

Abstract
Precise cell cycle regulation is critical for nervous system development. To assess the role of the cell cycle regulator, retinoblastoma (Rb) protein, in forebrain development, we studied mice with telencephalon-specific Rb deletions. We examined the role of Rb in neuronal specification and migration of diverse neuronal populations. Although layer specification occurred at the appropriate time in Rb mutants, migration of early-born cortical neurons was perturbed. Consistent with defects in radial migration, neuronal cell death in Rb mutants specifically affected Cajal–Retzius neurons. In the ventral telencephalon, although calbindin- and Lhx6-expressing cortical neurons were generated at embryonic day 12.5, their tangential migration into the neocortex was dramatically and specifically reduced in the mutant marginal zone. Cell transplantation assays revealed that defects in tangential migration arose owing to a cell-autonomous loss of Rb in migrating interneurons and not because of a defective cortical environment. These results revealed a cell-autonomous role for Rb in regulating the tangential migration of cortical interneurons. Taken together, we reveal a novel requirement for the cell cycle protein, Rb, in the regulation of neuronal migration.
Keywords: cell cycle, neurogenesis, neuronal differentiation, retinoblastoma, tangential migration
Introduction
Cell cycle regulation is essential for cortical neurogenesis, ensuring maintenance of the progenitor cell pool, production of the correct proportions of diverse cell types, and coordination of the timing of neuronal differentiation. During corticogenesis, cortical precursor cells located in the germinal zone of the dorsal telencephalon undergo multiple rounds of proliferation, between embryonic days (E) E10 and E17 (Takahashi et al, 1996). Following cell cycle withdrawal, newly born neurons initiate expression of early neuronal markers and commence migration into the developing cortical plate (CP). The first neurons generated, or the ‘pioneering neurons', are born around E10–11. Pioneering neurons give rise to the preplate, which is then split by subsequent neuronal cohorts into the superficial marginal zone (MZ) and the deeper subplate. The MZ, also known as layer I, is a heterogeneous population of neurons that includes Cajal–Retzius neurons, which provide important guidance cues for neuronal migration during CP formation. Cortical layers (lamina) II–VI form between the MZ and subplate in an inside-out pattern, such that earlier generated neurons reside in deep layers and later-born neurons give rise to more superficial layers (Takahashi et al, 1999). As cortical development proceeds, the subplate becomes separated from the germinal zone by the intermediate zone (IZ), a white matter tract containing afferent and efferent projections (Sidman and Rakic, 1973; Caviness, 1982).
In addition to the excitatory projection neurons that are born in the dorsal telencephalon, interspersed throughout the cortical layers are a population of GABAergic inhibitory interneurons that are generated in the ventral telencephalon and reach the cortex via tangential migration (Anderson et al, 1997; Lavdas et al, 1999; Sussel et al, 1999; Wichterle et al, 1999). Tangentially migrating interneurons follow very specific migratory routes, generally avoiding the developing striatum to form two distinct paths—either superficial to or underlying the striatal mantle (Marin et al, 2001). Superficially migrating neurons do not enter the CP, and instead migrate along the MZ, whereas interneurons following the deeper path travel through the lower IZ and subventricular zone (SVZ) (DeDiego et al, 1994; Lavdas et al, 1999; Denaxa et al, 2001). Once GABAergic interneurons have completed their tangential migration, they switch to a radial mode of migration to enter the CP (Polleux et al, 2002).
The time at which a newly generated neuron undergoes terminal mitosis and exits the cell cycle correlates highly with its eventual laminar fate and neuronal identity (McConnell and Kaznowski, 1991). The cell cycle dependence of laminar specification was best shown by a series of heterochronic transplantation studies in the ferret (McConnell and Kaznowski, 1991). Cells isolated at E29, which would normally give rise to layer VI, were [3H]thymidine-labeled in vitro and transplanted into post-natal hosts, in which layers II/III were currently being generated. It was shown that the majority of precursors transplanted during their S phase switched fates and migrated to layers II/III, thereby adopting the laminar fate appropriate for their new environment. In contrast, neurons that were in later cell cycle stages at the time of transplantation migrated to layer VI, maintaining the laminar identity appropriate for their birth date (McConnell and Kaznowski, 1991). These studies demonstrated that cells receive their environmental cues for correct laminar identity during terminal mitosis, and established the importance for proper cell cycle control in cortical development. Because of the strong correlation between neuronal subtype and time of generation, it is believed that precise cell cycle regulation and the determination of neuronal identity are intimately connected.
The retinoblastoma (Rb) protein is a key cell cycle regulator. First discovered as a tumor suppressor, Rb regulates the G1/S phase restriction point, thereby controlling entry into S phase (reviewed in Trimarchi and Lees, 2002). Studies with Rb-deficient embryos were the first to show that Rb has an important role in nervous system development. Rb-null mutants died by mid-gestation (E12–15) with massive apoptosis throughout the liver and nervous system, as well as ectopic mitoses (Clarke et al, 1992; Jacks et al, 1992; Lee et al, 1992, 1994). More recently, we and others have shown that Rb deficiency does not result in large-scale apoptosis in a cell-autonomous manner (Lipinski et al, 2001; Ferguson et al, 2002; MacPherson et al, 2003; Wu et al, 2003). Indeed, in the developing telencephalon, Rb deficiency is fully compatible with survival of the majority of neuronal populations (Ferguson et al, 2002). In contrast, Rb regulation of cell proliferation is cell-autonomous because telencephalon-specific Rb-deficient mutants exhibit ectopic cell divisions outside the germinal zones (Ferguson et al, 2002).
In this study, we examined the impact of deregulated cell cycle regulation resulting from the loss of Rb function in the developing telencephalon. We demonstrate that, despite defective exit from the cell cycle, Rb mutants appear to generate and specify diverse neuronal populations at the appropriate developmental time. Neuronal birthdating experiments, however, reveal that Rb mutants exhibit defective laminar patterning and impaired radial migration. Finally, by slice co-culture assays, we reveal a cell-autonomous defect in tangential migration. The results of our studies reveal a novel role for the tumor suppressor protein, Rb, in the regulation of neuronal migration during development.
Results
Telencephalon-specific Rb-deficient progenitor cells undergo ectopic cell divisions
In our initial examination of telencephalon-specific Rb knockouts, we demonstrated that these mutants retained the ectopic mitoses phenotype characteristic of Rb germline knockouts (Ferguson et al, 2002; Supplementary Figure 1B). Confocal analyses showing BrdU and βIII-tubulin co-labeling have previously identified the ectopically dividing cells as early neuroblasts, suggesting the ability to cycle after the initiation of neuronal differentiation (Ferguson et al, 2002). Here, we sought to examine the impact of deregulated cell cycle regulation resulting from Rb deficiency on laminar patterning, the timing of neuronal differentiation, and the regulation of distinct neuronal populations.
Laminar patterning is perturbed in the absence of Rb
As the precise timing of cell cycle exit is believed to be critical for proper generation of cortical layers (McConnell and Kaznowski, 1991; Takahashi et al, 1999), we questioned whether conditional Rb mutants might display defective laminar patterning. To ask whether layers were appropriately generated in the absence of Rb function, we first assessed the expression of layer-specific markers by in situ hybridization (Figure 1). Sections were examined at E15.5 to coincide with the peak occurrence of ectopic mitoses. At E15.5, only neurons in the deepest cortical layers, V and VI, have differentiated, whereas layer IV neurons are in the process of being born and layer II–III neurons are just beginning to withdraw from the cell cycle (Takahashi et al, 1999). Tbr1, a T-box transcription factor, is highly expressed in postmitotic glutamatergic projection neurons in layer VI (Rubenstein et al, 1999). We noted that Tbr1 was expressed in the Rb mutant CP at E15.5 but, unlike control embryos, strong Tbr1 labeling was also observed within the IZ (Figure 1C and D). CP expression of the pan-neuronal marker, SCG10 (Stein et al, 1988), in Rb mutant cortices was similar to littermate controls, but ectopic SCG10 expression was also detected within the IZ (Figure 1G and H), such that the boundary between the mutant CP and IZ lacked the clear definition observed in control embryos. The more dramatic SCG10 phenotype is likely due to its pan-neuronal expression as opposed to the more restricted expression of Tbr1 to deep-layer neurons. This pattern was further confirmed by in situ hybridization with the layer-restricted markers Id2, RORβ, and Otx1, which also displayed increased IZ expression in Rb mutants (data not shown). These results suggest either a requirement for Rb function in the establishment of a cortical laminar structure or in the specification of layer identities.
Figure 1.
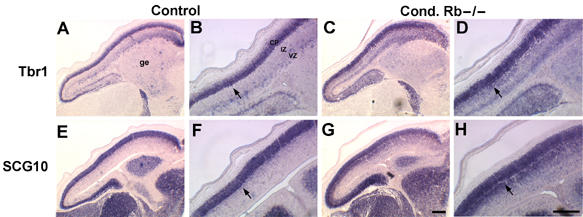
Laminar patterning is perturbed in the absence of Rb. In situ hybridization of E15.5 sagittal sections of mutant and control embryos demonstrates enhanced expression of neuronal markers, Tbr1 and SCG10, in the Rb mutants. Tbr1 labeling is slightly elevated within the mutant CP, and strongly upregulated within the IZ (arrows) (C, D), relative to control (A, B). Similarly, SCG10 expression is highly elevated within the mutant IZ (arrows) (G, H), relative to control (E, F). The boundary between the mutant CP and IZ lacks the clear definition observed in the control embryos (n=4 control; 5 cond. Rb−/−); bar=100 μm, ge=ganglionic eminence, MZ=marginal zone, CP=cortical plate, IZ=intermediate zone.
To distinguish between these possibilities, we examined whether the abnormal lamination in Rb mutants was the result of defective radial migration by conducting neuronal birthdating experiments (Figure 2). Pregnant females were injected with a single dose of BrdU at E13.5 and embryos were dissected at E18.5, when neurogenesis is complete but neuronal migration is still in progress. Examination of brightly labeled BrdU-positive cells revealed the location of neurons that exited the cell cycle at E13.5, as cells that continued to cycle would dilute the BrdU label. Quantification of BrdU-positive cells revealed an aberrant distribution of early-born neurons in the developing cortex of Rb mutants (Figure 2A and B). The number of BrdU-positive cells within the VZ was not significantly different between control and mutant brains. In control sections, an average of 33.3±9.2 BrdU-labeled cells was counted within the IZ as compared to 158.0±14.3 cells in the Rb mutant (Figure 2C; P<0.001). The substantial increase of BrdU-labeled cells within the mutant IZ was compensated for by a corresponding decrease in labeled cells within the CP (480.2±57.9 cells in controls and only 336.9±30.5 neurons in the Rb mutants) (Figure 2C; P<0.05). In contrast, the number of BrdU-positive cells within the VZ was not significantly different between control and mutant brains. Consistent with an increase in IZ cell number, total cell counts of E16.5 embryos revealed significantly increased cell numbers within the Rb mutant IZ (Supplementary Figure 1C–E). These results suggest that many of the Rb-deficient cells failed to reach their correct laminar destination within the cortex.
Figure 2.
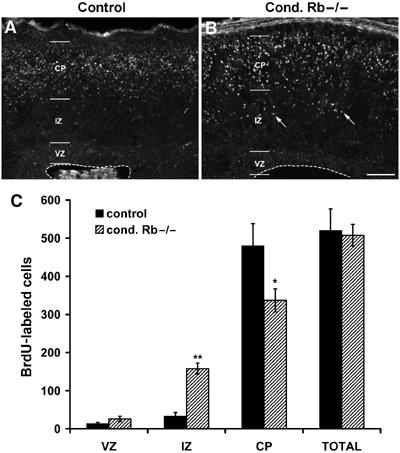
Rb-deficient cortical neurons exhibit delayed radial migration. Pregnant females at E13.5 of gestation were injected with a single dose of 20 μg/g body weight BrdU. Embryos were removed 5 days later at E18.5, fixed, and subjected to immunohistochemistry for BrdU (A, B). BrdU-labeled cells were counted across a 620-μm-wide section of dorsal cortex. In contrast to the control sections with only 33.3±9.2 cells, 158±14.3 labeled cells were counted within the IZ of Rb mutants, representing an almost five-fold increase (arrows) (C; P<0.001). This dramatic increase was compensated for by a corresponding decrease of labeled cells that had reached the CP. The controls had 480.2±57.9 cells within the CP compared to only 336.9±30.5 in the Rb mutants (C; P<0.05). These results indicate that although similar numbers of neurons were generated at E13.5 in control and mutant cortices, Rb-deficient cortical neurons are delayed in reaching their ultimate position within the CP. Error denotes standard error (n=3 control; n=4 cond. Rb−/−). Bar: 50 μm. MZ: marginal zone; CP: cortical plate; IZ: intermediate zone; VZ: ventricular zone.
Reduced number of Cajal–Retzius neurons in the Rb mutant MZ
We next examined whether there were defects in the generation or survival of layer I Cajal–Retzius cells, pioneering neurons born between E11 and E12 that synthesize and secrete Reelin, and which are essential to guide newly generated cortical neurons along radial glial fibers (Frotscher, 1998; Sarnat and Flores-Sarnat, 2002). To assess Cajal–Retzius cell number, immunohistochemistry with a Reelin (G10) antibody was performed on mutant and control E12.5 and E16.5 embryos (Figure 3). At E12.5, the number of Reelin-expressing cells in the cortical MZ was similar between mutant and control embryos (Figure 3A and D), with 126.2±12.3 cells in the control and 126.0±17.8 cells in the Rb mutant MZ (Figure 3G). In addition, total MZ cell counts at E13.5 demonstrated similar cell numbers between control and mutant littermates (Figure 3H). In contrast, at E16.5, quantification of Reelin-positive cells revealed a dramatic 50% decrease in Cajal–Retzius neurons in the mutant MZ, with an average of 143.8±11.8 cells in the controls and 67.5±6.9 cells in Rb mutants (Figure 3B, E, and I; P<0.001). To confirm that reduced Reelin labeling was due to a decrease in positive cells and not merely a downregulation of Reelin protein, we counted the total cell number within the MZ. Consistent with Reelin immunohistochemistry, total cell counts revealed that mutant cortices contained nearly 50% fewer MZ cells compared to control embryos (Figure 3J) (32±6 cells in the control and 18±3 cells in the mutant; P<0.05). These results suggest that Rb deficiency does not negatively impact upon the generation of Cajal–Retzius neurons, but does affect the survival of this specific neuronal population.
Figure 3.
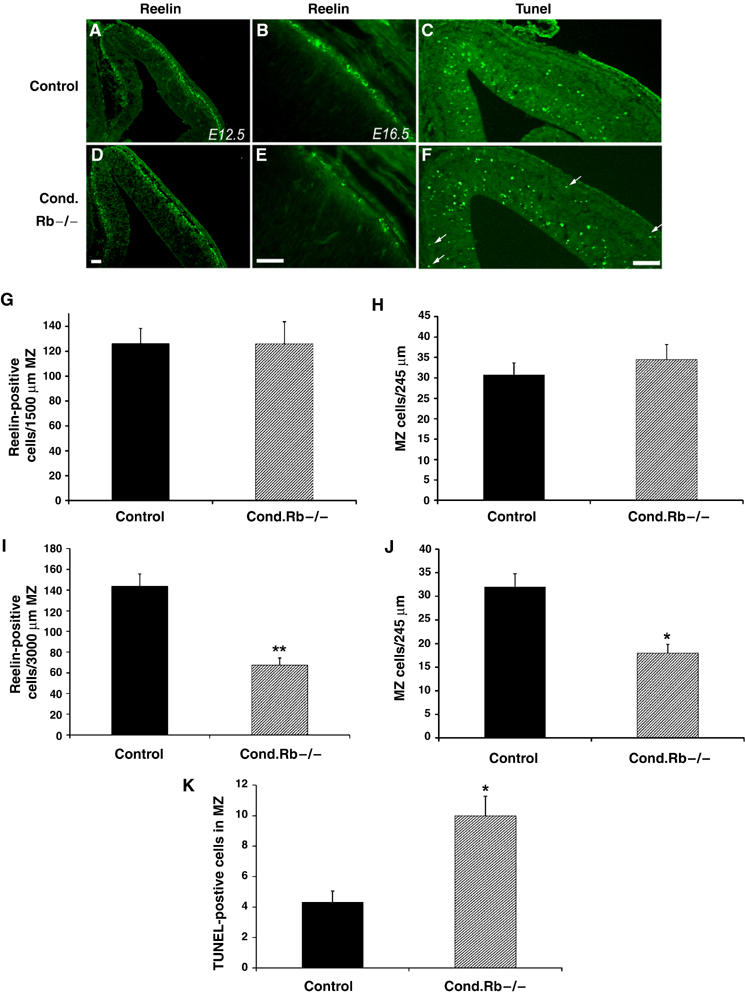
Rb is required for survival of Cajal–Retzius neurons. Coronal sections of control and Rb mutant embryos at E12.5 (A, D) and E16.5 (B, E) were subjected to immunohistochemistry with a Reelin (G10) antibody. Positive cells in each section were quantified along a 1500 μm (E12.5) or 3000 μm (E16.5) length of the MZ. At E12.5, Reelin expression in the cortical MZ appeared similar between mutant and control embryos (A, D, G) (n=3 control; n=3 cond. Rb−/−). Total MZ cells counted within a 245 μm length of the dorsal cortex of E13.5 embryos confirmed similar cell numbers between mutant and controls at this time (H; n=4 control; n=4 cond. Rb−/−). However, by E16.5, Rb mutants contained approximately 50% fewer Reelin-positive neurons as compared to control embryos (B, E, I, P<0.001) (n=5 controls; n=4 cond. Rb−/−). Total MZ cell number quantified within a 245 μm length of the dorsal cortex of E16.5 embryos resulted in a similar reduction in Rb mutants (J, P<0.05) (n=3 control; n=4 cond. Rb−/−). To detect cell death, E13.5 conditional mutant and control littermates were assayed for TUNEL labeling. On each section, positive cells were quantified within the MZ. Rb mutants exhibited significantly increased TUNEL labeling within the MZ (C, F, K, P<0.05; arrows point to representative cells) (n=4 control; n=5 cond. Rb−/−). Error denotes standard error. Bar: 25 μm. MZ: marginal zone.
Although telencephalon-specific Rb deficiency is not associated with the large-scale neuronal death characteristic of Rb germline knockouts, we previously reported a slight increase in TUNEL-positive cells within the mutant telencephalon at E13.5 (Ferguson et al, 2002). As Cajal–Retzius neurons are lost by E16.5 in Rb mutants, we questioned whether this was due to neuronal-specific apoptosis. To test this, we performed TUNEL labeling on mutant and control E13.5 and E16.5 embryos, and quantified positive cells within the germinal zones, IZ, CP, and along the MZ. At E13.5, control and Rb mutant sections contained similar numbers of TUNEL-positive cells within the germinal zones and IZ/CP (Supplementary Figure 2A). In contrast, Rb mutant sections showed elevated apoptosis within the MZ, with an average of 10.0±1.3 TUNEL-positive cells, as compared to 4.3±0.7 cells in control sections, representing a 2.3-fold increase (Figure 3K; P<0.05). At E16.5, TUNEL-positive cells in all quantified regions were not significantly different between mutant and control samples (Supplementary Figure 2B). Therefore, the specific increase in TUNEL labeling at E13.5 within the MZ of Rb mutants suggests a requirement for Rb in the survival of Cajal–Retzius neurons. Owing to the important role of Cajal–Retzius neurons in guiding radial migration of newly born cortical neurons, reduction of these cells by mid-neurogenesis may contribute to the aberrant neuronal migration we observed in Rb mutants.
Rb deficiency does not significantly impact neuronal specification
Owing to the defective terminal mitosis characteristic of Rb deficiency, we questioned whether the generation and/or specification of distinct neuronal populations may be altered in Rb mutants. We first examined the expression of cortical progenitor markers, including Ngn1, Ngn2, Pax6, and Emx1, by in situ hybridization at E15.5 in Rb mutant and control embryos. For all markers tested, no difference was observed between mutant and control sections (data not shown). Similarly, examination of ventral progenitor markers such as Hes1, Hes5, Nkx2.1, and Lhx7 did not reveal any defects in progenitor cell generation or specification in Rb mutants (data not shown). Given that post-mitotic cortical neurons were mislocalized within the Rb mutant cortex (Figure 1), we investigated whether Rb deficiency might also impact upon the positioning of ventrally derived interneurons. To test this, we examined expression of GABAergic interneuron markers early in neurogenesis and at mid-neurogenesis. At earlier time points, we did not observe any differences in the ventral populations with the examined markers, including calbindin (Figure 4A and B), Lhx6 (Figure 4C and D), GAD 65, and GAD 67 (Supplementary Figure 3A–D) in Rb mutant embryos. In contrast, at mid-neurogenesis, the distribution of calbindin and Lhx6 was noticeably perturbed in Rb mutant sections. Immunolabeling revealed that, although calbindin expression appeared normal in the ventral telencephalon, labeling was dramatically reduced in the temporal cortical MZ, to such an extent as to be almost absent (Figure 5B and D). Similarly, in situ hybridization with an Lhx6 riboprobe demonstrated defective expression in mutant embryos (Figure 5E–H). Although expression was similar in mutant and control embryos along the IZ/ SVZ migratory route, there was substantially reduced expression along the cortical MZ (Figure 5H). Furthermore, Lhx6 expression was reduced within the Rb mutant CP, suggesting that regardless of the migratory route, fewer Lhx6-positive interneurons were reaching the CP. In contrast, other interneuron markers were unaltered in Rb mutants, including calretinin, Lhx7, and Nkx2.1 (data not shown).
Figure 4.
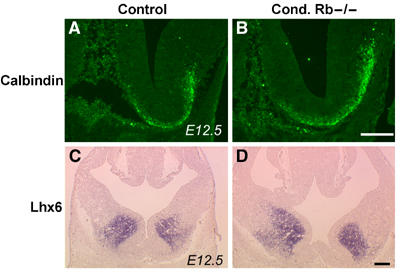
Rb deficiency does not impact interneuron specification or generation. To examine whether interneurons are properly generated in the absence of Rb, mutant and control E12.5 coronal sections were immunolabeled with a calbindin (D-28) antibody or subjected to in situ hybridization with an Lhx6 riboprobe. The generation of calbindin- (A, B) and Lhx6-positive (C, D) progenitors appeared similar in the mutant and control embryos (n=3 control; n=3 cond. Rb−/−). Bar: 50 μm.
Figure 5.
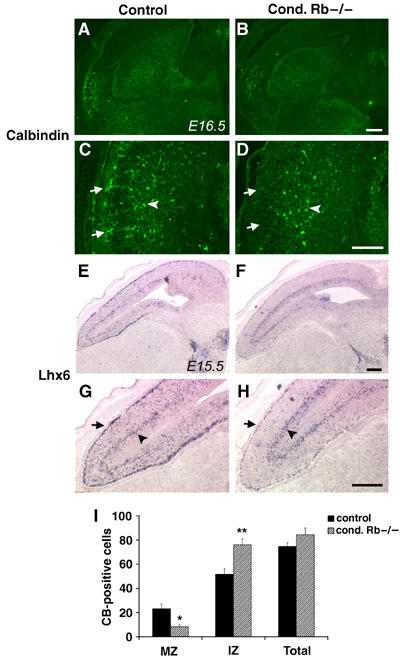
Cortical interneurons are mislocalized in Rb mutants. Rb mutant and control embryo sections were examined at mid-neurogenesis to determine whether specific interneuron populations may be impacted by Rb deficiency. E16.5 sections (coronal) were immunolabeled with a calbindin (D-28) antibody and E15.5 (sagittal) sections were subjected to in situ hybridization with an Lhx6 riboprobe. Although calbindin expression appeared normal in other telencephalic regions (A, B), these cells were dramatically reduced in the mutant MZ (C, D). Similarly, Lhx6 expression was normal along the SVZ/IZ migratory route, but was substantially reduced in the mutant CP and MZ (G, H; n=4 control, n=5 cond. Rb−/− (E15.5); n=5 control, n=5 cond. Rb−/− (E16.5). Calbindin-positive cells were quantified either within the MZ or deeper corresponding to the IZ (arrows point to MZ route; arrowheads denote IZ population). Although the total number of calbindin-positive cells does not differ, there is an approximately 50% reduction in cell number within the Rb mutant MZ (I, P<0.05). The decreased number of calbindin-positive neurons within the Rb mutant MZ is associated with a corresponding increase in these neurons within the mutant IZ (I, P<0.01). Error denotes standard error. Bar: 100 μm (A, B, E–H), 25 μm (C, D). MZ: marginal zone; IZ: intermediate zone.
Possible explanations for the mislocalization of Lhx6- and calbindin-expressing interneurons in the Rb mutant cortex are that they failed to properly migrate along their appropriate trajectories or that these neurons were lost by apoptosis. Calbindin-positive neurons were quantified in mutant and control sections along either the MZ or within the deeper IZ migratory route. Rb mutants showed a substantial 2.8-fold reduction in calbindin-positive neurons in the MZ, with 23.2±3.9 cells in the littermate controls and 8.3±1.7 cells in the mutant MZ (Figure 5I; P<0.05). This reduction in MZ neurons in the mutant appears to be accounted for by an increased IZ population. As compared to control sections with 51.6±4.9 cells within the IZ, Rb mutants exhibited significantly more calbindin-positive neurons along this migratory route with 76.1±5.1 cells (Figure 5I; P<0.01). Although the total number of calbindin-expressing neurons appeared slightly elevated in Rb mutants, the difference was not statistically significant (74.8±3.1 cells in controls and 84.4±5.6 in mutants). These results indicate that the dramatic reduction of these ventrally derived interneurons, specifically along the MZ migratory route, is not due to selective apoptosis, but instead suggests that these tangentially migrating neurons may be diverted from their normal MZ path into the deeper IZ trajectory.
A cell-autonomous requirement for Rb in interneuron migration
Cortical GABAergic interneurons are derived from the ventral ganglionic eminences (Anderson et al, 1997; Lavdas et al, 1999; Sussel et al, 1999; Wichterle et al, 1999). These neurons migrate tangentially through the ventral telencephalon to the cortex along distinct routes: a deep trajectory in the lower IZ/SVZ and a superficial route along the MZ. To determine whether Rb may be required to regulate tangential migration along the MZ route, we performed slice co-culture assays (Figure 6A). Mice were interbred to generate Rb mutants at a 25% frequency, with one parent additionally expressing green fluorescent protein (GFP) such that 50% of embryos would also be GFP-positive. Telencephalons of GFP-negative embryos were sectioned and plated onto coated filter-membrane inserts. Medial ganglionic eminence (MGE) explants were excised from GFP-positive littermates and placed directly on the area of the sections corresponding to the MGE. Co-cultures were grown in vitro for 72 h prior to fixation and immunolabeling for GFP. It has previously been shown that within 72 h, explanted cells will readily integrate into the slice and will migrate up to the dorsal cortex, along the appropriate MZ and IZ trajectories (Polleux et al, 2002). We assessed the migratory routes of GFP-positive cells, specifically focusing on the MZ trajectory, and classified each slice hemisphere into one of three categories: ‘cell integration', ‘MZ stops early', and ‘MZ migration'. ‘Cell integration' refers to the condition in which GFP-positive cells integrated into the section and initiated tangential migration in the appropriate ventrolateral direction; however, these cells failed to follow a distinct migratory route or reach the MZ. This was considered to be the most extreme form of failed migration (Figure 6B, E, and H). The second category, ‘MZ stops early', was considered to be a more moderate failure to migrate along the MZ and occurred when GFP-positive cells were detected in the MZ route, but did not reach the cortex (Figure 6C, F, and I). The third category, ‘MZ migration', included all sections in which a GFP-positive migratory route was observed along the MZ, reaching the dorsal cortex (Figure 6D, G, and J).
Figure 6.
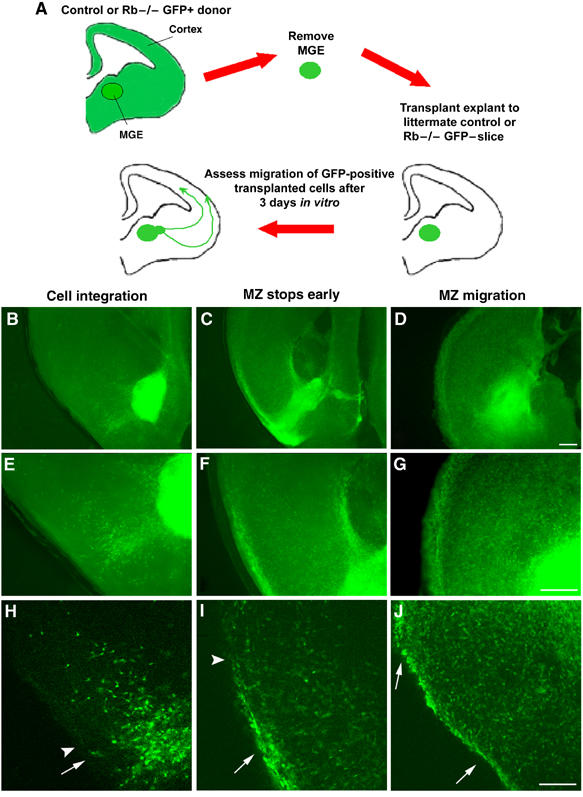
Slice co-cultures. To assess the requirement for Rb function in interneuron migration, we performed slice co-culture assays. (A) GFP-negative telencephalon sections were plated onto coated filter-membrane inserts in a six-well dish. MGE were removed from GFP-positive littermates and equal-sized pieces were placed directly on sections corresponding to the MGE. Co-cultures were grown in vitro for 72 h prior to fixation and GFP immunohistochemistry. The migratory routes of the GFP-positive cells, specifically the MZ trajectory, were analyzed and classified as follows: ‘cell integration', in which GFP-positive cells integrated into the section (arrow) but did not follow a distinct migratory route (arrowhead) or reach the MZ (B, E, H). ‘MZ stops early' occurred when GFP-positive cells formed an MZ route (arrow) but did not reach the dorsal cortex (arrowhead) (C, F, I) ‘MZ migration' included sections in which a GFP-positive MZ migratory route reaching the dorsal cortex was observed (arrows) (D, G, J). Bar: 100 μm (D, G), 50 μm (J). MGE: medial ganglionic eminence; MZ: marginal zone.
In addition to guiding radial migration, Cajal–Retzius neurons have recently been suggested to have a potential role in regulating the tangential migration of interneurons from the ventral telencephalon (Hack et al, 2002; Shinozaki et al, 2002; Morante-Oria et al, 2003; Stoykova et al, 2003). Because Rb mutants exhibit a 50% reduction in Cajal–Retzius neurons, we asked whether tangential migration in Rb mutants may be altered owing to defective environmental cues. To test this, we examined the MZ migratory routes formed by control MGE explants on either control or Rb mutant slices (Figure 7). When control explants were placed on control slices, 93.3% of sections revealed complete MZ migration (28/30), whereas only 7.1% failed to migrate and were classified as ‘cell integration' (2/30). Similarly, cells derived from control MGE explants placed on Rb mutant slices were able to appropriately migrate along the Rb-deficient MZ. Under control explant:mutant slice conditions, GFP cells displayed complete MZ migration, in all sections examined (19/19). These data demonstrate that, despite the reduced number of Cajal–Retzius neurons in the Rb mutants, control MGE-derived interneurons do not exhibit any defects in migrating along the MZ route in the Rb-null cortex.
Figure 7.
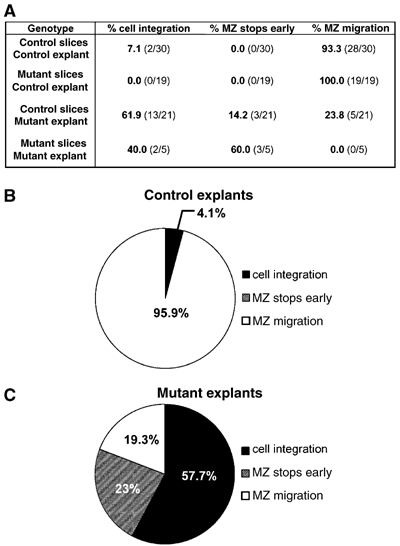
A cell-autonomous requirement for Rb in tangential migration. (A) The MZ migratory routes resulting from GFP-positive MGE explants were compared according to the previously described classifications (Figure 6). Control explants were able to complete MZ migration in 95.9% (47/49) of sections examined, regardless of whether they were placed on mutant or control slices (B). In contrast, only 19.2% (5/26) of mutant explants placed on either mutant or control slices displayed proper MZ migration (C). MZ: marginal zone.
We next questioned whether Rb may be required by migrating neurons in order to properly navigate the MZ migratory route. We compared the MZ migration of GFP cells derived from either control or mutant MGE explants on control slices. Although the large majority of control explants placed on control sections exhibited complete MZ migration (93.3%), Rb-deficient migrating neurons showed a dramatic failure to migrate along the MZ of the control slices. The majority of sections (61.9% or 13/21) exhibited the most severe form of failed migration in the form of ‘cell integration', whereas in 14.2% of sections, the MZ route stopped early (3/21), and only 23.8% of sections showed complete MZ migration (5/21). Furthermore, we examined the migratory capacity of Rb-deficient MGE-derived interneurons on Rb mutant slices. Under these conditions, 40% (2/5) of sections exhibited ‘cell integration', 60% (3/5) exhibited an MZ trajectory that stopped early, and none of the sections underwent complete MZ migration (Figure 7). These results demonstrate that Rb is essential for the MGE-derived interneurons to complete tangential migration along the MZ route. When pooled together, 95.9% (47/49) of sections with control explants showed complete MZ migration, whereas only 19.2% (5/26) of sections with mutant explants displayed proper MZ migration (Figure 7). These data comprise the first evidence indicating a cell-autonomous requirement for the cell cycle protein, Rb, in regulating neuronal migration during forebrain development.
Discussion
In this study, we examined the requirement for the cell cycle regulator, Rb, in telencephalic development. Although Rb mutants produced appropriate numbers of correctly specified cortical projection neurons and interneurons, the radial and tangential migration of these neuronal populations, respectively, were perturbed. In particular, neuronal birthdating and marker analyses revealed that the radial migration of early-born neurons within the cortex was impaired in the absence of Rb. Similarly, an examination of cortical interneuron markers and the use of slice co-culture assays revealed that Rb is essential for normal tangential migration. In conclusion, our results demonstrate a novel cell-autonomous function for the Rb tumor suppressor, in regulating neuronal migration during telencephalic development.
Cell cycle and cell fate
Proper timing of cell cycle exit and terminal mitosis is believed to be critical for the generation of specific neuronal cell types in the developing neocortex (McConnell and Kaznowski, 1991). We have previously shown the prevalence of ectopically dividing neuroblasts throughout the IZ and CP of Rb mutants (Ferguson et al, 2002). Despite the fact that Rb-deficient mutants exhibit defective terminal mitoses, we show here that cortical neuronal populations do not appear to be misspecified. First, all dorsal progenitor cell markers examined showed normal expression patterns in Rb mutants. Second, layer-specific markers were appropriately expressed in Rb mutants, with the exception of ectopic expression in the IZ at early developmental stages. While neuronal fate determination has been shown to be tightly coupled with the timing of terminal mitosis (McConnell, 1995; Waid and McLoon, 1995), the Rb-deficient mouse represents an anomaly in which these events become aberrantly uncoupled. Although Rb-deficient neurons fail to undergo terminal mitosis at the correct time, they are still able to generate the appropriate neuronal populations as in wild-type animals. This lack of correlation between neuronal gene expression and terminal mitosis suggests that terminal mitosis may not always be a prerequisite for the specification of the appropriate neuronal population.
Our neuronal birthdating experiments revealed that although similar numbers of early-born cortical neurons were generated in Rb mutants, they failed to reach their ultimate destination within the CP, suggestive of a role of Rb in radial migration. In contrast, later-born neurons were able to migrate to their appropriate layers, raising the possibility that Rb is required for the migration of specific neuronal subpopulations. We cannot, however, rule out the idea that the early-born cortical projection neurons migrate inappropriately because they are born in ectopic locations. Confocal microscopy has revealed that the vast majority of dividing cells in the IZ are committed neuroblasts co-expressing BrdU and the neuronal marker β-III tubulin (Ferguson et al, 2002). This is consistent with the interpretation that Rb-deficient neurons are committed to a specific fate before they leave the VZ but then continue to divide ectopically. At present, therefore, we cannot distinguish between whether (a) neurons born in the IZ fail to undergo appropriate radial migration as a consequence of their generation in an inappropriate location or (b) early-born projection neurons require Rb for their radial migration.
A key question when evaluating radial migration is whether any of the migrating cells, particularly those that fail to find their appropriate destination, undergo apoptosis. We have quantified apoptosis in each of the zones including VZ, IZ, CP, and MZ and have only found a significant increase in apoptosis in the MZ at E13.5 and no difference in any of these regions at E16.5. This suggests that Rb is not essential for the survival of CP neurons and that neurons born in the IZ do not appear to default to an apoptotic pathway. Our studies reveal however that Rb is required for the survival of Cajal–Retzius neurons in the MZ. It should be noted that mice carrying the conditional Rb mutation die at birth, hence we cannot comment on the long-term survival of these cortical neurons.
Selective loss of Cajal–Retzius neurons
Although Rb deficiency specific to the telencephalon does not induce the widespread apoptosis observed in germline knockouts, certain neuronal populations may require Rb for survival. We previously reported a small but significant increase in TUNEL-positive cells within the mutant telencephalon (Ferguson et al, 2002). We now demonstrate that the neuronal loss in the Rb mutants is dramatically increased within the cortical MZ, specifically affecting the Cajal–Retzius neurons. Although the initial generation of Reelin-positive Cajal–Retzius neurons is normal, by E16.5, these cells are reduced in number by nearly 50% in the Rb-deficient MZ. This raises the possibility that the reduction in Cajal–Retzius cell numbers could account, at least in part, for the defects in radial and tangential migration. Indeed, Cajal–Retzius neurons are known to be critical for guiding radial migration of newly generated cortical neurons (Frotscher, 1998; Sarnat and Flores-Sarnat, 2002), and the loss of these cells by mid-neurogenesis may be associated with the aberrant radial migration and defective laminar patterning observed in the Rb mutant cortex. A similar reduction of Cajal–Retzius neurons occurs in mice deficient for Emx2, in which the neurons appear to be properly generated but are subsequently lost. These mice exhibit defective radial migration and laminar patterning (Mallamaci et al, 2000; Shinozaki et al, 2002). Although a reduction of Cajal–Retzius neurons by mid-neurogenesis would not be expected to have a great effect on the positioning of early-born neurons, the loss may be responsible for more subtle defects.
Rb regulates tangential migration in a cell-autonomous manner
The specification and generation of GABAergic interneurons, specifically calbindin- and Lhx6-positive neurons, was not initially impaired; however, by mid-neurogenesis, expression of these cortical interneuron markers was dramatically reduced along the MZ, whereas there was a corresponding increase in the IZ. Thus, the total number of calbindin-positive neurons was similar between Rb mutant and control embryos, demonstrating that these interneurons were not lost owing to apoptosis or neurogenesis defects. Instead, we found that the reduction of cells within the MZ was associated with a corresponding increase in calbindin-positive cells within the IZ. These results support our interpretation that the specific MZ loss of interneurons is due to their failure to properly follow their MZ migratory route, and that these cells instead become re-routed toward the deeper IZ trajectory. We cannot, however, rule out that the impaired migration we observe could be the result of a failure of these cells to express the full complement of genes required for proper migration.
Our data show that control cells from MGE explants migrate equally well on control and mutant sections, indicating the migratory environment to be relatively inconsequential in terms of the defective migration observed in Rb mutants. Instead, we demonstrate that Rb is essential among certain MGE-derived interneurons in order to migrate along the MZ. Despite the dramatic reduction in MZ migration derived from Rb mutant explants, a small proportion of samples that included Rb-deficient interneurons (23.8%) demonstrated complete MZ migration. This may be explained by the heterogeneity of the MGE population, as Rb signaling may only be required in specific interneuron subsets.
What may account for the mechanism through which Rb could impact neuronal migration? Previous studies have shown that Rb interacts with a number of genes that can regulate differentiation. A well-established interaction is with the helix–loop–helix (HLH) family member, Id2, an important negative regulator of neuronal differentiation (Perk et al, 2005). By acting as a dominant inhibitor of proneural basic HLH factors, Id2 represses the transcription of neuron-specific genes. Rb has been shown to interact with Id2, thereby suppressing its activity (Lasorella et al, 2002). Several studies demonstrate the importance of the Rb interaction with Id2 in neural development. First, the neurological defects found in Rb knockouts are rescued by the absence of Id2 (Lasorella et al, 2000). Second, expression of Id2 in cortical progenitor cells was shown to inhibit the induction of neuron-specific genes, while this inhibition was alleviated by the co-expression of a constitutively active Rb (Toma et al, 2000).
The consequence of increased free Id2 activity in Rb deficiency could lead to inhibition of genes that impact on differentiation and migration. One such example is TrkB, which has previously been shown to be substantially reduced in Rb-deficient brains (Lee et al, 1994). In addition to its well-known role in neuronal survival (Atwal et al, 2000; Stucky et al, 2002), TrkB has also been shown to regulate radial and tangential neuron migration (Polleux et al, 2002; Medina et al, 2004). Recent studies have demonstrated that Id2 can also directly repress TrkB expression in neural cells (Liu et al, 2004). Thus, we envisage a model whereby the absence of Rb leads to deregulated Id2 activity causing inhibition of transcription of neuron-specific genes required for differentiation and migration, such as TrkB (Figure 8). Future studies exploring molecules that regulate neuronal migration in the Rb-deficient brain will be required to identify the specific pathways that are dependent on Rb.
Figure 8.
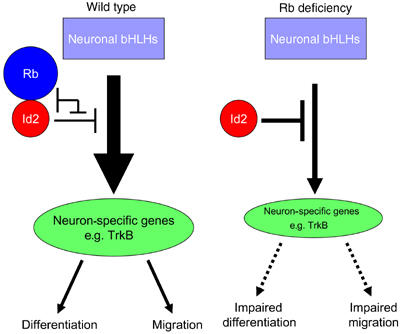
Proposed model of Rb-mediated regulation of neuronal differentiation and migration. In wild-type cells, Id2 is sequestered by Rb and is unable to inhibit basic HLH-mediated transcription of specific neuronal genes, such as TrkB, that are required for neuronal differentiation and migration. In the absence of Rb, Id2 activity is deregulated, allowing inhibition of TrkB transcription, which, in turn, leads to impaired neuronal migration and differentiation.
In conclusion, we demonstrate a cell type-specific requirement for Rb in the regulation of cortical development. Although the majority of cortical neurons survive in the absence of Rb, specific populations, including Reelin-positive Cajal–Retzius neurons, require Rb for survival. Furthermore, we reveal, for the first time, an essential role for the cell cycle protein, Rb, in regulating neuronal migration during cortical development.
Materials and methods
Mice
Telencephalon-specific Rb-deficient mice were generated by crossing Rb-F19 (Marino et al, 2000) and Foxg1-cre mice (Hebert and McConnell, 2000), as described previously (Ferguson et al, 2002). All experiments were approved by the University of Ottawa's Animal Care ethics committee adhering to the Guidelines of the Canadian Council on Animal Care.
Histology
Females at various stages of gestation were killed by a lethal injection of sodium pentobarbitol and embryos were removed and placed in 1 × PBS. Embryos were fixed in 4% paraformaldehyde/0.1 M phosphate buffer pH 7.4 for 1–2 days at 4°C. For frozen sections, tissue was subjected to sequential solutions of 12, 16, and 22% sucrose/0.1 M phosphate buffer for 1 day each at 4°C. Embryos were embedded in OCT (TissueTek 4583), frozen on liquid N2, and cut on a cryostat as 14 μm sections at −20°C and mounted on Superfrost slides (Fisher #12-550-15). For paraffin sections, fixed embryos were dehydrated in 60% ethanol for 1–2 days, embedded in paraffin wax, and sectioned at 6 μm thickness. Cresyl violet staining was performed on paraffin sections according to standard protocols.
Immunohistochemistry and in situ hybridization
Immunohistochemistry was performed on fixed frozen sections with the following primary antibodies: TuJ1 (mouse monoclonal hybridoma supernatant, 1:50, Dr David Brown, University of Ottawa), mouse monoclonal anti-Reelin G10 (1:500; Calbiochem, #553731), rabbit polyclonal anti-calbindin (D-28) (1:1000; Chemicon, AB1778), and mouse monoclonal anti-GAD 65 (1:100; BD Pharmingen, 559931/69221A). Sections were incubated in primary antibody overnight at 4°C, rinsed three times for 10 min each in PBS, and then incubated in the appropriate secondary antibody. For Reelin immunohistochemistry, sections were subjected to an antigen retrieval pretreatment: sections were brought to a boil in 10 mM sodium citrate buffer, pH 6.0, placed in an ice bath for 5 min, then the process was repeated twice more. For BrdU incorporation analyses, pregnant females were injected intraperitoneally with 20 or 50 μg BrdU/g body mass (Boehringer Mannheim #280879) and were processed as described previously (Ferguson et al, 2002). TUNEL staining was performed as described previously (Ferguson et al, 2002). Non-radioactive in situ hybridization and digoxygenin probe labeling was performed according to previously described protocols (Wallace and Raff, 1999). The following antisense riboprobes were used, as described previously: Tbr1, SCG10, Lhx6, and GAD 67. Riboprobe references are found in Supplementary data. Sections were examined with a Zeiss Axioskop 2 fluorescence microscope and visualized with a Sony Power HAD 3CCD color video camera with Northern Eclipse software.
Slice co-cultures
The conditional Rb mutant mice were bred such that one of the parents was additionally heterozygous for GFP. With this crossing, 25% of embryos would be expected to be Rb deficient while 50% of embryos should express GFP. Heterochronic slice co-cultures were performed on E16.5 litters, as previously described, with some modification (Polleux et al, 2002) (Supplementary data).
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We are indebted to Dr K Herrup for numerous helpful discussions. We thank J MacLaurin and L Jui for excellent technical assistance, and Drs W Hendelman, P Humphries, and L Maler for advice with anatomical examinations. This work was funded by a CIHR grant to RSS. KLF is a recipient of a CIHR studentship, KAM of OGS and SCN studentships, and JLV of an HSFC fellowship.
References
- Anderson SA, Eisenstat DD, Shi L, Rubenstein JL (1997) Interneuron migration from basal forebrain to neocortex: dependence on Dlx genes. Science 278: 474–476 [DOI] [PubMed] [Google Scholar]
- Atwal JK, Massie B, Miller FD, Kaplan DR (2000) The TrkB-Shc site signals neuronal survival and local axon growth via MEK and P13-kinase. Neuron 27: 265–277 [DOI] [PubMed] [Google Scholar]
- Caviness VS Jr (1982) Early events of neocortical assembly: experimental studies and human pathology. Int J Neurol 16–17: 102–109 [PubMed] [Google Scholar]
- Clarke AR, Maandag ER, van Roon M, van der Lugt NM, van der Valk M, Hooper ML, Berns A, te Riele H (1992) Requirement for a functional Rb-1 gene in murine development. Nature 359: 328–330 [DOI] [PubMed] [Google Scholar]
- DeDiego I, Smith-Fernandez A, Fairen A (1994) Cortical cells that migrate beyond area boundaries: characterization of an early neuronal population in the lower intermediate zone of prenatal rats. Eur J Neurosci 6: 983–997 [DOI] [PubMed] [Google Scholar]
- Denaxa M, Chan CH, Schachner M, Parnavelas JG, Karagogeos D (2001) The adhesion molecule TAG-1 mediates the migration of cortical interneurons from the ganglionic eminence along the corticofugal fiber system. Development 128: 4635–4644 [DOI] [PubMed] [Google Scholar]
- Ferguson KL, Vanderluit JL, Hebert JM, McIntosh WC, Tibbo E, MacLaurin JG, Park DS, Wallace VA, Vooijs M, McConnell SK, Slack RS (2002) Telencephalon-specific Rb knockouts reveal enhanced neurogenesis, survival and abnormal cortical development. EMBO J 21: 3337–3346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frotscher M (1998) Cajal–Retzius cells, Reelin, and the formation of layers. Curr Opin Neurobiol 8: 570–575 [DOI] [PubMed] [Google Scholar]
- Hack I, Bancila M, Loulier K, Carroll P, Cremer H (2002) Reelin is a detachment signal in tangential chain-migration during postnatal neurogenesis. Nat Neurosci 5: 939–945 [DOI] [PubMed] [Google Scholar]
- Hebert JM, McConnell SK (2000) Targeting of cre to the Foxg1 (BF-1) locus mediates loxP recombination in the telencephalon and other developing head structures. Dev Biol 222: 296–306 [DOI] [PubMed] [Google Scholar]
- Jacks T, Fazeli A, Schmitt EM, Bronson RT, Goodell MA, Weinberg RA (1992) Effects of an Rb mutation in the mouse. Nature 359: 295–300 [DOI] [PubMed] [Google Scholar]
- Lasorella A, Boldrini R, Dominici C, Donfrancesco A, Yokota Y, Inserra A, Iavarone A (2002) Id2 is critical for cellular proliferation and is the oncogenic effector of N-myc in human neuroblastoma. Cancer Res 62: 301–306 [PubMed] [Google Scholar]
- Lasorella A, Noseda M, Beyna M, Yokota Y, Iavarone A (2000) Id2 is a retinoblastoma protein target and mediates signalling by Myc oncoproteins. Nature 407: 592–598 [DOI] [PubMed] [Google Scholar]
- Lavdas AA, Grigoriou M, Pachnis V, Parnavelas JG (1999) The medial ganglionic eminence gives rise to a population of early neurons in the developing cerebral cortex. J Neurosci 19: 7881–7888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E, Hu N, Yuan SSF, Cox LA, Bradley A, Lee W, Herrup K (1994) Dual roles of the retinoblastoma protein in cell cycle regulation and neuron differentiation. Genes & Dev 8: 2008–2021 [DOI] [PubMed] [Google Scholar]
- Lee EY, Chang CY, Hu N, Wang YC, Lai CC, Herrup K, Lee WH, Bradley A (1992) Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature 359: 288–294 [DOI] [PubMed] [Google Scholar]
- Lipinski MM, Macleod KF, Williams BO, Mullaney TL, Crowley D, Jacks T (2001) Cell-autonomous and non-cell-autonomous functions of the Rb tumor suppressor in developing central nervous system. EMBO J 20: 3402–3413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Encinas M, Comella JX, Aldea M, Gallego C (2004) Basic helix–loop–helix proteins bind to TrkB and p21(Cip1) promoters linking differentiation and cell cycle arrest in neuroblastoma cells. Mol Cell Biol 24: 2662–2672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacPherson D, Sage J, Crowley D, Trumpp A, Bronson RT, Jacks T (2003) Conditional mutation of Rb causes cell cycle defects without apoptosis in the central nervous system. Mol Cell Biol 23: 1044–1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallamaci A, Mercurio S, Muzio L, Cecchi C, Pardini CL, Gruss P, Boncinelli E (2000) The lack of Emx2 causes impairment of Reelin signaling and defects of neuronal migration in the developing cerebral cortex. J Neurosci 20: 1109–1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin O, Yaron A, Bagri A, Tessier-Lavigne M, Rubenstein JL (2001) Sorting of striatal and cortical interneurons regulated by semaphorin–neuropilin interactions. Science 293: 872–875 [DOI] [PubMed] [Google Scholar]
- Marino S, Vooijs M, van Der Gulden H, Jonkers J, Berns A (2000) Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes Dev 14: 994–1004 [PMC free article] [PubMed] [Google Scholar]
- McConnell SK (1995) Constructing the cerebral cortex: neurogenesis and fate determination. Neuron 15: 761–768 [DOI] [PubMed] [Google Scholar]
- McConnell SK, Kaznowski CE (1991) Cell cycle dependence of laminar determination in developing neocortex. Science 254: 282–285 [DOI] [PubMed] [Google Scholar]
- Medina DL, Sciarretta C, Calella AM, Von Bohlen Und Halbach O, Unsicker K, Minichiello L (2004) TrkB regulates neocortex formation through the Shc/PLCgamma-mediated control of neuronal migration. EMBO J 23: 3803–3814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morante-Oria J, Carleton A, Ortino B, Kremer EJ, Fairen A, Lledo PM (2003) Subpallial origin of a population of projecting pioneer neurons during corticogenesis. Proc Natl Acad Sci USA 100: 12468–12473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perk J, Iavarone A, Benezra R (2005) Id family of helix–loop–helix proteins in cancer. Nat Rev Cancer 5: 603–614 [DOI] [PubMed] [Google Scholar]
- Polleux F, Whitford KL, Dijkhuizen PA, Vitalis T, Ghosh A (2002) Control of cortical interneuron migration by neurotrophins and PI3-kinase signaling. Development 129: 3147–3160 [DOI] [PubMed] [Google Scholar]
- Rubenstein JL, Anderson S, Shi L, Miyashita-Lin E, Bulfone A, Hevner R (1999) Genetic control of cortical regionalization and connectivity. Cereb Cortex 9: 524–532 [DOI] [PubMed] [Google Scholar]
- Sarnat HB, Flores-Sarnat L (2002) Cajal–Retzius and subplate neurons: their role in cortical development. Eur J Paediatr Neurol 6: 91–97 [DOI] [PubMed] [Google Scholar]
- Shinozaki K, Miyagi T, Yoshida M, Miyata T, Ogawa M, Aizawa S, Suda Y (2002) Absence of Cajal–Retzius cells and subplate neurons associated with defects of tangential cell migration from ganglionic eminence in Emx1/2 double mutant cerebral cortex. Development 129: 3479–3492 [DOI] [PubMed] [Google Scholar]
- Sidman RL, Rakic P (1973) Neuronal migration, with special reference to developing human brain: a review. Brain Res 62: 1–35 [DOI] [PubMed] [Google Scholar]
- Stein R, Mori N, Matthews K, Lo LC, Anderson DJ (1988) The NGF-inducible SCG10 mRNA encodes a novel membrane-bound protein present in growth cones and abundant in developing neurons. Neuron 1: 463–476 [DOI] [PubMed] [Google Scholar]
- Stoykova A, Hatano O, Gruss P, Gotz M (2003) Increase in reelin-positive cells in the marginal zone of Pax6 mutant mouse cortex. Cereb Cortex 13: 560–571 [DOI] [PubMed] [Google Scholar]
- Stucky CL, Shin JB, Lewin GR (2002) Neurotrophin-4: a survival factor for adult sensory neurons. Curr Biol 12: 1401–1404 [DOI] [PubMed] [Google Scholar]
- Sussel L, Marin O, Kimura S, Rubenstein JL (1999) Loss of Nkx2.1 homeobox gene function results in a ventral to dorsal molecular respecification within the basal telencephalon: evidence for a transformation of the pallidum into the striatum. Development 126: 3359–3370 [DOI] [PubMed] [Google Scholar]
- Takahashi T, Goto T, Miyama S, Nowakowski RS, Caviness VS Jr (1999) Sequence of neuron origin and neocortical laminar fate: relation to cell cycle of origin in the developing murine cerebral wall. J Neurosci 19: 10357–10371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Nowakowski RS, Caviness VS Jr (1996) The leaving or Q fraction of the murine cerebral proliferative epithelium: a general model of neocortical neuronogenesis. J Neurosci 16: 6183–6196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toma JG, El-Bizri H, Barnabe-Heider F, Aloyz R, Miller FD (2000) Evidence that helix–loop–helix proteins collaborate with retinoblastoma tumor suppressor protein to regulate cortical neurogenesis. J Neurosci 20: 7648–7656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trimarchi JM, Lees JA (2002) Sibling rivalry in the E2F family. Nat Rev Mol Cell Biol 3: 11–20 [DOI] [PubMed] [Google Scholar]
- Waid DK, McLoon SC (1995) Immediate differentiation of ganglion cells following mitosis in the developing retina. Neuron 14: 117–124 [DOI] [PubMed] [Google Scholar]
- Wallace VA, Raff MC (1999) A role for Sonic hedgehog in axon-to-astrocyte signalling in the rodent optic nerve. Development 126: 2901–2909 [DOI] [PubMed] [Google Scholar]
- Wichterle H, Garcia-Verdugo JM, Herrera DG, Alvarez-Buylla A (1999) Young neurons from medial ganglionic eminence disperse in adult and embryonic brain. Nat Neurosci 2: 461–466 [DOI] [PubMed] [Google Scholar]
- Wu L, de Bruin A, Saavedra HI, Starovic M, Trimboli A, Yang Y, Opavska J, Wilson P, Thompson JC, Ostrowski MC, Rosol TJ, Woollett LA, Weinstein M, Cross JC, Robinson ML, Leone G (2003) Extra-embryonic function of Rb is essential for embryonic development and viability. Nature 421: 942–947 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.