

Abstract
Type I restriction enzymes use two motors to translocate DNA before carrying out DNA cleavage. The motor function is accomplished by amino-acid motifs typical for superfamily 2 helicases, although DNA unwinding is not observed. Using a combination of extensive single-molecule magnetic tweezers and stopped-flow bulk measurements, we fully characterized the (re)initiation of DNA translocation by EcoR124I. We found that the methyltransferase core unit of the enzyme loads the motor subunits onto adjacent DNA by allowing them to bind and initiate translocation. Termination of translocation occurs owing to dissociation of the motors from the core unit. Reinitiation of translocation requires binding of new motors from solution. The identification and quantification of further initiation steps—ATP binding and extrusion of an initial DNA loop—allowed us to deduce a complete kinetic reinitiation scheme. The dissociation/reassociation of motors during translocation allows dynamic control of the restriction process by the availability of motors. Direct evidence that this control mechanism is relevant in vivo is provided.
Keywords: DNA translocation, molecular motor, restriction enzymes, SF2 helicase, single molecules
Introduction
For the linear motor enzymes—those that translocate along a polymer lattice—the protein complex must first associate with the track. A specific location of the initiation site can be critical for the enzyme function. The clearest examples of this are the large number of DNA-based motors. In these cases, the initiation site may be a specific DNA sequence, such as a promoter (Browning and Busby, 2004) or an origin of replication (Konieczny, 2003), or a specific nucleic acid structure, such as a double-strand DNA (dsDNA) break (Farah and Smith, 1997; Singleton et al, 2004). The motor must be loaded at the site so that translocation can proceed. In the classical examples, such as helicases and RNA polymerases, this requires that the motor binds single-stranded DNA. Although some proteins can self-load, for example, RecBCD at a dsDNA break (Roman and Kowalczykowski, 1989), others require a partner loading protein, for example, HPV E2 for E1 helicase (Stenlund, 2003). The loader may be involved in a number of key steps, such as sequence recognition, modulation of the initiation rate and/or initial strand separation. As there is a finite probability that the motor dissociates during subsequent translocation, the loader may also be required to facilitate reinitiation at the origin. Outside of the polymerase fields, the kinetics of initiation and reinitiation of DNA motors by loading proteins has not been studied in great detail, despite its clear importance. In part, this is because of difficulties in producing biologically relevant DNA substrates for in vitro studies (many assays rely on short synthetic DNA substrates) and because the motor is often analyzed in the absence of its loader.
In this study, we present a complete analysis of the initiation, termination and reinitiation kinetics of a complete superfamily 2 (SF2) helicase–loader complex, the Type I restriction–modification (RM) enzyme EcoR124I.
The Type I RM enzymes (Figure 1A) consist of three protein subunits: HsdR, HsdM and HsdS (Murray, 2000). No other protein components are believed to be required for their in vivo role in restricting the infectivity of phage DNA. HsdM and HsdS (HsdM2HsdS1) form an independently active methyltransferase (MTase), which specifically recognizes a bipartite DNA sequence. The HsdR motor subunit has amino-acid sequence and secondary structure motifs characteristic of both an SF2 DNA helicase and an endonuclease (Davies et al, 1999; Janscak et al, 1999b). However, for the correct functioning of the subunit, HsdR must bind DNA via the MTase. Only the full endonuclease complex (hereafter referred to as R2-complex) with a stoichiometry of HsdR2HsdM2HsdS1 (Dryden et al, 1997) has ATPase (Janscak et al, 1998), processive DNA translocase (Firman and Szczelkun, 2000; Seidel et al, 2004) and endonuclease properties (Janscak et al, 1998). During the DNA restriction reaction, the HsdR subunits, while remaining bound to the MTase, translocate adjacent DNA bidirectionally with a speed of several hundred base pairs (bp) per second (McClelland et al, 2005). Consequently, two DNA loops are extruded (Figure 1A; Yuan et al, 1980). Cleavage can occur thousands of base pairs away from the binding site (Szczelkun et al, 1997) and is thought to occur upon collision with a roadblock (Studier and Bandyopadhyay, 1988; Janscak et al, 1999a), which in vivo is most likely another Type I RM enzyme. Many purified Type I RM enzymes are found to dissociate under in vitro conditions into a partially assembled enzyme complex carrying only one HsdR (HsdR1HsdM2HsdS1, hereafter referred to as R1-complex) (Suri et al, 1984; Janscak et al, 1998). R1-complexes lack endonuclease activity, but are still able to translocate DNA (Firman and Szczelkun, 2000). For EcoR124I, it has been demonstrated that the translocation rate of an HsdR in an R1-complex is the same as in an R2-complex, although the processivity is reduced by more than three-fold (Seidel et al, 2004).
Figure 1.
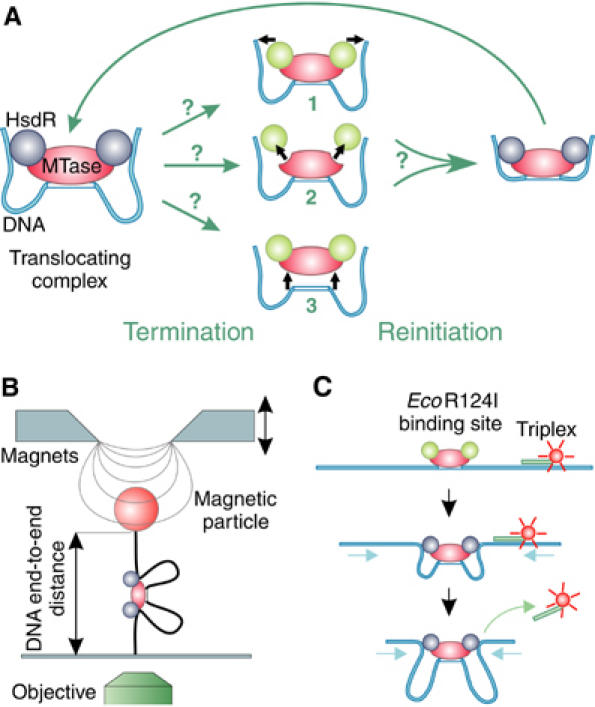
Schematic representation of termination and reinitiation of translocation by Type I RE and sketches of the two methods to measure translocation. (A) Three possible mechanisms for the termination of translocation by a loop-forming enzyme: (1) release of the translocated DNA by the HsdR subunits, with the enzyme complex remaining bound on the DNA; (2) disassembly of the enzyme by dissociation of the HsdR subunits from the MTase core unit; and (3) complete dissociation of the enzyme complex from the DNA. Following termination of translocation, reinitiation via the formation of an initial loop can occur; the mechanism utilized will depend on the termination route. Static HsdR molecules are drawn in green and translocating molecules in blue. (B) Magnetic tweezers. A DNA molecule with a single EcoR124I binding site is anchored between a magnetic bead and a glass slide. A pair of magnets is used to stretch the DNA molecule with different forces. Translocation of EcoR124I can be seen as a decrease in the DNA end-to-end distance, which is measured using video microscopy. (C) Triplex displacement assay. A tetramethylrhodamine-labeled triplex is attached at specific distances from the EcoR124I binding site. Translocation leads to displacement of the triplex resulting in an increase of fluorescence, which is monitored with a stopped-flow apparatus.
Although some of the translocation properties of EcoR124I and other Type I restriction enzymes (RE) have been well characterized, the processes accompanying initiation of translocation remain obscure. Recently, we showed that initiation results in the ATP-dependent formation of a small initial DNA loop ∼8 nm in length (van Noort et al, 2004). Furthermore, we have observed repeated reinitiation of the translocation activity, that is, following termination of translocation owing to limited processivity, motor activity can be readily re-established (Firman and Szczelkun, 2000; Seidel et al, 2004).
In the present study, we used bulk stopped-flow experiments and single-molecule magnetic tweezers technique to fully identify and quantify the individual steps during initiation, termination and reinitiation of translocation by EcoR124I. We demonstrate that translocation is terminated by disassembly of the enzyme complex: specifically, the translocating HsdR motor subunit dissociates from the core MTase, which itself remains bound to the DNA. In order to initiate a new translocation event, a new motor subunit has to bind. This means that the MTase core unit acts as a bona fide loader, repeatedly loading motor subunits from solution onto the adjacent DNA. The loading events of the two motors of EcoR124I occur in an independent manner. We further quantified how enzyme and ATP concentrations and applied force affected reinitiation, from which rates for motor binding and the subsequent initiation steps (including events such as ATP binding and initial loop formation) for the different stoichiometric forms of EcoR124I were obtained. This allowed us to present a kinetic scheme for the complete initiation, termination, reinitiation cycle (Figure 7). Both bulk and single-molecule experiments provided independent evidence for the identified reinitiation steps. Only the combination of the two techniques allowed the full characterization of each reaction rate in the reinitiation scheme. Finally, we discuss implications of the initiation scheme for in vivo restriction alleviation (RA) by Type I RE based on a preliminary characterization of an RA-deficient HsdR mutant.
Figure 7.
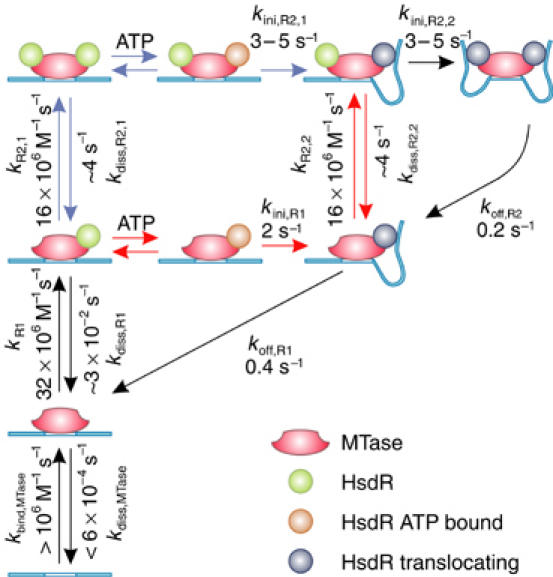
Dynamic cycling of initiation and termination of translocation by EcoR124I. For full details, see main text. Red and blue reaction arrows define the two different pathways how R2-events can be established.
The present study provides the first detailed picture of how an SF2 helicase DNA motor is loaded onto DNA. This insight might be helpful for investigations of other motors that translocate dsDNA like chromatin remodeling factors (Langst and Becker, 2004), but also helicases, which require a loading partner.
Results
Methods to characterize reinitiation
We applied single-molecule magnetic tweezers technique (Strick et al, 1998) and bulk stopped-flow experiments. Magnetic tweezers can repeatedly resolve single translocation events, as the end-to-end distance of a single stretched DNA molecule is monitored in real time (Figure 1B). With this technique, single EcoR124I translocation events can be seen as a transient decrease in the end-to-end distance owing to the formation of DNA loops (Figure 2A; Seidel et al, 2004). The time required to reinitiate translocation can then be derived from the time between two translocation events. In addition, magnetic tweezers give insight into the number of motor subunits bound. R1-complexes cause only events (R1-events) with a single translocation rate (Supplementary Figure 1), whereas R2-complexes can cause events (R2-events) with temporarily a doubled translocation rate (Seidel et al, 2004).
Figure 2.
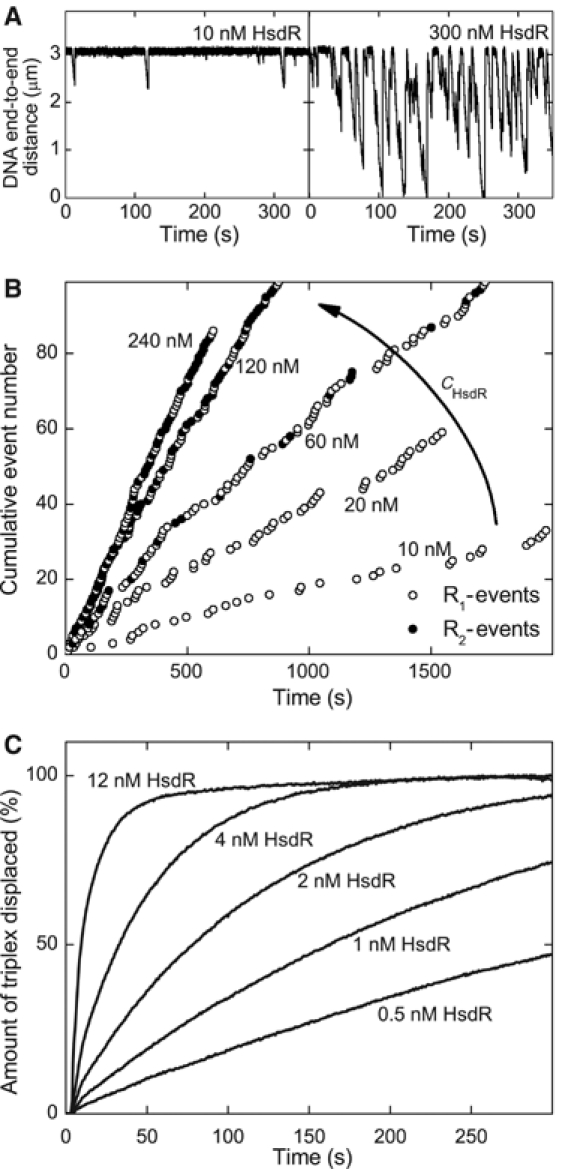
Dependence of the translocation activity on the motor subunit concentration. (A) Two time traces recorded in the magnetic tweezers at HsdR concentrations of 10 and 300 nM. The MTase concentration is 20 nM and the applied stretching force 0.8 pN. (B) The cumulative event number versus time of the occurrence of the event for various HsdR concentrations. The cumulative event number is obtained from time traces as in panel A by numbering every single translocation event consecutively. (C) Triple helix displacement profiles derived from stopped-flow experiments at low HsdR concentrations. Reactions were initiated by mixing preincubated triplex DNA, MTase and HsdR with an equal volume of reaction buffer with ATP (final concentration 4 mM). The triplex is 2047 bp away from the EcoR124I binding site. The final solution contains 30 nM MTase and 5 nM DNA, of which 2.5 nM carries one triplex. Therefore, only 25% of the translocation events can lead to triplex displacement, because there are 10 nM HsdR binding sites available (two per DNA-bound MTase).
An alternative way to measure translocation of Type I RE is through stopped-flow experiments. These utilize a fluorescently labeled triplex forming oligonucleotide (TFO) attached at a specific distance from the EcoR124I binding site (Figure 1C). Translocation by EcoR124I leads to dissociation of the triplex, which can be probed by monitoring the change in fluorescence upon displacement (Firman and Szczelkun, 2000; McClelland et al, 2005). Recorded displacement profiles (Figure 4C) are characterized by a lag time in which, owing to ongoing translocation, no triplex is displaced, followed by a rapid triplex displacement once the motors reached the triplex. Consequently, the lag time has a linear dependence on the distance between the triplex and the binding site of the enzyme. The overall shape of the displacement profile is determined by the rates for HsdR binding kbind,HsdR, initiation kini, translocation kstep, triplex displacement kTFO and termination of translocation koff (Firman and Szczelkun, 2000). For quantitative analysis of these rates, we modeled the recorded displacement profiles, based on the found reinitiation scheme (see Supplementary data).
Figure 4.
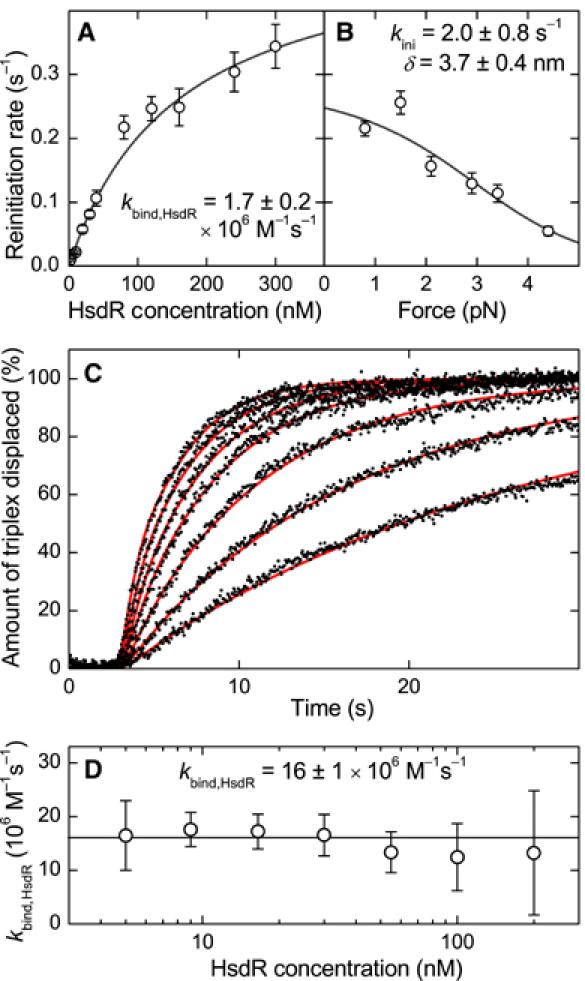
The kinetics of translocation reinitiation in single-molecule and stopped-flow bulk experiments. (A) Dependence of the reinitiation rate on the HsdR concentration obtained from the magnetic tweezers measurements depicted in Figure 2. By fitting the data with equation (2), an HsdR binding rate of kbind,HsdR=1.7±0.2 × 106 M−1 s−1 and an initiation rate at 0.8 pN of kini(0.8 pN)=0.3±0.2 s−1 is obtained. (B) Dependence of the reinitiation rate on the stretching force at 80 nM HsdR and 20 nM MTase. By fitting the data with equation (3) and using the previously determined binding rate, an initiation rate at zero force of kini=2.0±0.8 s−1 and a size for the initial DNA loop of δ=3.7±0.4 nm is obtained. The fit was weighted using the errors in the reinitiation rate. (C) Triplex displacement profiles for HsdR concentrations of (from right to left) 5, 9, 16.5, 30, 55, 100 and 200 nM (black dots). Reactions were initiated by mixing preincubated triplex DNA and MTase with an equal volume of reaction buffer with HsdR. ATP was present in both syringes at 4 mM. The spacing between triplex and EcoR124I site was 1409 bp. The final solution contains 30 nM MTase and 1 nM DNA, of which 0.5 nM carries the triplex. The more HsdR is added, the faster the reaction occurs. The red lines are fits to the data (see Supplementary data), which yield the HsdR binding rate at a given concentration and spacing. (D) HsdR binding rate to the MTase at different HsdR concentrations obtained from fitting the triplex displacement curves. For a given HsdR concentration, triplex displacement profiles were recorded for spacings between triplex and EcoR124I site of 479, 912, 1409 and 2047 bp. The plot shows the average binding rate for the four different spacings. As can be seen, the HsdR binding rate stays constant over the whole concentration range with an average kbind,HsdR=16±1 × 106 M−1 s−1.
The single-molecule measurements gave us detailed insight into the processes involved in reinitiation of translocation, which was necessary to understand the triplex displacement profiles and their dependence on reaction parameters. The bulk measurements themselves provided a possibility of rapid data acquisition under multiple different conditions. In the experiments described below, both methods independently measured the dependence of reinitiation on HsdR and MTase concentrations. Furthermore, magnetic tweezers yielded the force dependence of reinitiation and the concentration-dependent assembly state of the enzyme, whereas stopped-flow experiments measured the ATP dependence of initiation of translocation.
Termination and reinitiation of translocation activity
In order to investigate initiation/reinitiation of an enzyme motor, one needs to understand first the nature of translocation termination. Once DNA translocation by an EcoR124I enzyme is established, there are three possible mechanisms by which translocation can terminate (Figure 1A): (1) the DNA is simply released from the motor subunits, while the intact enzyme complex remains at its binding site; (2) the enzyme complex disassembles, that is, the motor subunits dissociate from the MTase core, which itself remains bound to the DNA; or (3) the complete enzyme complex dissociates from the DNA. In each case, different steps will be required to reinitiate translocation (Figure 1A). One way to discriminate between the different termination pathways is to measure how reinitiation depends on the relative subunit concentrations of EcoR124I. For model 1 (Figure 1A), reinitiation should be independent of subunit concentration, whereas for models 2 and 3, reinitiation should depend on second-order binding events. We can distinguish between models 2 and 3 by measuring whether reinitiation depends on the concentration of the complete enzyme (i.e., model 3) or of HsdR alone (i.e., model 2).
Motor subunit concentration and translocation activity
To distinguish between the different models for termination of translocation, we first studied how the HsdR motor subunit concentration influences reinitiation at a constant MTase concentration of 20 nM. Figure 2A shows two time traces recorded in the magnetic tweezers at 10 and 300 nM HsdR. Two striking differences between the traces can be seen: (1) the duration of the events is much longer at 300 nM than at 10 nM HsdR, which is due to the formation of R2-complexes at elevated HsdR concentrations (Janscak et al, 1998), which in turn exhibit a higher processivity (Seidel et al, 2004) and (2) the time interval between neighboring translocation events is much shorter at the higher HsdR concentration than at the lower one. This suggests that reinitiation strongly depends on the motor subunit concentration.
To investigate this further, translocation traces were recorded at additional HsdR concentrations. Motor events were identified (see Supplementary data), numbered consecutively and the event number was plotted against the time of its occurrence (Figure 2B). For a given HsdR concentration, the event number has a linear dependence on time. This shows that the observed level of enzyme activity was maintained over the whole measured time interval. Most significantly, we observe that the number of events per time interval increased as the HsdR concentration is increased. This strongly supports the previous conclusion that the translocation reinitiation is dependent on the motor subunit concentration. Therefore, rather than by simple DNA release from the enzyme (model 1 in Figure 1A), translocation must be terminated by a partial or complete dissociation of EcoR124I from the DNA (model 2 or 3).
In principle, a mixture of simple DNA release and dissociation could also account for a concentration-dependent mean reinitiation time. In that case, at lower HsdR concentrations, groups of translocation events should occur with short time intervals between single events (corresponding to DNA release), with these event groups separated by much longer times (corresponding to dissociation). This has for example been observed for FtsK (Saleh et al, 2004). However, no such event grouping was ever observed in our experiments (Figure 2B).
Independent evidence for the observed enzyme dissociation was obtained from the stopped-flow experiments. Displacement profiles on a DNA with a 2047 bp spacing between the EcoR124I site and the triplex were recorded for various HsdR concentrations between 0.5 and 12 nM, 30 nM MTase and 5 nM DNA (with 2.5 nM triplex bound). Under these conditions, the EcoR124I sites are saturated with MTase. The resulting displacement profiles at several HsdR concentrations are shown in Figure 2C. Even when the concentration of HsdR was significantly lower than the concentration of HsdR binding sites of 10 nM (two per DNA-bound MTase), efficient triplex displacement was observed. The amount of triplex displaced could exceed the input concentration of HsdR, which can only be explained if the enzyme, or a part of it, turns over; that is, there must be, at least partial, dissociation, which is in agreement with the magnetic tweezers measurements.
MTase core unit concentration and translocation activity
To further distinguish partial and complete dissociation (models 2 and 3; Figure 1A), we investigated how the translocation activity was affected by changes in MTase concentration. The following consecutive experiments were performed in the magnetic tweezers (Figure 3): (1) translocation activity was established by adding 20 nM MTase and 20 nM HsdR; (2) after 600 s, free MTase and free enzyme in solution were removed by flushing a solution containing only 20 nM HsdR; and (3) after 1200 s, the HsdR concentration was increased to 100 nM without the addition of further MTase. As one can see in Figure 3, the removal (after 600 s) of free MTase and in-solution-assembled enzyme did not affect translocation activity. Therefore, a translocation event cannot be terminated by dissociation of the whole enzyme, because in that case further translocation would immediately cease (rebinding would be very unlikely in our experimental setup). Even more strikingly, the event frequency increased after 1200 s solely due to increasing the HsdR concentration. Note also the occurrence of longer translocation events due to R2-complex formation. This shows that reinitiation of translocation is independent of free MTase or enzyme in solution, and is only dependent on the concentration of HsdR in solution, which demonstrates that the termination of a translocation event occurs via the partial disassembly of the enzyme complex (model 2; Figure 1A). Reinitiation of translocation must then occur via binding of a new HsdR subunit to the DNA-bound MTase. In the experiment depicted in Figure 3, translocation activity still continued for about 1800 s until the DNA molecule was lost (presumably owing to enzymatic cleavage; Seidel et al, 2004). Reinitiation events were repeatedly observed over long time periods in the absence of free MTase in solution and experiments always ended owing to loss of the DNA molecule. We can thus estimate a lower limit for the lifetime of the MTase on unmodified DNA of ∼1800 s from the longest recorded time traces (data not shown).
Figure 3.
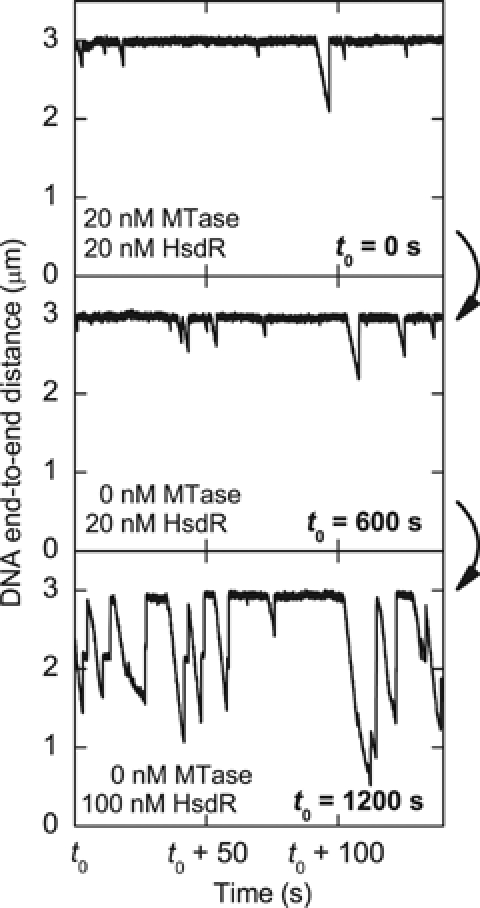
Dependence of the translocation activity on the free MTase in solution. The three time traces belong to consecutive experiments on the same DNA molecule. At t0=0 s, motor activity was initiated by adding 20 nM MTase and 20 nM HsdR into the flow cell. At t0=600 s, the free MTase in solution is removed by adding 250 μl (∼3-fold flow cell volume) of 20 nM HsdR only. After t0=1200 s, 250 μl of 100 nM HsdR is added. Translocation activity continued for another 1800 s and was terminated by the loss of the DNA molecule.
HsdR binding rate, initiation rate and initial loop formation from single-molecule measurements
After we have established that termination of translocation leads to disassembly of the enzyme complex, we can now focus on understanding initiation of translocation. From the experiments depicted in Figure 2A and B, we determined the mean time to reinitiate translocation 〈Treini〉, that is, to establish R1-activity (see Supplementary data for the analysis method). Figure 4A shows the corresponding reinitiation rate kreini=〈Treini〉−1 versus HsdR concentration. The relationship is linear for low HsdR concentrations and starts to saturate at higher concentrations. Thus, whereas at low concentrations HsdR binding is limiting, other steps in the initiation process become limiting at high concentrations. Discounting reverse reactions, the reinitiation process per HsdR binding site of the MTase can be written as follows:
![]()
where R0 is the non-translocating R1-complex, R1transl. is the translocating R1-complex, kbind,HsdR is the HsdR binding rate per binding site of the MTase and kini is the global initiation rate of translocation comprising all initiation steps following HsdR binding, such as ATP binding and initial loop formation. For this scheme, the mean reinitiation rate considering translocation in both directions can be written as (see Supplementary data)
![]()
where cHsdR denotes the free HsdR concentration in solution. The factor of 2 in equation (2) reflects the two HsdR binding sites per MTase, which opens two channels to initiate translocation. Fitting equation (2) to the experimental data (Figure 4A) yields kbind,HsdR=1.7±0.2 × 106 M−1 s−1 and kini(0.8 pN)=0.3±0.2 s−1 at the applied force of 0.8 pN.
Strictly speaking, the observed reaction rates are lower boundary values because we have not included any reverse reactions. However, initiation should be irreversible, as it is unlikely that the translocating complex would return to an initiation state (it would more likely dissociate and reset). Furthermore, the dissociation of the R1-complex is much slower than the subsequent initiation process and can thus be neglected (see below). Consequently, we believe that the application of the reaction scheme for the reinitiation of translocation (equation (1)) is justified. The above data fitting relies on the additional assumption that the free HsdR concentration in solution equals the input HsdR concentration, that is, that association with free MTase in solution does not occur in our experiments. For the following reasons, we believe that this is correct: (1) the reinitiation rate increases linearly even when cHsdR<cMTase; (2) triplex displacement profiles (Figure 2C) do not change when cMTase is varied from 30 to 120 nM (see Supplementary Figure 3); and (3) the association of free MTase and HsdR has previously been reported to be kinetically inhibited (Janscak et al, 1998).
The applied stretching force in the magnetic tweezers can affect rates of biochemical reactions, for example if the latter lead to a change in the end-to-end distance of the DNA. For example, the processivity of EcoR124I has been found to strongly depend on force (Seidel et al, 2004).
Previous atomic force microscopy (AFM) measurements revealed that binding of EcoR124I to DNA and initiation of translocation are associated with large conformational changes of the DNA at the binding site (van Noort et al, 2004). Upon binding of the MTase, the DNA is bent by about 50°, whereas subsequent HsdR binding does not affect the bending angle. During initiation of translocation, the DNA shortens by about 8 nm, which is thought to be due to the formation of an initial DNA loop. This step is found to depend on ATP binding, but not hydrolysis, by the HsdR subunit.
Therefore, one might expect that initial loop formation and not HsdR binding per se would be force dependent. Consistent with this, the reinitiation rate was independent of force for cHsdR<10 nM, where HsdR binding was limiting (not shown). For higher HsdR concentrations (cHsdR=80 nM), a strong decrease in the reinitiation rate with increasing forces was observed (Figure 4B), which indicates that the initial loop formation is limiting under these conditions. The force dependence of a chemical reaction k(F) with a rate constant k(F=0), in the absence of force, can be described by a force-dependent Arrhenius equation: k(F)=k(F=0)exp(−Fδ/kT), where δ denotes the size of a conformational change along the acting force F, which is required to overcome the reaction's energetic barrier (Howard, 2001). Combining this equation with equation (2), one can derive a simple relationship for the force dependence of the reinitiation rate:
![]()
Using the previously determined HsdR binding rate, a fit of the experimental data with equation (3) provides an initiation rate in the absence of force kini=2.0±0.8 s−1 and a contraction of δ=3.7±0.4 nm. The value for δ is in agreement with a large DNA contraction step upon initiation of translocation, that is, DNA loop formation. It is smaller than previously measured by AFM (van Noort et al, 2004). However, this may be because the contraction required to overcome the transition state barrier is smaller than that finally captured in the initial loop. We note that equation (3) is only correct if no other steps during initiation are rate limiting. This condition might not be fulfilled at limiting ATP concentrations (see below). Then, initiation must be split into a force-independent (ATP binding) and a force-dependent step (initial loop formation). The observation that only initial loop formation is limiting at saturating ATP is, however, supported by the fact that stopped-flow data give a similar value for the initiation rate (see below).
HsdR binding rate and ATP dependence of the initiation rate from bulk measurements
An independent quantitative description of the reinitiation process was obtained by using stopped-flow bulk experiments. In order to obtain an HsdR binding rate, triplex displacement profiles were recorded for various HsdR concentrations between 5 and 200 nM (Figure 4C) and four different distances between triplex and EcoR124I site (479–2047 bp). Traces were fit (see Supplementary data) by allowing variations in the value of kbind,HsdR and using fixed values for kstep=540 bp s−1, koff=0.2 s−1 (Seidel et al, 2004) and for kini and kTFO of 4.5 and 2.5 s−1 (obtained below). The overall shape of the concentration dependence of the displacement curves is well described by the fitting (red lines in Figure 4C). At each HsdR concentration, a single value for kbind,HsdR was obtained by averaging the binding rates obtained for the four different distances between triplex and EcoR124I site (Figure 4D). As one can see, the binding rate does not depend on the HsdR concentration (within experimental error), which validates the applied fitting procedure for the triplex displacement curves. By averaging over all HsdR concentrations used, a binding rate for the HsdR subunit per binding site of the MTase core unit of 16±1 × 106 M−1 s−1 is obtained. An identical binding rate is also obtained by fitting the displacement profiles in Figure 2C recorded at low HsdR concentrations of 0.5–12 nM (not shown). Thus, the obtained HsdR binding rate accurately describes the triplex displacement curves for the whole range of HsdR concentrations from 0.5 to 200 nM. This indicates that the HsdR binding rate does not depend on the conformation of the enzyme and must be very similar for R1- and R2-complex formation.
The HsdR binding rate obtained in the bulk measurements is almost an order of magnitude higher than the HsdR binding rate in the single-molecule experiments. We note that (1) the error in the concentration of active enzyme is low, as DNA cleavage assays were used to verify comparable enzymatic activity; (2) the obtained kbind,HsdR from the bulk measurements is rather insensitive to a different choice of fitting parameters; and (3) an even larger discrepancy in the rate of target site location (40- to 100-fold) between bulk (randomly coiled DNA) and single-molecule measurements (stretched DNA) has been recently reported for Type II RE (van den Broek et al, 2005). These authors suggested that in a random coil configuration of the DNA, an enzyme could find its target site more easily owing to three-dimensional transfers (jumping) between nonspecific sites (Halford and Marko, 2004), which would be inhibited in a stretched configuration. We therefore think that a similar effect causes the reduced HsdR binding rate in our single-molecule experiments. This implies that DNA–HsdR interactions may be a fundamental part of the pathway for location of the MTase core unit.
In previous AFM data, a requirement of ATP for the initial loop formation was found (van Noort et al, 2004). We therefore investigated how the ATP concentration influences initiation of translocation. Displacement profiles were recorded at six translocation distances for eight different ATP concentrations (see Figure 5A for the 479 bp spacing; other spacings are not shown). At each ATP concentration, values for kini, kstep and kTFO were obtained by fitting the profiles (see Supplementary data) using the bulk solution HsdR binding rate. The inset in Figure 5B shows the ATP dependence of the translocation rate. A Michaelis–Menten fit of the data provides kstep,max=576±2 bp s−1 and KM=74±4 μM in very close agreement with the values of 560±20 bp s−1 and 88±7 μM obtained from magnetic tweezers data (Seidel et al, 2004). Note that within the fitting procedure, kini and kTFO are indistinguishable (see Supplementary data). It is thus not possible to unambiguously assign which of the two rate constants obtained from the fit is kini and which is kTFO. We plotted the ATP dependence of both apparent rate constants obtained from the fit in Figure 5B. They both are strongly ATP dependent and provide similar values for the maximum rate at saturating ATP and for the Michaelis–Menten factor KM. For the first rate, a maximum kMax,1=5.2±0.3 s−1 and a KM,1=200±30 μM and, for the second rate, a kMax,2=3.3±0.1 s−1 and a KM,2=400±20 μM is obtained, which sets the limits for kini at saturating ATP to be between 3.2 and 5.5 s−1. The ATP dependence of the triplex displacement would be expected to be similar to that for kstep (Figure 5B, inset); however, it suggests that ATP binding at the triplex is weaker. This may be because both initiation and displacement of a roadblock are strained states, which alter the ATP binding pocket of the helicase domains. The initiation rate obtained at saturating ATP is similar to that from the single-molecule measurements. Given the different measurement techniques, it suggests that the initiation rates for R1- and R2-complexes are, within error, the same; the single-molecule measurements mainly probe R1-activity (i.e., the translocation of the first motor only), whereas the bulk measurements mainly probe R2-activity (reliable measurements are made at high HsdR, which favors formation of the R2-complex).
Figure 5.
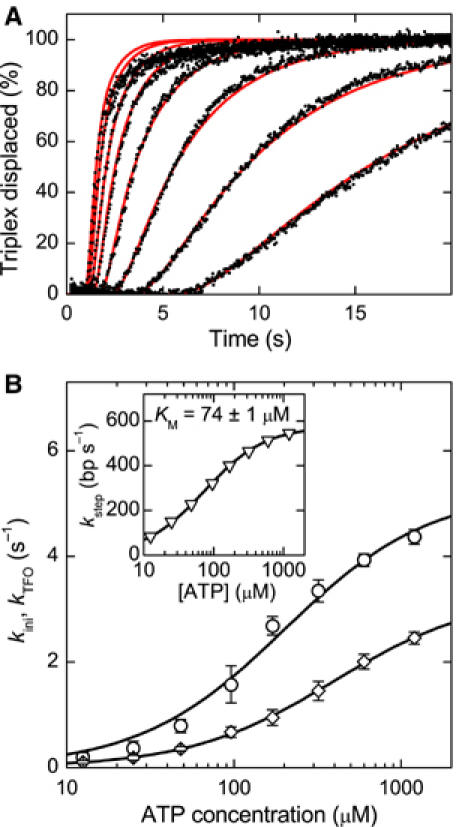
ATP dependence of the reinitiation rate kini. (A) Triple helix displacement profiles for ATP concentrations of (from right to left) 12.5, 25, 48, 96, 170, 320, 600 and 1200 μM (black dots). Reactions were initiated by mixing preincubated triplex DNA, MTase and HsdR with an equal volume of reaction buffer with ATP. The triplex is 479 bp away from the EcoR124I binding site. The final solution contains 30 nM MTase, 120 nM HsdR and 1 nM DNA, of which 0.5 nM carries the triplex. As the ATP concentration is increased, the profiles become faster. The red lines are fits to the data (see Supplementary data). (B) Initiation rate kini and triplex displacement rate kTFO versus ATP concentration as obtained from fitting the triplex displacement profiles. Note that the fitting procedure does not distinguish between kini and kTFO (see text and Supplementary data). From Michaelis–Menten fits, a maximum rate kMax,1=5.2±0.3 s−1 and a KM,1=200±30 μM is obtained for the first data set (open circle) and a kMax,2=3.3±0.1 s−1 and a KM,2=400±20 μM for the second data set (open diamonds). Thus, kini at saturating ATP is between 3.2 and 5.5 s−1. The inset shows the ATP dependence of the translocation rate. A maximum translocation rate of kstep,max=576±2 bp s−1 and a KM=74±1 μM is obtained from a Michaelis–Menten fit of the data.
Formation of R2-complexes
To gain insight into the assembly of the enzyme complex and the initiation pathway of R2-complexes, we analyzed from the magnetic tweezers data the occurrence of R1- and R2-events for the different HsdR concentrations measured. As can be seen in Figure 2B, the number of R2-events increases gradually with increasing HsdR concentrations, and R2- and R1-events are randomly distributed over time. The ratio between R2- and R1-events increases approximately linearly with HsdR concentration (Figure 6A). Our observation that R2-events occur randomly with respect to R1-events (Figure 2B) and with a probability that increases as more HsdR is added supports a dynamic assembly/disassembly model. Although one cannot rule out that some R1-events are caused by R2-complexes with only one translocating motor, the gradual increase in R2-events reflects that even at high HsdR concentrations, R1-complexes are present.
Figure 6.
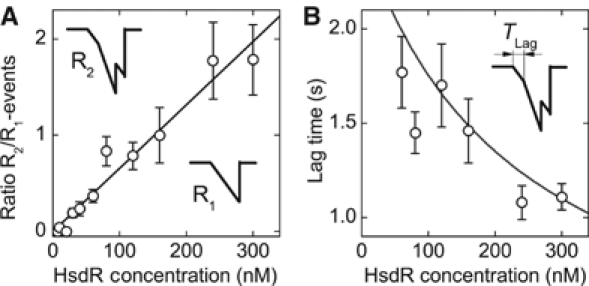
R2-complex formation and its dependence on the HsdR concentration. (A) Ratio between R2- and R1-events versus HsdR concentration. The insets in the graph illustrate the classification of each event type. The straight line in the graph is a linear fit to the data with a reciprocal slope of 152±8 nM. At this concentration, there are, on average, as many R1-events as R2-events. (B) Lag time versus HsdR concentration. The lag time is defined as the time it takes the second motor to establish translocation activity after the first motor started to translocate (illustrated in the inset). The solid line is plotted without adjustable parameters according to Tlag=(kR2,2cHsdR+koff,R1)−1 (see text).
In principle, there are two pathways for the formation of an R2-event: (1) by initiating translocation from a previously assembled R2-complex or (2) from an already translocating R1-complex to which a free HsdR molecule binds and initiates translocation. We analyzed how the time difference Tlag between initiation of the first motor and initiation of the second motor depends on the HsdR concentration (Figure 6B). One can see that Tlag decreases approximately two-fold at the applied range of concentrations. This dependence is not in agreement with initiation events occurring exclusively owing to previously assembled R2-events. The data strongly suggest that the association of a free HsdR molecule to an already translocating R1-complex can lead to R2-events. This demonstrates that binding and initiation of the two motors occur completely independently from each other.
Scheme for initiation, termination and reinitiation of translocation
The results of this study together with previous findings are summarized in the following model for initiation/reinitiation of translocation of EcoR124I (Figure 7):
Translocation activity is enabled by binding of MTase to its target site. As translocation events can be seen typically 30 s after adding the enzyme into the magnetic tweezers setup (data not shown), the binding rate for the MTase is at least kbind,MTase>106 M−1 s−1. As translocation activity is preserved for more than 30 min after removal of free MTase (see Results), dissociation of the MTase from an unmethylated target site must be very slow with kdiss,MTase<6 × 10−4 s−1.
R1-complexes are formed by HsdR binding to the MTase at a rate of kR1=32±2 × 106 M−1 s−1 for a randomly coiled DNA and at a rate of kR1=3.3±0.4 × 106 M−1 s−1 for stretched DNA. A dissociation rate of kdiss,R1∼3 × 10−2 s−1 is estimated from the measured dissociation constant Kdiss,R1∼1 nM (Janscak et al, 1998).
Initiation of translocation occurs in an ATP-dependent manner (van Noort et al, 2004; this study) with a KM of 200–400 μM.
At saturating ATP, the rate-limiting step in the initiation is the formation of an initial DNA loop for which a rate in the absence of force kini,R1=2.0±0.8 s−1 is found for the R1-complex. Initial loop formation is associated with a large DNA contraction step of 8 nm (van Noort et al, 2004).
Translocation of an R1-complex is terminated by dissociation of the HsdR from the MTase with an off-rate in the absence of force koff,R1=0.4 s−1 (Seidel et al, 2004).
R2-events, that is, translocation of both motors in an R2-complex, occur if the second HsdR establishes translocation before the first HsdR terminates translocation. The binding and initiation of motor activity for the second HsdR are independent of the first HsdR, that is, there is not a cooperative or ordered binding–initiation cycle. Furthermore, the associated rates for HsdR binding and initiation are largely not affected by the presence of an already bound HsdR subunit and its functional state (see Results). Thus, kbind,HsdR=kR2,1=kR2,2=16±1 × 106 M−1 s−1 and kini,R1∼kini,R2,1=kini,R2,2 between 3.2 and 5.5 s−1. From the equilibrium dissociation constant of an R2-complex of 240 nM (Janscak et al, 1998), we estimate a dissociation rate kdiss,R2,1=kdiss,R2,2∼4 s−1. The independence of the two HsdR subunits leads to two different pathways how R2-activity can be established (Figure 7).
R2-events are terminated by dissociation of one of the two translocating HsdR subunits. Given that the off-rate for a single HsdR motor in an R2-complex to terminate translocation is 0.1 s−1 (Seidel et al, 2004), the rate to terminate an R2-event is twice as high, koff,R2=0.2 s−1, because either HsdR subunit could dissociate.
Discussion
Dynamic disassembly and reassembly during translocation
In the present work, we found that reinitiation of translocation by the Type I RE EcoR124I depends strongly on the concentration of HsdR motor subunits in solution, whereas it is independent of the amount of MTase core unit in solution. These two observations provide evidence that EcoR124I disassembles between translocation events while the MTase core unit stays bound at the DNA. Although it is clear that HsdR dissociation is the final step in termination, it is not possible to rule out that it occurs first by DNA release from HsdR. Whatever the pathway, an obligate step for reinitiation of translocation is the binding of a new HsdR motor subunit from solution to the DNA-bound MTase. The MTase core unit serves, therefore, as a loader complex, which repeatedly loads the SF2 helicase motor onto the DNA.
The complete dissociation and rebinding of HsdR molecules has not been considered previously but is in agreement with earlier observations: (1) Type I RE do not turnover after cleavage, that is, it is necessary to add stoichiometric amounts of enzyme to DNA strands to get full cleavage of plasmid DNA carrying a single recognition site (Eskin and Linn, 1972). This is readily explained by the fact that the MTase remains bound to the DNA. (2) The direction of translocation between successive R1-complexes can switch (Firman and Szczelkun, 2000; Seidel et al, 2004), which can be readily explained by the HsdR dissociation and reassociation between translocation events. (3) It had been previously shown that R2-complexes are unstable and dissociate easily (Janscak et al, 1998). However, the commonly believed translocation model assumed a more static attachment of the HsdR subunits, because only equilibrium dissociation constants but no kinetic data were available. In contrast to previous measurements in the absence of ATP (Janscak et al, 1998), we found that R1-complexes readily undergo disassembly. Given the measured R1- and R2-complex formation rates (kR1=32 × 106 M−1 s−1, kR2=16 × 106 M−1 s−1) and the corresponding translocation off-rates (koff,R1=0.4 s−1, koff,R2=0.2 s−1 from Seidel et al, 2004), one can calculate a dissociation constant for the translocating R1-complex of ∼13 nM and for the translocating R2-complex of also ∼13 nM. This is different from the dissociation constants found in the absence of ATP of <1 nM for the R1-complex and 240 nM for the R2-complex (Janscak et al, 1998). The altered dissociation constants in the presence of ATP suggest that there is a different pathway of enzyme disassembly in the absence of ATP, which is most likely due to structural rearrangements of the enzyme during initiation of translocation.
The scheme for initiation, reinitiation and termination of translocation (Figure 7) should allow the prediction of any measurable quantity of the translocation process. To test the robustness of the model, we derived the R2/R1 ratio and the dependence of the lag time on the HsdR concentration in the magnetic tweezers (Figure 6; Supplementary data). This yields R2/R1≈kR2,2cHsdR/koff,R1=230 nM (with kR2,2=kbind,HsdR=1.7 × 106 M−1 s−1 for stretched DNA), in reasonable agreement with the experimentally determined value of 150 nM (Figure 6A), and Tlag≈(kR2,2cHsdR+koff,R1)−1, which describes closely the measured dependence of the lag time on the HsdR concentration (Figure 6B). Accordingly, it is also possible to obtain the calculated dependencies for randomly coiled DNA for which HsdR binding is faster.
In vivo implications
In vivo, Type I RE do not cleave the bacterial chromosome even if unmethylated sites are present (Makovets et al, 1999). This phenomenon, called restriction alleviation (RA), has only been clearly demonstrated for the Type I family of RE. It is still not understood how the RE can distinguish between ‘self' DNA and foreign viral DNA independently of the methylation status of the DNA. Recently, it has been proposed that the condensed state of the bacterial nucleoid prevents translocation and thus cutting of chromosomal DNA (Keatch et al, 2004). Extensive genetic investigations have identified that classical RA has an enzymatic basis in the form of the protease ClpXP (Murray, 2000). However, although Type IA and Type IB RE require ClpXP for RA (Makovets et al, 1999), the Type IC enzymes, including EcoR124I, and the Type ID enzymes do not need any additional cofactor (Makovets et al, 2004). For EcoR124I, single amino-acid substitutions in the HsdR subunit were identified that could inhibit RA. Owing to their location in the C-terminal domain, it was suggested that the mutations might alter the stability of the HsdR–MTase interface and prevent motor dissociation. It was speculated that such an increased complex stability might be the driving force for RA by EcoR124I.
In the present study, we confirm that HsdR disassembly is a key feature of the translocation mechanism of EcoR124I. To rule out the possibility that the results are a peculiarity of the in vitro-reconstituted enzyme used here, we carried out additional experiments using in vivo-assembled EcoR124I; these unambiguously prove disassembly and reassembly during translocation for in vivo-assembled enzyme (see Supplementary data).
To gain further insight into whether the disassembly of EcoR124I is the driving force for RA, we undertook a preliminary characterization of one of the RA-deficient mutants, HsdR(A957V), identified by Makovets et al (2004). Surprisingly, we find dissociation/reassembly similar to wild-type (wt)HsdR (see Supplementary data). Strikingly, however, similar levels of translocase and nuclease activities are reached at ∼4-fold reduced HsdR concentrations as compared to wtHsdR. This increased efficiency is not due to an altered processivity, but rather due to faster HsdR binding/initiation, as revealed by an increased number of R1- and R2-events per given time in single-molecule traces (see Supplementary data). These observations strongly suggest that the balance between disassembly and reassembly is used to tune restriction activity in vivo by the amount of available HsdR. By keeping the HsdR expression levels low, the cell is able to prevent formation of R2-events and thus cleavage-proficient R2-complexes. However, HsdR(A957V) can more easily assemble translocating R2-complexes, which, in turn, leads directly to the increased cleavage observed in vitro and in vivo. The essential restriction of viral DNA might be accomplished by membrane-bound EcoR124I complexes (Holubova et al, 2004).
Materials and methods
Proteins
EcoR124I RE was purified, reconstituted and tested as previously described (Seidel et al, 2004; McClelland et al, 2005).
DNA constructs
For magnetic tweezers experiments, singly nicked DNA constructs with a single EcoR124I site were prepared from SpeI- and BamHI-digested pSFV1 plasmid (Seidel et al, 2004). Constructs for triple helix-based translocation assays were prepared according to McClelland et al (2005).
Magnetic tweezers measurements
Magnetic tweezers experiments using 2.8 μm magnetic beads (Dynal) were carried out as described previously (Seidel et al, 2004) in reaction Buffer R (50 mM Tris–HCl pH 8.0, 10 mM MgCl2, 1 mM dithiothreitol) supplemented with 4 mM ATP. Unless otherwise stated, the MTase concentration was 20 nM and the applied stretching force 0.8 pN. All measurements were carried out at 25°C.
Triple helix displacement assays
Preparation of fluorescently labeled triplex DNA and analysis by stopped flow were carried out as described previously (McClelland et al, 2005) with the following modifications. Tetramethylrhodamine-labeled TFOs (MWG) were used, which allowed increased sensitivity down to 0.5 nM triplex (data not shown). λex was 547 nm (6 nm bandwidth) with a 570 nm band-pass filter placed between the sample chamber and photomultiplier tube. Reactions were recorded using a continuous time base. All reactions were carried out in Buffer R at 25°C.
Data analysis
Time traces recorded with the magnetic tweezers were fit and treated as described previously (Seidel et al, 2004). Reinitiation times were extracted from the fitted data as explained in Supplementary data.
Triple helix displacement profiles were processed according to McClelland et al (2005). Kinetic fitting to the data was carried out using numerical integration as described in Supplementary data.
Supplementary Material
Supplementary Information
Acknowledgments
We acknowledge the financial support of the European Commission through the Mol Switch project (IST-2001-38036), the Nederlandse Organisatie voor Wetenschappelijk Onderzoek (NWO) and the Wellcome Trust. MDS is a Wellcome Trust Senior Research Fellow in Basic Biomedical Science. We thank C van der Scheer and T van der Heijden for helpful discussions and P Janscak for the kind gift of R.EcoR124I.
References
- Browning DF, Busby SJW (2004) The regulation of bacterial transcription initiation. Nat Rev Microbiol 2: 57–65 [DOI] [PubMed] [Google Scholar]
- Davies GP, Martin I, Sturrock SS, Cronshaw A, Murray NE, Dryden DTF (1999) On the structure and operation of type I DNA restriction enzymes. J Mol Biol 290: 565–579 [DOI] [PubMed] [Google Scholar]
- Dryden DTF, Cooper LP, Thorpe PH, Byron O (1997) The in vitro assembly of the EcoKI type I DNA restriction/modification enzyme and its in vivo implications. Biochemistry 36: 1065–1076 [DOI] [PubMed] [Google Scholar]
- Eskin B, Linn S (1972) Deoxyribonucleic acid modification and restriction enzymes of Escherichia coli B. 2. Purification, subunit structure, and catalytic properties of the restriction endonuclease. J Biol Chem 247: 6183–6191 [PubMed] [Google Scholar]
- Farah JA, Smith GR (1997) The RecBCD enzyme initiation complex for DNA unwinding: enzyme positioning and DNA opening. J Mol Biol 272: 699–715 [DOI] [PubMed] [Google Scholar]
- Firman K, Szczelkun MD (2000) Measuring motion on DNA by the type I restriction endonuclease EcoR1241 using triplex displacement. EMBO J 19: 2094–2102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halford SE, Marko JF (2004) How do site-specific DNA-binding proteins find their targets? Nucleic Acids Res 32: 3040–3052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holubova I, Vejsadova I, Firman K, Weiserova M (2004) Cellular localization of type I restriction–modification enzymes is family dependent. Biochem Biophys Res Commun 319: 375–380 [DOI] [PubMed] [Google Scholar]
- Howard J (2001) Mechanics of Motor Proteins and the Cytoskeleton. Sunderland, MA: Sinauer Associates [Google Scholar]
- Janscak P, Dryden DTF, Firman K (1998) Analysis of the subunit assembly of the type IC restriction–modification enzyme EcoR124I. Nucleic Acids Res 26: 4439–4445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janscak P, MacWilliams MP, Sandmeier U, Nagaraja V, Bickle TA (1999a) DNA translocation blockage, a general mechanism of cleavage site selection by type I restriction enzymes. EMBO J 18: 2638–2647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janscak P, Sandmeier U, Bickle TA (1999b) Single amino acid substitutions in the HsdR subunit of the type IB restriction enzyme EcoAI uncouple the DNA translocation and DNA cleavage activities of the enzyme. Nucleic Acids Res 27: 2638–2643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keatch SA, Su TJ, Dryden DTF (2004) Alleviation of restriction by DNA condensation and non-specific DNA binding ligands. Nucleic Acids Res 32: 5841–5850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konieczny I (2003) Strategies for helicase recruitment and loading in bacteria. EMBO Rep 4: 37–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langst G, Becker PB (2004) Nucleosome remodeling: one mechanism, many phenomena? Biochim Biophys Acta 1677: 58–63 [DOI] [PubMed] [Google Scholar]
- Makovets S, Doronina VA, Murray NE (1999) Regulation of endonuclease activity by proteolysis prevents breakage of unmodified bacterial chromosomes by type I restriction enzymes. Proc Natl Acad Sci USA 96: 9757–9762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makovets S, Powell LM, Titheradge AJB, Blakely GW, Murray NE (2004) Is modification sufficient to protect a bacterial chromosome from a resident restriction endonuclease? Mol Microbiol 51: 135–147 [DOI] [PubMed] [Google Scholar]
- McClelland SE, Dryden DTF, Szczelkun MD (2005) Continuous assays for DNA translocation using fluorescent triplex dissociation: application to type I restriction endonucleases. J Mol Biol 348: 895–915 [DOI] [PubMed] [Google Scholar]
- Murray NE (2000) Type I restriction systems: sophisticated molecular machines (a legacy of Bertani and Weigle). Microbiol Mol Biol Rev 64: 412–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roman LJ, Kowalczykowski SC (1989) Characterization of the helicase activity of the Escherichia-coli RecBCD enzyme using a novel helicase assay. Biochemistry 28: 2863–2873 [DOI] [PubMed] [Google Scholar]
- Saleh OA, Perals C, Barre FX, Allemand JF (2004) Fast, DNA-sequence independent translocation by FtsK in a single-molecule experiment. EMBO J 23: 2430–2439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidel R, van Noort J, van der Scheer C, Bloom JGP, Dekker NH, Dutta CF, Blundell A, Robinson T, Firman K, Dekker C (2004) Real-time observation of DNA translocation by the type I restriction modification enzyme EcoR124I. Nat Struct Mol Biol 11: 838–843 [DOI] [PubMed] [Google Scholar]
- Singleton MR, Dillingham MS, Gaudier M, Kowalczykowski SC, Wigley DB (2004) Crystal structure of RecBCD enzyme reveals a machine for processing DNA breaks. Nature 432: 187–193 [DOI] [PubMed] [Google Scholar]
- Stenlund A (2003) Initiation of DNA replication: lessons from viral initiator proteins. Nat Rev Mol Cell Biol 4: 777–785 [DOI] [PubMed] [Google Scholar]
- Strick TR, Allemand JF, Bensimon D, Croquette V (1998) Behavior of supercoiled DNA. Biophys J 74: 2016–2028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studier FW, Bandyopadhyay PK (1988) Model for how type-I restriction enzymes select cleavage sites in DNA. Proc Natl Acad Sci USA 85: 4677–4681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suri B, Shepherd JCW, Bickle TA (1984) The EcoA restriction and modification system of Escherichia coli 15T: enzyme structure and DNA recognition sequence. EMBO J 3: 575–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczelkun MD, Janscak P, Firman K, Halford SE (1997) Selection of non-specific DNA cleavage sites by the type IC restriction endonuclease EcoR124I. J Mol Biol 271: 112–123 [DOI] [PubMed] [Google Scholar]
- van den Broek B, Noom MC, Wuite GJL (2005) DNA-tension dependence of restriction enzyme activity reveals mechanochemical properties of the reaction pathway. Nucleic Acids Res 33: 2676–2684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Noort J, van der Heijden T, Dutta CF, Firman K, Dekker C (2004) Initiation of translocation by Type I restriction–modification enzymes is associated with a short DNA extrusion. Nucleic Acids Res 32: 6540–6547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan R, Hamilton DL, Burckhardt J (1980) DNA translocation by the restriction enzyme from Escherichia coli K. Cell 20: 237–244 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information