

Abstract
Ribosomal protein L7/12 is crucial for the function of elongation factor G (EF-G) on the ribosome. Here, we report the localization of a site in the C-terminal domain (CTD) of L7/12 that is critical for the interaction with EF-G. Single conserved surface amino acids were replaced in the CTD of L7/12. Whereas mutations in helices 5 and 6 had no effect, replacements of V66, I69, K70, and R73 in helix 4 increased the Michaelis constant (KM) of EF-G·GTP for the ribosome, suggesting an involvement of these residues in EF-G binding. The mutations did not appreciably affect rapid single-round GTP hydrolysis and had no effect on tRNA translocation on the ribosome. In contrast, the release of inorganic phosphate (Pi) from ribosome-bound EF-G·GDP·Pi was strongly inhibited and became rate-limiting for the turnover of EF-G. The control of Pi release by interactions between EF-G and L7/12 appears to be important for maintaining the conformational coupling between EF-G and the ribosome for translocation and for timing the dissociation of the factor from the ribosome.
Keywords: EF-G, GTP-binding proteins, GTPase, rapid kinetics, translation
Introduction
In the ribosomal elongation cycle, elongation factor G (EF-G) promotes the translocation step. EF-G is a large GTPase that consists of five domains (Ævarsson et al, 1994; Czworkowski et al, 1994). The GTPase activity of EF-G, which is very low intrinsically (<10−5 s−1; Parmeggiani and Sander, 1981), is stimulated by several orders of magnitude upon binding of EF-G·GTP to the ribosome (Rodnina et al, 1997). GTP hydrolysis by EF-G is stimulated by isolated L7/12 (Savelsbergh et al, 2000b) and the GTPase activity of EF-G on the ribosome is decreased about 1000-fold when ribosomes are depleted of L7/12 (Mohr et al, 2002; Diaconu et al, 2005), indicating that a major contribution to GTPase activation on the ribosome comes from protein L7/12. In comparison, the rate of translocation on ribosomes lacking L7/12 is reduced only 50-fold, that is, to the rate observed when there is no GTP hydrolysis (Mohr et al, 2000, 2002; Katunin et al, 2002). On native ribosomes containing L7/12, the steps following GTP hydrolysis, including tRNA–mRNA translocation and the release of inorganic phosphate (Pi) from EF-G·GDP·Pi, are much slower than GTP hydrolysis and delayed by about 30 ms. The rate-limiting step for both translocation and Pi release is a rearrangement of the ribosome (‘unlocking') that is induced by the binding of EF-G·GTP to the ribosome and promoted by GTP hydrolysis (Savelsbergh et al, 2003). Translocation is much less efficient, and has different activation parameters, with non-hydrolyzable GTP analogs or GDP, compared to GTP, indicating different reaction pathways (Katunin et al, 2002). The active form of EF-G in promoting unlocking and, thereby, translocation is EF-G·GDP·Pi, indicating that the retention of Pi in the nucleotide-binding pocket following GTP hydrolysis is important for conformational coupling between EF-G and the ribosome (Savelsbergh et al, 2003). On the other hand, the turnover of EF-G is very slow with GTP analogs (Belitsina et al, 1976; Kaziro, 1978; Katunin et al, 2002), indicating that the release of Pi is required for EF-G to dissociate from the ribosome.
The L7/12 stalk of the large ribosomal subunit forms part of the binding site of EF-G on the ribosome. The stalk comprises protein L10 and, in Escherichia coli, four copies of protein L7/12. Protein L7/12 comprises two domains (Dey et al, 1995; Wahl et al, 2000). The N-terminal domain consists of two α helices (α1, 2) and is responsible for dimer formation and for anchoring the L7/L12 dimers to helix α8 of protein L10 on the 50S subunit (Diaconu et al, 2005). N- and C-terminal domains are connected by a long hinge assigned as helix α3 in the crystal structure (Wahl et al, 2000). The hinge is likely to be unstructured in solution (Bocharov et al, 2004; Mulder et al, 2004) and confers mobility to the C-terminal domain (CTD) (Dey et al, 1995; Wahl et al, 2000; Diaconu et al, 2005). The globular CTD, which is the site of factor binding, consists of three α helices (α4–6) and a double-stranded β sheet (Leijonmarck and Liljas, 1987). Mutagenesis and kinetic studies suggested that helices 4 and 5 in the CTD of L7/12 are involved in the initial contact with helix D of elongation factor Tu (EF-Tu), promoting ternary complex binding to the ribosome (Kothe et al, 2004). Except for the partial functional analysis of a single mutant, K70A, in helix 4 of the L7/12 CTD (Savelsbergh et al, 2003), the interactions between the CTD of L7/12 and EF-G that are involved in regulating EF-G functions on the ribosome have not been characterized in detail and the location of the binding site of EF-G in the CTD of L7/12 is not known. In the present work, the interaction between the CTD of protein L7/12 and EF-G was studied in the E. coli system by introducing amino-acid changes in the CTD of L7/12. Ribosomes reconstituted with mutant L7/12 were studied with respect to EF-G functions on the ribosome, including GTP hydrolysis, translocation, and Pi release.
Results
Effects of amino-acid replacements in ribosomal protein L7/12 on multiple-turnover reactions of EF-G
Single amino acids were exchanged at conserved surface positions in the CTD of E. coli L7/12 (Figure 1). Mutagenesis was performed and mutant L7/12 expressed and purified as described in Materials and methods. Ribosomes were depleted of L7/12 by NH4Cl/ethanol treatment and reconstituted with mutant or wild-type (wt) L7/12 (Mohr et al, 2002). Under conditions where L7/12 depletion was complete, only trace amounts of L10 and no other ribosomal proteins were lost. In agreement with earlier reports (Kischa et al, 1971; Hamel et al, 1972; Sander et al, 1975), ribosome-dependent GTP hydrolysis by EF-G was strongly inhibited by the removal of L7/12, and full activity was restored by adding back excess wt L7/12 to depleted cores (Figure 2).
Figure 1.
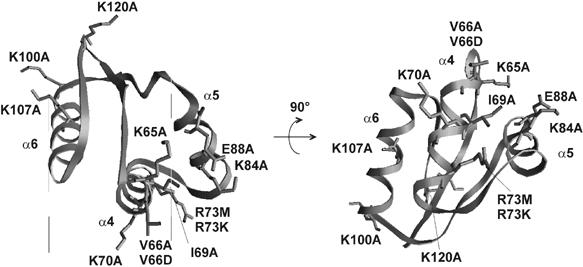
Positions of mutations in the CTD of protein L7/12 from E. coli. Amino acids are numbered according to the sequence of the protein as in the crystal structure (Leijonmarck and Liljas, 1987).
Figure 2.
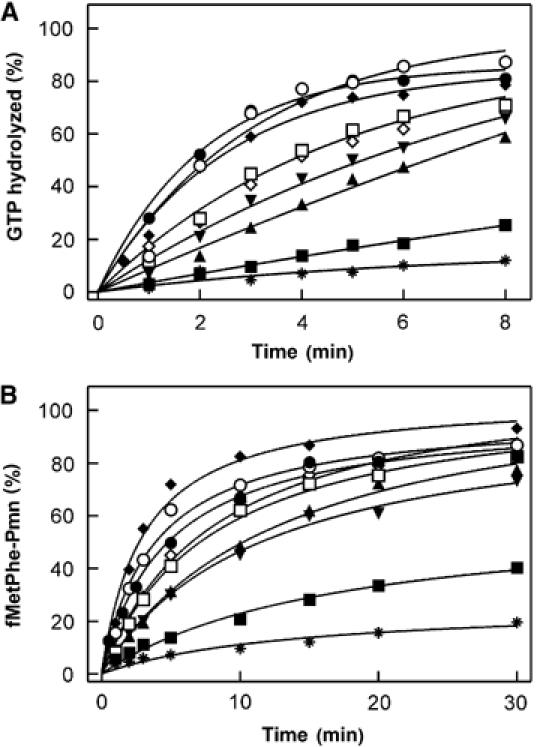
Effect of L7/12 mutations on turnover reactions of EF-G. (A) GTP hydrolysis. Time courses were measured with native ribosomes (•), ribosomes reconstituted with wt L7/12 (○), R73K (⧫), V66A (□), R73 M (⋄), I69A (▾), K70A (▴), or V66D (▪), and ribosome cores lacking L7/12 (*). EF-G (0.04 μM) was incubated with [γ-32P]GTP (20 μM) and ribosomes (0.2 μM) at 37°C. (B) Translocation. Pretranslocation complex carrying fMetPhe-tRNAPhe in the A site (0.2 μM) was incubated with EF-G (0.5 nM) and GTP (1 mM) at 37°C. At the indicated times, puromycin (Pmn; 1 mM) was added and the reaction stopped after 10 s. The extent of fMetPhe-Pmn formation is given relative to the initial amount of pretranslocation complex. Symbols as in panel A.
Ribosomes reconstituted with mutant L7/12 were first assayed for their ability to stimulate GTP hydrolysis by EF-G under conditions of multiple turnover, using catalytic amounts of EF-G. Amino-acid replacements at four positions in helix 4 of the CTD of L7/12 (V66A/D, I69A, K70A, and R73M) reduced the rates of EF-G-dependent turnover GTP hydrolysis (Figure 2A). Two other mutations in helix 4, R73K and K65A, had no effect (data not shown). Alanine substitutions in positions K84, E88, K107, K100, and K120 did not affect turnover GTP hydrolysis (data not shown). Helix 4 mutations that had an effect on the activity of EF-G on the ribosome were studied in more detail.
To quantify the effects of helix 4 mutations on kcat and KM of GTP hydrolysis, initial rates were measured at various concentrations of ribosomes added in excess over EF-G. The mutations lowered kcat up to 15-fold and increased KM up to about three-fold, resulting in an up to 50-fold decrease of the catalytic efficiency, kcat/KM (Table I). The strongest effect (50-fold decrease of kcat/KM) was observed when valine at position 66 was replaced with the charged amino acid aspartate (V66D). The more conservative V66A exchange decreased kcat/KM about five-fold. A 4- to 10-fold decrease of kcat/KM resulted from the replacements I69A, K70A, and R73M.
Table 1.
Effect of mutations in the CTD of protein L7/12 on EF-G turnover during GTP hydrolysis and translocation on the ribosome
| L7/12 | GTP hydrolysis |
Translocation |
||||
|---|---|---|---|---|---|---|
| kcat (s−1) | KM (μM) | kcat/KM (μM−1 s−1) | kcat (s−1) | KM (μM) | kcat/KM (μM−1 s−1) | |
| wt | 3.0±0.1 | 0.07±0.02 | 43±12 | 1.6±0.1 | 0.07±0.01 | 20±4 |
| V66A | 1.3±0.1 | 0.17±0.04 | 8±2 | 0.7±0.1 | 0.16±0.05 | 4±1 |
| V66D | 0.2±0.1 | 0.22±0.06 | 0.9±0.5 | 0.4±0.1 | 0.47±0.06 | 1.0±0.3 |
| I69A | 0.9±0.1 | 0.08±0.04 | 11±6 | 0.7±0.1 | 0.13±0.03 | 5±1 |
| K70A | 0.7±0.1 | 0.17±0.07 | 4±2 | 0.8±0.1 | 0.12±0.03 | 6±1 |
| R73K | 1.9±0.2 | 0.05±0.02 | 38±15 | n.d. | n.d. | n.d. |
| R73M |
1.6±0.3 |
0.20±0.04 |
8±2 |
0.7±0.1 |
0.17±0.02 |
8±1 |
| wt, wild type; n.d., not determined. | ||||||
Independent evidence for the effect of mutations in L7/12 on EF-G function was obtained when the translocation of fMetPhe-tRNAPhe was studied under multiple-turnover conditions, using catalytic amounts of EF-G. In this assay, the replacement R73K had no effect, whereas the mutations R73M, V66A, I69A, K70A, and particularly V66D, substantially reduced the turnover rate of translocation (Figure 2B). To quantify the effects on the kinetic parameters of multiple-turnover translocation, Michaelis–Menten titrations were performed with pretranslocation complexes prepared from ribosomes carrying mutant L7/12 (Table I). The mutations decreased kcat and increased KM values for EF-G-dependent translocation in a similar fashion as observed for GTP hydrolysis (Table I). Again the strongest effect was observed for the V66D mutant (20-fold decrease of kcat/KM), whereas V66A, I69A, K70A, and R73M had moderate effects (two- to five-fold decrease of kcat/KM).
The two turnover assays yielded values for kcat and KM that were similar within a factor of two (Table I). Thus, on both vacant ribosomes (GTP hydrolysis assay) and pretranslocation complexes (translocation assay), the turnover of EF-G is controlled by an interaction between L7/12 and EF-G, which involves specific amino-acid residues that are all located on one side of helix 4 of the CTD of L7/12.
Single-round GTP hydrolysis and translocation
To study the functional importance of the interaction between L7/12 and EF-G for specific functions of EF-G, we measured the effects of helix 4 mutations on GTP hydrolysis, Pi release, and tRNA translocation under single-round conditions. Single-round GTP hydrolysis was measured at saturating EF-G concentration (Figure 3A). Although the presence of L7/12 is required to stimulate rapid GTP hydrolysis by EF-G on the ribosome (Mohr et al, 2002), there was no systematic effect of the mutations in helix 4 on the first round of rapid GTP hydrolysis, as GTP hydrolysis rates observed for ribosomes reconstituted with mutant L7/12 were higher or lower by at most a factor of two compared to ribosomes with wt L7/12 (Figure 3A and B). Notably, ribosomes containing mutant L7/12(V66D), which had the strongest inhibitory effect on EF-G turnover (Table I), stimulated GTP hydrolysis at the same rate as ribosomes containing wt L7/12. We concluded that none of the mutated amino-acid side chains in L7/12 is essential for ribosome-induced rapid GTP hydrolysis by EF-G and that GTP hydrolysis was invariably much faster than the following steps (see below).
Figure 3.
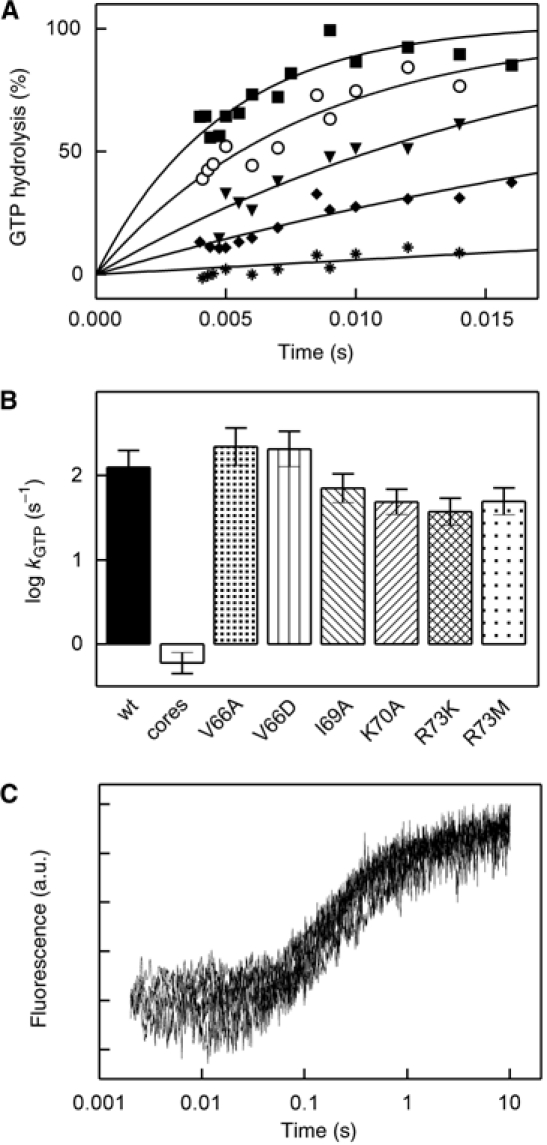
Effect of L7/12 mutations on single-round GTP hydrolysis and translocation catalyzed by EF-G. (A) Time courses of GTP hydrolysis. EF-G and [γ-32P]GTP were rapidly mixed with ribosomes (Materials and methods). Unlabeled GTP (1 mM) was added together with the ribosomes in order to limit [γ-32P]GTP hydrolysis to a single round (Rodnina et al, 1997). Symbols as in Figure 2. (B) Rate constants of GTP hydrolysis. Time courses shown in panel A were evaluated by exponential fitting to yield rate constants as indicated. (C) Time courses of translocation as monitored by the fluorescence of fMetPhe-tRNAPhe(Prf16/17). Overlaid stopped-flow traces are shown for ribosomes reconstituted with wt L7/12, V66A, V66D, I69A, K70A, R73M, and R73K.
The extent of translocation was not affected by any of the mutations in L7/12 (Figure 3C), consistent with the previous observation that even in the absence of L7/12 translocation was complete (Mohr et al, 2002). Also the rate of translocation, as measured by fluorescence stopped-flow, was not significantly altered by any of the helix 4 mutations (Figure 3C), as shown previously for the K70A mutation (Savelsbergh et al, 2003). This result indicates that the contact of L7/12 with EF-G is not important for translocation and that the inhibition observed under conditions of multiple turnover is not due to an inhibition of translocation.
Release of inorganic phosphate
The release of Pi from ribosome-bound EF-G·GDP·Pi was monitored by the fluorescence of MDCC-labeled phosphate-binding protein (PBP) that strongly increases upon binding of Pi (Brune et al, 1994; Savelsbergh et al, 2003) (Figure 4A). Pi release took place in two phases: The first round of Pi release gave rise to a rapid burst, which was followed by a slower phase due to turnover; the curves leveled off after a few seconds (not shown in Figure 4A) due to the saturation of PBP with Pi and the uptake of Pi by the ‘Pi mop' (Materials and methods). Exponential fitting yielded values for both rates and amplitudes of the burst as well as of the turnover reaction. Burst rates were in the range of 20±10 s−1 for wt and mutant L7/12, that is, there was no systematic influence of the mutations on the burst rate. However, the burst amplitudes were decreased with the L7/12 mutants, except R73K and R73M (Figure 4B). Compared to the amplitude of GTP hydrolysis, the amplitude of Pi release was lower by 30–40% (I69A, V66A) or 75–85% (K70A, V66D).
Figure 4.
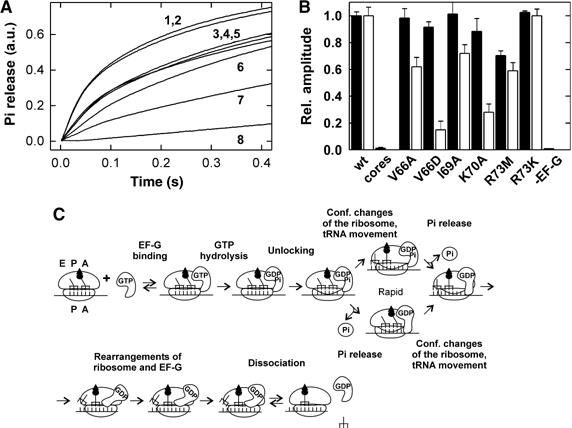
Pi release from EF-G after GTP hydrolysis. (A) Time courses of Pi release. EF-G (3 μM) and GTP (30 μM) were mixed with ribosomes (0.5 μM) in the stopped-flow apparatus and the liberation of Pi was monitored by the fluorescence change of MDCC-labeled PBP (Materials and methods). Traces are shown for ribosomes reconstituted with wt L7/12 (1), R73K (2), R73M (3), V66A (4), I69A (5), K70A (6), V66D (7) and for ribosome cores depleted of L7/12 (8). Exponential fitting yielded burst rates of 20±10 s−1 in all cases, except for cores depleted of L7/12 where the burst phase is lacking. The rates of the slower turnover phase were 3–4 s−1 (wt, R73M, R73K), 2 s−1 (V66A, I69A), or 1 s−1 (V66D, K70A). The final level of all time courses was identical within 10% and was normalized to 1.0. (B) Relative burst amplitudes of single-round GTP hydrolysis (closed bars) and Pi release (open bars). The extent of [γ-32P]GTP hydrolysis was determined either from quench-flow data (Figure 3) or from the amount of [γ-32P]GTP hydrolyzed after 10 s at conditions where multiple turnover was suppressed by the addition of unlabeled GTP. Amplitudes of Pi release were estimated from the data in panel A. Burst amplitudes for GTP hydrolysis and Pi release determined for ribosomes reconstituted with wt L7/12 were set to 1.0 and relative numbers are given for cores and reconstituted mutant ribosomes. (C) Kinetic scheme of translocation used for the evaluation of rate constants of Pi release (Savelsbergh et al, 2003). Binding of EF-G to the ribosome is followed by rapid GTP hydrolysis (50–200 s−1, cf. Figure 3B); a conformational change (‘unlocking'; 10–35 s−1; Savelsbergh et al, 2003) precedes and limits (fully or partially) the following tRNA–mRNA translocation. Translocation (or a structural rearrangement accompanying tRNA movement) and Pi release are parallel and independent of one another. The rate of Pi release is decreased to 1–2 s−1 by mutations at positions 66, 69, and 70 of the L7/12 CTD. Subsequent conformational changes of the ribosome and EF-G are important for EF-G dissociation from the ribosome.
The observation of an up to 85% decrease of the burst amplitude with no change of the burst rate can only be explained by a branched model of translocation of the type we have derived from kinetic analyses (Savelsbergh et al, 2003) (Figure 4C). Alternative models, such as the early model suggested by Spirin (1985) according to which tRNA translocation preceded GTP hydrolysis or a recent model that postulates a rate-limiting nucleotide exchange on EF-G on the ribosome (Zavialov et al, 2005), do not contain a branch and no explicit Pi release step and are, therefore, unsuitable to interpret the present observations. Furthermore, these models are inconsistent with the kinetic data that define the timing of EF-G binding, GTP hydrolysis, and tRNA–mRNA movement during translocation (Rodnina et al, 1997; Savelsbergh et al, 2000a, 2000b, 2003; Seo et al, 2004).
According to the branched model (Figure 4C), following ribosome unlocking translocation may occur first, followed by Pi release (upper branch), or, alternatively, Pi release may take place first, followed by translocation (lower branch). On native ribosomes, the latter two reactions are intrinsically rapid and their rates are determined by the unlocking step; therefore, rate constants of translocation and Pi release cannot be determined in the wt situation, and the observed burst amplitude of Pi release reflects the sum of the upper and lower branches of the reaction. However, when Pi release becomes slower than unlocking, while translocation remains fast, the lower branch will be disfavored and the amplitude of the rapid burst of Pi release will decrease, due to partitioning between the lower and upper branches of the scheme (for mathematical treatment of partitioning, see Fersht, 1985). The fact that the burst amplitude of Pi release was decreased by mutations in L7/12, while the amplitude of rapid GTP hydrolysis was not (Figure 4), indicated that the mutations disfavored Pi release relative to the parallel reaction. On vacant ribosomes (used in the present experiments), where there is no translocation, the parallel reaction presumably consists in a conformational rearrangement of the ribosome that follows unlocking (and accompanies translocation on translating ribosomes). Vacant ribosomes can be used for the analysis of Pi release, because kinetic studies have revealed that the rates of first-round and multiple-turnover Pi release on translocating and vacant ribosomes are about the same (Savelsbergh et al, 2003; data not shown), indicating that Pi release is not affected by the functional state of the ribosome. Depending on the actual rates, the burst rate of Pi release is given either by the sum of the rates of the two competing reactions, Pi release and ribosome rearrangement (or translocation), or is determined by the rate of the unlocking step that precedes both reactions (Savelsbergh et al, 2003). The rate of the unlocking step was not affected by the mutations, as the rate of translocation was not changed significantly by any of the mutations (Figure 3C). Consequently, the apparent rate of the residual burst of Pi release also should not be affected by the mutations, which is exactly what was observed (Figure 4A).
After the burst phase, a slower phase of Pi release is observed (Figure 4A), which on wt ribosomes reflects the turnover of EF-G. However, on ribosomes carrying mutant L7/12, part of the amplitude of slow Pi release is due to first-round Pi release, which was slowed down by the mutations. For example, the V66D mutation, which had the strongest effect on turnover, reduced the burst amplitude of Pi release to 15%, compared to the native situation (100%), whereas the rate of translocation was not changed (Figure 3C). Thus, in this case, 85% of the reaction followed the upper branch of the kinetic scheme (Figure 4C), and 85% of Pi was released at the rate of the following multiple-turnover reaction, which was about 1 s−1 for the V66D mutant, comparable to the numbers measured by turnover GTP hydrolysis and translocation (Table I). Compared to the burst rate of Pi release on native ribosomes, 30 s−1, the V66D mutation lowered the apparent rate of Pi release about 30-fold. The other mutations (V66A, I69A, K70A, and R73M) inhibited Pi release about 15-fold, to around 2 s−1, again comparable to the other two turnover assays (Table I). This suggests that the L7/12 mutations slowed down Pi release to such an extent that it became rate-limiting for the turnover of EF-G, that is, kcat of the turnover reaction(s) represented the rate of Pi release. On core ribosomes lacking L7/12, there was no burst of Pi release, as GTP hydrolysis was very slow (0.6 s−1), and turnover Pi release was slow as well (Figure 4A). In this case, GTP hydrolysis is rate-limiting for Pi release and EF-G turnover (Mohr et al, 2002).
To examine whether the effect of L7/12 mutations on Pi release was specific for EF-G, we performed measurements with EF-Tu. Mutations in helices 4 and 5 of L7/12 (K65A, V66A, V66D, I69A, K70A, R73M, and K84A) reduced the rate of association of the ternary complex EF-Tu·GTP·aa-tRNA with the ribosome, but had no effect on the rate constant of GTP hydrolysis by EF-Tu (Kothe et al, 2004; Diaconu et al, 2005). Helix 4 mutations lowered the apparent rate of the first round of Pi release from EF-Tu about three- to four-fold (multiple rounds were not observed due to very slow nucleotide exchange on EF-Tu) (Figure 5). However, this inhibition is fully explained by the reduction of the association rate constant caused by the mutations, from 100 μM−1 s−1 (wt L7/12) to about 20–30 μM−1 s−1 (mutant L7/12) (Kothe et al, 2004; Diaconu et al, 2005). Thus, mutations in L7/12 do not directly affect Pi release from EF-Tu, suggesting that the control of Pi release from EF-G by contacts with helix 4 of L7/12 is specific for EF-G.
Figure 5.
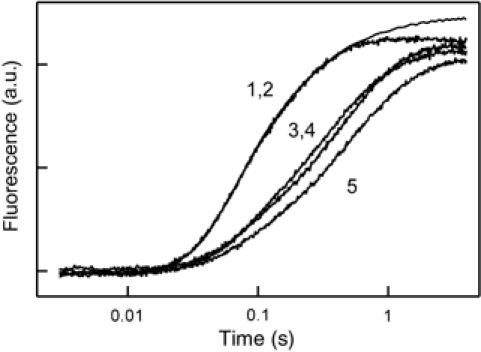
Time courses of Pi release from EF-Tu. Purified ternary complex EF-Tu·GTP·Phe-tRNAPhe (1 μM) was rapidly mixed with poly(U)-programmed ribosomes (1 μM) containing AcPhe-tRNAPhe in the P site. Traces are shown for native ribosomes (1) and ribosomes reconstituted with wt L7/12 (2), R73M (3), K70A (4), or K84A (5).
Discussion
The presence of protein L7/12 is important for the function of GTP-binding translation factors on the ribosome. The highly mobile CTDs of L7/12 reach out from the ribosome and promote the recruitment of the factors to the ribosome (Diaconu et al, 2005). Furthermore, L7/12 contributes to the stimulation of the GTPase activity of ribosome-bound factors by inducing or stabilizing their active conformation (Mohr et al, 2002; Diaconu et al, 2005). Sequence alignment of bacterial L7/12 proteins has revealed highly conserved patches in the CTD, in particular in helices 4 and 5, suggesting a site of interaction with translation factors (Leijonmarck and Liljas, 1987; Wahl et al, 2000; Wieden et al, 2001). Further evidence suggesting a role of this region of L7/12 in factor function came from the observations that the E82K mutation led to altered translational accuracy, decreased growth rates in vivo (Kirsebom and Isaksson, 1985; Kirsebom et al, 1986), and reduced translational efficiency in vitro (Bilgin et al, 1988). Chemical modification of R73 of L7/12 decreased the translational activity of the ribosomes (Koteliansky et al, 1977; Hernandez et al, 1984). Finally, amino-acid exchanges in helices 4 and 5 in the CTD of L7/12 decreased the rate of ternary complex binding to the ribosome, most likely by compromising interactions between L7/12 and helix D in the G domain of EF-Tu (Kothe et al, 2004).
The present data show that hydrophobic residues (V66, I69) as well as positively charged residues (K70, R73) in helix 4 of L7/12 are involved in the interaction with EF-G. All four mutations resulted in increased KM values for the multiple-turnover reactions of EF-G on the ribosome, suggesting that helix 4 is (part of) the binding site and that the mutated residues may be directly involved in the interaction. The four residues are located on the surface of L7/12 and are either strictly conserved (K70, R73) or appear to allow only conservative replacements (66, I/V/L; 69, I/L) (Wieden et al, 2001). The presence of a basic residue at position 73 seems to be important, because the replacement R73K had essentially no effect on activity of EF-G on the ribosome, in contrast to the significant effect of the mutation R73M. In the crystal structure of L7/12, these residues form a ridge–groove–ridge surface, where the ridges are formed by charged amino acids and the bottom of the groove by hydrophobic amino acids. The dimensions of the surface would allow an α helix to bind along the groove, with charged residues positioned at opposite sides of the helix.
The region of EF-G that is involved in binding to L7/12 has not been characterized so far. Three-dimensional reconstructions of EF-G–ribosome complexes from cryo-electron microscopic images indicate that there is a contact between protein L7/12 and the G′ domain of EF-G (Valle et al, 2003; Diaconu et al, 2005), which is located at a considerable distance from the phosphate side of the nucleotide-binding pocket. Taking into account the nature of the amino-acid side chains in L7/12 that according to the present analysis appear to be involved in the interaction, the complementary surface in EF-G is expected to comprise both negatively charged and hydrophobic residues. Of the three helices in the G′ domain, helix BG′ contains the appropriate side chains (Ævarsson et al, 1994; Czworkowski et al, 1994) and, thus, is a likely candidate to form (part of) the interaction site. Support for this contention comes from the observation that mutations in helix BG′ strongly affect the function of EF-G on the ribosome (A Savelsbergh, unpublished results).
L7/12 is required for the full stimulation of the GTPase activity of EF-G, as removal of L7/12 reduces the rate of ribosome-stimulated GTP hydrolysis about 1000-fold (Mohr et al, 2002; Diaconu et al, 2005). Replacements of several conserved amino acids in the CTD of L7/12, including the single conserved arginine (R73), had little or no effect on single-round GTP hydrolysis by EF-G on the ribosome (Savelsbergh et al, 2000b; present results), indicating that none of these amino acids is directly involved in GTP hydrolysis. Rather, L7/12 appears to bring about the GTPase activation of EF-G by inducing a particular conformation of the factor on the ribosome that is required for rapid GTP hydrolysis, and the induction of the active GTPase conformation is not impaired by any of the mutations tested.
As shown by the present results, L7/12 plays a crucial role in Pi release from EF-G. Mutations of V66, I69, or K70 in helix 4 of L7/12 decrease the rate of the first round of Pi release such that it becomes rate-limiting for the turnover reaction. Rates of Pi release on mutant ribosomes are 1–2 s−1, that is, 15–30 times lower than the rate of Pi release on native ribosomes, which is ∼30 s−1 (Savelsbergh et al, 2003). This suggests that the nucleotide-binding pocket of the ribosome-bound factor, including the γ-phosphate site, closes upon binding of EF-G·GTP to the ribosome, in keeping with the increased affinity of the nucleotide to ribosome-bound EF-G (Baca et al, 1976), and is opened by interactions (or a change of interactions) between the CTD of L7/12 and EF-G, allowing Pi to dissociate. The contact of the L7/12 CTD with EF-G that is established early and leads to GTPase activation may be retained until Pi is released or EF-G·GDP dissociates from the ribosome. GTP hydrolysis enables EF-G to promote a rearrangement of the ribosome (unlocking), which is rate-limiting for both translocation and Pi release, indicating that the active form of the factor is EF-G·GDP·Pi (Savelsbergh et al, 2003). The retention of Pi in the nucleotide-binding pocket is important for EF-G function, because if Pi release were uncontrolled, the conformational coupling between EF-G and the ribosome would probably be disturbed or interrupted, interfering with the ribosome rearrangement required for translocation. Thus, controlling GTP hydrolysis and Pi retention (or release) by interactions between EF-G and L7/12 provides a device for timing the functions of EF-G on the ribosome.
Materials and methods
Buffers and reagents
Buffer A: 20 mM Tris–HCl, pH 7.5, 0.6 M NH4Cl, 20 mM MgCl2, and 5 mM 2-mercaptoethanol; buffer B: 50 mM Tris–HCl, pH 7.5, 30 mM KCl, 7 mM MgCl2, and 25% glycerol; buffer C: 50 mM Tris–HCl, pH 7.5, 70 mM NH4Cl, 30 mM KCl, and 7 mM MgCl2; buffer D: 50 mM Tris—HCl, pH 7.5, 70 mM NH4Cl, 30 mM KCl, 10 mM MgCl2, and 2 mM DTT. GTP, phosphoenolpyruvate, and pyruvate kinase were from Roche Diagnostics. Radioactive compounds were from ICN. All other chemicals were from Merck.
Ribosomes, mRNAs, tRNAs, and factors
Ribosomes from E. coli MRE 600 were prepared as described (Rodnina and Wintermeyer, 1995). fMet-tRNAfMet, [14C]Phe-tRNAPhe, MFTI-mRNA, EF-Tu, EF-G, and initiation factors were prepared and purified as described (Rodnina et al, 1994a, 1994b, 1997, 1999).
Preparation of L7/12 mutants
The plasmid used as template for mutagenesis, pGEX-5x-3-L7/12, contained the gene for glutathione S-transferase (GST) fused to the gene of L7/12 (Savelsbergh et al, 2000b). Mutations were introduced by PCR mutagenesis using Pfu polymerase and verified by DNA sequencing. Mutant proteins were expressed in E. coli BL21 as GST fusion proteins and purified by affinity chromatography on glutathione-Sepharose 4B as described (Savelsbergh et al, 2000b), except that cleavage of the GST fusion proteins by factor Xa (Novagen) was carried out directly on the affinity matrix. The purity of the resulting preparations of L7/12 protein was >90% according to SDS–PAGE. Wt L7/12 was expressed and purified in the same way as the L7/12 mutants.
Depletion and reconstitution of ribosomes
Ribosomes were practically completely depleted of proteins L7/12 by NH4Cl/ethanol treatment (Tokimatsu et al, 1981) with some modifications as described previously (Mohr et al, 2002). A 450 pmol portion of purified ribosomes was incubated in 450 μl of buffer A on ice for 10 min. The solution was mixed with 250 μl of cold ethanol and gently stirred on ice. After 10 min, another 250 μl of ethanol was added and the mixture stirred for another 5 min. The mixture was centrifuged at 15 000 g for 30 min, and the ribosomal pellet was dissolved in buffer B. According to immunoblot analysis with anti-L7/12 antibodies (provided by AG Tonevitski, Moscow State University), the NH4Cl/ethanol-treated ribosomes contained no detectable L7/12, that is, the L7/12 content was <2%. The removal of ribosomal proteins other than L7/12 was controlled by precipitating proteins from the NH4Cl/ethanol supernatant with acetone; according to SDS–PAGE, the supernatant contained, besides L7/12, trace amounts of L10, and no other protein. For reconstitution, ribosomes depleted of L7/12 (‘cores') were incubated with a five-fold excess of purified wt or mutant L7/12 for 30 min at 37°C; the extent of reconstitution was >80%, as determined by immunoblot analysis.
GTP hydrolysis
To measure ribosome-stimulated multiple-turnover GTP hydrolysis by EF-G, ribosomes (0.2 μM) were mixed with EF-G (0.04 μM) in buffer C at 37°C and the reaction was initiated by the addition of [γ-32P]GTP (20 μM). Steady-state kinetic parameters, kcat and KM, were determined under conditions of initial velocity. Incubation was for 1.5 min (wt L7/12, R73K, and R73M) or 2.5 min (all other mutants). Samples (30 μl) were quenched by adding 1 M HClO4/3 mM potassium phosphate (30 μl) and analyzed by thin-layer chromatography on PEI-cellulose in 0.5 M potassium phosphate (pH 3.5). The extent of hydrolysis was quantified using a phosphoimager (Bio-Rad).
Single-round GTP hydrolysis by EF-G was measured by mixing native or reconstituted ribosomes (1 μM) with EF-G (3 μM) and [γ-32P]GTP (30 μM) in a quench-flow apparatus (KinTek Corp., Austin, TX, USA). Reactions were quenched with 1 M HClO4/3 mM potassium phosphate and analyzed by extraction of 32P-labeled inorganic phosphate into ethyl acetate in the presence of sodium molybdate (Leder and Bursztyn, 1966; Rodnina et al, 1997).
Dissociation of inorganic phosphate
Pi release from EF-G after GTP hydrolysis was measured by the fluorescence change of MDCC-labeled phosphate-binding protein (MDCC-PBP) (Brune et al, 1994) in buffer C at 37°C (Mohr et al, 2000; Savelsbergh et al, 2000a). Ribosomes and EF-G at the indicated concentrations were mixed with GTP (30 μM), MDCC-PBP (2.5 μM), purine nucleoside phosphorylase (0.1 U/ml), and 7-methylguanosine (200 μM) (the latter two components serving as ‘Pi mop' to take up trace amounts of contaminating Pi) (Brune et al, 1994) in a stopped-flow apparatus (Applied Photophysics), and the fluorescence change of MDCC-PBP upon binding of Pi was monitored. Pi release from EF-Tu was studied in buffer D at 20°C, using ternary complex that was purified by gel filtration on Superdex 75 (Rodnina et al, 1994a). Ribosomes were programmed with poly(U) and AcPhe-tRNAPhe in the P site and purified by ultracentrifugation through a sucrose cushion (Gromadski and Rodnina, 2004). Fluorescence was excited at 425 nm and measured after passing a cutoff filter (KV 450, Schott).
Translocation
To prepare initiation complexes, ribosomes (0.5 μM) were incubated with a three-fold molar excess of MFTI-mRNA (GGCAAGGAGGUAAAUAAUGUUUCACGAUU; codons used for the incorporation of M (fMet) and F (Phe) are underlined) and a 1.5-fold excess each of IF1, IF2, IF3, and f[3H]Met-tRNAfMet in buffer C containing GTP (1 mM) for 30 min at 37°C. The ternary complex EF-Tu·GTP·[14C]Phe-tRNAPhe was prepared by first incubating EF-Tu (2 μM) with GTP (1 mM), phosphoenolpyruvate (3 mM), and pyruvate kinase (0.5 mg/l) for 30 min at 37°C and then with [14C]Phe-tRNAPhe (1 μM) for an additional 5 min. Equal volumes each of initiation complex and ternary complex were mixed and incubated for 5 min at 20°C to form pretranslocation complex carrying tRNAfMet in the P site and fMetPhe-tRNAPhe in the A site.
Multiple-turnover translocation was induced by adding EF-G (final concentration 0.5 nM) to pretranslocation complexes present in increasing amounts (0.05–0.7 μM). Steady-state kinetic parameters (kcat, KM) were determined under conditions of initial velocity. Incubation was for 2 min (wt L7/12, R73K, and R73M), 5 min (V66D), or 4 min (all other mutants). The extent of translocation was measured by reaction with puromycin (Pmn; 1 mM, 10 s, 37°C), monitoring the formation of fMetPhe-Pmn (Rodnina et al, 1997). Single-round translocation was measured by fluorescence stopped-flow (Savelsbergh et al, 2003), using pretranslocation complex containing fluorescent fMetPhe-tRNAPhe (Prf16/17) (0.5 μM), saturating amounts of EF-G (3 μM), and GTP (1 mM).
Acknowledgments
We thank Yuri Semenkov and Vladimir Katunin (Petersburg Nuclear Physics Institute, Russia) for gifts of purified tRNA; Alexander Tonevitski (Moscow State University, Russia) for providing antibodies; and Petra Striebeck, Carmen Schillings, Astrid Böhm, and Simone Möbitz for expert technical assistance. This work was supported by the Deutsche Forschungsgemeinschaft, the European Union, the Alfried Krupp von Bohlen und Halbach-Stiftung, and the Fonds der Chemischen Industrie. UK was supported by a fellowship of the Studienstiftung des deutschen Volkes.
References
- Ævarsson A, Brazhnikov E, Garber M, Zheltonosova J, Chirgadze Y, al-Karadaghi S, Svensson LA, Liljas A (1994) Three-dimensional structure of the ribosomal translocase: elongation factor G from Thermus thermophilus. EMBO J 13: 3669–3677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baca OG, Rohrbach MS, Bodley JW (1976) Equilibrium measurements of the interactions of guanine nucleotides with Escherichia coli elongation factor G and the ribosome. Biochemistry 15: 4570–4574 [DOI] [PubMed] [Google Scholar]
- Belitsina NV, Glukhova MA, Spirin AS (1976) Stepwise elongation factor G-promoted elongation of polypeptides on the ribosome without GTP cleavage. J Mol Biol 108: 609–613 [DOI] [PubMed] [Google Scholar]
- Bilgin N, Kirsebom LA, Ehrenberg M, Kurland CG (1988) Mutations in ribosomal proteins L7/L12 perturb EF-G and EF-Tu functions. Biochimie 70: 611–618 [DOI] [PubMed] [Google Scholar]
- Bocharov EV, Sobol AG, Pavlov KV, Korzhnev DM, Jaravine VA, Gudkov AT, Arseniev AS (2004) From structure and dynamics of protein L7/L12 to molecular switching in ribosome. J Biol Chem 279: 17697–17706 [DOI] [PubMed] [Google Scholar]
- Brune M, Hunter JL, Corrie JE, Webb MR (1994) Direct, real-time measurement of rapid inorganic phosphate release using a novel fluorescent probe and its application to actomyosin subfragment 1 ATPase. Biochemistry 33: 8262–8271 [DOI] [PubMed] [Google Scholar]
- Czworkowski J, Wang J, Steitz TA, Moore PB (1994) The crystal structure of elongation factor G complexed with GDP, at 2.7 Å resolution. EMBO J 13: 3661–3668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dey D, Oleinikov AV, Traut RR (1995) The hinge region of Escherichia coli ribosomal protein L7/L12 is required for factor binding and GTP hydrolysis. Biochimie 77: 925–930 [DOI] [PubMed] [Google Scholar]
- Diaconu M, Kothe U, Schlünzen F, Fischer N, Harms JM, Tonevitski AG, Stark H, Rodnina MV, Wahl MC (2005) Structural basis for the function of the ribosomal L7/12 stalk in factor binding and GTPase activation. Cell 121: 991–1004 [DOI] [PubMed] [Google Scholar]
- Fersht A (1985) Enzyme Structure and Mechanism. New York: Freeman WH and Company [Google Scholar]
- Gromadski KB, Rodnina MV (2004) Kinetic determinants of high-fidelity tRNA discrimination on the ribosome. Mol Cell 13: 191–200 [DOI] [PubMed] [Google Scholar]
- Hamel E, Koka M, Nakamoto T (1972) Requirement of an Escherichia coli 50 S ribosomal protein component for effective interaction of the ribosome with T and G factors and with guanosine triphosphate. J Biol Chem 247: 805–814 [PubMed] [Google Scholar]
- Hernandez F, de No C, Palacian E (1984) Functional implication of the sole arginine residue of ribosomal proteins L7/L12. Mol Biol Rep 10: 75–78 [DOI] [PubMed] [Google Scholar]
- Katunin VI, Savelsbergh A, Rodnina MV, Wintermeyer W (2002) Coupling of GTP hydrolysis by elongation factor G to translocation and factor recycling on the ribosome. Biochemistry 41: 12806–12812 [DOI] [PubMed] [Google Scholar]
- Kaziro Y (1978) The role of guanosine 5′-triphosphate in polypeptide chain elongation. Biochim Biophys Acta 505: 95–127 [DOI] [PubMed] [Google Scholar]
- Kirsebom LA, Amons R, Isaksson LA (1986) Primary structures of mutationally altered ribosomal protein L7/L12 and their effects on cellular growth and translational accuracy. Eur J Biochem 156: 669–675 [DOI] [PubMed] [Google Scholar]
- Kirsebom LA, Isaksson LA (1985) Involvement of ribosomal protein L7/L12 in control of translational accuracy. Proc Natl Acad Sci USA 82: 717–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kischa K, Möller W, Stoffler G (1971) Reconstitution of a GTPase activity by a 50S ribosomal protein and E. coli. Nat New Biol 233: 62–63 [DOI] [PubMed] [Google Scholar]
- Koteliansky VE, Domogatsky SP, Gudkov AT, Spirin AS (1977) Elongation factor-dependent reactions of ribosomes deprived of proteins L7 and L12. FEBS Lett 73: 6–11 [DOI] [PubMed] [Google Scholar]
- Kothe U, Wieden HJ, Mohr D, Rodnina MV (2004) Interaction of helix D of elongation factor Tu with helices 4 and 5 of protein L7/12 on the ribosome. J Mol Biol 336: 1011–1021 [DOI] [PubMed] [Google Scholar]
- Leder P, Bursztyn H (1966) Initiation of protein synthesis II. A convenient assay for the ribosome-dependent synthesis of N-formyl-C14-methionylpuromycin. Biochem Biophys Res Commun 25: 233–238 [DOI] [PubMed] [Google Scholar]
- Leijonmarck M, Liljas A (1987) Structure of the C-terminal domain of the ribosomal protein L7/L12 from Escherichia coli at 1.7 Å. J Mol Biol 195: 555–579 [DOI] [PubMed] [Google Scholar]
- Mohr D, Wintermeyer W, Rodnina MV (2000) Arginines 29 and 59 of elongation factor G are important for GTP hydrolysis or translocation on the ribosome. EMBO J 19: 3458–3464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohr D, Wintermeyer W, Rodnina MV (2002) GTPase activation of elongation factors Tu and G on the ribosome. Biochemistry 41: 12520–12528 [DOI] [PubMed] [Google Scholar]
- Mulder FA, Bouakaz L, Lundell A, Venkataramana M, Liljas A, Akke M, Sanyal S (2004) Conformation and dynamics of ribosomal stalk protein L12 in solution and on the ribosome. Biochemistry 43: 5930–5936 [DOI] [PubMed] [Google Scholar]
- Parmeggiani A, Sander G (1981) Properties and regulation of the GTPase activities of elongation factors Tu and G, and of initiation factor 2. Mol Cell Biochem 35: 129–158 [DOI] [PubMed] [Google Scholar]
- Rodnina MV, Fricke R, Wintermeyer W (1994a) Transient conformational states of aminoacyl-tRNA during ribosome binding catalyzed by elongation factor Tu. Biochemistry 33: 12267–12275 [DOI] [PubMed] [Google Scholar]
- Rodnina MV, Savelsbergh A, Katunin VI, Wintermeyer W (1997) Hydrolysis of GTP by elongation factor G drives tRNA movement on the ribosome. Nature 385: 37–41 [DOI] [PubMed] [Google Scholar]
- Rodnina MV, Savelsbergh A, Matassova NB, Katunin VI, Semenkov YP, Wintermeyer W (1999) Thiostrepton inhibits turnover but not GTP hydrolysis by elongation factor G on the ribosome. Proc Natl Acad Sci USA 96: 9586–9590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodnina MV, Semenkov YP, Wintermeyer W (1994b) Purification of fMet-tRNA(fMet) by fast protein liquid chromatography. Anal Biochem 219: 380–381 [DOI] [PubMed] [Google Scholar]
- Rodnina MV, Wintermeyer W (1995) GTP consumption of elongation factor Tu during translation of heteropolymeric mRNAs. Proc Natl Acad Sci USA 92: 1945–1949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander G, Marsh RC, Voigt J, Parmeggiani A (1975) A comparative study of the 50S ribosomal subunit and several 50S subparticles in EF-T-and EF-G-dependent activities. Biochemistry 14: 1805–1814 [DOI] [PubMed] [Google Scholar]
- Savelsbergh A, Katunin VI, Mohr D, Peske F, Rodnina MV, Wintermeyer W (2003) An elongation factor G-induced ribosome rearrangement precedes tRNA–mRNA translocation. Mol Cell 11: 1517–1523 [DOI] [PubMed] [Google Scholar]
- Savelsbergh A, Matassova NB, Rodnina MV, Wintermeyer W (2000a) Role of domains 4 and 5 in elongation factor G functions on the ribosome. J Mol Biol 300: 951–961 [DOI] [PubMed] [Google Scholar]
- Savelsbergh A, Mohr D, Wilden B, Wintermeyer W, Rodnina MV (2000b) Stimulation of the GTPase activity of translation elongation factor G by ribosomal protein L7/12. J Biol Chem 275: 890–894 [DOI] [PubMed] [Google Scholar]
- Seo HS, Kiel M, Pan D, Raj VS, Kaji A, Cooperman BS (2004) Kinetics and thermodynamics of RRF, EF-G, and thiostrepton interaction on the Escherichia coli ribosome. Biochemistry 43: 12728–12740 [DOI] [PubMed] [Google Scholar]
- Spirin AS (1985) Ribosomal translocation: facts and models. Prog Nucleic Acid Res Mol Biol 32: 75–114 [DOI] [PubMed] [Google Scholar]
- Tokimatsu H, Strycharz WA, Dahlberg AE (1981) Gel electrophoretic studies on ribosomal proteins L7/L12 and the Escherichia coli 50 S subunit. J Mol Biol 152: 397–412 [DOI] [PubMed] [Google Scholar]
- Valle M, Zavialov A, Sengupta J, Rawat U, Ehrenberg M, Frank J (2003) Locking and unlocking of ribosomal motions. Cell 114: 123–134 [DOI] [PubMed] [Google Scholar]
- Wahl MC, Bourenkov GP, Bartunik HD, Huber R (2000) Flexibility, conformational diversity, and two dimerization modes in complexes of ribosomal protein L12. EMBO J 19: 174–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieden HJ, Wintermeyer W, Rodnina MV (2001) A common structural motif in elongation factor Ts and ribosomal protein L7/12 may be involved in the interaction with elongation factor Tu. J Mol Evol 52: 129–136 [DOI] [PubMed] [Google Scholar]
- Zavialov AV, Hauryliuk VV, Ehrenberg M (2005) Guanine-nucleotide exchange on ribosome-bound elongation factor G initiates the translocation of tRNAs. J Biol 4: 9. [DOI] [PMC free article] [PubMed] [Google Scholar]