Figure 6A and B; blue), we next incorporated molecular models of the NBDs and of TMDs 1 and 2 of SUR1 (Figure 6C; red) with the TMDs in the upper half of the structure and the NBDs below (as indicated by the orientation of Kir6.2). We first tried a series of models of SUR1 based on each of the available structures of MsbA (Chang and Roth, 2001; Chang, 2003; Reyes and Chang, 2005), a prokaryotic ABC transporter that corresponds to one TMD and one NBD of SUR1 fused together. However, the density map of the KATP channel could not satisfactorily accommodate any of the three MsbA structures in the correct orientation (see Supplementary data). In contrast, a good fit was found with a model of SUR1 that was based on a model of P-gp (Stenham et al, 2003). This was modelled on individual domains derived from a variety of sources, but constructed using EM and crosslinking data from P-gp itself (Stenham et al, 2003). When the NBDs of this model were aligned with the bilobed densities in the lower half of the KATP channel structure (presumed to be NBDs), the TMDs were completely encapsulated by density at 2σ (Figure 6C). We then positioned three additional SUR models into the density in corresponding positions.
Figure 6.
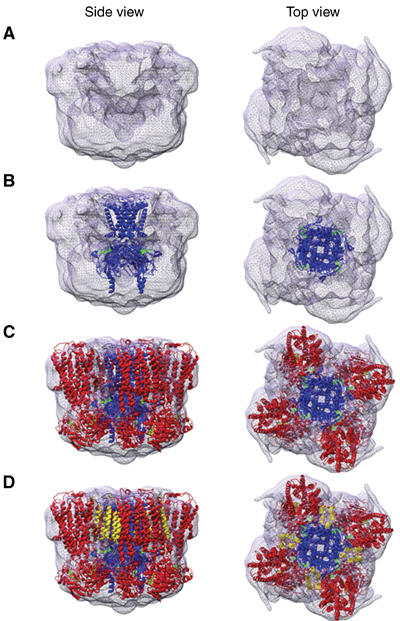
Location of molecular models within the 3D structure. (A–D) Top and side views, respectively, of the EM density with models of Kir6.2 (blue), SUR1 minus TMD0 (red), and TMD0 (yellow) inserted sequentially from (B) to (D). ATP molecules are shown in green.
The NBDs fit naturally into the lower bilobed densities (Figures 5A and 6C). Assuming that the upper density corresponds to the TMDs of SUR1, there are two possible positions of the TMDs relative to the NBDs. If the bilobed density corresponds to NBDs 1 and 2 of a single SUR1 subunit, then the molecule must adopt a 30° tilt in order to fit the TMDs into the upper density. While a global tilt of this order has been observed previously in membrane proteins (Toyoshima and Mizutani, 2004), it is not common. An alternative configuration is obtained if the bilobed density corresponds to NBDs 1 and 2 of adjacent SUR1 subunits. In this case, the NBDs lie directly below the corresponding density in the upper half of the complex (Figure 5A). This configuration is more harmonious with crystallographic studies of prokaryotic ABC transporters (Locher et al, 2002; Chen et al, 2003; Higgins and Linton, 2004; Reyes and Chang, 2005) and functional studies of CFTR (Vergani et al, 2005), which suggest that the NBDs lie further apart in the ligand-free structure than when nucleotide is bound, and that they move closer together on binding nucleotide. The distance between NBDs 1 and 2 in this interpretation (Figure 5A) is consistent with that found for the NBDs of MalK when crystallized in the absence of nucleotide (Chen et al, 2003). Thus, as our EM structure was obtained in the absence of ligand, we favour the idea that bilobed densities correspond to NBD1 and NBD2 of adjacent subunits. It is also worth noting that the proximity of the NBDs in adjacent subunits suggests that they might interact structurally and/or functionally: for example, the ATPase activity of the NBDs in one subunit could be influenced by the adjacent SUR subunit.
Unlike most other ABC proteins, SUR1 possesses an additional set of TMDs (TMD0; Figure 1). There is no crystal structure available for homology modelling of this domain and its location within the KATP channel complex has been a mystery. We therefore searched for unallocated density in the upper half of the EM structure that might correspond to TMD0. The most likely candidate density extends from the boundary of Kir6.2 between TMDs 1 and 2 of adjacent SUR1 subunits (Figure 6C). TMD0 was modelled as a bundle of five helices (yellow) of approximately the same length of those found in the SUR1 and oriented so as to be approximately perpendicular to the presumed location of the membrane. This five-helix bundle could be accommodated within the candidate density (Figure 6D), a location that is consistent with previous studies demonstrating physical and functional interactions between Kir6.2 and TMD0, which imply that they lie in close proximity (Babenko and Bryan, 2002; Chan et al, 2003). It is important to note that a significant amount of protein (∼800 residues in the whole complex; see Supplementary data) is not included in the models, as no structural information is available for modelling these residues.
When viewed from slightly above the upper half of the complex, a narrow cleft is observed that extends between adjacent SUR1 subunits towards the centre (Figure 7A). At this threshold and resolution, only one opening per subunit is found in the whole complex. In cross-section, the cleft runs from just below the membrane to the predicted ATP-binding site, providing a potential route for ATP access (Figure 7B). The location of the ATP-binding site in the Kir6.2 model is supported by site-directed mutagenesis (Antcliff et al, 2005) and the fact that mutations causing neonatal diabetes lie within the putative ATP-binding site and impair KATP channel inhibition by ATP (Proks et al, 2004; Ashcroft, 2005).
Figure 7.
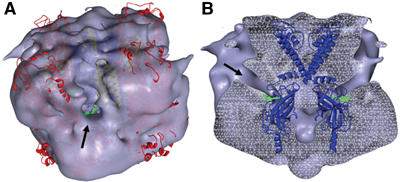
Location of molecular models within the 3D structure. (A, B) Surface and transverse section views, respectively, of the EM density with models of Kir6.2 (blue), TMD0 (yellow), and SUR1 minus TMD0 (red) inserted, showing clefts (arrows) through which ATP (green) could access its binding site on Kir6.2. The Kir6.2 model only is shown in (B).
Discussion
Our studies provide the first step towards a structural understanding of the KATP channel complex. A feature that emerges strongly from the data is the intimate packing of the eight subunits in the 3-D structure, especially in the region assigned to the cytoplasmic domains. The close juxtaposition of Kir6.2 and SUR1 subunits in both transmembrane and cytosolic domains provides for extensive intersubunit interactions and is consistent with earlier studies demonstrating physical interactions between SUR1 and Kir6.2 within both transmembrane and cytosolic domains (Giblin et al, 1999; Mikhailov et al, 2000; Schwappach et al, 2000; Babenko and Bryan, 2002; Chan et al, 2003; Bryan et al, 2004; Rainbow et al, 2004). It further suggests that movement of the NBDs of SUR1 produced by nucleotide binding/hydrolysis (Higgins and Linton, 2004; Vergani et al, 2005) may influence the opening and closing of the pore by direct interaction with the cytosolic domains of Kir6.2, as well as via the TMDs. A further novel finding is that the SUR1 subunits pack so tightly that they appear to interact with one another, both within the membrane and at the level of the NBDs. This affords the possibility of allosteric interactions between drug-binding sites, or nucleotide-binding sites, on different subunits. The structure also provides a wide cytosolic docking platform, formed mainly by the NBDs of SUR1, with which cytosolic proteins may interact: an increasing number of proteins have been shown to regulate KATP channel activity via binding to the NBDs, including creatine kinase (Crawford et al, 2002b), lactate dehydrogenase (Crawford et al, 2002a), and syntaxin (Cui et al, 2004).
An ER retention signal (RKR) is present in both the Kir6.2 and SUR1 subunits of the KATP channel complex and prevents these proteins from being trafficked independently to the plasma membrane (Zerangue et al, 1999). When both subunits are coexpressed, the RKR motifs are masked, allowing surface expression. Although our model of Kir6.2 is truncated at residue 358 and therefore lacks the RKR motif (residues 369–371), it is evident from the tight packing of the subunits in the EM density that access to Kir6.2 will be restricted by the cytosolic domains of SUR1.
The compact nature of the structure, with extensive cytosolic interfacial contacts between subunits, necessitates a specific pathway by which ATP can access its binding site on the buried Kir6.2 subunit. We observed a candidate for such a pathway in the structure, which runs between the adjacent SURs in our model of the KATP complex (Figures 6 and 7). If ATP approaches its binding site via a cleft surrounded by SUR, this may explain how the sulphonylurea receptor enhances the affinity of Kir6.2 for ATP (Tucker et al, 1997). This situation is reminiscent of a similar cleft in the acetylcholine receptor, lying at the interface between subunits, which permits access of acetylcholine to its binding site (Unwin, 2005).
The EM structure provides the first structural insight into the location of TMD0 of SUR1 in the KATP channel complex. Among mammalian ABC transporters, this domain is found only in SUR and MRP proteins: a similar domain is observed in the yeast cadmium factor YCF1 (Szczypka et al, 1994) and in the plant ABC transporter AtMRP5 (Lee et al, 2004), but has not been reported to date in bacterial ABC proteins. As there is no structural information on TMD0 (since it is not present in any ABC protein that has been crystallized), it was modelled as a simple five-helix bundle. Initial speculations placed TMD0 on the outside of the KATP channel complex (Zerangue et al, 1999; Schwappach et al, 2000), but subsequent studies showing that Kir6.2 and TMD0 interact both physically and functionally (Babenko and Bryan, 2002; Chan et al, 2003) led to the proposal that TMD0 is sandwiched between Kir6.2 and TMDs 1/2 of SUR1 (Bryan et al, 2004). However, insertion of homology models of Kir6.2 and SUR1 (TMD1 to NBD2) into the EM density reveals that there is no room for TMD0 in this position (Figure 6). Instead, it is likely that the five TMD0 helices lie between adjacent SUR subunits, and interface with the outer transmembrane (TM) helix (TM1) of Kir6.2. In this position, residues 76–78 in TM1 of Kir6.2, which specify physical assembly with SUR1 (Schwappach et al, 2000), are well placed to interact with TMD0. Likewise, the positioning of TMD0 in this location is also consistent with functional studies (Babenko and Bryan, 2002, Chan et al, 2003) showing that the cytosolic loop (L0) between TMDs 0 and 1 of SUR1 lies in close proximity to the N-terminus of Kir6.2.
The bilobed densities observed in the lower part of the structure have been assigned to the NBDs of SUR1, but there is some ambiguity as to whether these correspond to NBDs 1 and 2 of the same, or adjacent, subunits. As described above, we favour the latter possibility because the SUR1F–Kir6.2 protein was purified in the absence of nucleotide. Under this condition, the NBDs are expected to lie further apart (Chen et al, 2003; Higgins and Linton, 2004; Yamada and Kurachi, 2004; Vergani et al, 2005). The tight packing of the subunits within the SUR1F–Kir6.2 complex means that the NBDs will then lie in closer proximity to those of adjacent SUR1 subunits. The location of the densities in the KATP channel structure is thus consistent with a model in which the NBDs may adopt one of two stable states, in which NBD1 interacts with NBD2 of the same subunit when ligand is bound, but with NBD2 of the adjacent subunit in the ligand-free state. Consequently, conformational changes induced by Mg-nucleotide interactions with one SUR1 molecule may influence binding/hydrolysis at the others.
Our studies also provide the first measurements of the ATPase activity of intact SUR. The maximal rate of ATP hydrolysis (110 nmol Pi/min/mg protein) and the Km (0.4 mM) are similar to those found for purified CFTR (Rosenberg et al, 2004) and MRP1 (Chang et al, 1998; Mao et al, 1999). Although the ATPase activity of the SUR1–Kir6.2 tetramer and monomeric SUR1 has not been measured previously, it has been determined for the isolated NBDs, by fusing them to maltose-binding protein (Masia et al, 2005). For NBD2, which showed a higher ATPase activity, the Ki was 20 mM and the maximal rate of ATP hydrolysis was ∼20 nmol Pi/min/mg protein. The turnover rate for the purified SUR1–Kir6.2 tetramer was 1.7 nmol Pi/s/nmol protein (calculated using a molecular weight of 229 kDa for the monomeric fusion protein, which includes affinity tags and linkers). This is 65 times greater than the calculated turnover rate for NBD2 (0.026 nmol Pi/s/nmol protein). The higher affinity and greater turnover rate found for the ATPase activity of the intact KATP channel complex probably reflects the requirement for heterodimerization of the NBDs for concerted binding of nucleotide (Chen et al, 2003; Higgins and Linton, 2004; Vergani et al, 2005). Additionally, it may be due to Kir6.2 influencing the ATPase activity of the NBDs of SUR1, or due to cooperative interactions between the SUR1 subunits within the tetramer. Whatever the mechanism underlying the difference in ATPase activity between SUR1–Kir6.2 and the isolated NBDs, the demonstrated activity of the purified KATP channel complex means that structure–function relationships can be studied in the future.
Conclusion
In conclusion, our results provide the first view of the 3-D structure of the KATP channel. The addition of molecular models of SUR1 and Kir6.2, using known domains of K+ channels and ABC proteins, evokes specific suggestions about subunit–subunit interactions that provide a solid basis for future experimental tests. Furthermore, the sequence similarity between members of the Kir6.x, and SURx, families (Seino and Miki, 2003) is such that, at 18 Å resolution, cardiac, skeletal, and vascular smooth muscle KATP channels will share structural features similar to that of the β-cell SUR1–Kir6.2 channel described here.
Finally, this structure is one of the few obtained for large mammalian plasma membrane protein complexes, and is unusual in being derived from a recombinant fusion protein. The tandem-fusion strategy helps anchor the subunits together and might be useful for structural studies of other membrane protein complexes having low levels of expression or weaker subunit interactions that are disrupted during protein purification.
Materials and methods
Purification of recombinant Kir6.2–SUR1 complexes
A fusion protein, SUR1–Kir6.2, was made by linking the C-terminus of rat SUR1 (Genbank L40624) to the N-terminus of mouse Kir6.2 (Genbank D50581) by a SerAlaSerAlaSerAla linker (Mikhailov et al, 1998). To facilitate purification, a 3-FLAG tag (MetAspTyrLysAspHisAspTyrLysAspIleAspTyrLysAspAspAspAspLys) was inserted between TMs 12 and 13 of SUR1 (SUR1F). This construct was inserted into the transfer vector pAcYM1. We used Spodoptera frugiperda (Sf21) cells for generation of the recombinant baculoviruses and Sf9 cells for protein expression, because it was easier to obtain recombinant baculoviruses in Sf21 cells and Sf9 cells gave higher protein expression. Sf21 cells were cotransfected with SUR1F–Kir6.2 transfer vector and Autographa californica nuclear polyhedrosis virus DNA (AcNPV PAK6). Recombinant baculoviruses were obtained and amplified in Sf21 insect cells. Sf9 insect cells were grown to a density of 2 × 106 cells/ml and infected with recombinant baculoviruses in a ratio of 1:5. Cells were harvested 72 h postinfection, centrifuged (3000 g, 10 min), washed with phosphate-buffered saline (PBS), and resuspended in 50 mM Tris–HCl, pH 8.8, 50 mM NaCl containing a mix of protease inhibitors (Complete™, Roche). Cells were disrupted using a Stansted TC5W Homogeniser (Stansted Fluid Power Ltd) at a pressure of 10 000 psi, centrifuged at 200 g for 10 min, and loaded on a step sucrose gradient (10%/46%). After centrifugation at 100 000 g for 1 h, the intermediate phase was collected and diluted four times with 50 mM Tris–HCl, pH 8.8, 200 mM NaCl. For protein solubilization, dodecylmaltoside (DDM) was added to a final concentration of 0.2% and the mix incubated for 30 min on ice. The solution was then centrifuged at 12 000 g for 20 min and the supernatant collected. Anti-FLAG M2 affinity gel (Sigma) was added to the solution and incubated for 2 h at 4°C. The suspension was applied to an empty column and washed with 50 vol of 50 mM Tris–HCl, pH 8.8, 200 mM NaCl, and 0.1% DDM. Purified proteins were eluted with 100 μM 3-FLAG peptide (Sigma) and 0.05% 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) in 50 mM Tris, pH 8.8, 150 mM NaCl, and 0.1% DDM.
An 8% SDS–PAGE gel was used for analysis of purified proteins. The molecular mass of the entire KATP channel complex was estimated from native PAGE (5% separating gel, 4% stacking gel), using the Perfluorooctanoate (PFO)-PAGE method as described by Ramjeesingh et al (1999). PFO detergent was obtained from Fluorochem p.l.c., Glossop, UK.
Immunogold labelling
For immunolabelling studies, an anti-FLAG M2 affinity gel with attached SUR1F–Kir6.2 protein was prepared as described above and incubated for 2 h at 4°C with an equal amount of anti-NBD1 antiserum (Mikhailov and Ashcroft, 2000) diluted 1:10 in 50 mM Tris–HCl, pH 8.8, 200 mM NaCl, and 0.1% DM buffer. The suspension was washed with 10 vol of 50 mM Tris–HCl, pH 8.8, 200 mM NaCl, and 0.1% DM, and then incubated for 2 h at 4°C with an equal amount of 5 nm gold-labelled anti-rabbit antiserum diluted 1:10 in 50 mM Tris–HCl, pH 8.8, 200 mM NaCl, and 0.1% DM buffer. The suspension was washed with 10 vol of 50 mM Tris–HCl, pH 8.8, 200 mM NaCl, and 0.1% DM. The resulting complex of SUR1–Kir6.2∼anti-NBD1∼anti-rabbit-5 nm gold was eluted with 100 μM 3X-FLAG peptide. The labelled purified protein was negatively stained with 4% w/v uranyl acetate and examined by electron microscopy at room temperature.
Cryonegative staining
For structure determination, samples were not labelled with antibody, but were embedded in a trehalose-ammonium molybdate film and examined under cryo conditions, in order to preserve the protein structure more faithfully. Samples were incubated briefly (20 s) with freshly glow-discharged C-coated 400 mesh/in copper electron microscopy grids, and then immediately transferred to a droplet of 1% trehalose, 6% ammonium molybdate, pH 7, with a 30 s incubation. After blotting on Whatman #4 filter paper, the grids were transferred to a Philips CM200 electron microscope, fitted with a field emission gun and an Oxford Instruments cryoholder, and cooled with liquid nitrogen to <100 K. Data were recorded on Kodak SO163 electron microscopy film at a calibrated magnification of × 37 800 and under low-dose conditions. A defocus range of −2 to −4 μm was sampled for the 55 micrographs used in the data set. Films were developed in double-strength Kodak D-19 developer solution for 12 min. Micrographs were digitized at 2000 dpi using a UMAX3000 densitometer, giving a resolution at the specimen level of 3.35 Å/pixel.
Image processing
Image processing was carried out with EMAN software (Ludtke et al, 1999). Particles were selected manually, using a box size of 80 pixels (270 Å). Automatic particle picking was precluded because of the relatively low contrast of the particles when embedded in trehalose, and because some aggregation was observed (aggregation was minimized by using freshly purified protein and by the presence of lipid). Particles (7413) were selected from 55 micrographs, and, after CTF correction and iterative centering, 5996 particles were selected for further analysis. Classification of particles low pass filtered to 25 Å using the multi-reference alignment algorithm of EMAN (startnrclasses) yielded ∼100 evenly populated classes. A subset of well-centred and high signal:noise classes (30) representing a range of different projections of the complex was used to generate a preliminary 3D model using the cross-common lines approach, followed by 3D reconstruction by backprojection. Symmetry (C2 or C4) was either applied or not applied (C1) at this point. Evenly spaced projections were generated from the preliminary structures (and from subsequent refinements of them) and each selected raw particle was compared against all the projections and sorted into the three projection classes that it best matched. The projections were then used to re-align all the particles in that class prior to averaging them to produce a new projection class average. A new 3-D model was then calculated as before. After several such rounds of iterative refinement, convergence to the final structures for each symmetry was reached, as judged by no further improvement in the Fourier shell correlation (FSC) between 3-D models generated from contiguous iterations in the procedure. As a test for resolution, the data set was split into two halves and two separate 3-D structures calculated. This was repeated for each symmetry (C1, C2, C4). The FSC between the two substructures was calculated, and the spatial frequency at which the FSC dropped below 0.5 was taken as an estimate of resolution. The structure refined with C4 symmetry had the best resolution and back projections from this structure had the best correlation with the unsymmetrized class averages. Therefore, C4 symmetry was used for all further analysis. Density maps were displayed using Chimera software (Pettersen et al, 2004).
To ensure that the final structure was not dependent on the initial model, we repeated the above steps starting with a featureless sphere of density. The resulting structure was consistent with that achieved with a well-defined starting model and had a resolution of 18 Å. We also tested whether the refinement algorithm might generate artefacts in our structure by refining ∼3000 boxes selected at random: the resulting structure was a featureless spherical density.
Functional studies
Electrophysiology. The SUR1F–Kir6.2 fusion protein was expressed in Xenopus occytes and currents were recorded from inside-out membrane patches (Gribble et al, 1997). The pipette solution contained (mM): 140 KCl, 1.2 MgCl2, 2.6 CaCl2, and 10 HEPES (pH 7.4 with KOH). The internal (bath) solution contained (mM): 107 KCl, 10 EGTA, 10 HEPES (pH 7.2 with KOH; final [K+] ∼140 mM). ATP concentration–response curves were fit with the Hill equation (equation (1)):
![]()
where [ATP] is the ATP concentration, IC50 is the ATP concentration at which inhibition is half-maximal, and h is the Hill coefficient. To control for possible rundown, Ic was taken as the mean of the current in control solution before and after ATP application.
[3H]glibenclamide binding. Sf9 cells were suspended at a density of 5 × 105 cells/ml in PBS (Sigma) and incubated for 30 min at room temperature with 1 nM [3H]glibenclamide and different concentrations of unlabeled glibenclamide in a final volume of 400 μl. The incubation was stopped by rapid separation on Whatman GF/C filters, presoaked for 30 min in PBS. After washing the filters, the amount of bound [3H]glibenclamide was determined. Nonspecific [3H]glibenclamide binding to the filter and mock-transfected cells alone was always <5–10 × 103 molecules per cell (n>20) and therefore not subtracted. Bound [3H]glibenclamide (A) was estimated from (equation (2)):
![]()
where Amax is the maximal [3H]glibenclamide binding.
ATP hydrolysis. ATPase activity was measured using a colorimetric assay for liberated inorganic phosphate (Chifflet et al, 1988). Briefly, 30 μl of purified protein was incubated in reaction buffer (150 mM NH4Cl, 5 mM MgSO4, 50 mM Tris–HCl, pH 7.4) in the presence of 0–4 mM Na2ATP for 3 h at 37°C. Final reaction volumes were 50 μl. The reaction was terminated by addition of 50 μl 12% SDS. To control for contaminating phosphate present in ATP stocks, denatured protein was incubated with 0–4 mM Na2ATP and used as blanks for the corresponding experimental samples. ATPase activity was plotted as a function of ATP concentration and the Michaelis–Menten equation was fitted to the data. Experiments were carried out in duplicate on three separate preparations of freshly purified SUR1F–Kir6.2.
86Rb+ uptake assay. Purified SUR1F–Kir6.2 was reconstituted into liposomes containing 450 mM KCl, 10 mM Hepes, 4 mM NMG, pH 7.4 (Heginbotham et al, 1998) at a ratio of 2–3 μg protein per mg lipid. Lipids consisted of 1-palmitoyl-2-oleoyl phosphatidylethanolamine (POPE), phosphatidylglycerol (POPG) (Avanti Polar Lipids), and PIP2 (Echelon Biosciences) in a ratio of 15:5:1. 86Rb+uptake was measured as described by Heginbotham et al (1998). Briefly, uptake was initiated by mixing 80–100 μl liposomes with 900 μl uptake buffer (400 mM sorbitol, 10 mM Hepes, 4 mM NMG, 50 μM KCl, pH 7.4) containing 0.5 μCi/ml 86Rb+. At each time-point, a 100 μl sample of the reaction mixture was passed through a 1.5 ml Dowex cation exchange column, in the NMG+ form, to remove extra-liposomal 86Rb+, and collected into a scintillation vial. Samples were mixed with 12 ml scintillation fluid and counted using a liquid scintillation counter (Beckman). After the final time-point, valinomycin was added to a final concentration of ∼1 μg/ml to determine maximal 86Rb+ uptake at equilibrium. Lipsomes that did not contain SUR1F–Kir6.2 were used as a control.
Supplementary Material
Supplementary Information 1
Supplementary Information 2
Supplementary Information 3
Acknowledgments
We thank Liang Zhang and Martyn Hulley for help with PFO Page and Vicky Ball for assistance with the flux assay, Drs Nigel Unwin, Peiyi Wang, Catherine Venien-Bryan, and Svetla Stoilova-McPhie for technical assistance with EM, and Dr Shozeb Haider for the Kir6.2 model. We thank Diabetes UK, the Wellcome Trust, and the Royal Society for support. FMA is the Royal Society GlaxoSmithKline Research Professor. JC holds a Wellcome Trust Structural Biology studentship and a Canadian National Scholarship at Linacre College.
References
- Antcliff JF, Haider S, Proks P, Sansom MS, Ashcroft FM (2005) Functional analysis of a structural model of the ATP-binding site of the KATP channel Kir6.2 subunit. EMBO J 24: 229–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashcroft FM (2005) ATP-sensitive potassium channelopathies: focus on insulin secretion. J Clin Invest 115: 2047–2058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashcroft FM, Gribble FM (2000) New windows on the mechanism of action of KATP channel openers. Trends Pharmacol Sci 21: 439–445 [DOI] [PubMed] [Google Scholar]
- Babenko AP, Bryan J (2002) SUR-dependent modulation of KATP channels by an N-terminal KIR6.2 peptide. Defining intersubunit gating interactions. J Biol Chem 277: 43997–44004 [DOI] [PubMed] [Google Scholar]
- Beis K, Collins RF, Ford RC, Kamis AB, Whitfield C, Naismith JH (2004) Three-dimensional structure of Wza, the protein required for translocation of group 1 capsular polysaccharide across the outer membrane of Escherichia coli. J Biol Chem 279: 28227–28232 [DOI] [PubMed] [Google Scholar]
- Bryan J, Vila-Carriles WH, Zhao G, Babenko AP, Aguilar-Bryan L (2004) Toward linking structure with function in ATP-sensitive K+ channels. Diabetes 53 (Suppl 3): S104–S112 [DOI] [PubMed] [Google Scholar]
- Campbell JD, Sansom MSP, Ashcroft FM (2003) Potassium channel regulation. Structural insights into the function of the nucleotide-binding domains of the human sulphonylurea receptor. EMBO Rep 4: 1038–1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan KW, Zhang H, Logothetis DE (2003) N-terminal transmembrane domain of the SUR controls trafficking and gating of Kir6 channel subunits. EMBO J 22: 3833–3843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang G (2003) Structure of MsbA from Vibrio cholera: a multidrug resistance ABC transporter homolog in a closed conformation. J Mol Biol 330: 419–430 [DOI] [PubMed] [Google Scholar]
- Chang G, Roth CB (2001) Structure of MsbA from E. coli: a homolog of the multidrug resistance ATP binding cassette (ABC) transporters. Science 293: 1793–1800 [DOI] [PubMed] [Google Scholar]
- Chang XB, Hou YX, Riordan JR (1998) Stimulation of ATPase activity of purified multidrug resistance-associated protein by nucleoside diphosphates. J Biol Chem 273: 23844–23848 [DOI] [PubMed] [Google Scholar]
- Chen J, Lu G, Lin J, Davidson AL, Quiocho FA (2003) A tweezers-like motion of the ATP-binding cassette dimer in an ABC transport cycle. Mol Cell 12: 651–661 [DOI] [PubMed] [Google Scholar]
- Chifflet S, Torriglia A, Chiesa R, Tolosa SA (1988) Method for the determination of inorganic phosphate in the presence of labile organic phosphate and high concentrations of protein: application to lens ATPases. Anal Biochem 168: 1–4 [DOI] [PubMed] [Google Scholar]
- Clement JP IV, Kunjilwar K, Gonzalez G, Schwanstecher M, Panten U, Anguliar-Bryan L, Bryan J (1997) Association and stoichiometry of KATP channel subunits. Neuron 18: 827–838 [DOI] [PubMed] [Google Scholar]
- Crawford RM, Bundas GR, Jovanovic S, Ranki HJ, Wilson TJ, Davies AM, Jovanovic A (2002a) M-LDH serves as a sarcolemmal KATP channel subunit essential for cell protection against ischemia. EMBO J 21: 3936–3948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford RM, Ranki HJ, Botting CH, Budas GR, Jovanovic A (2002b) Creatine kinase is physically associated with the cardiac ATP-sensitive K+ channel in vivo. FASEB J 16: 102–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui N, Kang Y, He Y, Leung YM, Xie H, Pasyk EA, Gao X, Sheu L, Hansen JB, Wahl P, Tsushima RG, Gaisano HY (2004) H3 domain of syntaxin 1A inhibits KATP channels by its actions on the sulfonylurea receptor 1 nucleotide-binding folds-1 and -2. J Biol Chem 279: 53259–53265 [DOI] [PubMed] [Google Scholar]
- Fan Z, Makielski JC (1997) Anionic phospholipids activate ATP-sensitive potassium channels. J Biol Chem 272: 5388–5395 [DOI] [PubMed] [Google Scholar]
- Giblin JP, Leaney JL, Tinker A (1999) The molecular assembly of ATP-sensitive potassium channels. Determinants on the pore forming subunit. J Biol Chem 274: 22652–22659 [DOI] [PubMed] [Google Scholar]
- Gribble FM, Ashfield R, Ammala C, Ashcroft FM (1997) Properties of cloned ATP-sensitive K+ currents expressed in Xenopus oocytes. J Physiol 498: 87–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribble FM, Reimann F (2003) Sulphonylurea action revisited: the post-cloning era. Diabetologia 46: 875–891 [DOI] [PubMed] [Google Scholar]
- Heginbotham L, Kolmakova-Partensky L, Miller C (1998) Functional reconstitution of a prokaryotic K+ channel. J Gen Physiol 111: 741–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins CF, Linton KJ (2004) The ATP switch model for ABC transporters. Nat Struct Mol Biol 11: 918–926 [DOI] [PubMed] [Google Scholar]
- Kane GC, Liu XK, Yamada S, Olson TM, Terzic A (2005) Cardiac KATP channels in health and disease. J Mol Cell Cardiol 38: 937–943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo A, Gulbis JM, Antcliff JF, Rahman T, Lowe ED, Zimmer J, Cuthbertson J, Ashcroft FM, Ezaki T, Doyle DA (2003) Crystal structure of the potassium channel KirBac1.1 in the closed state. Science 300: 1922–1926 [DOI] [PubMed] [Google Scholar]
- Lee EK, Kwon M, Ko JH, Yi H, Hwang MG, Chang S, Cho MH (2004) Binding of sulfonylurea by AtMRP5, an Arabidopsis multidrug resistance-related protein that functions in salt tolerance. Plant Physiol 134: 528–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis HA, Buchanan SG, Burkley SK, Conners K, Dickey M, Dorwart M, Fowler R, Gao X, Guggino WB, Hendrickson WA, Hunt JF, Kearins MC, Lotimer D, Maloney PC, Post KW, Rajashankar KR, Rutter ME, Sauder JM, Shriver S, Thibodeau PH, Thomas PJ, Zhang M, Zhao W (2004) Structure of nucleotide-binding domain 1 of the cystic fibrosis transmembrane conductance regulator. EMBO J 23: 282–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locher KP, Lee AT, Rees DC (2002) The E. coli BtuCD structure: a framework for ABC transporter architecture and mechanism. Science 296: 1091–1098 [DOI] [PubMed] [Google Scholar]
- Ludtke SJ, Baldwin PR, Chiu W (1999) EMAN: semiautomated software for high-resolution single-particle reconstructions. J Struct Biol 128: 82–97 [DOI] [PubMed] [Google Scholar]
- Mao Q, Leslie EM, Deeley RG, Cole SP (1999) ATPase activity of purified and reconstituted multidrug resistance protein MRP1 from drug-selected H69AR cells. Biochim Biophys Acta 1461: 69–82 [DOI] [PubMed] [Google Scholar]
- Masia R, Enkvetchakul D, Nichols CG (2005) Differential nucleotide regulation of KATP channels by SUR1 and SUR2A. J Mol Cell Cardiol (advance online publication 10 May 2005; doi:10.1016/j.yjmcc.2005.03.009) [DOI] [PubMed] [Google Scholar]
- Mikhailov MV, Ashcroft SJ (2000) Interactions of the sulfonylurea receptor 1 subunit in the molecular assembly of beta-cell K(ATP) channels. J Biol Chem 275: 3360–3364 [DOI] [PubMed] [Google Scholar]
- Mikhailov MV, Mikhailova EA, Ashcroft SJ (2000) Investigation of the molecular assembly of beta-cell KATP channels. FEBS Lett 482: 59–64 [DOI] [PubMed] [Google Scholar]
- Mikhailov MV, Mikhailova EA, Ashcroft SJ (2001) Molecular structure of the glibenclamide binding site of the beta-cell K(ATP) channel. FEBS Lett 499: 154–160 [DOI] [PubMed] [Google Scholar]
- Mikhailov MV, Proks P, Ashcroft FM, Ashcroft SJ (1998) Expression of functionally active ATP-sensitive K-channels in insect cells using baculovirus. FEBS Lett 429: 390–394 [DOI] [PubMed] [Google Scholar]
- Nishida M, MacKinnon R (2002) Structural basis of inward rectification: cytoplasmic pore of the G protein-gated inward rectifier GIRK1 at 1.8 A resolution. Cell 111: 957–965 [DOI] [PubMed] [Google Scholar]
- Pegan S, Arrabit C, Zhow W, Kwiatkowski W, Collis A, Slesinger PA, Choe S (2005) Cytoplasmic domain structures of Kir2.1 and Kir3.1 show sites for modulating gating and rectification. Nat Neurosci 8: 279–287 [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF chimera—a visualization system for exploratory research and analysis. J Comput Chem 25: 1605–1612 [DOI] [PubMed] [Google Scholar]
- Proks P, Antcliff JF, Lippiat J, Gloyn AL, Hattersley AT, Ashcroft FM (2004) Molecular basis of Kir6.2 mutations associated with neonatal diabetes or neonatal diabetes plus neurological features. Proc Natl Acad Sci USA 101: 17539–17544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainbow RD, James M, Hudman D, AlJobi M, Singh H, Watson PJ, Ashmole I, Davies NW, Lodwick D, Norman RI (2004) Proximal C-terminal domain of sulphonylurea receptor 2A interacts with pore-forming Kir6 subunits in KATP channels. Biochem J 379: 173–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramjeesingh M, Huan LJ, Garami E, Bear CE (1999) Novel method for evaluation of the oligomeric structure of membrane proteins. Biochem J 342: 119–123 [PMC free article] [PubMed] [Google Scholar]
- Reyes CL, Chang G (2005) Structure of the ABC transporter MsbA in complex with ADP vanadate and lipopolysaccharide. Science 308: 1028–1031 [DOI] [PubMed] [Google Scholar]
- Rosenberg MF, Kamis AB, Aleksandrov LA, Ford RC, Riordan JR (2004) Purification and crystallization of the cystic fibrosis transmembrane conductance regulator (CFTR). J Biol Chem 279: 39051–39057 [DOI] [PubMed] [Google Scholar]
- Rosenberg MF, Kamis AB, Callaghan R, Higgins CF, Ford RC (2003) Three-dimensional structures of the mammalian multidrug resistance P-glycoprotein demonstrate major conformational changes in the transmembrane domains upon nucleotide binding. J Biol Chem 278: 8294–8299 [DOI] [PubMed] [Google Scholar]
- Samso M, Wagenknecht T, Allen PD (2005) Internal structure and visualization of transmembrane domains of the RyR1 calcium release channel by cryo-EM. Nat Struct Mol Biol 12: 539–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwappach B, Zerangue N, Jan YN, Jan LY (2000) Molecular basis for K(ATP) assembly: transmembrane interactions mediate association of a K+ channel with an ABC transporter. Neuron 26: 155–167 [DOI] [PubMed] [Google Scholar]
- Seino S, Miki T (2003) Physiological and pathophysiological roles of ATP-sensitive K+ channels. Prog Biophys Mol Biol 81: 133–176 [DOI] [PubMed] [Google Scholar]
- Stenham DR, Campbell JD, Sansom MS, Higgins CF, Kerr ID, Linton KJ (2003) An atomic detail model for the human ATP binding cassette transporter P-glycoprotein derived from disulfide cross-linking and homology modeling. FASEB J 17: 2287–2289 [DOI] [PubMed] [Google Scholar]
- Szczypka MS, Wemmie JA, Moye-Rowley WS, Thiele DJ (1994) A yeast metal resistance protein similar to human cystic fibrosis transmembrane conductance regulator (CFTR) and multidrug resistance-associated protein. J Biol Chem 269: 22853–22857 [PubMed] [Google Scholar]
- Toyoshima C, Mizutani T (2004) Crystal structure of the calcium pump with a bound ATP analogue. Nature 430: 529–535 [DOI] [PubMed] [Google Scholar]
- Tucker SJ, Gribble FM, Zhao C, Trapp S, Ashcroft FM (1997) Truncation of Kir6.2 produces ATP-sensitive K+ channels in the absence of the sulphonylurea receptor. Nature 387: 179–183 [DOI] [PubMed] [Google Scholar]
- Unwin N (2005) Refined structure of the nicotinic acetylcholine receptor at 4A resolution. J Mol Biol 346: 967–989 [DOI] [PubMed] [Google Scholar]
- Vergani P, Lockless SW, Nairn AC, Gadsby DC (2005) CFTR channel opening by ATP-driven tight dimerization of its nucleotide-binding domains. Nature 433: 876–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada M, Ishii M, Hibino H, Kurachi Y (2004) Mutation in nucleotide-binding domains of sulfonylurea receptor 2 evokes Na-ATP-dependent activation of ATP-sensitive K+ channels: implication for dimerization of nucleotide-binding domains to induce channel opening. Mol Pharmacol 66: 807–816 [DOI] [PubMed] [Google Scholar]
- Yamada M, Kurachi Y (2004) The nucleotide-binding domains of sulfonylurea receptor 2A and 2B play different functional roles in nicorandil-induced activation of ATP-sensitive K+ channels. Mol Pharmacol 65: 1198–1207 [DOI] [PubMed] [Google Scholar]
- Zerangue N, Schwappach B, Jan YN, Jan LY (1999) A new ER trafficking signal regulates the subunit stoichiometry of plasma membrane K(ATP) channels. Neuron 22: 537–548 [DOI] [PubMed] [Google Scholar]
- Zingman LV, Alekseev AE, Bienengraeber M, Hodgson D, Karger AB, Dzeja PP, Terzic A (2001) Signaling in channel/enzyme multimers: ATPase transitions in SUR module gate ATP-sensitive K+ conductance. Neuron 31: 233–245 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information 1
Supplementary Information 2
Supplementary Information 3