

Abstract
NF-κB activation is an important mechanism of mammalian UV response to protect cells. UV-induced NF-κB activation depends on the casein kinase II (CK2) phosphorylation of IκBα at a cluster of C-terminal sites, but how it is regulated remains unclear. Here we demonstrate that β-arrestin2 can function as an effective suppressor of UV-induced NF-κB activation through its direct interaction with IκBα. CK2 phosphorylation of β-arrestin2 blocks its interaction with IκBα and abolishes its suppression of NF-κB activation, indicating that the β-arrestin2 phosphorylation is critical. Moreover, stimulation of β2-adrenergic receptors, a representative of G-protein-coupled receptors in epidermal cells, promotes dephosphorylation of β-arrestin2 and its suppression of NF-κB activation. Consequently, the β-arrestin2 suppression leads to promotion of UV-induced cell death, which is also under regulation of β-arrestin2 phosphorylation. Thus, β-arrestin2 is identified as a phosphorylation-regulated suppressor of UV response and this may play a functional role in the response of epidermal cells to UV.
Keywords: β-arrestin2, CK2, NF-κB
Introduction
The UV response in mammalian cells is mediated through activation of several transcription factors, including NF-κB (Shaulian et al, 2000). In resting cells, NF-κB is predominantly sequestered in the cytoplasm through association with specific inhibitors IκB (reviewed by Silverman and Maniatis, 2001). The binding of IκB to NF-κB masks the nuclear localization sequences (NLS) of the latter and thus traps NF-κB in the cytoplasm. UV irradiation of mammalian cells leads to phosphorylation of IκB by casein kinase II (CK2) and triggers the ubiquination-dependent degradation of IκB (Bender et al, 1998; Kato et al, 2003). NF-κB is then released and translocated to the nucleus and promotes target genes transcription through binding to specific promoters (Israel, 2000; Karin and Ben-Neriah, 2000). NF-κB pathway is under various regulations. For example, Gao et al (2004) have shown that β-arrestin2, one of the major regulatory and signaling molecules in G-protein-coupled receptor (GPCR) signal transduction, directly interacts with IκBα and prevents phosphorylation and degradation of IκBα. Similar results are also obtained from Witherow et al (2004). It has been shown that deficiency of the murine NF-κB results in embryonic lethality as a result of fetal hepatocyte apoptosis (Beg and Baltimore, 1996), indicating that NF-κB plays a critical role in regulation of apoptosis. NF-κB exerts its antiapoptotic effect through inducing various antiapoptotic genes, such as Bcl-xL, X-IAP, IAP1 and 2, IEX-1L, and Bfl-1, following treatment with diverse stimuli, including UV irradiation, inflammatory cytokines, and chemotherapeutic drugs (Barkett and Gilmore, 1999; Baldwin, 2001).
GPCRs comprise a large gene family of up to 1000 members that mediate many distinct physiological functions as diverse as phototransduction, olfaction, vascular tone, cardiac output, digestion, and pain (Oakley et al, 2000). Recently, increasing evidence indicates the potential role of GPCRs in epidermal functions (Takahashi et al, 1996; Schallreuter et al, 1997; Denda et al, 2003; Gillbro et al, 2004; Steckelings et al, 2004). β2AR, one of the well-studied GPCRs, are ubiquitously expressed in the epidermis and function to maintain homeostasis in normal physiological settings as well as pathological states in epidermis (Koizumi et al, 1991; Steinkraus et al, 1996; Denda et al, 2003). Epinephrine and other catecholamines, which stimulate β2AR, mediate signals not only in autocrine regulation among epidermal cells but also from the sympathetic nervous system (Steckelings et al, 2004). However, the molecular mechanism underlying β2AR regulation of epidermis remains to be investigated.
β-Arrestins are ubiquitous multifunctional adaptor proteins that regulate numerous aspects of GPCR functions and have been recently shown to serve as mediators that link the receptors to other signaling pathways (Goodman et al, 1996; Luttrell et al, 1999; McDonald et al, 2000; Perry et al, 2002; Chen et al, 2003). Recent studies showed that β-arrestins are subjected to several post-translational modifications. Upon stimulation with β2AR, β-arrestin2 is rapidly and transiently ubiquitinated through its association with Mdm2, and the ubiquitination of β-arrestin2 is required for the GPCR internalization (Shenoy et al, 2001). The functions of β-arrestins can also be regulated by phosphorylation. Cytosolic β-arrestin1 is constitutively phosphorylated at Ser412 by ERKs, and stimulation of β2AR leads to dephosphorylation of β-arrestin1 on the plasma membrane, which is essential for targeting receptor to clathrin-coated pits and for binding to c-Src to induce ERK activation (Lin et al, 1997; Luttrell et al, 1999). β-Arrestin2 is also reported to be constitutively phosphorylated by CK2, predominantly at residues Ser361 and Thr383, and β2AR stimulation promotes dephosphorylation of β-arrestin2 (Kim et al, 2002; Lin et al, 2002). However, the functional role of β-arrestin2 phosphorylation remains unclear.
Results
β-Arrestin2 suppresses UV-induced NF-κB activation
Previous studies have shown that β-arrestin2 could inhibit NF-κB activation induced by activators such as TNFα (Gao et al, 2004, Witherow et al, 2004), LPS, and IL-1β (our unpublished data). As UV irradiation also induced NF-κB activation (Li and Karin, 1998; Shaulian et al, 2000), we first investigated whether β-arrestin2 had a similar function in UV pathway. β-Arrestin-null murine embryonic fibroblasts (MEFs) and wild-type MEFs were exposed to UV irradiation for 1 min (10 J/m2), and then incubated for up to 6 h. Cells were harvested at the indicated time points and the level of IκBα was detected by immunoblot. As shown in Figure 1A, UV irradiation led to a time-dependent IκBα degradation in β-arrestin-null MEFs, which was much less in wild-type MEFs. Next, UV-induced NF-κB p65 nuclear translocation, which follows IκBα degradation, was examined. UV irradiation caused a much stronger nuclear translocation of NF-κB p65 and NF-κB DNA-binding activity in β-arrestin-null MEFs than that in their wild-type counterparts (Figure 1A). The purity of nuclear and cytoplasmic fractions was demonstrated with antibodies specific for Sp1, a known nuclear protein (Figure 1A), and tubulin, known to be cytoplasmic (data not shown). The specificity of NF-κB DNA-binding activity was ascertained as reported previously (Gao et al, 2004).
Figure 1.
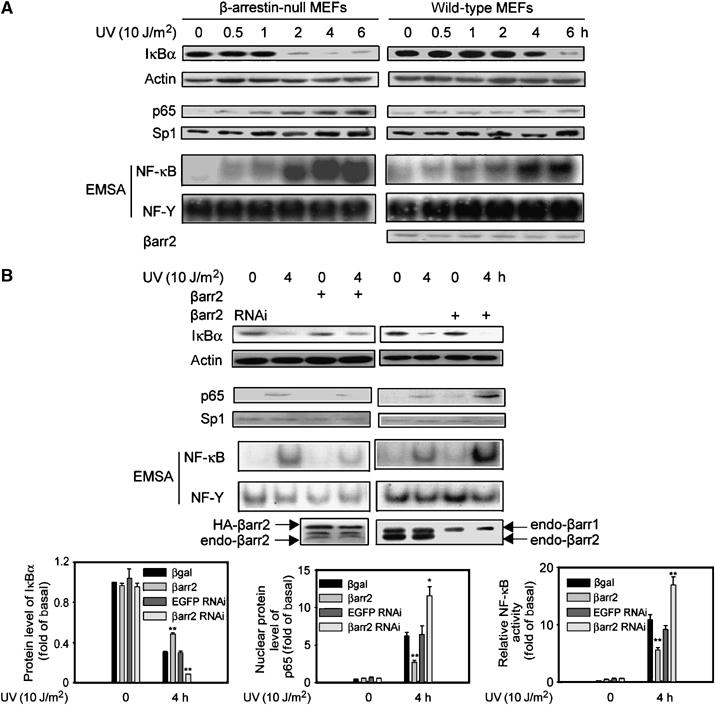
β-arrestin2 suppresses UV-induced NF-κB activation. (A) Time course of UV-induced IκBα degradation measured by immunoblot, nuclear translocation of NF-κB p65 by immunoblot, and NF-κB DNA-binding activities by EMSA using nuclear extracts in β-arrestin-null MEFs and wild-type MEFs. (B) UV-induced degradation of IκBα was measured by immunoblot in A431 cells expressed with β-arrestin2 or β-arrestin2 siRNA. UV-induced nuclear translocation of NF-κB p65 was measured by immunoblot and UV-induced NF-κB DNA-binding activities by EMSA using nuclear extracts of A431 cells expressed with β-arrestin2 or β-arrestin2 siRNA. The actual increase (upon overexpression) is four-fold of basal and decrease (with siRNA) is about 95% of basal β-arrestin2 expression. Data are representative of at least three independent experiments and shown as mean±s.e. *P<0.05, and **P<0.01 versus control. βarr2: β-arrestin2.
To further analyze β-arrestin2's function in UV-induced NF-κB activation, human epidermal carcinoma A431 cells were transiently transfected with β-arrestin2 or β-arrestin2 siRNA and irradiated with UV. The IκBα degradation, p65 nuclear translocation, and NF-κB DNA-binding activity were all decreased by expression of β-arrestin2, while they were all augmented by application of β-arrestin2 siRNA (Figure 1B). As a control, AP1 transcription activity was unaffected under similar conditions (data not shown).
Next we analyzed whether β-arrestin2 stabilized IκBα after UV irradiation via its direct binding to IκBα as in the case of TNFα stimulation (Gao et al, 2004). Previous work had shown that deletion of amino acids 1–19 from the N-terminus or 186–409 from the C-terminus of β-arrestin2 resulted in complete loss of its ability to bind to IκBα, while deletion of amino acids 240–409 did not. These results indicated that β-arrestin2 possessed two binding sites to IκBα (one among the amino acids 1–19 and another 186–240) (Figure 2A), and positively charged amino acids in β-arrestin2 were important for its interaction with other proteins like GPCR and Mdm2 (data not shown). The mutations of the positively charged amino acids in those two fragments were screened, and the mutations of Lys11, Lys12, Lys230, and Lys231 to Ala of β-arrestin2 (β-arrestin2 4M) resulted in nearly complete loss of its ability to bind IκBα (Figure 2B).
Figure 2.
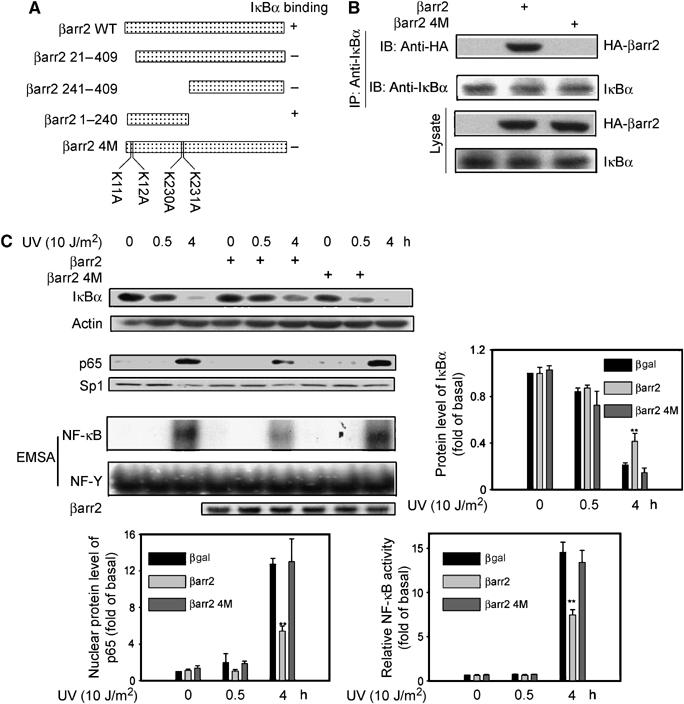
Interaction of β-arrestin2 with IκBα is required for its suppression on NF-κB activation. (A) Schematic representation of β-arrestin2 and its mutants. (B) No interaction between β-arrestin2 4M and IκBα. Cell lysates from β-arrestin-null MEFs expressed with β-arrestin2 or β-arrestin2 4M were subjected to immunoprecipitation and then immunoblot with antibody as indicated. (C) No effects of β-arrestin2 4M on IκBα degradation, NF-κB p65 translocation, and NF-κB DNA-binding activity. β-Arrestin-null MEFs expressed with β-arrestin2 or β-arrestin2 4M were exposed to UV irradiation and IκBα degradation by immunoblot, nuclear translocation of NF-κB p65 by immunoblot, and NF-κB DNA-binding activity by EMSA were examined. Data are representative of at least three independent experiments, and are shown as mean±s.e. *P<0.05 and **P<0.01 versus control. IP, immunoprecipitated; IB, immunoblot.
We then compared the effect of β-arrestin2 and the 4M mutant on NF-κB signaling pathway by re-expression in β-arrestin-null MEFs. β-Arrestin2 re-expression decreased degradation of IκBα, NF-κB p65 nuclear translocation, and NF-κB DNA-binding activity upon UV irradiation (Figure 2C), further confirming β-arrestin2's function in UV pathway. In contrast to wild-type β-arrestin2, β-arrestin2 4M did not rescue the suppressive function in β-arrestin-null MEFs, as it did not change any of these responses to UV irradiation. These results strongly suggest that β-arrestin2 suppresses UV-induced NF-κB activation via its interaction with IκBα.
β-Arrestin2 phosphorylation regulates its suppression of NF-κB activation
β-Arrestin2 is constitutively phosphorylated by CK2 at the C-terminus, but its functions remain unclear up to now (Lin et al, 2002). Moreover, CK2 kinase activity towards IκBα is increased after UV irradiation (Kato et al, 2003). Therefore, we first examined whether β-arrestin2 was further phosphorylated under UV irradiation. β-Arrestin2 re-introduced into β-arrestin-null MEFs with or without UV irradiation was immunoprecipitated and then analyzed by antibody against p-Ser and p-Thr to detect its phosphorylation status. Figure 3A shows that both Ser and Thr phosphorylation of β-arrestin2 were increased upon UV irradiation. Increased β-arrestin2 phosphorylation upon UV irradiation was also found in A431 by using metabolical labeling of [32P]orthophosphate followed by immunoprecipitation (Figure 3B). Furthermore, treatment of the cells with a p38 inhibitor (SKF86002, 20 μM for 90 min) significantly reduced the level of phosphorylation, suggesting that p38 was involved in UV-induced β-arrestin2 phosphorylation. We then examined the effect of CK2 phosphorylation of β-arrestin2 on its interaction with IκBα. β-Arrestin-null MEFs were transfected with plasmids encoding β-arrestin2, CK2, or IKKβ, and the cells were incubated with 10 μM MG132 for 4 h to inhibit IκBα degradation. Under these conditions, β-arrestin2 was phosphorylated by CK2 but not IKKβ. Cell lysates were prepared and used for coimmunoprecipitation to detect β-arrestin2–IκBα interaction. Figure 3C shows that β-arrestin2–IκBα interaction was significantly reduced by expression of CK2 but not IKKβ, suggesting that CK2 phosphorylation of β-arrestin2 may regulate its binding to IκBα. Moreover, UV irradiation of β-arrestin-null MEFs with re-expression of β-arrestin2 also reduced the β-arrestin2–IκBα interaction, further confirming our conclusion (Figure 3D). MG132 was also used to inhibit IκBα degradation in these experiments.
Figure 3.
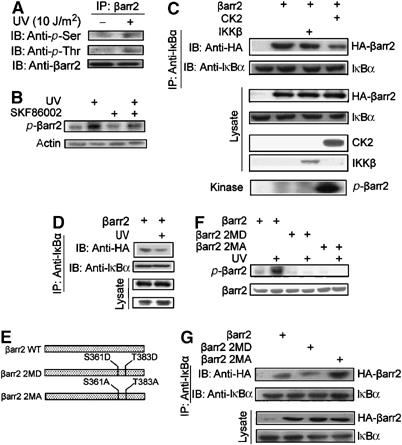
CK2 phosphorylation of β-arrestin2 blocks its interaction with IκBα. (A) Increased phosphorylation of β-arrestin2 after UV irradiation. β-Arrestin-null MEFs re-introduced with β-arrestin2 were irradiated with UV for 1 h and cell lysates were subjected to immunoprecipitation and immunoblot with antibody as indicated. (B) In vivo phosphorylation of β-arrestin2. A431 cells treated with or without SKF86002 were metabolically labeled with [32P]orthophosphate for 90 min and irradiated with UV for 1 h. Cell lysates were subjected to immunoprecipitation and autoradiography. (C) Blockage of β-arrestin2 and IκBα interaction by CK2. Cell lysates from β-arrestin-null MEFs coexpressed with β-arrestin2, and CK2 or IKKβ were subjected to immunoprecipitation and then immunoblot with antibody as indicated. MG132 (10 μM) for 4 h was used to inhibit IκBα degradation. (D) Blockage of β-arrestin2 and IκBα interaction by UV irradiation. β-Arrestin-null MEFs re-introduced with β-arrestin2 were irradiated with UV for 1 h and cell lysates were subjected to immunoprecipitation and immunoblot with antibody as indicated. MG132 (10 μM) for 4 h was used to inhibit IκBα degradation. (E) Schematic representation of β-arrestin2 phosphorylation mutants. (F) Phosphorylation of β-arrestin2 and phosphorylation site mutants with or without UV irradiation in β-arrestin-null MEFs was measured using in vivo phosphorylation. (G) IκBα interaction with phosphorylation site mutants of β-arrestin2. Phosphorylation sites of β-arrestin2 were mutated into Asp (β-arrestin2 2MD) or Ala (β-arrestin2 2MA) as indicated. Cell lysates from β-arrestin-null MEFs expressed with β-arrestin2, β-arrestin2 2MD, or β-arrestin2 2MA were subjected to immunoprecipitation and then immunoblot with antibody as indicated. Data are representative of at least three independent experiments and are shown as mean±s.e. *P<0.05 and **P<0.01 versus control.
β-Arrestin2 is phosphorylated mainly at Ser361 and Thr383 by CK2 (Lin et al, 2002). Next we generated two β-arrestin2 mutants in which Ser361 and Thr383 were substituted with Asp (β-arrestin2 2MD) or Ala (β-arrestin2 2MA) (Figure 3E). Phosphorylation of these mutants upon UV irradiation was then analyzed after their re-expression in β-arrestin-null MEFs. Phosphorylation of both β-arrestin2 2MD and 2MA mutants was significantly reduced compared to the wild-type β-arrestin2, and their level of phosphorylation was no longer increased by UV irradiation (Figure 3F), confirming that Ser361 and Thr383 are the predominant phosphorylation sites in β-arrestin2. Furthermore, coimmunoprecipitation experiments showed that β-arrestin2 2MD mutant, which mimics the phosphorylated β-arrestin2, exhibited significantly reduced interaction with IκBα, while β-arrestin2 2MA, which mimics the dephosphorylated β-arrestin2, displayed much stronger interaction with IκBα than β-arrestin2 (Figure 3G). These results suggested that phosphorylation of β-arrestin2 regulates its interaction with IκBα. Consistent with this, coexpression of CK2 or UV irradiation could no longer decrease the interaction of β-arrestin2 2MD or 2MA mutant with IκBα (Supplementary data) since they could not be phosphorylated by CK2 (Lin et al, 2002).
To assess whether β-arrestin2 phosphorylation and consequent changes in its interaction with IκBα regulate its suppressive function in IκBα degradation, the phosphorylation mutants of β-arrestin2 were expressed in β-arrestin-null MEFs. As shown in Figure 4, β-arrestin2 2MD mutant was not as effective as the wild-type β-arrestin2 in its suppressive function for UV-induced IκBα degradation, NF-κB p65 nuclear translocation, and NF-κB DNA-binding activity. In contrast, β-arrestin2 2MA mutant showed significantly increased suppression of these responses of NF-κB pathway to UV irradiation compared to wild-type β-arrestin2. Taken together, these data indicate that β-arrestin2 phosphorylation negatively regulates its suppression on NF-κB activation through interfering its interaction with IκBα.
Figure 4.
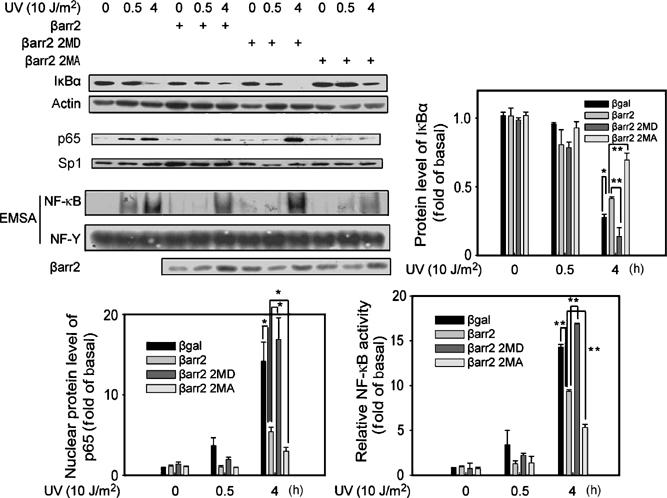
β-Arrestin2 phosphorylation regulates its suppression on NF-κB activation. Regulation of UV-induced IκBα degradation, NF-κB p65 nuclear translocation, and NF-κB DNA-binding activity by β-arrestin2 phosphorylation. β-Arrestin-null MEFs expressed with β-arrestin2 WT, β-arrestin2 2MD, or β-arrestin2 2MA were exposed to UV irradiation and IκBα degradation, NF-κB p65 nuclear translocation, and NF-κB DNA-binding activity were examined. Data are representative of at least three independent experiments and are quantified as mean±s.e. *P<0.05 and **P<0.01 versus control.
β-Arrestin2 suppression is stimulated by β2AR
As a major regulatory molecule in GPCR signal transduction, β-arrestin2's functions are stimulated by GPCRs (Luttrell et al, 1999; Wang et al, 2003), and β2AR promotes dephosphorylation of β-arrestin2 at Ser361 and Thr383 (Lin et al, 2002). To evaluate a potential role of β2AR in the regulation of β-arrestin2's function in UV-induced NF-κB activation, we first confirmed that β2AR promoted β-arrestin2 dephosphorylation in our system. As shown in Figure 5A, Iso stimulation reduced β-arrestin2 phosphorylation in A431 cells, while phosphatase inhibitor okadaic acid (50 nM for 30 min) abolished this effect. As a result, β-arrestin2–IκBα interaction was enhanced by β2AR stimulation (Figure 5B). In contrast, the interaction of β-arrestin2 phosphorylation mutants with IκBα was not affected by β2AR stimulation, indicating that GPCR stimulation of β-arrestin2–IκBα interaction is through promoting dephosphorylation of β-arrestin2.
Figure 5.
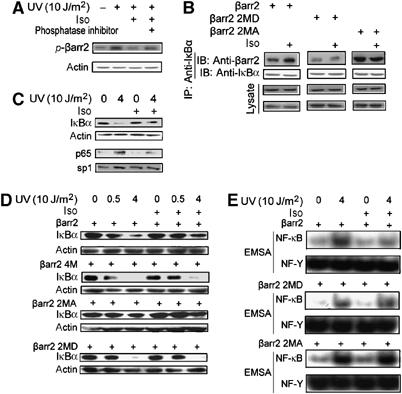
β-arrestin2 suppression is stimulated by β2AR. (A) β2AR promoted β-arrestin2 dephosphorylation. A431 cells were exposed to UV irradiation after stimulation with or without Iso (10 μM for 1 min) and phosphorylation of β-arrestin2 was measured using in vivo phosphorylation. Okadaic acid (50 nM) for 30 min was used before Iso stimulation or not. (B) Enhancement of β-arrestin2 and IκBα interaction by β2AR stimulation. β-Arrestin-null MEFs coexpressed with β2AR, and β-arrestin2, β-arrestin2 2MD, or β-arrestin2 2MA were stimulated with or without Iso (10 μM) for 1 min. Cell lysates were then subjected to immunoprecipitation and immunoblot with antibodies as indicated. (C) Enhancement of β-arrestin2 effects on IκBα degradation and NF-κB p65 nuclear translocation by β2AR stimulation. A431 cells were exposed to UV irradiation after stimulation with or without Iso (10 μM for 1 min), and then IκBα degradation and NF-κB p65 nuclear translocation were examined by immunoblot. (D, E) Dependence of β2AR stimulation on β-arrestin2 phosphorylation. β-Arrestin-null MEFs coexpressed with β2AR and β-arrestin2, β-arrestin2 2MD, or β-arrestin2 2MA were exposed to UV irradiation after stimulation with or without Iso (10 μM for 1 min) and then cell lysates were used to examine IκBα degradation by immunoblot (D). Nuclear extracts of the cells were separated to examine the NF-κB DNA-binding activity by EMSA (E). Data are representative of at least three independent experiments and quantified as mean±s.e. *P<0.05 and **P<0.01 versus control. IP, immunoprecipitated; IB, immunoblot.
Next we examined whether β-arrestin2 suppression of NF-κB activation induced by UV was stimulated by β2AR. As shown in Figure 5C, treatment of β2AR agonist (Iso) significantly suppressed the degradation of IκBα and NF-κB p65 nuclear translocation in A431 cells with endogenous β2AR, and pretreatment with β2AR antagonist (Pro) totally abolished the Iso effect (data not shown). We then examined the effect of β2AR stimulation in β-arrestin-null MEFs that had been transfected with β2AR and wild-type β-arrestin2 or its mutants. We found that β-arrestin2 suppression of UV-induced IκBα degradation and NF-κB DNA-binding activity were further enhanced by β2AR stimulation in β-arrestin-null MEFs with re-expression of β-arrestin2, but not those with re-expression of the 2MD or 2MA mutants (Figure 5D and E). Moreover, β2AR stimulation did not alter UV-induced NF-κB activation in β-arrestin-null MEFs with re-expression of β-arrestin2 4M, which did not bind to IκBα or suppress NF-κB activation. These results clearly demonstrate that β2AR functionally crosstalks to UV pathway and that this crosstalk is mediated by its stimulation of β-arrestin2–IκBα interaction.
β-Arrestin2 functions to promote UV-induced cell death
UV-induced IκBα degradation leads to the activation of NF-κB (Kato et al, 2003), which is an important antiapoptotic transcription factor via inducing expression of various antiapoptotic target genes, such as bfl-1 and bcl-xl (Barkett and Gilmore, 1999; Baldwin, 2001). We investigated the potential physiological consequences of β-arrestin2's suppression of the NF-κB activation by examining UV-induced cell death in our systems. As shown in Figure 6A and B, UV-induced expression of bfl-1 and bcl-xl was decreased at mRNA level as determined by real-time PCR after expression of β-arrestin2 in either A431 cells or β-arrestin-null MEFs. Conversely, knockdown of β-arrestin2 in A431 cells by RNAi significantly increased UV-induced expression of bfl-1 and bcl-xl (Figure 6A).
Figure 6.
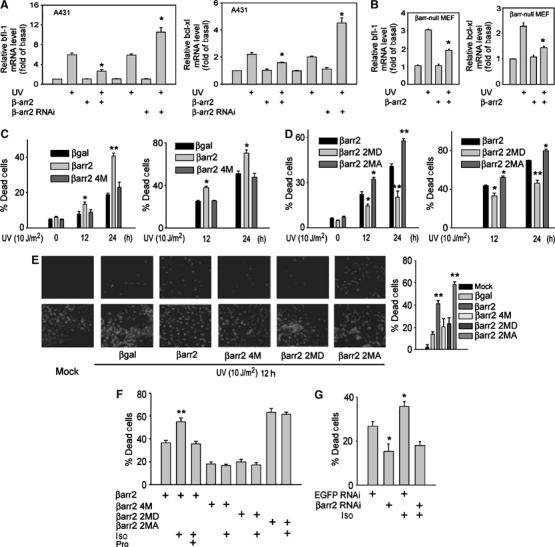
β-arrestin2 functions to promote UV-induced cell death. (A) A431 cells expressed with β-arrestin2 or β-arrestin2 siRNA were subjected to RT-qPCR for detection of bfl-1 and bcl-xl expression after UV irradiation for 12 h. (B) β-Arrestin-null MEFs expressed with β-arrestin2 were subjected to RT-qPCR for detection of bfl-1 and bcl-xl expression after UV irradiation for 12 h. (C, D) β-Arrestin-null MEFs expressed with β-arrestin2, β-arrestin2 4M (C), β-arrestin2 2MD (D), or β-arrestin2 2MA (D) were exposed to UV irradiation for the indicated times, and cell death was measured using MTT and FACS assay. (E) β-Arrestin-null MEFs expressed with β-arrestin2 or its mutants were exposed to UV irradiation, and cell death was measured using TUNEL assay. Data are representative of at least three independent experiments. (F) β-Arrestin-null MEFs coexpressed with β2AR and β-arrestin2, β-arrestin2 4M, β-arrestin2 2MD, or β-arrestin2 2MA were exposed to UV irradiation after stimulation with or without Iso (10 μM for 1 min). Pro (10 μM) was added 5 min before Iso. Cell death was measured using TUNEL assay. (G) A431 cells expressed with β-arrestin2 siRNA were exposed to UV irradiation after stimulation with or without Iso (10 μM for 1 min). Cell death was measured using TUNEL assay. Data were quantified as mean±s.e. *P<0.05, and **P<0.01 versus control.
Consistent with the effects of β-arrestin2 on the expression of these antiapoptotic genes, re-expression of β-arrestin2 in β-arrestin-null MEFs increased the sensitivity of these cells to UV-induced cell death as measured by both MTT and fluorescence-activated cell sorting (FACS) assays (Figure 6C). In contrast, re-expression of β-arrestin2 4M mutant, which does not bind to IκBα, did not alter UV-induced cell death in these cells. Similar results were obtained in p53-null Saos2 cells, which excluded the possible involvement of Mdm2/p53 pathway in which β-arrestin2 also plays a functional role (Wang et al, 2003) (Supplementary data). Analysis of the β-arrestin2 phosphorylation site mutants further supported a functional role of β-arrestin2's suppression on NF-κB activation in the regulation of UV-induced cell death. Consistent with their effects on NF-κB activation, β-arrestin2 2MD mutant was less effective than the wild-type β-arrestin2 on its promotion of UV-induced cell death. In contrast, β-arrestin2 2MA mutant showed much higher effect on this cell death as compared to wild-type β-arrestin2 in both β-arrestin-null MEFs (Figure 6D) and p53-null Saos2 cells (Supplementary data). Lastly, the effect of β-arrestin2 and its various mutants on UV-induced cell death was further confirmed by TUNEL assays (Figure 6E).
We also examined the effect of β2AR in UV-induced cell death. As shown in Figure 6F, β-arrestin2 promotion on UV-induced cell death was further enhanced by β2AR stimulation only in the expression of β-arrestin2 but not its mutants 2MD and 2MA, and pretreatment with β2AR antagonist (Pro) totally abolished the Iso effect. Moreover, β2AR stimulation did not alter UV-induced cell death in the expression of β-arrestin2 4M, which did not bind to IκBα or suppress NF-κB activation (Figure 6F). Finally, we found that knockdown of endogenous β-arrestin2 by β-arrestin2 RNAi in human epidermal carcinoma A431 cells greatly suppressed UV-induced cell death and that the stimulatory effect of β2AR activation by Iso in UV-induced cell death was also lost (Figure 6G).
Discussion
UV response in mammalian cells mainly involves NF-κB activation, which protects epidermis against apoptosis (reviewed by Baldwin, 1996). NF-κB activation induced by UV irradiation differs from that induced by other stimuli such as TNFα and LPS: it makes use of CK2 instead of IKK to phosphorylate IκBα. Our current work clearly showed that β-arrestin2, the multifunctional signal molecule, functions as a GPCR-stimulated suppressor of UV-induced NF-κB activation and its antiapoptotic function. Previously, we have found that β-arrestin2 can modulate TNFα-induced NF-κB activation and subsequent expression of NF-κB target genes (Gao et al, 2004). Thus, activation of NF-κB by a wide variety of biological stimuli, mediated by either IKK or CK2, is negatively regulated by β-arrestin2, demonstrating that β-arrestin2 functions as an intrinsic suppressor of NF-κB pathway.
In TNFα activation of NF-κB pathway, β-arrestin2–IκBα interaction remains unchanged after TNFα stimulation. In UV responses, however, β-arrestin2–IκBα interaction is regulated by β-arrestin2's phosphorylation status upon UV irradiation. A working model depicting the role of β-arrestin2 in cellular responses to UV is proposed in Figure 7. Upon UV irradiation, CK2 phosphorylates β-arrestin2, diminishes its interaction with IκBα, and releases IκBα for degradation. This model suggests that β-arrestin2, which binds to phosphorylated signal molecules and thus regulates their functions, is itself regulated by phosphorylation. Our current work provides further support for the functional role of β-arrestin2 phosphorylation, as reported previously in the case of β-arrestin2 binding to clathrin (Kim et al, 2002; Lin et al, 2002). This study also proposes a new model of one kinase in a signal cascade, which can phosphorylate both the downstream signal molecule (i.e. IκBα) and its suppressor (i.e. β-arrestin2) in a synchronized way to more dedicatedly and accurately transduce the signal.
Figure 7.
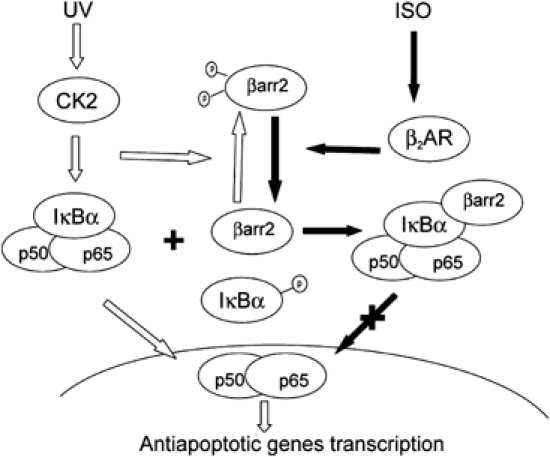
Model illustration that β-arrestin2 functions as a phosphorylation-regulated suppressor to UV-induced NF-κB activation.
Although β-arrestin2 and β-arrestin1 share similar functional roles in many circumstances like GPCR signal transduction, accumulating evidence also revealed potential functional differences between them (Luttrell and Lefkowitz, 2002). For example, β-arrestin1 binds some GPCRs such as β2AR, MOR, and endothelin type receptor with lower affinity than β-arrestin2, and is less efficient in membrane translocation upon agonist stimulation, while β-arrestin1 and β-arrestin2 bind other receptors, including angiotensin II type 1A receptor, neurotensin 1 receptor, and substance P receptor, with similar high affinities and translocate to the membrane with similar efficiencies (Oakley et al, 2000). Further, it has been reported that β-arrestin2 is predominantly distributed in the cytoplasm, while β-arrestin1 is localized equally in the cytoplasm and nucleus (Scott et al, 2002). Our current study also provides evidence that, although β-arrestin1 showed a similar ability to modulate UV-induced NF-κB activation as β-arrestin2, β-arrestin1 Q394L mutant, which is predominantly distributed in the cytoplasm, did not modulate NF-κB activation, suggesting that β-arrestin1 may exert its effect through different mechanisms in the nucleus. Our unpublished data show that β-arrestin1 could modulate histone H4 acetylation and transcription of genes like p27 and c-fos, indicating that epigenetic regulation of gene expression is one function of β-arrestin1 in the nucleus. Thus, β-arrestin1 may exert its inhibitory function through epigenetic histone modification. The precise mechanism underlying β-arrestin1 modulation of UV-induced NF-κB activation is worthy of further investigation.
Arrestins, besides their well-established extensive interactions with GPCRs, are recently displayed to functionally interact with growth factor receptors (e.g. insulin-like growth factor receptor; Lin et al, 1998) and transforming growth factor-β receptor (Chen et al, 2003), thus indicating their potential role in regulation of cellular proliferation and apoptosis. Previous studies have revealed that the irreversible association of Drosophila arrestin to the activated rhodopsin leads to retinal cell apoptosis (Kiselev et al, 2000), and the GPCR-stimulated binding of β-arrestin2 to Mdm2 enhances the p53-mediated apoptosis (Wang et al, 2003). Our current study further demonstrates that β-arrestin2 effectively modulates UV-induced NF-κB activation and consequently suppresses the antiapoptotic function of NF-κB, revealing its general role in proapoptosis in different signal pathways. However, it seems that the function of arrestins is not limited to proapoptosis, since they are also reported to be involved in the prevention of apoptosis (Povsic et al, 2003; Revankar et al, 2004).
The epidermis is at the forefront of protection for the organism against multiple environmental insults. One of such insults is UV light, which is considered to be a potent and complete carcinogen causing the most common form of cancers, nonmelanoma skin cancers (Quinn, 1997; Brash and Ponten, 1998; Wikonkal and Brash, 1999). Various studies have shown that the UV insult in epidermis induces three major signal pathways, namely AP1, p53, and NF-κB. While activation of AP1 and p53 is proapoptotic, activation of NF-κB serves an antiapoptotic function (Kato et al, 2003). It is generally believed that the skin cancers relevant to UV irradiation are related to the imbalance of proapoptotic and antiapoptotic functions of these signal pathways. The current work showing that effective suppression of UV-induced NF-κB activation by β-arrestin2-mediated GPCR signaling indicates stimulation of GPCRs such as β2AR may promote UV-induced apoptosis and thus repress the carcinogenesis in epidermis. If this is the case, some of the GPCRs in epidermis might be used as a potential target for prevention and treatment of UV-relevant skin cancers.
Materials and methods
Materials and reagents
Anti-β-arrestins rabbit polyclonal antibody (A1CT) is a generous gift from Dr Robert J Lefkowitz (Duke University Medical Center, Durham). Antibodies directed against IκBα (rabbit) and p65 (rabbit) were obtained from Santa Cruz Biotechnology, anti-GG from Covance, anti-HA, Flag, actin, and sp1 from Sigma. RhTNFα was from R&D, and phosphatase inhibitor (okadaic acid), p38 inhibitor (SKF86002), MG132, Iso, and Pro from sigma. NF-κB activation inhibitor (sn50 peptide) and CK2 inhibitor (5,6-dichloro-1-β-D-ribofuranosylbenzimidazole) were from Calbiochem. The peak emission wavelength of the UV-C used was 254 nm.
Cell transfection and plasmids
Transient transfection of Saos2 cells was performed using the calcium phosphate method as described previously (Wang et al, 2003). Wild-type and β-arrestin-null MEF cell lines were kindly provided by Dr Robert J Lefkowitz, and were transfected by LipofectAMINE (Invitrogen). A431 cells was also transfected by LipofectAMINE. For all transfection experiments, CMV-β-Gal (for expression) and EGFP RNAi (for siRNA) were used as a control. Full length of human IκBα was cloned into modified pcDNA3 vector in-frame with Flag at the N-terminus. Plasmids cDNA encoding β-arrestin2 and its mutants were generated as described (Wang et al, 2003) and were cloned into modified pcDNA3 vector in-frame with HA at the C-terminus. Full length of human IKKβ was cloned into modified pcDNA3 vector in-frame with Myc at the N-terminus. The GG tagged CK2α plasmid was kindly provided by Dr Lin Li (Shanghai Institutes for Biological Sciences). The authenticity of the DNA sequences was confirmed by sequencing. Construction of siRNA plasmid for β-arrestin2 was described previously (Sun et al, 2002; Wang et al, 2003; Gao et al, 2004).
Immunoprecipitation and immunoblot
For transient transfections, endogenous IκBα was immunoprecipitated with anti-IκBα (rabbit) and protein A-Sepharose beads (Amersham Biosciences). After extensive washing and heating in the sample buffer, the complexes were subjected to immunoblot (Wang et al, 2003).
Nuclear extracts preparation
After serum starvation for 12 h, cells were exposed to UV irradiation (10 J/m2) for a certain time after stimulation with or without Iso (10 μM for 1 min). Nuclear extracts were prepared by the method as reported previously (Gao et al, 2004), with minor modifications with 6 μl of NP-40 10% (v/v) for MEFs and 5 μl of NP-40 10% (v/v) for A431 cells.
Electrophoretic mobility shift assay (EMSA)
EMSAs were performed using nuclear extracts obtained at various times before or after exposure to UV irradiation (10 J/m2) with or without Iso stimulation (10 μM for 1 min). Double-stranded gel shift probes (50 ng) corresponding to the human consensus NF-κB sequences (5′-AGT TGA GGG GAC TTT CCC AGG C-3′, Promega), the consensus AP1 sequences (5′-CGC TTG ATG ACT CAG CCG GAA-3′, Promega), and the consensus NF-Y probes (5′-AGA CCG TAC GTG ATT GGT TAA TCT CTT-3′, Santa Cruz Biotechnology) were end-labeled with γ-32P-ATP and T4 polynucleotide kinase. The reaction mixtures (20 μl) containing about 10 μg nuclear extracts and binding buffer (Promega) were incubated at room temperature for 10 min and 0.5 ng 32P-labeled DNA probes were added, followed by another 20-min incubation. Samples were subjected to electrophoresis in 10% nondenaturing polyacrylamide gels. In competition or antibody supershift experiments, the reaction mixtures were preincubated at room temperature with competing cold probes (50-fold excess) for 10 min or with specific antibody for 1 h before addition of the 32P-labeled probes.
In vitro kinase assays
Expressed myc-IKKβ or GG-CK2 was immunoprecipitated using anti-myc or anti-GG antibody. The beads were extensively washed and resuspended in 30 μl of kinase buffer with 10 μM ATP and 2 μCi of γ-32P-ATP, containing recombinant GST-β-arrestin2. The kinase reaction was performed at 30°C for 30 min and stopped by addition of 6 μl of 6 × SDS sample buffer. The samples were subjected to SDS–PAGE (10%), electroblot, and autoradiography.
In vivo phosphorylation
For metabolic labeling, cells were starved in phosphate-free Dulbecco's modified Eagle's medium (Life Technologies, Inc.) for 1 h, labeled for 90 min in the same medium containing [32P]orthophosphate (0.2 mCi/ml), and harvested for β-arrestin2 immunoprecipitation.
Reverse transcription quantitative real-time PCR
Total RNAs were extracted with TRIzol (Invitrogen) according to the manufacturer's instructions. Reverse transcription of purified RNA was performed using oligo(dT) priming and superscript II reverse transcriptase (Invitrogen). The quantification of all gene transcripts was performed by qPCR, using Brilliant SYBR Green QPCR Master Mix and a Light Cycler apparatus (Stratagene).
MTT
After transfection, MEFs or Saos2 cells were cultivated in a 96-well chamber at 1000–10 000 cells per well, and exposed to UV irradiation (10 J/m2) for various times. MTT reagent was then added and cells were incubated for 4 h. After adding the detergent reagent for 10 h in the dark, absorbance in 570 nm was recorded.
Fluorescence-activated cell sorting
After exposure to UV irradiation (10 J/m2) for various times, at least 106 cells were fixed in 70% ethanol for 1 h. Then, propidium iodide staining solution (PI solution) together with Rnase A were added and the sub-G1 peak of cells was analyzed by passage through a FACSCalibur flow cytometer.
DNA fragmentation/fluorescein staining (TUNEL)
Cells were grown on glass coverslips; 48 h after split, the cells were exposed to UV irradiation (10 J/m2) for a certain time and fixed with 4% polyformaldehyde for 15 min. Cells were washed with PBS and incubated in PBS containing 0.2% BSA and 0.2% Tween for 15 min. After fluorescein staining with TdT end-labeling cocktail (TdT buffer, Biotin-dUTP, and TdT) and avidin-FITC, DNA fragmentation was analyzed on a Leica TCS SP2 laser confocal fluorescence microscope.
Statistical analysis
Data were analyzed by Student's t-test for comparison of independent means, with pooled estimates of common variances. For all tests, P<0.05 was considered significant.
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Acknowledgments
We thank Dr Robert J Lefkowitz (Duke University Medical Center, Durham) for providing anti-β-arrestin antibody (A1CT) and MEF cell lines and thank Dr Jun-Lin Guan (Department of Molecular Medicine, Cornell University), Dr Lan Ma (National Laboratory of Medical Neurobiology, Fudan University Medical Center, Shanghai), and Dr Jianguo Geng (Institute of Biochemistry and Cell Biology, Shanghai) for constructive suggestions. We also thank Dr Hua Gao, Dr Yue Sun, Yaya Wang, and Shunmei Xin for their technical assistance, Peihua Wu for kind help, and Dr Yanxiang Ni for helpful discussion. This research was supported by grants from the Ministry of Science and Technology (2003CB515405, 2004AA235061, and 2005CB522406), the National Natural Science Foundation of China (30021003), and Shanghai Municipal Commission for Science and Technology (03DZ19213).
References
- Baldwin AS Jr (1996) The NF-κB and IκB proteins: new discoveries and insights. Annu Rev Immunol 14: 649–683 [DOI] [PubMed] [Google Scholar]
- Baldwin AS (2001) Control of oncogenesis and cancer therapy resistance by the transcription factor NF-κB. J Clin Invest 107: 241–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkett M, Gilmore TD (1999) Control of apoptosis by Rel/NF-κB transcription factors. Oncogene 18: 6910–6924 [DOI] [PubMed] [Google Scholar]
- Beg AA, Baltimore D (1996) An essential role for NF-κB in preventing TNF-α-induced cell death. Science 274: 782–784 [DOI] [PubMed] [Google Scholar]
- Bender K, Gottlicher M, Whiteside S, Rahmsdorf HJ, Herrlich P (1998) Sequential DNA damage-independent and -dependent activation of NF-κB by UV. EMBO J 17: 5170–5181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brash DE, Ponten J (1998) Skin precancer. Cancer Surv 32: 69–113 [PubMed] [Google Scholar]
- Chen W, Kirkbride KC, How T, Nelson CD, Mo J, Frederick JP, Wang XF, Lefkowitz RJ, Blobe GC (2003) β-Arrestin 2 mediates endocytosis of type III TGF-β receptor and down-regulation of its signaling. Science 301: 1394–1397 [DOI] [PubMed] [Google Scholar]
- Denda M, Fuziwara S, Inoue K (2003) β2-Adrenergic receptor antagonist accelerates skin b-arrestinier recovery and reduces epidermal hyperplasia induced by b-arrestinier disruption. J Invest Dermatol 121: 142–148 [DOI] [PubMed] [Google Scholar]
- Gao H, Sun Y, Wu Y, Luan B, Wang Y, Qu B, Pei G (2004) Identification of β-arrestin2 as a G-protein-coupled receptor-stimulated regulator of NF-κB pathways. Mol Cell 14: 303–317 [DOI] [PubMed] [Google Scholar]
- Gillbro JM, Marles LK, Hibberts NA, Schallreuter KU (2004) Autocrine catecholamine biosynthesis and the β-adrenoceptor signal promote pigmentation in human epidermal melanocytes. J Invest Dermatol 123: 346–353 [DOI] [PubMed] [Google Scholar]
- Goodman OB Jr, Krupnick JG, Santini F, Gurevich VV, Penn RB, Gagnon AW, Keen JH, Benovic JL (1996) β-Arrestin acts as a clathrin adaptor in endocytosis of the β2-adrenergic receptor. Nature 383: 447–450 [DOI] [PubMed] [Google Scholar]
- Israel A (2000) The IKK complex: an integrator of all signals that activate NF-κB? Trends Cell Biol 10: 129–133 [DOI] [PubMed] [Google Scholar]
- Karin M, Ben-Neriah Y (2000) Phosphorylation meets ubiquitination: the control of NF-κB activity. Annu Rev Immunol 18: 621–663 [DOI] [PubMed] [Google Scholar]
- Kato T, Delhase M, Hoffmann A, Karin M (2003) CK2 is a C-terminal IκB kinase responsible for NF-κB activation during the UV response. Mol Cell 12: 829–839 [DOI] [PubMed] [Google Scholar]
- Kim YM, Barak LS, Caron MG, Benovic JL (2002) Regulation of arrestin-3 phosphorylation by casein kinase II. J Biol Chem 277: 6837–6846 [DOI] [PubMed] [Google Scholar]
- Kiselev A, Socolich M, Vinos J, Hardy RW, Zuker CS, Ranganathan R (2000) A molecular pathway for light-dependent photoreceptor apoptosis in Drosophila. Neuron 28: 139–152 [DOI] [PubMed] [Google Scholar]
- Koizumi H, Yasui C, Fukata T, Ohkawara A, Ueda T (1991) β-Adrenergic stimulation induces intracellular Ca++ increase in human epidermal keratinocytes. J Invest Dermatol 96: 234–237 [DOI] [PubMed] [Google Scholar]
- Li N, Karin M (1998) Ionizing radiation and short wavelength UV activate NF-κB through two distinct mechanisms. Proc Natl Acad Sci USA 95: 13012–13017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin FT, Chen W, Shenoy S, Cong M, Exum ST, Lefkowitz RJ (2002) Phosphorylation of β-arrestin2 regulates its function in internalization of β2-adrenergic receptor. Biochemistry 41: 10692–10699 [DOI] [PubMed] [Google Scholar]
- Lin FT, Daaka Y, Lefkowitz RJ (1998) β-Arrestins regulate mitogenic signaling and clathrin-mediated endocytosis of the insulin-like growth factor I receptor. J Biol Chem 273: 31640–31643 [DOI] [PubMed] [Google Scholar]
- Lin FT, Krueger KM, Kendall HE, Daaka Y, Fredericks ZL, Pitcher JA, Lefkowitz RJ (1997) Clathrin-mediated endocytosis of the β2-adrenergic receptor is regulated by phosphorylation/dephosphorylation of β-arrestin1. J Biol Chem 273: 31051–31057 [DOI] [PubMed] [Google Scholar]
- Luttrell LM, Ferguson SS, Daaka Y, Miller WE, Maudsley S, Della Rocca GJ, Lin F, Kawakatsu H, Owada K, Luttrell DK, Caron MG, Lefkowitz RJ (1999) β-Arrestin-dependent formation of β2-adrenergic receptor-Src protein kinase complexes. Science 283: 655–661 [DOI] [PubMed] [Google Scholar]
- Luttrell LM, Lefkowitz RJ (2002) The role of β-arrestins in the termination and transduction of G-protein-coupled receptor signals. J Cell Sci 115 (Part 3): 455–465 [DOI] [PubMed] [Google Scholar]
- McDonald PH, Chow CW, Miller WE, Laporte SA, Field ME, Lin FT, Davis RJ, Lefkowitz RJ (2000) β-Arrestin2: a receptor-regulated MAPK scaffold for the activation of JNK3. Science 290: 1574–1577 [DOI] [PubMed] [Google Scholar]
- Oakley RH, Laporte SA, Holt JA, Caron MG, Barak LS (2000) Differential affinities of visual arrestin, β-arrestin1, and β-arrestin2 for G protein-coupled receptors delineate two major classes of receptors. J Biol Chem 275: 17201–17210 [DOI] [PubMed] [Google Scholar]
- Perry SJ, Baillie GS, Kohout TA, McPhee I, Magiera MM, Ang KL, Miller WE, McLean AJ, Conti M, Houslay MD, Lefkowitz RJ (2002) Targeting of cyclic AMP degradation to β2-adrenergic receptors by β-arrestins. Science 298: 834–836 [DOI] [PubMed] [Google Scholar]
- Povsic TJ, Kohout TA, Lefkowitz RJ (2003) Arrestin1 mediates insulin-like growth factor 1 (IGF-1) activation of phosphatidylinositol 3-kinase (PI3K) and anti-apoptosis. J Biol Chem 278: 51334–51339 [DOI] [PubMed] [Google Scholar]
- Quinn AG (1997) Ultraviolet radiation and skin carcinogenesis. Br J Hosp Med 58: 261–264 [PubMed] [Google Scholar]
- Revankar CM, Vines CM, Cimino DF, Prossnitz ER (2004) Arrestins block G protein-coupled receptor-mediated apoptosis. J Biol Chem 279: 24578–24584 [DOI] [PubMed] [Google Scholar]
- Schallreuter KU, Pittelkow MR, Swanson NN, Beazley WD, Korner C, Ehrke C, Buttner G (1997) Altered catecholamine synthesis and degradation in the epidermis of patients with atopic eczema. Arch Dermatol Res 289: 663–666 [DOI] [PubMed] [Google Scholar]
- Scott MG, Le Rouzic E, Perianin A, Pierotti V, Enslen H, Benichou S, Marullo S, Benmerah A (2002) Differential nucleocytoplasmic shuttling of β-arrestins. Characterization of a leucine rich nuclear export signal in β-arrestin2. J Biol Chem 277: 37693–37701 [DOI] [PubMed] [Google Scholar]
- Shaulian E, Schreiber M, Piu F, Beeche M, Wagner EF, Karin M (2000) The mammalian UV response: c-Jun induction is required for exit from the p53-imposed checkpoint. Cell 103: 897–907 [DOI] [PubMed] [Google Scholar]
- Shenoy SK, McDonald PH, Kohout TA, Lefkowitz RJ (2001) Regulation of receptor fate by ubiquitination of activated β2-adrenergic receptor and β-arrestin. Science 294: 1307–1313 [DOI] [PubMed] [Google Scholar]
- Silverman N, Maniatis T (2001) NF-κB signaling pathways in mammalian and insect innate immunity. Genes Dev 15: 2321–2342 [DOI] [PubMed] [Google Scholar]
- Steckelings UM, Wollschlager T, Peters J, Henz BM, Hermes B, Artuc M (2004) Human skin: source of and target organ for angiotensin II. Exp Dermatol 13: 148–154 [DOI] [PubMed] [Google Scholar]
- Steinkraus V, Mak JC, Pichlmeier U, Mensing H, Ring J, Barnes PJ (1996) Autoradiographic mapping of β-adrenoceptors in human skin. Arch Dermatol Res 288: 549–553 [DOI] [PubMed] [Google Scholar]
- Sun Y, Cheng Z, Ma L, Pei G (2002) β-Arrestin2 is critically involved in CXCR4-mediated chemotaxis, and this is mediated by its enhancement of p38 MAPK activation. J Biol Chem 277: 49212–49219 [DOI] [PubMed] [Google Scholar]
- Takahashi H, Kinouchi M, Tamura T, Iizuka H (1996) Decreased β 2-adrenergic receptor-mRNA and loricrin-mRNA, and increased involucrin-mRNA transcripts in psoriatic epidermis: analysis by reverse transcription-polymerase chain reaction. Br J Dermatol 134: 1065–1069 [PubMed] [Google Scholar]
- Wang P, Gao H, Ni Y, Wang B, Wu Y, Ji L, Qin L, Ma L, Pei G (2003) β-Arrestin2 functions as a G-protein-coupled receptor-activated regulator of oncoprotein Mdm2. J Biol Chem 278: 6363–6370 [DOI] [PubMed] [Google Scholar]
- Wikonkal NM, Brash DE (1999) Ultraviolet radiation induced signature mutations in photocarcinogenesis. J Investig Dermatol Symp Proc 4: 6–10 [DOI] [PubMed] [Google Scholar]
- Witherow DS, Garrison TR, Miller WE, Lefkowitz RJ (2004) β-Arrestin inhibits NF-κB activaty by means of its interaction with the NF-κB inhibitor IκBα. Proc Natl Acad Sci USA 101: 8603–8607 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5