

Abstract
The large majority of histidine kinases (HKs) are multifunctional enzymes having autokinase, phosphotransfer and phosphatase activities, and most of these are transmembrane sensor proteins. Sensor HKs possess conserved cytoplasmic phosphorylation and ATP-binding kinase domains. The different enzymatic activities require participation by one or both of these domains, implying the need for different conformational states. The catalytic domains are linked to the membrane through a coiled-coil segment that sometimes includes other domains. We describe here the first crystal structure of the complete cytoplasmic region of a sensor HK, one from the thermophile Thermotoga maritima in complex with ADPβN at 1.9 Å resolution. The structure reveals previously unidentified functions for several conserved residues and reveals the relative disposition of domains in a state seemingly poised for phosphotransfer. The structure thereby inspires hypotheses for the mechanisms of autophosphorylation, phosphotransfer and response-regulator dephosphorylation, and for signal transduction through the coiled-coil segment. Mutational tests support the functional relevance of interdomain contacts.
Keywords: crystal structure, PhoQ, phosphotransfer, selenomethionyl MAD, two-component systems
Introduction
Nearly all living cells use phosphorylation-mediated signal transduction mechanisms in responding to metabolic, environmental and cell-cycle stimuli. ‘Two-component' regulatory systems involving His-Asp phosphorelays predominate for signal transduction in prokaryotes and are commonplace in fungi and plants (reviewed by Stock et al, 2000). The paradigmatic two-component system consists of two basic protein units: a sensor histidine kinase (HK) and a response regulator (RR). The former acts as the signal receptor and possesses an autokinase activity that promotes phosphorylation of a histidine residue in a conserved domain. The phosphoryl group is then transferred to an aspartate residue of the RR (usually a transcription factor), triggering the cellular response. The response is proportional to the degree of RR phosphorylation, which depends not only on the efficiency of the autokinase and transfer reactions but also in many cases on an intrinsic autophosphatase activity in the RR and/or destabilization of the aspartyl phosphate bond by the cognate HK (termed regulated phosphatase activity). Signals typically mediate responses by influencing the HK autokinase and/or phosphatase activity (Russo and Silhavy, 1993).
The large majority of HKs, labeled class I HKs (Bilwes et al, 1999), are homodimeric membrane proteins in which each subunit contains a short amino-terminal cytoplasmic segment followed by a transmembrane α helix (TM1) and an extracellular (or periplasmic) sensing domain that is connected via a second membrane-spanning α helix (TM2) to a carboxy-terminal cytoplasmic kinase domain (Stock et al, 2000). The extracellular sensing domains are variable in sequence, reflecting the wide range of environmental signals to which HKs respond. Conversely, the cytoplasmic portion typically includes a conserved catalytic core of approximately 250 residues, which contains a set of characteristic sequence motifs, labeled the H, N, G1, F and G2 boxes (Parkinson and Kofoid, 1992). This core portion of class I HKs can be dissected into two distinct functional domains: an N-terminal dimerization and histidine phosphotransfer (DHp) domain and a C-terminal catalytic and ATP-binding (CA) domain. The DHp domain, which contains the autophosphorylation site (H box), forms a stable dimer and can be phosphorylated in the presence of ATP by the CA domain (Stock et al, 2000). The isolated CA domain is monomeric and encompasses the conserved N, G1, F, and G2 boxes.
The segment that connects TM2 to the catalytic core in class I HKs is variable in length and sequence, but it typically includes a common structural element called the HAMP (histidine kinase, adenylyl cyclase, methyl-accepting chemotaxis proteins and phosphatase) or P-type linker (Aravind and Ponting, 1999; Williams and Stewart, 1999). HAMP linkers are variable in length (40–180 residues) and have a predicted topology of two amphipathic helices separated by a loop region. They have been hypothesized to transmit signals between the external input domain and the cytoplasmic output module (Fabret et al, 1999; Williams and Stewart, 1999).
Atomic structures have been reported for the isolated DHp and CA domains of some HK sensors. Structures of both dissected domains have been determined by NMR spectroscopy for the osmosensor EnvZ from Escherichia coli (Tanaka et al, 1998; Tomomori et al, 1999), and CA domain structures have been determined by X-ray crystallography for the Thermotoga maritima CheA, E. coli PhoQ and NtrB CA domains (Bilwes et al, 1999; Marina et al, 2001; Song et al, 2004). The EnvZ DHp domain consists of two α helices that dimerize to form a four-helix bundle in which the histidine phospho-acceptors protrude from helices into the solvent (Tomomori et al, 1999). The four CA domains assume a mixed α/β sandwich fold made from five β strands and three α helices. These kinase domains are structurally related to the ATP-binding domains of the GHL ATPase family (GyraseB, Hsp90 and MutL); thereby these ATPase become the GHKL superfamily (Dutta and Inouye, 2000).
The three enzymatic activities (autokinase, phosphotransfer and phosphatase) associated with the cytoplasmic region of the HK each require the participation of one or both of the DHp and CA domains (Tanaka et al, 1991; Hsing et al, 1998), suggesting that these domains can exist in different conformational states with respect to one another. Structural characterization of these signaling states has been thwarted in the dissection approach. Here we have analyzed an intact cytoplasmic domain from a HK sensor protein, that of T. maritima TM0853. The resulting structure inspires testable hypotheses about the mechanism of signal transduction in HKs.
Results
Characterization and structure determination
In a survey aimed at expressing the cytoplasmic portions of HK sensors, we succeeded to clone, express and purify a fragment of a protein from T. maritima (ORF TM0853). This is a putative HK sensor by virtue of sequence similarities that are especially striking in the catalytic domain (Figure 1). Much of the full-length protein partitioned into the soluble fraction when expressed in E. coli, but the membrane-associated fraction increased with temperature consistent with membrane localization in the natural thermophilic host (Table I). We defined its cytoplasmic portion to comprise residues 233–489 and produced the corresponding fragment (HK853-CD). The purified protein is dimeric, as estimated by gel filtration chromatography and crosslinking assays (data not shown). HK853-CD was shown to possess an ATP-dependent autokinase activity in vitro (Figure 2A) and to support phosphotransfer to both PhoP and OmpR RRs (Figure 2B). Crystals of HK853-CD grown in the presence of the inert ATP analog AMPPNP diffracted beyond 2 Å Bragg spacings. Despite the amino-acid sequence similarity (Figure 1), efforts to solve the HK853-CD structure by molecular replacement based on models of either the PhoQ or EnvZ catalytic subdomains were unsuccessful. Consequently, the structure was determined from a selenomethionine-substituted protein by the MAD method.
Figure 1.
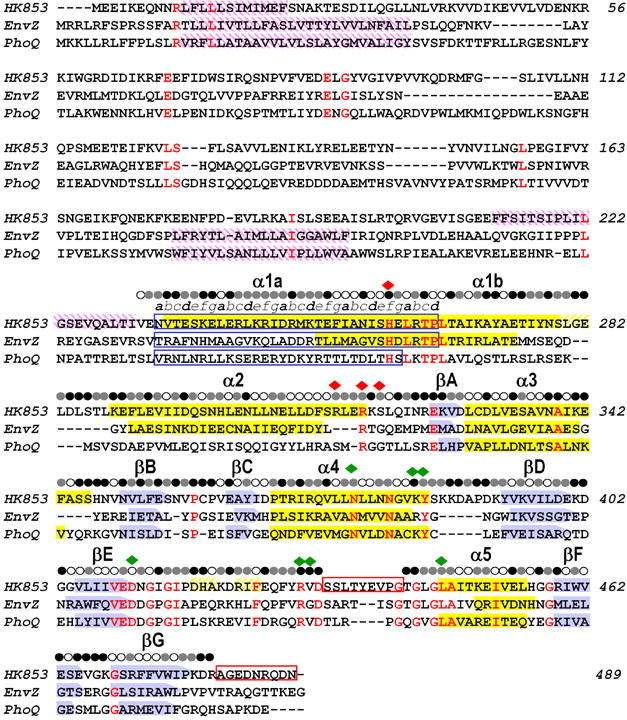
Sequence alignment of TM0853, EnvZ and PhoQ HKs. The three amino-acid sequences are aligned based on their structures. β sheets are shown as blue arrows and α helices as yellow filled boxes. Transmembrane regions predicted by the DAS program (Cserzo et al, 1997) are shown as purple intermediate shading and coiled-coil motifs predicted by LEARNCOIL program (Singh et al, 1998) are enclosed in blue boxes showing the helical position from a to g. Disordered regions are enclosed in red boxes. Residues identical in all three sequences are colored in red. The solvent accessibility of the HK853–CD is indicated for each residue by an open circle if the fraction solvent accessibility is >0.4, a half-filled circle if it is 0.1–0.4 and a filled circle if it is <0.1. Residues that interact with the ADPβN and the sulfate ion in HK853–CD are indicated by green and red diamonds, respectively.
Table 1.
Expression of full-length HK853 in E. coli
| T (°C) | Distribution of HK853 between cellular compartments (%) |
HK853 portion of total protein within each cellular compartment (%) |
||
|---|---|---|---|---|
| Membrane | Soluble | Membrane | Soluble | |
| 20 | 5.0±0.8 | 95.0±0.8 | 31.5±16.1 | 32.0±6.4 |
| 25 | 8.5±1.2 | 91.5±1.2 | 57.7±6.5 | 33.0±4.5 |
| 30 | 14.6±2.7 | 85.4±2.7 | 60.9±5.5 | 26.3±3.8 |
| 37 |
25.9±7.7 |
74.1±7.7 |
70.4±5.9 |
26.1±10.3 |
| Results are averages of two independent experiments, each of which was quantified twice (two independent gels). Possible inclusion bodies were eliminated before analysis. | ||||
Figure 2.
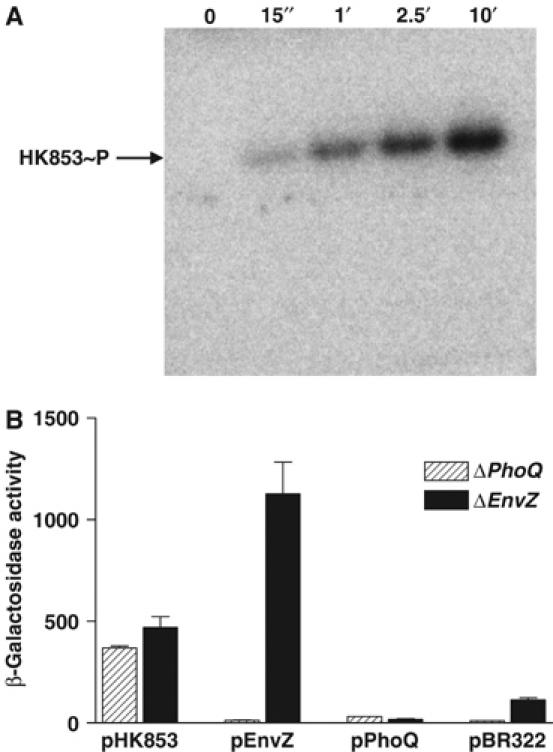
In vitro and in vivo HK853 activity. (A) Time course of in vitro autophosphorylation of HK853–CD with [γ-32P]ATP. In all, 1–2 μM of HK853–CD was incubated in reaction buffer and samples were removed at indicated time points, reaction stopped by addition of SDS–PAGE sample buffer, subjected to gel electrophoresis, and phosphorylated protein was visualized by phosphorimaging. (B) Activation of the PhoQ–PhoP and EnvZ–OmpR HK–RR systems. E. coli reporter strains containing a PhoP-activated lacZ, but devoid of PhoQ (ΔPhoQ), or an OmpR-activated lacZ, but devoid of EnvZ (ΔEnvZ), were transformed with the pBR322-derived plasmids pHK853, pEnvZ, pPhoQ or pBR322 (expressing the respective full-length sensor kinases or a negative control). Response was assayed by β-galactosidase activity. The low activation by PhoQ was due to repressing divalent cations in culture media.
An atomic model of HK853-CD was built and refined to 1.9 Å resolution. The electron densities for the nine C-terminal residues and internal-loop residues 433–441 were extremely weak or absent, and these regions were presumed to be disordered. The refined structure contains 240 residues, a hydrolyzed AMPPNP molecule, one sulfate ion and 179 water molecules, and it has good stereochemistry with no Ramachandran outliers (Table II).
Table 2.
Diffraction and structure determination statistics
| HK853-CD | Selenomethionyl I370M/V373M HK853–CD |
||||
|---|---|---|---|---|---|
| Native | Selow | Seedge | Sepeak | Sehigh | |
| Diffraction data | |||||
| Wavelength (Å) | 0.9678 | 0.9918 | 0.9794 | 0.9788 | 0.9678 |
| Spacing limit (Å) | 1.9 | 2.1 | 2.1 | 2.1 | 2.1 |
| Unique reflections | 21 892 | 16 406 | 16 405 | 16 416 | 16 434 |
| Rmerge (%)a | 6.1 (26.8) | 4.6 | 4.5 | 4.6 | 4.8 |
| I/σ | 13.8 (2.7) | 11.9 | 12.3 | 12.1 | 11.3 |
| Completeness (%) | 98.0 (99.7) | 97.7 | 95.5 | 96.8 | 97.7 |
| Redundancy | 13.4 (5.2) | 7.3 | 6.2 | 6.9 | 7.3 |
| MAD phasing | |||||
| Phasing powerb (ΔΔλ/Δ±h) | 2.64/2.09 | 3.04/3.13 | 0.91/2.46 | ||
| Rcullisc (ΔΔλ/Δ±h) | 0.51/0.79 | 0.48/0.58 | 0.68/0.71 | ||
| Overall FOM (acentric/centric)d | 0.68/0.50 | ||||
| Refinement | |||||
| Bragg spacings (Å) | 20–1.9 | ||||
| Re/Rfreef (%) | 24.7/27.5 (27.9/32.0) | ||||
| Number of protein atoms | 1960 | ||||
| Number of solvent atoms | 179 | ||||
| Number of non-protein atoms | 33 | ||||
| Average B factor (Å2) | 35.6 | ||||
| R.m.s. bonds (Å), angles (deg) | 0.011/1.7 | ||||
| Ramachandran analysisg | |||||
| Favored/outlier (%) |
97.7/0.0 |
|
|
|
|
| Values in parentheses refer to the highest resolution shell (2.0–1.9). | |||||
| aRmerge=∑∣I−〈I〉∣/∑I, where I is the observed intensity and 〈I〉 the average intensity. | |||||
| bPhasing power=root-mean-square (Fh/E), where Fh=heavy atom structure factor amplitude and E=residual lack of closure error. ΔΔλ is for dispersive differences relative to Selow. Δ±h is for Bijvoet differences. | |||||
| cRcullis=∑∣∣Fh(obs)-∣Fh(calc)∣∣/∑∣Fh(obs)∣, where Fh(obs) and Fh(calc) are the observed and calculated heavy atoms structural factor amplitudes, respectively. | |||||
| dFigure of merit=∣F(hkl)best∣/F(hkl). | |||||
| eR=∑∣∣Fo∣−∣Fc∣∣/∑∣Fo∣. | |||||
| fRfree is calculated as R, but on 5.2% of all reflections that are never used in crystallographic refinement. | |||||
| gAnalysis from http://kinemage.biochem.duke.edu. | |||||
Overall structure
There is one HK853-CD subunit in the asymmetric unit of the crystal, and it exploits a two-fold symmetry axis of the lattice to generate a homodimer, as expected from our solution studies and results on homologous systems (Yang and Inouye, 1991; Ninfa et al, 1993). Each protomeric subunit consists of two distinct domains, an N-terminal helical hairpin domain and a C-terminal α/β domain, which are connected by a short linker (residues 318–322) (Figure 3). The dimer interface is exclusively between helical-hairpin domains and the diad axis runs parallel with the helices such that the N-termini are adjacent, as if poised to emanate from the membrane.
Figure 3.
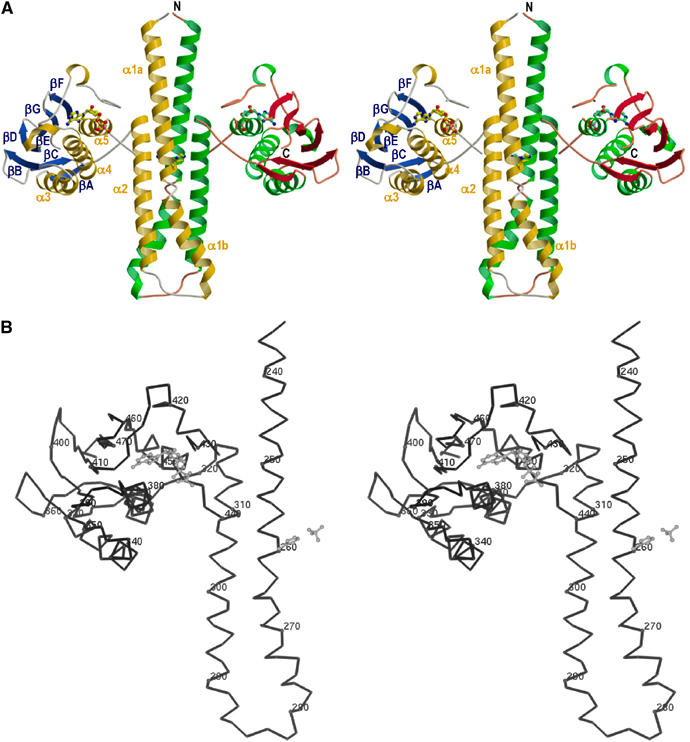
Molecular structure of the cytoplasmic portion of TM0853. (A) Ribbon representation of the crystallographic dimer of HK853–CD, including ADPβN. The α helices are labeled α1–α5, and colored gold (subunit A) and green (subunit B), β strands are labeled βA–βF, and colored blue (subunit A) and red (subunit B). The positions of N and C termini are labeled in subunit B. The ADPβN molecule and the phospho-acceptor residue (His260) are shown in ball-and-stick representation. The membrane would be located on the top of N-terminal residues. (B) Stereo Cα trace of the gold and blue protomer. Every tenth Cα is indicated as a sphere and numbered. The ADPβN molecule, the His260 and the sulfate ion coordinated with His260 are drawn in a gray ball-and-stick representation. The orientation is as in panel (A).
The helical-hairpin domain comprises residues 232–317 and has its two antiparallel helices connected by a nine-residue turn (residues 279–287). The first helix extends for about 75 Å from the N-terminus (232) to residue 278, but it has a pronounced kink induced by Pro265 such that we designate two parts; helix α1a includes the His260 phosphorylation site and helix α1b makes helix–bundle contacts with helices α2 and α2′, the symmetry mate. Helix α2 (residues 288–317) is shorter (∼50 Å).
The C-terminal domain (residues 323–489) assumes an α/β sandwich fold: one layer comprises a mixed five-strands β sheet (βB, βD–βG), which is nearly orthogonal to the helical-hairpin structure, and the other layer consists of three α helices (α3–α5). In addition, this domain contains a pair of short antiparallel β strands (βA and βC) and one disulfide bridge (Cys330–Cys359) linking the N-terminal segment of α3 (just following βA) with βC.
Dimeric association of helical-hairpin domains
The dimer of helical hairpin domains has two parts, a coiled-coil portion composed of the 22 N-terminal residues of helix α1a and a four-helix bundle portion composed of the rest (Figures 3 and 4). C-terminal segments of the α1 helices each interact with both α2 helices in an antiparallel manner with a left-handed twist of about 25°, the most favored assembly for a four-helix bundle (Chou et al, 1988). The four-helix bundle portion corresponds to the DHp domain and the coiled-coil extension includes the HAMP or P-type linker. His 260, the H-box histidine and presumed site of phosphorylation, is on the surface of this four-helix bundle. There is a large interface of association, burying 2100 Å2 of solvent-accessible surface area from each protomer. The majority of this interface is in the four-helix bundle (1500 Å2), but the coiled-coil interface is also substantial (600 Å2).
Figure 4.
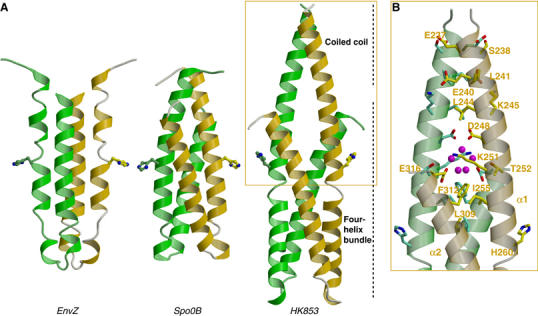
Comparison of DHp domains of EnvZ HK, Spo0B phosphotransferase and TM0853 HK. (A) Ribbon diagrams of the three DHp domains in their dimeric forms (protomers colored in green and gold). The DHp domains have been oriented with the plane containing the phospho-accepting histidines, which are shown in stick representation, and the principal helix axes parallel to the page. (B) Detail of the HK853 coiled-coil motif. Residues interacting in this motif are shown as stick representation and labeled on one protomer. Solvent molecules (magenta) are shown in the cavity generated at the juncture between the coiled coil of α1a helices and the four-helix bundle.
Several hydrophobic residues at the four-helix bundle interface are conserved among HKs. This interface also includes hydrophilic interactions, notably two hydrogen bonds (Thr252–Glu316′ and Arg263–Asn307′) and one salt bridge (Lys270–Glu303′), although of these only Arg263 is conservative. At the apex of the bundle, the connection between α1b and α2 helices is rather extended and consists of alternately exposed polar and buried hydrophobic residues. This segment is intrinsically flexible, as judged by elevated B factors. The α2 helices splay apart C-terminally (from 11 Å along the bundle to 18 Å at the open end), and the α1a helices emerge from between the separated α2 helices to meet in the coiled coil.
The transition from antiparallel bundle to parallel coiled-coil interactions generates a small cavity filled with water molecules (Figure 4B). Ile255 (from α1 helices), and Leu309 and Phe312 (from α2 helices) form the bottom portion of this cavity and are conserved in class I HKs. In contrast, the middle and upper portions are composed of polar (Thr252) and charged (Lys251 and Glu316) residues that interact with waters of the cavity. A hydrogen bond between carboxylate Oδ1 atoms of Asp248 and its symmetry mate Asp248′ (one presumably protonated) closes the top of the cavity and initiates the N-terminal coiled-coil interactions. The Oδ2 atoms of Asp248 and Asp248′ are both hydrogen bonded to the same water molecule of the cavity, which makes additional hydrogen bounds with other cavity waters. Side chains of Leu241 and Leu244 pack against their symmetry-related residues to form part of the hydrophobic core of the coiled-coil N-terminal segment.
The four-helix bundle domain of HK853 differs from the NMR-derived structure of the isolated DHp domain from E. coli EnvZ (Tomomori et al, 1999) in two significant respects. Firstly, the twist angle of the HK853 four-helix bundle (∼25°) is higher than that of EnvZ (∼10°), which is unusually parallel for this topological class (Dutta et al, 1999). Secondly, the connections between hairpin helices are crossed in the two models, that is, α2 and α2′ are interchanged. This is a surprising difference for members of the same sequence family, especially as the four-helix bundle topology of the more distantly related Bacillus subtilis Spo0B histidine phosphotransferase (Varughese et al, 1998) is the same as that observed for HK853 (Figure 4A). The connecting segment between helices was reported as unstructured in EnvZ, raising concern about the linkage geometry, but the use of mixtures of labeled and unlabeled EnvZ protein in the NMR analysis should have distinguished interchain from intrachain interactions. The helical-hairpin connector also has elevated atomic mobility in the HK853 structure, but the path of connecting electron density is unambiguous. This topological distinction, if indeed real, necessarily has mechanistic implications. The HK853 structure is consistent with trans-phosphorylation, as is observed (Yang and Inouye, 1991; Ninfa et al 1993; Qin et al, 2000), whereas the alternative would seem to be only consistent with phosphorylation on the same chain unless the DHp to CA linkage is radically different in EnvZ. It may also be that the truncated EnvZ DHp domain has an unnatural conformation in the absence of its kinase and coiled-coil neighbors.
Nucleotide binding in the kinase domain
The CA domain in HK853-CD is similar in structure to the corresponding kinase domains isolated from PhoQ (Marina et al, 2001), CheA (Bilwes et al, 1999), EnvZ (Tanaka et al, 1998) and NtrB (Song et al, 2004). Superimpositions of 125 Cα atoms of the PhoQ, NtrB and CheA domains with CA of HK853 give r.m.s. deviations of 1.4, 1.3 and 1.6 Å2, respectively. Although the protein was crystallized in presence of the inert ATP analog AMPPNP and MgCl2, no electron density was observed for the γ phosphate or for Mg2+ ions. We subsequently discovered that the AMPPNP nucleotide was hydrolyzed in the crystallization buffer (Na cacodylate (pH 6.5)+LiSO4) into a distinct product similar to ADP, which we identify as ADPβN (ADP-NH2 as designed by Yount et al, 1971) (see Materials and methods). Surprisingly, ATP is stable under these conditions. In PhoQ and CheA structures, the Mg2+ ion bridges the three nucleotide phosphates (Bilwes et al, 2001; Marina et al, 2001), suggesting that the absence of the Mg2+ ion in the HK853-CD structure could be due to the loss of γ-phosphate interaction. The nucleotide in the ADP–CheA complex (Bilwes et al, 2001), which also lacks the metal ion, is positioned similarly to ADPβN in HK853.
The segment that joins the conserved F and G2 boxes of HKs has the flexibility to adopt different conformations. In PhoQ and CheA it covers the nucleotide (Bilwes et al, 2001; Marina et al, 2001); hence, it is called the ATP lid. In HK853-CD, residues 433–441 of the ATP lid are disordered, suggesting high mobility in absence of the γ phosphate and the Mg2+. Similarly, the ATP lid was not observable in CheA and NtrB structures solved with empty nucleotide sites (Bilwes et al, 1999; Song et al, 2004). The ATP-lid segment of HK853 is anchored at each end by hydrophobic residues, the F-box namesake Phe425 and conservative Leu446, which interact with one another and with other hydrophobic residues (Ile424 and Ile460 in HK853) as in PhoQ and CheA counterparts (Marina et al, 2001). This supports the generality of this hydrophobic patch motif in ATP-lid attachment.
We have previously described two major groups of HKs, typified by PhoQ and CheA, based on nucleotide-binding geometry and mechanistic roles of key residues (Marina et al, 2001). The HK853 kinase belongs to the predominant PhoQ group, which also includes EnvZ (Figure 1), and nucleotide-binding residues in HK853 are disposed as they are in PhoQ (Figure 5). Structurally, an aromatic residue (Tyr393 in PhoQ, Tyr384 in HK853) is sandwiched between the adenine base, with which it stacks, and aliphatic portions of a basic residue (Lys392 in PhoQ, Lys 383 in HK853). Both functional groups make hydrogen bonds with nucleotide phosphates (γ in PhoQ, β in HK853). In addition, this sequence group has a conserved arginine or glutamine in the ATP lid, and both in PhoQ and in HK853 these residues interact with nucleotide β phosphates, suggesting a catalytic role for Arg430 of HK853 analogous to that found for Arg434 of PhoQ. Overall, the HK853 kinase structure supports our previous suggestions on histidine-kinase classification and catalytic mechanism.
Figure 5.
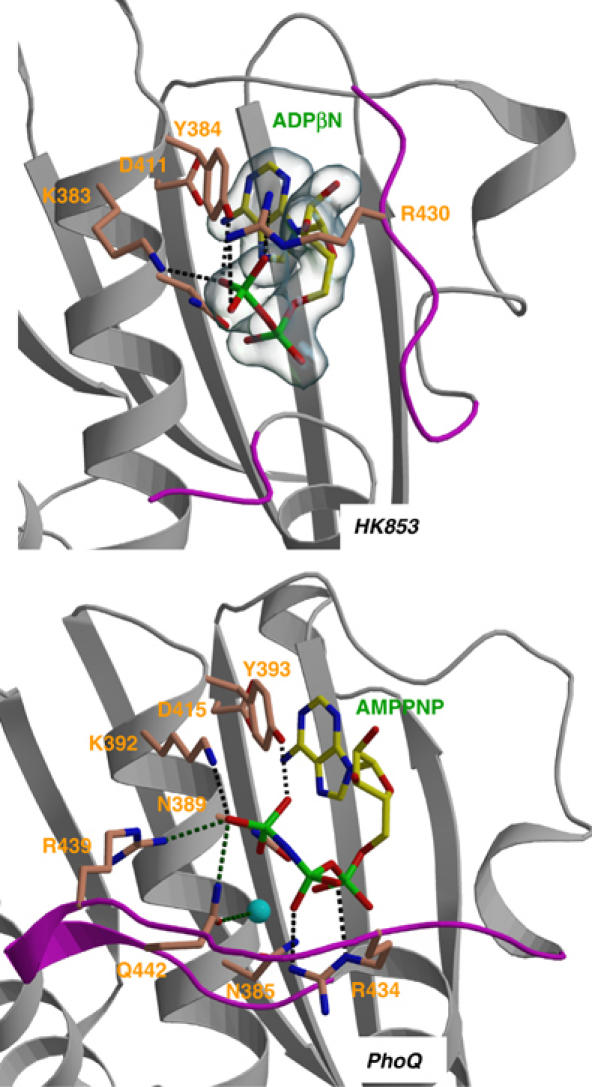
Comparison of the nucleotide-binding site of the TM0853 and PhoQ HKs. Secondary structures surrounding the ATP-binding site are drawn as gray ribbons and the ATP lids are in magenta. The nucleotides and the residues interacting with their phosphates are depicted as sticks and labeled. The Mg2+ ion of PhoQ is drawn as a cyan sphere. Hydrogen bonds are shown as dotted lines. Electron density of the HK853 nucleotide is contoured as a semitransparent blue surface at a level of 1σ.
Environment of the histidine phosphoacceptor site
His260 in HK853 corresponds to the absolutely conserved histidine, at which phosphorylation has been established to occur in representative HKs. Its side chain is fully exposed on helix α1 near the α1a–α1b kink provoked by Pro265 (Figure 4). An electron-dense feature, consistent with a sulfate ion coming from the 1.3 M SO42− present in the crystallization mixture, is located nearby, with oxygen atom O1 of this sulfate ion 2.6 Å from Nɛ of His260. The Nɛ atom on the imidazole ring is a more stable phosphorylation site than Nδ (Hultquist et al, 1996), and NMR studies confirmed it as the phosphorylation site in CheA (Zhou and Dahlquist, 1997). Based on these features and chemical similarities between sulfate and phosphate, it seems reasonable to conclude that the histidyl sulfate interaction may be mimicking phosphorylation of the histidine. This His260-associated sulfate ion also interacts with Arg317′, Arg314′ and Ser319′ of the neighboring protomer in the dimer (Figure 6), making hydrogen bonds to the side chains of Arg317′ (O1–Nη2 at 3.0 Å and O2–Nη1 at 2.9 Å), Arg314′ (O1–Nɛ at 3.0 Å) and Ser319′ (O3–Oγ at 2.6 Å). These three residues reside in a weakly conserved motif, termed X region (Hsing et al, 1998), which comprises the C-terminal end of helix α2 and the interdomain linker in the HK853-CD structure. The X region has been implicated in regulating phosphatase activity (see Discussion).
Figure 6.
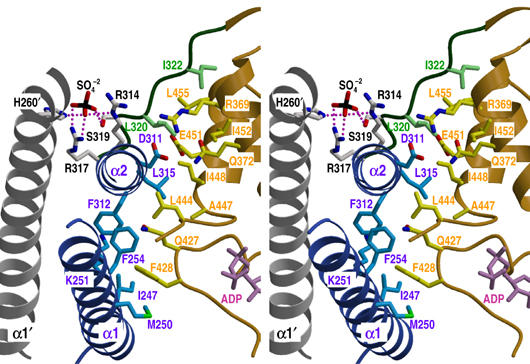
Interactions between DHp and CA domains. Stereoview of the structural elements involved in interdomain contacts and sulfate ion interactions. The DHp domain, CA domain and interdomain-connecting loop are represented in blue, gold and green ribbon diagrams, respectively. Additionally, the α1′ helix, which presents His260′ as a sulfate ligand, is shown in gray. The interacting side chains are shown as sticks with the same carbon atom color as the corresponding domain, except the sulfate-interacting residues that are in gray. Nitrogen, oxygen, sulfur and nucleotide molecule are drawn in blue, red, black and magenta, respectively. Residue labels take the colors of their domains. Hydrogen bonds and salt bridges between the sulfate ion and interacting residues are indicated by purple dots.
Interactions between the two cytoplasmic domains
This structure provides the first picture of interdomain contacts between the catalytic and DHp domains of a histidine-kinase sensor. The domains are connected by an extended linker (residues 318–322) in this state of the molecule, but a substantial interface between domains (1250 Å2) is nevertheless buried from solvent exposure. Contacts are formed principally from conserved hydrophobic residues and they are exclusively within a promoter (Figure 6). The DHp domain contributes 525 Å2 to the buried surface area, involving residues from helices α1a and α2 helices at the open end of the helical hairpin; the catalytic domain buries 475 Å2 from the α3 helix, the conserved G2 box (N-terminus of helix α4) and the conserved F box; and residues from the linker segment add the remaining 250 Å2.
The interaction interface can be divided into two patches grouped around each helix of the DHp domain. The α2 helix interacts with the α3 and α4 (G2 box) helices of the catalytic domain and with the connecting loop. In detail, there is a cluster of hydrophobic side chains, contributed by Leu315 and Phe312 in α2 (DHp), Leu320 and Ile322 (linker), and Leu444, Ala447 and Ile448 in α4 (CA), that is flanked on each side by hydrophilic residues exposing the aliphatic portion of their side chains to this cluster. Those at one side, Arg369 and Gln372 from α4 and Asp311 in α2, are hydrogen bonded together in a unique triple residue interaction. The second region is characterized by the projection of one of the conserved F box phenylalanines (Phe428) towards the α1 helix, where it is accommodated in a hydrophobic pocket formed by Ile247, Met250, Phe254 and the aliphatic portion of the Lys251 side chain. The two buried areas are connected through an interaction between Phe254 in α1 and Phe312 in α2.
Mutational tests for functional relevance of interdomain contacts
The structure invites the hypothesis that contacts seen between DHp and CA domains may support a labile association, under control of the sensor domain, to be released for autophosphorylation and maintained for phosphatase and phosphotransferase activities. To examine this hypothesis, we designed a series of 15 mutant variants, incorporating mutations at seven sites, and tested them for functional relevance in our autokinase assay. These mutations are in three classes: changes at interfacial hydrophobic residues, introductions of candidates for disulfide formation and proline substitutions in the interdomain linker segment. Figure 7 compares the kinetics of autophosphorylation with wild-type activity for several of these variants, and Supplementary Table SII records the initial rates for all. Most of the mutations affect autokinase activity significantly, usually in the initial rate of phosphorylation and also, often differentially, in the achieved or projected equilibrium level. Equilibrium levels may reflect differences in rates of counteracting intrinsic dephosphorylation, so we concentrate our analysis on the initial rates.
Figure 7.
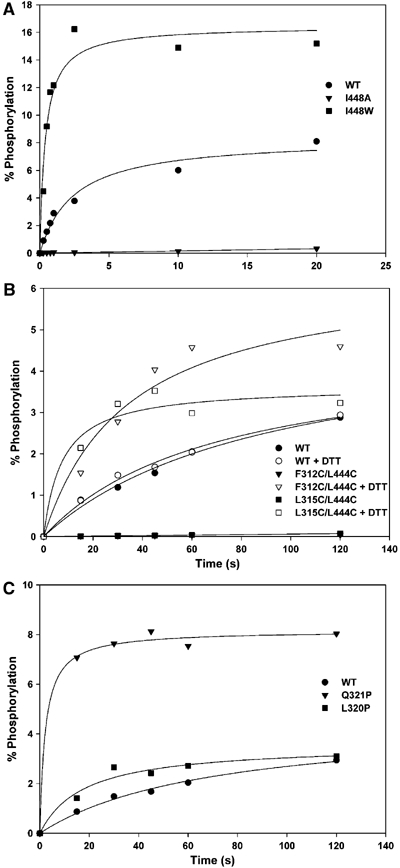
Autokinase activity of interfacial mutant variants. (A) Mutations at the interfacial residue Ile448. (B) Double-cysteine mutations when reduced by DTT (open symbols) and when oxidized (filled symbols), where the negligible activity is overlapping. (C) Mutations to proline in the linker segment.
The mutational analysis provides compelling evidence for the importance of this interface in controlling autokinase activity. Activity is sensitive to mutation at all tested interfacial hydrophobic residues (Supplementary Table SII) and most strikingly so at Ile448 (Figure 7A); I448A had a six-fold rate increase, but I448W had negligible activity despite good solution properties. Activities for variants L315W and F428E were also strongly affected (∼3-fold increases). The double cysteine mutations, F312C/L444C and L315C/L444C, further corroborate the functional significance of the interface (Figure 7B). There is no activity under conditions conducive to disulfide coupling of the domains, but activity increases to rates consistent with single mutations at Leu315 and Leu444 when the cysteines are reduced. The greatest increase of all (>12 fold) came with the conformation-restricting linker mutation L320P (Figure 7C).
Discussion
HKs are multifunctional enzymes that participate in autokinase, phosphotransferase and phosphatase reactions, and their phospho-accepting histidines typically have roles in all three activities (Hsing et al, 1998). The kinase and/or phosphatase activities are regulated by sensor input and are dependent on the presence of nucleotide (reviewed in Stock et al, 2000). These observations, together with the large separation between ATP and the phosphoacceptor histidine in this structure of HK853-CD, suggest that the cytoplasmic portions of HK sensor must access multiple conformational states, some of which are critical for catalytic action.
We present a structure-based scheme for the multiple activities of HKs in Figure 8. This scheme is based on the structure in the state found here (Figure 3) and on models for other relevant states (Figure 9). In unphosphorylated state A, a kinase domain loaded with ATP is poised to phosphorylate the acceptor histidine. Upon sensor input, if needed, this kinase domain is freed to adopt a conformation appropriate for forming the A–B transition state with the phosphoacceptor histidine on the opposite protomer. In phosphorylated state B, the kinase domain is relaxed to a conformation whereby the site surrounding the phosphorylated histidine is available for interaction in transition state B–A* with a cognate RR. Phosphotransfer to the acceptor aspartyl residue can then ensue. In unphosphorylated state A*, which may depend on sensor input, the interdomain conformation permits interaction with a cognate phosphorylated regulatory domain in the A*–A intermediate, and this accelerates aspartate dephosphorylation.
Figure 8.
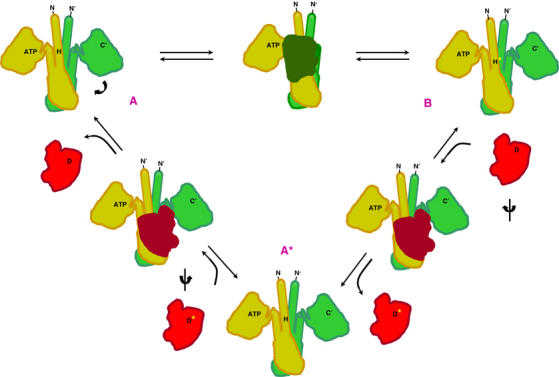
Structure-based schematic of the reactions catalyzed by HK sensors. The kinase autophosphorylation (A → B), phosphotransferase (B → A*) and phosphatase (A* → A) activities are shown on projected outlines of the enzyme and protein–substrate models. Positions of N and C termini, ATP and the phospho-accepting histidine (H) are indicated on an HK dimer (orange and green). Position of phospho-accepting aspartate (D) is indicated on a RR (red). The transferred phosphoryl group is indicated as a yellow asterisk.
Figure 9.
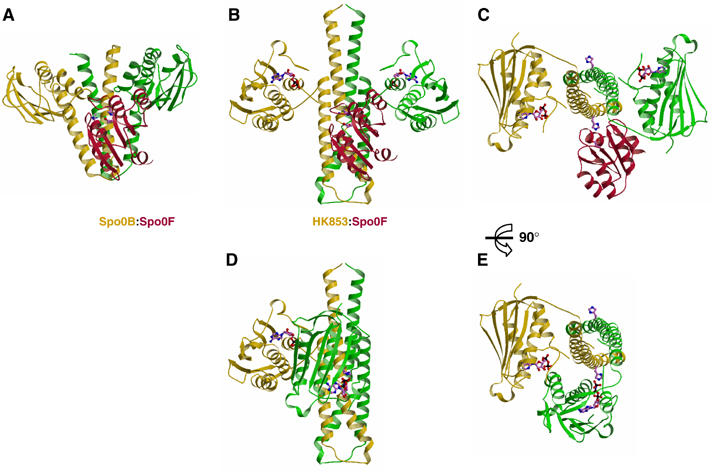
Models of complexes for the phosphotransferase and kinase reactions catalyzed by HK853-CD. (A) Ribbon representation of the experimental complex (Zapf et al, 2000) between Spo0B (green and yellow) and Spo0F (red). For clarity, only one Spo0F molecule is drawn. (B) The Spo0F RR (red) docked onto the HK853–CD dimer (green and yellow) as a model of the phosphotransferase complex (see text). The catalytic histidine and aspartate residues and the nucleotide are shown as stick models. (C) Orthogonal view of (B). (D) Model of HK853–CD poised for the autokinase reaction. The catalytic domain of one protomer has been moved to align the γ phosphate of its ATP moiety with the phosphoaccepting histidine of the other promoter to permint trans-phosphorylation. The histidine and nucleotide are shown in stick representation. (E) Orthogonal view of (D).
The scheme depicted in Figure 8 has symmetric ground states and asymmetric intermediate structures, but our modeling (Figure 9) is also consistent with symmetric intermediates where both phospho-accepting histidines are equivalently engaged. Asymmetry is observed in the auto-phosphorylation and phosphatase reactions of NtrB (Jiang et al, 2000; Pioszak and Ninfa, 2003), but both EnvZ–OmpR and FixL–FixJ form 2:2 HK–RR phosphotransfer complexes (Miyatake et al, 1999; Yoshida et al, 2002).
Implications of the HK853-CD structure for catalysis and regulation
Our crystal structure of HK853-CD has attributes appropriate for a model of class I HK sensors in all ground states (A, B and A*) and for intermediates in the phosphotransfer and phosphatase reactions. It is, however, obviously inappropriate for the autokinase reaction since the imidazole of His260 and the β-phosphate position of the nucleotide are separated by 25 Å and wrongly oriented for interaction. How then might this crystal structure relate to the states depicted in the cycle of catalytic reactions depicted in Figure 8? Two features of the structure seem relevant in this regard. First, although this is an unphosphorylated protein molecule, we propose that the ordered sulfate ion near the phosphorylatable histidine mimics the phosphate group in the phosphohistididyl protein. This sulfate ion is hydrogen bonded to the Nɛ atom of His260 and also to side chains of conservative Arg317′ and other groups from the opposing protomer. In that sense the structure may represent a model for ground state B, which catalyzes phosphotransfer to the RR.
The second structural feature relevant to the reaction scheme is concerned with the interdomain contacts. The C-terminal half of helix α2 is a central participant in this interaction, and the corresponding sequence is part of the weakly conserved X motif identified in a mutational analysis of EnvZ (Hsing et al, 1998). Most mutations that eliminate phosphatase activity without diminishing kinase activity (K+P−) mapped to this region. The position of Tyr287 in EnvZ (Leu315 in HK853) seems especially critical since it was affected in multiple isolates from the mutational screen. Leu315 has a central place in the interdomain interface (Figure 6), making contacts with catalytic domain residues Leu444, Ala447 and Ile448, consistent with a pivotal role in stabilizing phosphatase state A*. Mutations at nearby EnvZ residues, including Arg289 (sulfate ligand Arg317 in HK853), also confer a K+P− phenotype. The X motif mutation L288P had no effect on the residual phosphatase activity of the isolated DHp domain from EnvZ (Zhu et al, 2000), however, consistent with the dependence of regulated phosphatase action on interdomain contacts. Key residues from the catalytic-domain side of the interface, including Phe428 and Leu444, are near the nucleotide within the ATP-lid segment (Figure 6), consistent with NtrB mutations (Pioszak and Ninfa, 2003) and the dependence of phosphatase activity on the presence of nucleotide (Keener and Kustu, 1988; Jung and Altendorf, 1998).
We expect that regulatory signals act to control interfacial stability. Signals from the external sensor domain, presumably transduced through the coiled-coil segment to the four-helix bundle, must ultimately affect the viability of the interface, destabilizing it for kinase action and stabilizing it for phosphatase action. This interface clearly must give way to permit the catalytic domain to swing around and perform the trans-histidine phosphorylation. Indeed, our mutational disruptions of the interface do accelerate autokinase activity, whereas activity is blocked upon interface stabilization. Signal-dependent conformational changes within the four-helix bundle may further distinguish states A, B and A*.
Model for the autophosphorylation reaction of HK853-CD
Since the conformation of HK853-CD in our crystals is inappropriate for the autokinase reaction, we have modeled the disposition of domains needed for the autophosphorylation reaction. This was done by docking an isolated catalytic domain (residues 322–489) onto dimeric DHp domains (residues 232–317) in a manner consistent with phosphotransfer from ATP to histidine, and then considering constraints imposed in reconnecting the linker peptide (residues 317–322). ATP was modeled into the catalytic domain by adding γ-phosphate to ADPβN as in the AMPPNP of CheA (Bilwes et al, 2001), and the Oβ1–Pγ bond of this ATP was aligned with the Cβ–Cγ bond of His260 in the DHp domain with the catalytic domain, separated such that the Oβ1(ATP)–Nɛ2(His260) distance would be appropriate for the kinase transition state (∼4.5 Å). Such models were constructed for His260 in each of the three major conformations for this residue, m, t and p (χ1 at minus 60° (49% abundance), trans (32%) and plus 60° (13%) following Lovell et al, 2000), and for rigid-body rotations at 10° intervals about the axis defined by Oβ1(ATP)–Cβ(His260). Each docking was tested for cis and trans connectivity between domains.
All models generated in the m histidine conformation of the crystal structure have serious steric clashes or present infeasible linkages. Models without steric problems can be generated in the p conformation for both cis- and trans-autophosphorylation reactions, but this conformation is unfavored. His260 is most exposed when in the t conformation, which is as observed in the Spo0B–Spo0F complex (Zapf et al, 2000). A small range of t models can be connected in favorable linker peptide conformations for the trans mode of reaction. One of these, presenting optimal shape complementarity free of steric conflicts, is shown in Figure 9. Conformational changes to achieve this model are in linker residues 317–320 and, in keeping with this, the conformation-restricted mutant L320P is hyperactive.
Model for the interaction of HK853-CD with its RR
In the current absence of structural information on complexes between DHp and RR domains, we have modeled this interaction for HK853 by analogy with the cognate complex between Spo0B and Spo0F, phosphorelay components for sporulation in B. subtilis (Zapf et al, 2000). Spo0B is evolutionarily and structurally (Figure 4) related to HKs, and it can freely transfer a phosphoryl group to or from the intermediate RR Spo0F. It does this via a histidine residue that corresponds to the phosphoacceptor histidine in the kinase homologs, and this residue is in proximity with the phosphorylatable aspartate in the Spo0B–Spo0F complex. We constructed an HK853–RR complex by superimposing the phospho-accepting histidine helices of Spo0B and HK853–CD, thereby orienting Spo0F into a hypothetical phosphotransfer complex with HK853 (Figure 9).
This Spo0B–Spo0F-based model has properties appropriate for the HK853–RR complex. HK853–CD readily accommodates Spo0F with only minor clashes and with burial of substantial surface into the interface (2250 Å2 in total). The most significant steric clash is between the sensitive X region of HK853–CD, discussed above, and the loop between β4 and α4 of Spo0F (Figure 9C), which undergoes structural changes upon RR activation (Birck et al, 1999; Lewis et al, 1999). The surface of HK853–CD that is buried into the phosphotransfer interface covers much of helix α1b downstream of the phospho-accepting histidine (Figure 9B), consistent with NMR titration experiments of the OmpR receiver domain interacting with the isolated EnvZ DHp domain (Tomomori et al, 1999). Finally, although His260 is poorly oriented for phosphotransfer in its HK853-CD m conformation, a productive t conformation similar to that observed in the Spo0B–Spo0F complex (Zapf et al, 2000) is achieved by a simple χ1 rotation, whereby Nɛ of His260 comes to lie an appropriate 5.1 Å from an Oδ of Asp54 in Spo0F.
Implications of the coiled-coil region for signal transduction
The coiled-coil portion of HK853-CD (residues 232–253 of helix α1a) extends into the DHp domain and we expect it to emerge directly from the transmembrane, four-helix bundle of this sensor protein. It must thereby play a role in signal transduction. Comparable coiled-coil segments are predicted to exist in this and other HK sensors (Figure 1); indeed, Singh et al (1998) identified putative coiled-coil helical structures preceding the phospho-accepting histidine in 76% of 189 class I HKs. The HAMP domain linkers that typically connect the transmembrane domains of these and other sensors to their catalytic domains have been predicted to consist of two amphipathic helices separated by a loop region (Butler and Falke, 1998; Williams and Stewart, 1999). The linker segment sometimes includes whole additional domains, such as the cysteine-cluster domain of NarX (Stewart, 2003) and the PAS domain of DcuS (Golby et al, 1999). Typically, the second HAMP helix corresponds to the coiled-coil segment found in HK853–CD and predicted for most others. The HAMP segment of chemotactic receptors is stably folded (Butler and Falke, 1998) and has been modeled as a continuous coiled coil that also extends through the membrane and into the periplasmic sensor domain (Kim et al, 1999; Falke and Hazelbauer, 2001). This is analogous to the topology in HK853 and may be typical, perhaps with extra loops and domains bulging as gall-like extrusions from the coiled-coil stem.
The coiled coil in HK853–CD has hydrophilic residues occupying several of the normally hydrophobic a and d contact positions of the canonical heptad repeats (Figures 1 and 4B), and the predicted coiled coils in related sensor proteins also show a weak hydrophobic character of the a–d core (Tao et al, 2002). This property may confer an interfacial plasticity of importance in signal transmission. Mutations that increase the hydrophobicity of the predicted coiled-coil core tend to bias various HKs toward one or the other signaling state (Kalman and Gunsalus, 1990; Tokishita et al, 1992; Tao et al, 2002). Included among these are substitutions and short deletions in the HAMP domain of EnvZ that alter the ratio of kinase-to-phosphatase activity (Park and Inouye, 1997).
Signal transduction by class I HK sensors begins with changes in the sensor domains induced by ligands or other stimuli. Conformational changes are transduced through the transmembrane four-helix bundle into the cytoplasmic domain of the dimeric receptor. These changes ultimately affect the kinase and/or phosphatase activities mediated by the catalytic domains. Two transduction models have been proposed: (i) a rotational movement of the helices with respect to one another (Cochran and Kim, 1996) and (ii) a piston-like movement of one or two helices with respect to the other helices in the bundle (reviewed by Falke and Hazelbauer, 2001), favored by the preponderance of evidence from chemotactic receptors. The coiled-coil linker domains may serve to modulate and perhaps amplify these movements, but in any case they must transmit the signal. It is apparent from the structure and mutational analysis of HK853–CD that even subtle changes could affect the latch between the helical-hairpin and kinase domains and the disposition of the phospho-accepting histidine residue. Just as signals transduced through coiled coils from across the membrane may effect these changes in class I kinases, regulatory factors that modulate other kinases may act on comparable interfaces.
Materials and methods
Cloning and protein production
ORF TM0853 was cloned from genomic T. maritima DNA for recombinant expression in E. coli. Vectors were designed to produce the putative sensor HK, both histidine-tagged in full length for localization assays (His-HK853), and as the predicted cytoplasmic domain (residues 232–489) for structure analysis (HK853–CD). Plasmid pHK853 was constructed by cloning TM803 into a pBR322-derived plasmid designed for in vivo phosphotransfer assays (Regelmann et al, 2002). Mutant variants of HK853–CD were designed for a battery of functional tests and to add methionine residues for Se MAD phasing. HK853–CD was purified by ammonium sulfate fractionation, ion-exchange chromatography and size exclusion chromatography. Predicted size and complete Se incorporation were confirmed by mass spectrometry.
Cellular and biochemical characterization
Cellular localization of the full-length sensor kinase was determined, as a function of temperature, by ultracentrifugal separation of soluble and membrane fractions followed by pulldown on His-HK853 on Ni-chelating beads and staining on SDS–PAGE gels. Autokinase activity was assayed, as described before (Marina et al, 2001), by following the incorporation of radiolabel from [γ-32P]ATP into purified wild-type or mutant HK853–CD as separated on SDS–PAGE gels. Phosphotransfer to PhoP or OmpR in vivo was assayed by measuring β-galactosidase activity from E. coli strains harboring PhoP-activated phoN-lacZ (Waldburger and Sauer, 1996) or OmpR-activated ompC-lacZ (Hsing and Silhavy, 1997) fusions, respectively, after transformation by pHK853 or control plasmids. Nucleotides were identified by FPLC separations from washed crystals or from nucleotide hydrolysis reactions in comparison with retention times of various adenosine nucleotides.
Crystallographic analysis
Crystals were grown from 1.25 M Li2SO4 and 50–200 mM ammonium acetate at pH 6.5. They are in space group C2221 with unit cell dimensions a=79.3 Å, b=162.1 Å and c=42.5 Å. Cryopreservation was achieved in mother liquor plus 7.5% ethylene glycol and 15% sucrose. The structure was solved at 2.1 Å resolution from a MAD experiment based on selenomethionyl I370M/V373M HK853–CD and refined at 1.9 Å resolution against wild-type native HK853–CD. All diffraction data were measured at NSLS beamline X4A. Results are deposited with PDB accession code 2C2A.
Supplementary Material
Supplementary Material
Acknowledgments
We thank C Mott for assistance in plasmid production, J Escolano and I Esmorís for help in purifying mutant proteins, and members of the Hendrickson and Waldburger laboratories for helpful discussions. This work was supported in part by NIH grants GM34102 (WAH) and AI41566 (CDW), and by Ministerio de Ciencia y Tecnología grant BIO2002-03709 (AM) in Spain. Beamline X4A at the National Synchrotron Light Source (NSLS), a DOE facility, is supported by the New York Structural Biology Center.
References
- Aravind L, Ponting CP (1999) The cytoplasmic helical linker domain of receptor histidine kinase and methyl-accepting proteins is common to many prokaryotic signalling proteins. FEMS Microbiol Lett 176: 111–116 [DOI] [PubMed] [Google Scholar]
- Bilwes AM, Alex LA, Crane BR, Simon MI (1999) Structure of CheA, a signal-transducing histidine kinase. Cell 96: 131–141 [DOI] [PubMed] [Google Scholar]
- Bilwes AM, Quezada CM, Croal LR, Crane BR, Simon MI (2001) Nucleotide binding by the histidine kinase CheA. Nat Struct Biol 8: 353–360 [DOI] [PubMed] [Google Scholar]
- Birck C, Mourey L, Gouet P, Fabry B, Schumacher J, Rousseau P, Kahn D, Samama JP (1999) Conformational changes induced by phosphorylation of the FixJ receiver domain. Struct Fold Des 7: 1505–1515 [DOI] [PubMed] [Google Scholar]
- Butler SL, Falke JJ (1998) Cysteine and disulfite scanning reveals two amphiphilic helices in the linker region of the aspartate chemoreceptor. Biochemistry 37: 10746–10756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou K, Maggiora GM, Nemethy G, Scheraga HA (1988) Energetics of the structure of the four-α-helix bundle in proteins. Proc Natl Acad Sci USA 85: 4295–4299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochran AG, Kim PS (1996) Imitation of Escherichia coli aspartate receptor signaling in engineered dimers of the cytoplasmic domain. Science 271: 1113–1116 [DOI] [PubMed] [Google Scholar]
- Cserzo M, Wallin E, Simon I, von Heijne G, Elofsson A (1997) Prediction of transmembrane alpha-helices in procariotic membrane proteins: the Dense Alignment Surface method. Prot Eng 10: 673–676 [DOI] [PubMed] [Google Scholar]
- Dutta R, Inouye M (2000) GHKL, an emergent ATPase/kinase superfamily. Trends Biochem Sci 25: 24–28 [DOI] [PubMed] [Google Scholar]
- Dutta R, Qin L, Inouye M (1999) Histidine kinase: diversity of domain organization. Mol Microbiol 34: 633–640 [DOI] [PubMed] [Google Scholar]
- Fabret C, Feher VA, Hoch JA (1999) Two-component signal transduction in Bacillus subtilis: how one organism sees its world. J Bacteriol 181: 1975–1983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falke JJ, Hazelbauer GL (2001) Transmembrane signaling in bacterial chemoreceptors. Trends Biochem Sci 26: 257–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golby P, Davies S, Kelly DJ, Guest JR, Andrews SC (1999) Identification and characterization of a two-component sensor-kinase and response-regulator system (DcuS–DcuR) controlling gene expression in response to C-4-dicarboxylates in Escherichia coli. J Bacteriol 181: 1238–1248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsing W, Russo FD, Bernd KK, Silhavy TJ (1998) Mutations that alter the kinase and phosphatase activities of the two-component sensor EnvZ. J Bacteriol 180: 4538–4546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsing W, Silhavy TJ (1997) Function of conserved histidine-243 in phosphatase activity of EnvZ, the sensor of porin osmoregulation in Escherichia coli. J Bacteriol 179: 3729–3735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hultquist DE, Moyer RW, Boyer PD (1996) The preparation and characterization of 1-phosphohistidine and 3-phosphohistidine. Biochemistry 5: 322–331 [DOI] [PubMed] [Google Scholar]
- Jiang P, Peliska JA, Ninfa AJ (2000) Asymmetry in the autophosphorylation of the two-component regulatory system transmitter protein nitrogen regulator II of Escherichia coli. Biochemistry 39: 5057–5065 [DOI] [PubMed] [Google Scholar]
- Jung K, Altendorf K (1998) Truncation of amino acids 12–128 causes deregulation of the phosphatase activity of the sensor kinase KdpD of Escherichia coli. J Biol Chem 273: 17406–17410 [DOI] [PubMed] [Google Scholar]
- Kalman LV, Gunsalus RP (1990) Nitrate-independent and molybdenum-independent signal transducction mutations in narX that alter regulation of anaerobic respiratory genes in Escherichia coli. J Bacteriol 172: 7049–7056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keener J, Kustu S (1988) Protein kinase and phosphoprotein phosphatase activities of nitrogen regulatory proteins NTRB and NTRC of enteric bacteria: roles of the conserved amino-terminal domain of NTRC. Proc Natl Acad Sci USA 85: 4976–4980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KK, Yokota H, Kim SH (1999) Four-helical-bundle structure of the cytoplasmic domain of a serine chemotaxis receptor. Nature 400: 787–792 [DOI] [PubMed] [Google Scholar]
- Lewis RJ, Barnnigan JA, Muchova K, Barak I, Wilkinson AJ (1999) Phosphorylated aspartate in the structure of a response regulator protein. J Mol Biol 294: 9–15 [DOI] [PubMed] [Google Scholar]
- Lovell SC, Word JM, Richardson JS, Richardson DC. (2000) The penultimate rotamer library. Proteins 40: 389–408 [PubMed] [Google Scholar]
- Marina A, Mott C, Auyzenber A, Hendrickson WA, Waldburger CD (2001) Structural and mutational analysis of PhoQ histidine kinase catalytic domain. J Biol Chem 276: 41182–41190 [DOI] [PubMed] [Google Scholar]
- Miyatake H, Mukai M, Adachi S, Nakamura H, Tamura K, Iizuka T, Shiro Y, Strange RW, Hasnain SS (1999) Iron coordination structures of oxygen sensor FixL characterized by Fe K-edge extended x-ray absorption fine structure and resonance Raman spectroscopy. J Biol Chem 274: 23176–23184 [DOI] [PubMed] [Google Scholar]
- Ninfa EG, Atkinson MR, Kamberov ES, Ninfa AJ (1993) Mechanism of autophosphorylation of Escherichia coli nitrogen regulator II (NRII or NtrB): trans-phosphorylation between subunits. J Bacteriol 175: 7024–7032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, Inouye M (1997) Mutational analysis of the linker region of the EnvZ, an osmosensor in Escherichia coli. J Bacteriol 179: 4382–4390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkinson JS, Kofoid EC (1992) Communication modules in bacterial signaling proteins. Annu Rev Genet 26: 71–112 [DOI] [PubMed] [Google Scholar]
- Pioszak AA, Ninfa AJ (2003) Mechanism of the PII-activated phosphatase activity of Escherichia coli. NRII (NtrB): how the different domains of NRII collaborate to act as a phosphatase. Biochemistry 42: 8885–8899 [DOI] [PubMed] [Google Scholar]
- Qin L, Dutta R, Kurokawa H, Ikura M, Inouye M (2000) A monomeric histidine kinase derived from EnvZ, an Escherichia coli osmosensor. Mol Microbiol 36: 24–32 [DOI] [PubMed] [Google Scholar]
- Regelmann AG, Lesley JA, Mott C, Stokes L, Waldburger CD (2002) Mutational analysis of the Escherichia coli PhoQ sensor kinase: differences with the Salmonella enterica serovar Typhimurium PhoQ protein and in the mechanism of Mg2+ and Ca2+ sensing. J Bacteriol 184: 5468–5478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo FD, Silhavy TJ (1993) The essential tension: opposed reactions in bacterial two-component regulatory systems. Trends Microbiol 1: 306–310 [DOI] [PubMed] [Google Scholar]
- Singh M, Berger B, Kim PS, Berger J, Cochran A (1998) Computational learning reveals coiled coil-like motifs in histidine kinase linker domains. Proc Natl Acad Sci USA 95: 2738–2743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, Peisach D, Pioszak AA, Xu Z, Ninfa AJ (2004) Crystal structure of the C-terminal domain of the two-component system transmitter protein nitrogen regulator II (NRII; NtrB), regulator of nitrogen assimilation in Escherichia coli. Biochemistry 43: 6670–6678 [DOI] [PubMed] [Google Scholar]
- Stewart V (2003) Nitrate- and nitrite-responsive sensors NarX and NarQ of proteobacteria. Biochem Soc Trans 31: 1–10 [DOI] [PubMed] [Google Scholar]
- Stock AM, Robinson VL, Goudreau PN (2000) Two component signal transduction. Annu Rev Biochem 69: 183–215 [DOI] [PubMed] [Google Scholar]
- Tanaka T, Kawata M, Mukai K (1991) Altered phosphorylation of Bacillus subtilis DegU caused by single amino acid changes in DegS. J Bacteriol 173: 5507–5515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Saha SK, Tomomori C, Ishima R, Liu D, Tong KI, Park H, Dutta R, Swindells MB, Yamazaki T, Ono AM, Kainosho M, Inouye M, Ikura M (1998) NMR structure of the histidine kinase domain of the E. coli osmosensor EnvZ. Nature 396: 88–92 [DOI] [PubMed] [Google Scholar]
- Tao W, Malone CL, Ault AD, Deschenes RJ, Fassler JS (2002) A cytoplasmic coiled-coil domain is required for histidine kinase activity of the yeast osmosensor, SLN1. Mol Microbiol 43: 459–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokishita S, Kojima A, Mizuno T (1992) Transmembranes signal tranduction and osmoregulation in Escherichia coli: functional importance of the transmembrane regions of membrane-located protein kinase EnvZ. J Biochem 111: 707–713 [PubMed] [Google Scholar]
- Tomomori C, Tanaka T, Dutta R, Park H, Saha SK, Zhu Y, Ishima R, Liu D, Tong KI, Kurokawa H, Qian H, Inouye M, Ikura M (1999) Solution structure of the homodimeric core domain of Escherichia coli histidine kinase EnvZ. Nat Struct Biol 6: 729–734 [DOI] [PubMed] [Google Scholar]
- Varughese KI, Madhusudan Zhou XZ, Whiteley JM, Hoch JA (1998) Formation of a novel four-helix bundle and molecular recognition sites by dimerization of a response regulator phosphotransferase. Mol Cell 2: 485–493 [DOI] [PubMed] [Google Scholar]
- Waldburger CD, Sauer RT (1996) Signal detection by the PhoQ sensor-transmitter. Characterization of the sensor domain and a response-impaired mutant that identifies ligand-binding determinants. J Biol Chem 271: 26630–26636 [DOI] [PubMed] [Google Scholar]
- Williams SB, Stewart V (1999) Functional similarities among two-component sensors and methyl-accepting chemotaxis proteins suggest a role for linker region amphipathic helices in transmembrane signal transduction. Mol Microbiol 33: 1093–1102 [DOI] [PubMed] [Google Scholar]
- Yang Y, Inouye M (1991) Intermolecular complementation between two defective mutant signal-transducing receptors of Escherichia coli. Proc Natl Acad Sci USA 88: 11057–11061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida T, Qin L, Inouye M (2002) Formation of the stoichiometric complex of EnvZ, a histidine kinase, with its response regulator, OmpR. Mol Microbiol 46: 1273–1282 [DOI] [PubMed] [Google Scholar]
- Yount RG, Babcock D, Ballantyne W, Ojala D (1971) Adenylyl imidodiphosphate, an adenosine triphosphate analog containing a P--N--P linkage. Biochemistry 10: 2484–2489 [DOI] [PubMed] [Google Scholar]
- Zapf J, Sen U, Madhusudan, Hoch JA, Varughese KI (2000) A transient interaction between two phosphorelay proteins trapped in a crystal lattice reveals the mechanism of molecular recognition and phosphotransfer in signal transduction. Structure 8: 851–862 [DOI] [PubMed] [Google Scholar]
- Zhou H, Dahlquist FW (1997) Phosphotransfer site of the chemotaxis-specific protein kinase CheA as revealed by NMR. Biochemistry 36: 699–710 [DOI] [PubMed] [Google Scholar]
- Zhu Y, Qin L, Yoshida T, Inouye M (2000) Phosphatase activity of histidine kinase EnvZ without kinase catalytic domain. Proc Natl Acad Sci USA 97: 7808–7813 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material