

Abstract
Nociceptors, or pain-sensitive receptors, are unique among sensory receptors in that their sensitivity is increased by noxious stimulation. This process, called sensitization or hyperalgesia, is mediated by a variety of proinflammatory factors, including bradykinin, ATP and NGF, which cause sensitization to noxious heat stimuli by enhancing the membrane current carried by the heat- and capsaicin-gated ion channel, TRPV1. Several different mechanisms for sensitization of TRPV1 have been proposed. Here we show that NGF, acting on the TrkA receptor, activates a signalling pathway in which PI3 kinase plays a crucial early role, with Src kinase as the downstream element which binds to and phosphorylates TRPV1. Phosphorylation of TRPV1 at a single tyrosine residue, Y200, followed by insertion of TRPV1 channels into the surface membrane, explains most of the rapid sensitizing actions of NGF.
Keywords: membrane trafficking, neurotrophic factor, pain, sensory transduction, tyrosine kinase
Introduction
The survival of an animal depends on its ability to detect and avoid damaging levels of heat, a task which is carried out by heat-sensitive primary afferent neurons, or nociceptors (Bessou and Perl, 1969). The heat threshold of nociceptors is around 43°C under normal conditions, corresponding to the temperature at which the heat sensation in normal human subjects changes from pleasant warmth to painful heat. Following injury or inflammation the threshold for sensing heat pain becomes lower, a phenomenon known as heat hyperalgesia or sensitization (Besson and Chaouch, 1987). Sensitization is not intrinsic to nociceptors, but is caused by the release by tissue damage or stress of extracellular inflammatory mediators, which increase the sensitivity to heat of nociceptive neurons (Cesare and McNaughton, 1996; Galoyan et al, 2003). Some of the best-studied inflammatory mediators are bradykinin, ATP and NGF.
The heat-gated ion channel TRPV1 (Caterina et al, 1997) is an important detector of noxious levels of heat, though it is clear that other mechanisms also contribute (Caterina et al, 1999, 2000; Davis et al, 2000; Woodbury et al, 2004). TRPV1 does, however, appear to be the principal mechanism by which the sensitivity to heat is enhanced by inflammatory mediators, because heat hyperalgesia is largely absent in animals from which the gene for TRPV1 has been deleted (Caterina et al, 2000; Davis et al, 2000). The membrane current carried by TRPV1 in expression systems was found to be increased by bradykinin, ATP and NGF (Premkumar and Ahern, 2000; Chuang et al, 2001; Tominaga et al, 2001; Vellani et al, 2001; Prescott and Julius, 2003), in agreement with results obtained in primary nociceptive neurons (Cesare and McNaughton, 1996; Shu and Mendell, 1999; Tominaga et al, 2001; Vellani et al, 2001; Bonnington and McNaughton, 2003). The molecular mechanism by which these mediators, and in particular NGF, cause sensitization of TRPV1 has, however, proven controversial. Some evidence supports PKA (Shu and Mendell, 2001) or PI3 kinase (Bonnington and McNaughton, 2003; Zhuang et al, 2004) as members of signalling pathways activated by the binding of NGF to TrkA and leading to TRPV1 sensitization. Other work supports the idea that endogenous PIP2 in the nociceptor membrane inhibits TRPV1, and that activation of PLCγ by TrkA leads to metabolism of PIP2 and relief of TRPV1 from inhibition (Chuang et al, 2001; Prescott and Julius, 2003). We have investigated this question in more detail in the present study. The results show that NGF, acting on the TrkA receptor, activates a signalling pathway in which PI3 kinase plays a crucial early role, with Src kinase as the downstream element that binds to and phosphorylates TRPV1. Phosphorylation at a single tyrosine residue explains most of the rapid effects of NGF on TRPV1.
Previous work has shown that the sensitivity to capsaicin is enhanced by PKC activation with no change in maximum current, at least in the short term (Vellani et al, 2001), and that the temperature dependence of heat-gated current is shifted to lower temperatures following exposure to bradykinin (Cesare and McNaughton, 1996). These observations can only be explained by a change in channel properties, and neither is consistent with a simple increase in the number of TRPV1 channels in the membrane. Other evidence, however, suggests that TRPV1 channels can be inserted into the membrane by regulated exocytosis (Morenilla-Palao et al, 2004; Van Buren et al, 2005). In the present study, we show that this latter mechanism is the most important contributor to sensitization by NGF.
Results
TRPV1 sensitization by NGF is reproduced in an expression system
We used HEK293 cells transfected with plasmids containing cDNAs coding for TRPV1 and for the TrkA receptor for NGF as a model system to investigate the sensitization of TRPV1. Capsaicin was initially used as an experimentally convenient surrogate for heat stimuli to activate TRPV1, a reasonable strategy because capsaicin activates only TRPV1 (Caterina et al, 2000; Davis et al, 2000), and because the TRPV1 ion current, whether activated by heat or by capsaicin, is sensitized by inflammatory mediators in a similar manner (Premkumar and Ahern, 2000; Vellani et al, 2001). Transfected cells were loaded with the intracellular calcium indicator fluo-4 and imaged in a confocal microscope. The increase in calcium-dependent fluorescence (ΔF) caused by applying a subsaturating concentration of capsaicin was used as an index of the activation of TRPV1 (Figure 1A).
Figure 1.

NGF enhances TRPV1 function and total phosphorylation. (A) Calcium-dependent fluorescence, F, relative to maximal fluorescence, Fmax, as a function of time from a single HEK293 cell stably transfected with TrkA and transiently transfected with hTRPV1. Pulses of capsaicin (100 nM, applied as shown at top) elicited submaximal increases in [Ca]i. Exposure to NGF (100 ng/ml, see top) enhanced the capsaicin-induced calcium increase. Arrows show responses used for calculation of sensitization ratio (see B). (B) Frequency distributions of ratios between ΔF for arrowed capsaicin applications in (A), with NGF (black bars) and without NGF (open bars). Ratios greater than 8 collected at right. Continuous curve shows Gaussian function fitted to data without NGF. Arrow shows the upper 95% confidence limit of Gaussian. Cells with ratios above this value were categorized as sensitized. The mean ΔF ratio enhancement following NGF application, averaged over all capsaicin-responsive cells, was 3.30±0.38 (mean±s.e.m., n=125). (C) Percentage of cells sensitized without (first bar) and with NGF (100 ng/ml), and with an inhibitor of PI3 kinase (wortmannin, 100 nM) and of PKA (H89, 10 μM). The final bar shows sensitization with S502A/S801A TRPV1. Error bars show s.e.m.; significance levels (see Materials and methods) given relative to the data in NGF (second bar). (D) Time course of increase in total phosphorylation. Mean ratios (n=2) between TRPV1 phosphor-specific (ProQ Diamond) and total protein (SyPro Ruby) fluorescence intensities. Ratios normalized to that before exposure to NGF. (E) Comparison between phosphor-specific fluorescence increase following exposure to NGF (100 ng/ml, 40 min) in WT TRPV1, and in S502A/S801A and Y200F mutant TRPV1. Pro-Q Diamond fluorescence at the top, restained with SyPro Ruby for total protein at the bottom. Arrows show the position of the main TRPV1 band. TRPV1 appears as multiple bands because of variable glycosylation (Jahnel et al, 2001). (F) NGF-dependent phosphor-specific fluorescence increase, relative to control, in experiments similar to that shown in (E), for WT TRPV1 and TRPV1 mutants S502A/S801A and Y200F (n=3).
Application of NGF increased ΔF in a subpopulation of transfected cells expressing high levels of TrkA (see Supplementary Figure 1). We calculated the ratio between the values of ΔF when sensitization had reached a peak, and that immediately before exposure to NGF. Without NGF the distribution of these ratios was well fitted by a Gaussian (Figure 1B). We calculated the percentage of cells in which the ratio exceeded the 95% confidence limit of the control distribution as an index of the sensitization of TRPV1 following exposure to NGF (see Figure 1B). Sensitization was markedly inhibited by the PI3 kinase inhibitor wortmannin, but was little affected by the PKA inhibitor H89 (Figure 1C), as was found in nociceptive neurons (Bonnington and McNaughton, 2003).
TRPV1 sensitization is associated with phosphorylation
Bradykinin causes sensitization of TRPV1 by activating PKCɛ (Cesare et al, 1999), which potentiates gating by phosphorylating rat TRPV1 at serine residues 502 and 800 in rat TRPV1 (Numazaki et al, 2002). We therefore investigated the possibility that sensitization of TRPV1 by NGF is also caused by phosphorylation. We quantified the total phosphorylation at serine, threonine and tyrosine groups by using ProQ Diamond, a fluorescent dye that binds to all phosphorylated residues (Martin et al, 2003). TRPV1 was observed to be phosphorylated in the basal state, and following exposure to NGF the normalized fluorescence increased by approximately 20% (Figure 1D and E). However, mutating serines 502 and 801 (the hTRPV1 residues corresponding to those for rTRPV1) had only a small effect on either the increased level of phosphorylation (Figure 1E and F) or on the functional enhancement following exposure to NGF (Figure 1C, last bar). NGF must therefore sensitize TRPV1 by a pathway different from bradykinin. We show below that phosphorylation of tyrosine 200 is critical for NGF-induced sensitization, and mutation of this residue was found to substantially reduce the NGF-induced increase in total phosphorylation (Figure 1E and F).
An antiphosphotyrosine antibody was next used to probe for specific phosphorylation at tyrosine residues in TRPV1. Tyrosine residues in TRPV1 were to some extent phosphorylated in the basal state (see Supplementary Figure 2), and a significant enhancement in phosphorylation was observed following exposure to NGF (Figure 2A), with a time course similar to the increase in total phosphorylation (Figure 1D) but somewhat slower than that of the functional sensitization (Figure 1A).
Figure 2.

Src kinase and tyrosine phosphorylation of TRPV1. (A) Increase in tyrosine phosphorylation (pY) of TRPV1 on exposure to NGF (100 ng/ml). TRPV1 immunoprecipitated with anti-V5 antibody (IP: TRPV1) and probed for tyrosine phosphorylation with 4G-10 antibody (IB: pY). Blot then stripped and reprobed with anti-V5 to quantify total TRPV1 (IB: TRPV1). (B) Basal and NGF-induced pY are inhibited by the Src inhibitor PP2. Cells pretreated with 10 μM PP2 for 1 h before application of NGF (100 ng/ml, 30 min). Densities of bands in the top gel, relative to the first band, are 1.00, 1.32, 0.45, and 0.54. (C) Basal and NGF-induced pY or TRPV1 are enhanced by cotransfection of c-Src (lanes 3 and 4) and inhibited by cotransfection of dominant-negative Src (lanes 5 and 6). Band densities 1.00, 1.31, 1.39, 1.67, 0.64, and 0.61. (D) Src tyrosine kinase activity increases following NGF exposure. Cells transfected with TrkA and Src exposed to NGF as indicated. In vitro tyrosine kinase assay using acid-denatured enolase as substrate (see Materials and methods). Blot stripped and reprobed with anti-Src to quantify total Src (lower). (E) Src-dependent tyrosine phosphorylation of TRPV1 is reduced by SHP-1 and enhanced by dominant-negative SHP-1 C455S. Cells transfected with hTRPV1 and c-Src, and with SHP-1 constructs as indicated. (F) The tyrosine phosphatase inhibitor pervanadate (250 μM for 20 min) enhanced TRPV1 tyrosine phosphorylation (lanes 1 and 2). The effect is greater when either TrkA (lanes 3 and 4) or Src (lanes 5 and 6) is cotransfected. (G) Functional enhancement of TRPV1-dependent Ca influx by NGF is inhibited by the Src inhibitor PP2 (10 μM) or by dominant-negative Src. Pervanadate (1 mM for 4 min) enhances TRPV1 gating. See Figure 1A–C for other details. Significance levels (see Materials and methods) relative to NGF (second bar) except for pervanadate data, which are relative to the bar without NGF.
Src binds to and phosphorylates TRPV1
There is some evidence that Src family kinases may regulate members of the TRP family (Xu et al, 2003; Jin et al, 2004; but see Vriens et al, 2004). We therefore investigated the possibility that Src may be responsible for phosphorylating TRPV1. We found that the specific Src inhibitor PP2 reduced basal tyrosine phosphorylation and abolished the NGF-induced enhancement in phosphorylation (Figure 2B). Transfection with src enhanced both the basal and the NGF-stimulated tyrosine phosphorylation, while transfection with dominant-negative src greatly reduced both the basal and the NGF-stimulated tyrosine phosphorylation (Figure 2C). Src kinase activity is increased by NGF with a time course similar to that of TRPV1 tyrosine phosphorylation (Figure 2D). Functional experiments also support a role for Src in sensitization by NGF, because exposure to PP2 or transfection of dominant-negative src considerably reduced sensitization (Figure 2G).
Substrates that are efficiently phosphorylated by Src kinase are in turn good substrates for the SHP-1 tyrosine phosphatase (Frank et al, 2004). Tyrosine phosphorylation of TRPV1 by Src was reversed by cotransfecting the tyrosine phosphatase SHP-1, and conversely was enhanced by cotransfection of dominant-negative SHP-1C455S (Figure 2E). The tyrosine phosphatase inhibitor pervanadate (Zhao et al, 1996) enhanced phosphorylation of TRPV1 by both endogenous and transfected Src (Figure 2F). Figure 2G (last bar) shows that pervanadate also enhanced the response of TRPV1 to capsaicin in functional experiments.
Other lines of evidence show that Src binds to and directly phosphorylates TRPV1. Tyrosine phosphorylation of TRPV1 was seen when TRPV1 and Src were cotransfected in the absence of TrkA, indicating that the kinase activity of Src alone was sufficient for TRPV1 tyrosine phosphorylation (see Figures 2E and 4A). Src and TRPV1 coimmunoprecipitate, and the binding is promoted by TrkA activation with a time course similar to that of the increase in phosphorylation of TRPV1 (Figure 3A). Finally, purified Src can be shown to directly phosphorylate purified TRPV1 in vitro (Figure 3B).
Figure 4.
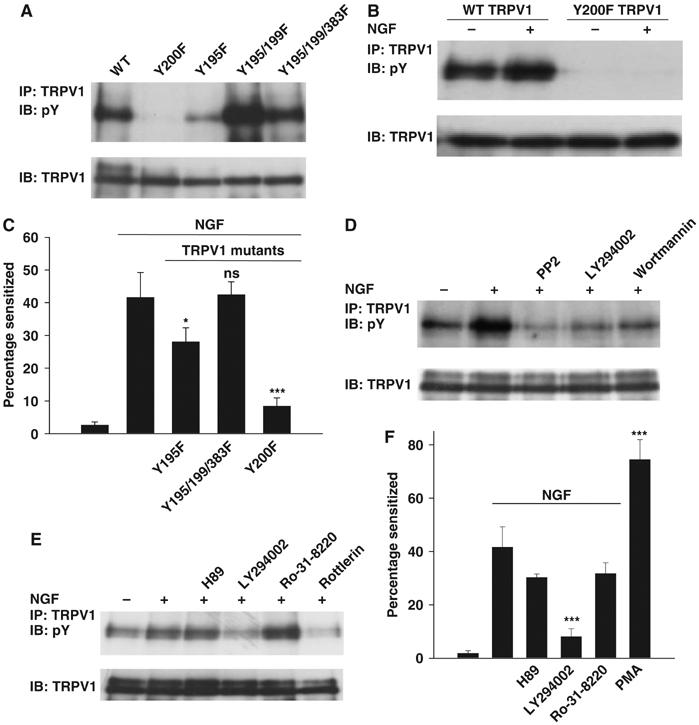
Pathways and target sites leading to TRPV1 functional enhancement. (A) Target sites on TRPV1 for Src-dependent tyrosine phosphorylation. Cells transfected with TRPV1 and c-Src (no TrkA). (B) Y200F TRPV1 mutation abolishes both basal and NGF-induced tyrosine phosphorylation. Cells transfected with TRPV1 and TrkA (no c-Src). (C) Effect of TRPV1 tyrosine residue mutations on functional enhancement of TRPV1 by NGF. See Figure 1A for experimental details. (D) Effect of Src inhibitor PP2 (10 μM) and PI3 kinase inhibitors LY294002 (25 μM) and wortmannin (100 nM) on NGF-stimulated tyrosine phosphorylation of TRPV1. (E) Effect of inhibitors as shown on tyrosine phosphrylation of TRPV1. H-89, 10 μM; LY294002, 25 μM; Ro-31-8220, 2 μM; rottlerin, 10 μM. Relative band densities 1.00, 1.27, 1.76, 0.79, 2.35, and 0.58. (F) Inhibition of functional enhancement of TRPV1 by H89, 10 μM; LY294002, 25 μM; Ro-31-8220, 500 nM. The last bar shows the effect of PMA, 1 μM. Significance levels in C and F relative to second bar (see Materials and methods).
Figure 3.

Src binds to and phosphorylates TRPV1. (A) Association between endogenous Src and TRPV1 is promoted by NGF. Src immunoprecipitated with B-12 antibody and TRPV1 association probed with anti-V5. Blot reprobed with anti-Src (middle blot). Lower blot shows total cell lysate (TCL) probed with anti-V5 for TRPV1 expression. (B) Src phosphorylates TRPV1 in vitro. See details in Materials and methods. (C) SH3 domain of Src binds to TRPV1. GST-tagged fragments of Src (as shown) used to precipitate TRPV1 from TCL. Src domains were 150–247 (SH2) and 83–144 (SH3). (D) Src binds to a proline-rich domain in the N-terminal region of TRPV1. TRPV1 phosphorylation probed with 4G-10 antibody (upper blot) following transfection of cells with DNA constructs as shown. Lower blot shows TRPV1. (E) Src binds to N- and C-terminal fragments of TRPV1. GST-tagged fragments of TRPV1 (N-TRPV1: residues 1–433; C-TRPV1: residues 681–839) used to precipitate Src from the TCL of Src-transfected HEK293 cells. Blot probed with anti-Src B-12 antibody (upper) and reprobed with anti-GST antibody (lower). Locations of Src- and GST-tagged TRPV1 fragments shown with arrows. (F) Effect of deletions from TRPV1 on basal tyrosine phosphorylation. ND: Δ1–433; CD: Δ681–839; CD 777–820: Δ777–820. Locations of TRPV1 and its mutants are shown with arrows.
Src binds to target proteins either via its SH2 domain, which recognizes phosphotyrosine, or via the SH3 domain, which binds proline-rich regions containing the motif PXXP (Pawson et al, 2001). To determine which Src domains directly bind to TRPV1, glutathione S-transferase (GST)–Src–SH2 and GST–Src–SH3 fusion protein were produced and in vitro pulldown assays were performed. As shown in Figure 3C, the isolated SH3 domain of Src binds strongly to TRPV1, while the binding of the adjacent SH2 domain was not above the nonspecific background binding of GST. Binding of the SH3 domain of Src to a PXXP motif in the N-terminal tail of TRPV1 (residues 35–38) as a prerequisite for phosphorylation of TRPV1 is demonstrated by experiments in which these two prolines were mutated to alanines, and Src-dependent tyrosine phosphorylation of TRPV1 was found to be largely abolished (Figure 3D).
We next investigated binding of Src to the N- and C-terminal tails of TRPV1 by constructing GST-coupled fragments of these regions. Binding of Src to the N-terminal fragment was observed, as would be expected on the basis of the experiments outlined above, but the C-terminal tail of TRPV1 was also found to bind Src (Figure 3E). We also examined the effect of removing the N- and C-terminal tails of TRPV1 on the ability of Src to phosphorylate TRPV1. Removal of the N-terminal cytoplasmic region almost completely abolished phosphorylation of TRPV1 (Figure 3F), consistent with the evidence outlined above that Src binds to and phosphorylates the N-terminal region. Removal of the entire C-terminal cytoplasmic region, however, increased tyrosine phosphorylation of TRPV1 (Figure 3F), consistent with the idea that Src binds to both the N- and the C-terminal tails and that these two regions compete for the available active Src, but that only binding to the N-terminal tail leads to phosphorylation of TRPV1.
Removal of amino acids 777–820 in the C-terminal domain enhances the sensitivity of TRPV1 and abolishes sensitization by NGF, one of the main lines of evidence behind the idea that PIP2 inhibits TRPV1 channel activity by binding to this domain (Prescott and Julius, 2003). In Figure 3F, however, we found that removal of the 777–820 domain promoted tyrosine phosphorylation of TRPV1 as potently as removal of the entire C-terminal. The enhanced tyrosine phosphorylation caused by the 777–820 deletion is at the crucial N-terminal Y200 site (see below), because mutation of the Y200 site largely abolished the increased phosphorylation caused by the 777–820 deletion (last lane in Figure 3F). Mutations in the 777–820 region of the C-terminal therefore have an indirect modulatory effect on phosphorylation by Src of tyrosine 200 in the N-terminal tail of TRPV1.
TRPV1 tyrosine residues involved in sensitization
To identify specific sites involved in sensitization by NGF, we mutated tyrosine residues in TRPV1. We showed above that deletion of the entire N-terminal domain abolished tyrosine phosphorylation, and we therefore focussed on N-terminal tyrosines. Mutation of Y195, Y199, Y375, Y383 and Y402 to the similarly sized phenylalanine, either singly or in combination, did not abolish the NGF-induced enhancement in tyrosine phosphorylation, although some reduction was observed with mutations of Y195, Y199 or Y383 (Figure 4A, and other results not shown). The Y195F mutant and a triple Y195F/Y199F/Y383F mutant were tested in functional experiments similar to those shown in Figure 1A, but both exhibited sensitization by NGF similar to that of wild-type (WT) TRPV1 (Figure 4C), showing that any changes of tyrosine phosphorylation at these residues do not have major functional consequences. However, when Y200 was mutated, there was a substantial reduction in Src-dependent tyrosine phosphorylation (Figure 4A), and the additional phosphorylation induced by NGF was completely abolished (Figure 4B). In functional experiments, the Y200F mutant was found to be markedly less sensitive to capsaicin than WT TRPV1 (ΔF/Fmax=0.2629±0.0189 in response to 100 nM capsaicin, compared with ΔF/Fmax=0.4182±0.0252 for WT TRPV1, difference significant, P=7.1 × 10−4, t-test). The Y200F mutation also largely removed functional sensitization following NGF application (Figure 4C).
Signalling intermediates leading to TRPV1 sensitization
We used measures of tyrosine phosphorylation of TRPV1, in combination with functional experiments, to probe for signalling intermediates leading to TRPV1 sensitization. NGF-induced tyrosine phosphorylation was greatly reduced by the PI3 kinase inhibitors LY294002 and wortmannin (Figure 4D and E). Functional sensitization was reduced to a similar extent by both inhibitors (Figures 1C and 4F). Transfection with dominant-negative PI3 kinase abolished the NGF-induced increase in TRPV1 tyrosine phosphorylation (see Supplementary Figure 2). These experiments confirm that PI3 kinase is an important element in the signalling chain leading to sensitization of TRPV1 by NGF.
Experiments in sensory neurons have shown that the broad-spectrum PKC inhibitor bisindolylmaleimide (BIM) abolishes the sensitization of TRPV1 by NGF (Bonnington and McNaughton, 2003). We found, however, that another broad-spectrum PKC inhibitor, Ro-31-8220, did not inhibit either NGF-dependent phosphorylation of TRPV1 (Figure 4E) or functional sensitization (Figure 4F). A difference between the two PKC inhibitors is that Ro-31-8220 is less effective against PKCδ (Shah and Catt, 2002), suggesting that the action of BIM may be due to inhibition of PKCδ. Consistent with this idea, the PKCδ-specific inhibitor rottlerin was found to inhibit NGF-dependent phosphorylation of TRPV1 (Figure 4E) and functional sensitization of TRPV1 by NGF in mouse sensory neurons (Figure 5D).
Figure 5.

TrkA residues initiating sensitization. (A) Effect of rat TrkA mutations on basal tyrosine phosphorylation of TRPV1. (B) TrkA Y760F mutation abolishes basal and NGF-stimulated TRPV1 tyrosine phosphorylation. Relative band densities 1.00, 1.32, 0.20, and 0.20. (C) Effect of TrkA tyrosine residue mutations on functional enhancement of TRPV1 by NGF. The final two bars show the effect of Y794F and Y760F TrkA mutants with S502A/S801A TRPV1 (DM). (D) Functional enhancement of TRPV1 in mouse DRG neurons by NGF inhibited by PP2 (10 μM) and rottlerin (10 μM). (E) Activation of TRPV1 by pH 5.5. Capsaicin sensitivity tested at the end. Other details as in Figure 1A. (F) Activation of TRPV1 by protons potentiated by NGF but inhibited with Y200F TRPV1 or Y760F TrkA with S502A/S801A TRPV1. Other details as in Figures 4C and 5C. Significance levels in C, D and F relative to second bar (see Materials and methods).
Nonspecific activation of PKC using PMA caused a large sensitization (Figure 4F), which can be attributed in part to activation of PKCɛ and direct phosphorylation of TRPV1 as described previously (Cesare et al, 1999; Numazaki et al, 2002). The evidence described above, however, suggests that a component of the sensitization by PMA can also be attributed to activation of PKCδ and consequent activation of Src.
Sites on TrkA initiating sensitization
Two phosphorylated tyrosine residues on TrkA have been proposed to activate different signalling cascades: Y499 activates the PI3 kinase and ras/Erk pathways, while Y794 activates PLCγ (Kaplan and Miller, 2000). However, mutation of either of these residues, or both together, enhanced rather than abolished tyrosine phosphorylation of TRPV1 (Figure 5A and other results not shown). Mutation of Y499 had little effect on functional sensitization (Figure 5C). Mutation of Y794, however, did reduce sensitization (Figure 5C), a result which has been interpreted by Chuang et al (2001) as showing that PLCγ initiates the pathway leading to TRPV1 sensitization (Chuang et al, 2001). However, another possible explanation is that mutation of the Y794 site improves access to a third site responsible for activating PI3 kinase, leading to constitutive activation of the PI3 kinase pathway. The consequent basal phosphorylation of TRPV1, as seen in Figure 5A, would then reduce the sensitizing effect of NGF, because few further sites are available to be phosphorylated.
The Y760 site on TrkA has been implicated in activation of PI3K (Obermeier et al, 1993) and we found that mutation of this site abolished tyrosine phosphorylation of TRPV1 (Figure 5A and B) and substantially reduced sensitization by NGF (Figure 5C). The Y760F TrkA mutant exhibited normal activity in other respects (see Supplementary Figure 3). The remaining sensitization seen with the Y760F TrkA mutant can be attributed to parallel activation of PKCɛ, because it is abolished by a double mutation of S502 and S801, the sites phosphorylated by PKCɛ (Figure 5C, and see below). These experiments show that the Y760 site is the principal means by which activated TrkA causes tyrosine phosphorylation and sensitization of TRPV1, at least on the time scale examined here.
Two pathways mediate sensitization
The Y760 mutant of TrkA completely lesioned tyrosine phosphorylation of TRPV1 (Figure 5A), but did not completely abolish sensitization in response to NGF (Figure 5C). This result, together with earlier data showing that some residual sensitization remains following block of PI3K or Src (Figures 1C and 2G), or removal of the Y200 site on TRPV1 (Figure 4C), suggests that another pathway activated by TrkA may be able to cause a low level of sensitization. One possibility is that the PKCɛ pathway activated by bradykinin (Cesare et al, 1999) could also be recruited as a result of the ability of TrkA to activate PLCγ (Kaplan and Miller, 2000), and could sensitize TRPV1 by phosphorylating serines 502 and 801. Mutating both these TRPV1 serine residues to alanine had only a slight effect on sensitization by NGF (see last bar in Figure 1C), as expected if the major pathway for sensitization depends on phosphorylation of Y200. However, transfecting TrkA mutated at Y760, to lesion the pathway leading to tyrosine phosphorylation of TRPV1, together with TRPV1 mutated at S502 and S801, to lesion serine phosphorylation of TRPV1 arising from activation of the PKCɛ pathway, completely abolished sensitization (Figure 5C), showing that these two pathways account for all of the sensitization of TRPV1 by NGF. Note also that the S502A/S801A mutation of TRPV1 does not affect sensitization caused by the Y794 mutant of TrkA (Figure 5C), consistent with the hypothesis advanced above that signalling to Src with this TrkA mutant is intact, but that functional sensitization is lower than with WT TrkA because of basal hyperphosphorylation of TRPV1.
Sensitization of TRPV1 in neurons
Selected experiments were repeated in mouse dorsal root ganglion (DRG) neurons. As in the HEK293 expression system, the specific Src inhibitor PP2 was found to largely but not completely abolish sensitization by NGF (Figure 5D). The PKCδ inhibitor rottlerin, which inhibited tyrosine phosphorylation of TRPV1 (Figure 4E), also inhibited sensitization by NGF in neurons (Figure 5D), consistent with an involvement of PKCδ in activating Src (see above).
Sensitization of TRPV1 is agonist-independent
Protons activate TRPV1 by binding to a site remote from the capsaicin-binding site (Jordt et al, 2000), and are therefore a suitable stimulus to test the generality of our findings. In cells transfected with TRPV1, a robust increase in [Ca]i was observed on lowering the pH to 5.5 (Figure 5E). The increase in [Ca]i in response to low pH was sensitized by NGF in a manner similar to that of capsaicin (Figure 5E and F). As was found with capsaicin as agonist (see above), the Y200F TRPV1 mutant was found to be markedly less sensitive to low pH than WT TRPV1 (ΔF/Fmax=0.1698±0.0213 in response to pH 5.5, compared with ΔF/Fmax=0.4032±0.0239 for WT TRPV1, significant difference, P=7.9 × 10−5, t-test). The TRPV1 Y200F mutant largely abolished sensitization by NGF (Figure 5F). A TrkA Y760F mutation, in combination with mutation of the S502 and S801 sites on TRPV1 (see above), completely abolished sensitization. These results are identical within experimental error to those obtained using capsaicin to activate TRPV1. We note in addition that Galoyan et al (2003) showed that the membrane current activated by heat stimuli in neurons is sensitized by NGF.
NGF increases expression of TRPV1 in the surface membrane
Sensitization of TRPV1 could occur because the gating of existing channels is enhanced, or because new channels are inserted into the surface membrane. Figure 6A shows that there is an increase in surface membrane TRPV1 following exposure to NGF, with a time course similar to that of TRPV1 phosphorylation (Figures 1D and 2A). The TRPV1 Y200F mutation reduces the basal level of surface membrane expression of TRPV1, consistent with the lower sensitivity of this mutant to both capsaicin and low pH (see above), and with the lower level of basal tyrosine phosphorylation observed in this mutant (Figure 4A and B). The Y200F mutation also abolishes the increase in membrane TRPV1 following exposure to NGF (Figure 6A). Consistent with the effect of inhibitors of PI3 kinase or Src in abolishing the sensitizing actions of NGF, inhibitors of these signalling intermediates also abolished NGF-dependent membrane insertion of TRPV1 (Figure 6B).
Figure 6.
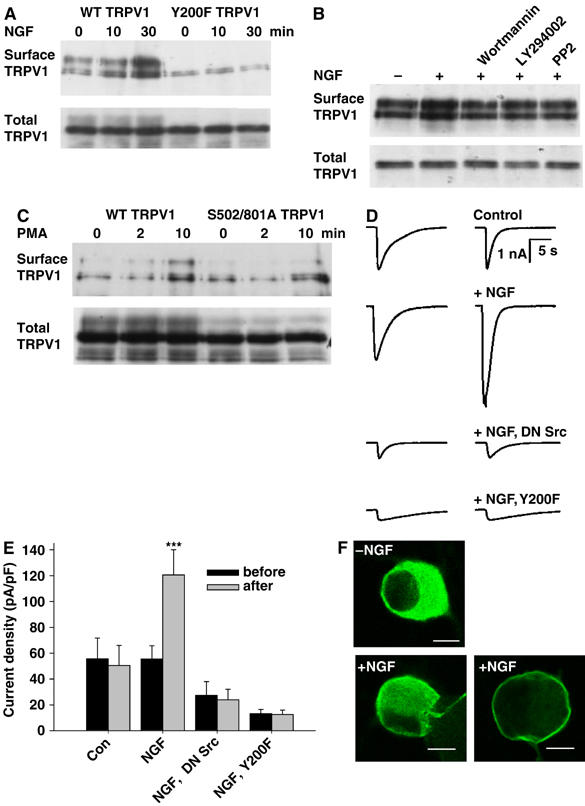
NGF enhances surface membrane expression of TRPV1. (A) Surface membrane expression of WT TRPV1 is enhanced by NGF, but Y200F TRPV1 expression is low and not affected by NGF. Surface TRPV1 isolated as in Materials and methods. Increase in WT surface membrane TRPV1 1.6±0.22-fold after 10 min (P<5%) and 2.4±0.18-fold after 30 min (P<0.1%, n=4). (B) Wortmannin, LY294002 and PP2 block NGF-dependent increase in surface membrane TRPV1. Relative band densities 1.00, 1.54, 1.08, 1.02, and 1.05. (C) PMA enhances surface membrane TRPV1, and enhancement is not inhibited by mutations S502A/S801A. Relative band densities 1.00, 0.77, 1.64, 1.18, 0.80, and 1.70. (D) Inward currents activated by 1-s pulses of 10 μM capsaicin in HEK293 cells cotransfected with TrkA and TRPV1. Traces shown for each condition were obtained 10 min apart with NGF (100 ng/ml) applied between pulses (apart from in first trace). Trace 3 with DN Src, trace 4 with Y200F TRPV1. (E) Collected results of patch-clamp experiments carried out as in (D). Each bar is the mean of at least seven experiments. (F) NGF promotes membrane insertion of TRPV1 in mouse DRG neurons. TRPV1 immunoreactivity imaged as described in Materials and methods. Scale bar 10 μm.
Figure 6C shows that an increase in surface membrane TRPV1 is also observed 10 min after application of the nonselective PKC activator PMA. The S501A/S801A mutation, which abolishes the sensitizing effect of agents acting through PKCɛ, does not abolish the increased membrane insertion of TRPV1 caused by application of PMA. This result suggests that membrane insertion of TRPV1 following PMA treatment may be mediated by activation of PKCδ followed by activation of Src, for which evidence has been obtained in other experiments (Figures 4E and 5D).
Whole-cell patch clamp was used to confirm that NGF increases expression of functional TRPV1 ion channels in the surface membrane. Figure 6D shows experiments in which we applied a saturating concentration of capsaicin (10 μM) in order to activate all TRPV1 channels maximally. NGF caused a substantial increase in current, which was absent in cells cotransfected with dominant-negative Src or in cells transfected with TRPV1 mutated at the Y200 Src phosphorylation site (lower two traces). The results are summarized in Figure 6E. Note also that the maximal membrane current density is lower both with cotransfection of DN Src and with Y200TRPV1, in support of the idea that phosphorylation of TRPV1 at Y200 regulates the basal level of membrane expression of TRPV1 as well as the increase caused by NGF.
Translocation of TRPV1 to the membrane following NGF application is also seen in DRG neurons (Figure 6F). Following application of NGF, TRPV1 was observed to be either partially or completely translocated to the membrane in a subset of neurons (lower part of Figure 6F). Translocation was observed in 17.5±1.9% of TRPV1-positive neurons following exposure to NGF, compared with 5.9±0.3% in control conditions.
The experiments described here show that a major effect of NGF is to cause a translocation of TRPV1 to the cell surface membrane. We note, however, that after 10 min NGF had stimulated a 1.6-fold increase in membrane TRPV1 (see legend to Figure 6A), while the mean functional sensitization after 8-min exposure to NGF was 3.3-fold (see legend to Figure 1B). These results suggest that an additional mechanism is active, particularly at early times. The PKCɛ–S502/S801 pathway outlined above contributes to this early sensitization, because mutation of the S502 and S801 residues, together with inhibition of Y200 phosphorylation by mutation of the Y760 site of TrkA, abolishes sensitization at all times (Figure 5C and F). A possible additional involvement of tyrosine phosphorylation in modulating gating of TRPV1 is not, however, excluded by the present experiments.
Discussion
The work reported here shows that phosphorylation by Src of a single tyrosine residue, Y200, is the major mechanism by which NGF causes rapid sensitization of TRPV1. The Y200 site is well conserved among rat, human, mouse, guinea-pig, rabbit, dog and chicken TRPV1, suggesting that this tyrosine may be an important common regulatory site for TRPV1. We also note that this site is also conserved across TRPV1–4 (see Figure 7A), suggesting a wider role in modulating TRPV function.
Figure 7.
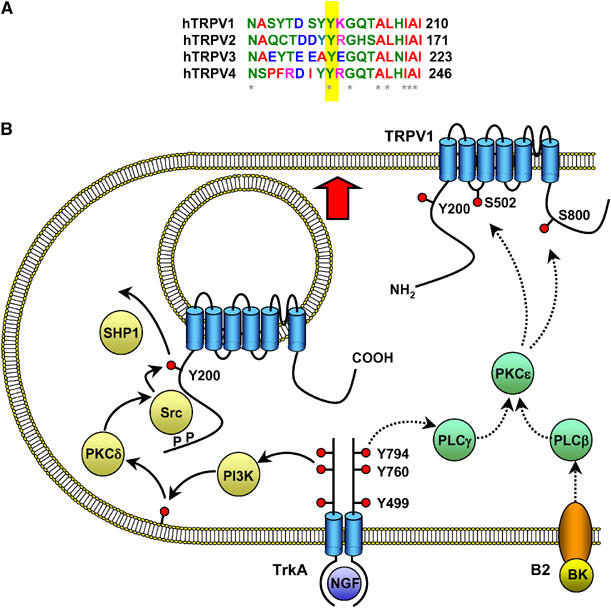
(A) Alignment of sequences adjacent to Y200 for human TRPV1–4. The bottom line shows conserved residues (*), which include the TRPV1 Y200 site (yellow bar). (B) Schematic diagram of the signaling pathways important in sensitization of TRPV1 by TrkA. The functionally most significant pathway is shown at the left (yellow, solid arrows). A smaller component of sensitization following exposure to NGF is mediated by phosphorylation of TRPV1 at residues S502 and S801, probably by the PLCγ/PKCɛ pathway (green, dashed arrows). PKCɛ is a crucial intermediate in sensitization of TRPV1 by bradykinin (pathway shown at the lower right of the diagram).
Figure 7B summarizes the signalling pathways connecting activated TrkA to sensitization of TRPV1 by NGF, as elucidated in the present study. The major pathway (left) promotes insertion of TRPV1 into the surface membrane. This pathway is initiated by autophosphorylation of the Y760 site of TrkA, followed by activation of PI3 kinase, and with Src as the downstream element directly responsible for phosphorylating TRPV1 at Y200. Other intermediate components are also likely to be involved in this pathway. We have obtained evidence for a role for PKCδ in activating Src, but other downstream members of the signalling pathways activated by PI3 kinase, such as Akt and PDK1, are possible additional intermediates. A second pathway, in which TrkA activates PKCɛ, leading to phosphorylation of TRPV1 at S502 and S801 (right), plays a more minor role in sensitization by NGF, but is the major mechanism by which bradykinin and ATP sensitize TRPV1 (Cesare et al, 1999; Numazaki et al, 2002).
We have found no evidence in the present study for involvement of PKA (Shu and Mendell, 2001) or PIP2 (Chuang et al, 2001; Prescott and Julius, 2003) in sensitization of TRPV1 by NGF. Evidence for a role for PIP2 rested in part on the observation that lesion of the PLCγ activation site on TrkA reduced sensitization (Chuang et al, 2001). The effect of mutating this TrkA site can, however, be explained by basal activation of the PI3K–Src pathway (see above and Figure 5A), without the need to invoke a pathway leading to PIP2 metabolism. A second line of evidence in support of a role for PIP2 was the observation that TRPV1 function is enhanced, and sensitization is reduced, by removal of a supposed PIP2-binding cassette between residues 777 and 820 in the C-terminal tail (Prescott and Julius, 2003). Our finding that removal of this domain promotes phosphorylation of the critical N-terminal Y200 site on TRPV1, probably because an unidentified site in the 777–820 domain binds to and competes for Src, provides an alternative explanation for the effects of mutations in the C-terminal 777–820 domain.
A role for ERK has also been proposed (Zhuang et al, 2004). We note that the ERK inhibitor UO126 had no effect on rapid potentiation of TRPV1 by NGF in DRG neurons (Bonnington and McNaughton, 2003) and that lesion of the Y499 site on TrkA, which is the major pathway for ERK activation (Kaplan and Miller, 2000), also had no significant effect on sensitization (see Figure 5C). An involvement of ERK on a longer time scale is, however, not excluded by the work reported in the present study, which is limited to the period 10–20 min after NGF application.
Tyrosine phosphorylation has been demonstrated to regulate the trafficking of ion channels (Wang et al, 2002), transporters (Shiratori et al, 1997) and receptors (Hayashi and Huganir, 2004; Cho et al, 2005). We present here evidence that increased tyrosine phosphorylation at the Y200 site also promotes the trafficking of the TRPV1 channel to the surface membrane. Trafficking following tyrosine phosphorylation is a novel means of regulation of the heat- and capsaicin-gated ion channel, TRPV1.
Materials and methods
Cell culture and transfection
HEK293 cells were cultured as described (Vellani et al, 2001). Cells were transiently transfected using PolyFect reagent (Qiagen) in accordance with the manufacturer's instructions. To create a stable cell line expressing the TrkA receptor, rat trkA cDNA was incorporated into HEK293 cells using the Superfect transfection reagent (Qiagen) as per the manufacturer's instructions. Clones were selected under G418. The stable clone was used for all calcium-imaging experiments, except those for TrkA mutants.
Imaging of intracellular calcium
Calcium imaging of transfected HEK293 cells was performed as described previously (Bonnington and McNaughton, 2003). Briefly, cells were plated on coverslips and loaded with fluo-4 AM (Molecular Probes). Coverslips were then transferred to a BioRad confocal microscope and images were collected. Pulses of capsaicin were applied at 4 min intervals and 100 ng/ml NGF was added after the sixth pulse. Neither capsaicin nor NGF released significant amounts of calcium from intracellular stores (see Supplementary Figure 1). A pH 5.5 solution was also used to activate TRPV1 in some experiments. Endogenous ASIC1a response in HEK293 cells was inactivated by lowering pH to 6.7 (Vellani et al, 2001). Inhibitors were applied (where appropriate) for 30 min before addition of NGF. Significance was tested using one-way analysis of variance (ANOVA) with Bonferroni's post hoc test (SPSS for Windows). Significance levels in figures are given as: NS, not significant, >5%; *, P<5%; **, P<1%; ***, P<0.1%. All errors shown as bars in figures or quoted in the text are ±s.e.m.
Isolation and culture of neurons
Neurons from the dorsal root ganglia of 3–5-day-old mice (TO strain) were isolated and cultured for 3 days as described (Bonnington and McNaughton, 2003). Neurons were cultured in medium with 50 ng/ml NGF for 1 day, followed by 2 days in 0NGF. For 12 h before the experiment, anti-NGF antibody was added to remove residual NGF.
Plasmids and DNA mutation
c-Src and DN-Src were from Dr Mark S Shapiro (Gamper et al, 2003), rat trkA and its mutants (Y499F and Y794F trkA) from Dr David Julius (Chuang et al, 2001), SHP-1 and SHP-1 C455S from Dr Frank D Böhmer (Frank et al, 2004) and dominant-negative PI3-kinase ΔP85α from Dr Julian Downward (Wennstrom and Downward, 1999). The Y499F/Y794F TrkA double mutant was constructed by combination of the Y499F site mutant with the Y794F site mutant using BmgBI and BsiWI. The Y760F TrkA mutant was constructed by using QuickChange (Stratagene) site-directed mutagenesis. TRPV1 mutants were made using megaprimer-based PCR as described (Barik and Galinski, 1991). All DNA constructs were confirmed by DNA sequencing.
Immunoprecipitation and immunoblotting
The following antibodies were used: anti-pY (4G-10, Upstate Biotechnology), anti-V5 (Invitrogen), anti-TrkA (Trk(C-14)), and anti-Src (B-12) from Santa Cruz Biotechnology. Following treatment, transfected cells were solubilized in lysis buffer (20 mM Tris, pH 8.0, 150 mM NaCl, 1 mM EDTA, 2 mM EGTA, 1% NP-40, 1 mM PMSF, 50 mM NaF, 1 mM sodium orthophosphate, 20 mM β-glycerophosphate, 10 mM sodium pyrophosphate, and protease inhibitors (Roche)), cell lysates were centrifuged at 12 000 r.p.m. for 10 min, cleared supernatant was mixed with anti-V5, to precipitate TRPV1, or anti-Src antibody, to precipitate Src, and protein A-agarose (Santa Cruz), and was incubated for 3 h. Immunocomplexes were collected by centrifugation, washed with lysis buffer, followed by boiling in SDS–PAGE sample buffer and loading on 7.5% polyacrylamide gels. Proteins were transferred from the gels to Hybond-P membrane. Blots were blocked and incubated with primary antibody. Blots were washed prior to addition of HRP-conjugated sheep anti-mouse (for 4G-10, B-12 and V5) or donkey anti-rabbit secondary antibody (for Trk(C-14)). After washing, blots were developed using ECL chemiluminescent reagent (Amersham Biosciences) prior to being exposed to X-ray film. All blots shown in figures are typical of at least two and in most cases three similar results.
Total phosphorylation
TRPV1 protein immunoprecipitation with anti-V5 was performed as described above. Total phosphoprotein in polyacrylamide gel was stained using Pro-Q™ Diamond phosphoprotein gel stain (Molecular Probes) in accordance with the manufacturer's instructions. The stained gel was visualized on a Typhoon 9400 scanner (Amersham Biosciences) using excitation at 532 nm and a 580-nm bandpass emission filter. To normalize the protein bands, the gel was destained in 10% ethanol and 7% acetic acid solution, and total protein was restained with SYPRO® Ruby total protein gel stain (Molecular Probes), and visualized on the scanner using excitation at 457 nm and a 610-nm bandpass emission filter. The phosphorylation level of each protein band was determined by calculation of the ratio of Pro-Q Diamond dye to SYPRO Ruby dye signal intensities.
Kinase assay
Src kinase activity was determined by an in vitro kinase assay. Briefly, endogenous Src was precipitated with anti-Src antibody and washed with lysis buffer. Followed by two washes with kinase buffer (20 mM HEPES pH 7.4, 10 mM MgCl2, 10 mM MnCl2, 0.2 mM sodium orthovanadate, 1 mM DTT), the kinase reaction was initiated by adding 5 μg acid-denatured enolase and 50 μM ATP at 30°C for 10 min. The reaction was then stopped by mixing with sample buffer and loaded onto 12% SDS–PAGE gel. Tyrosine phosphorylation of enolase was evaluated by the 4G-10 anti-pY antibody as described above.
For direct in vitro phosphorylation of TRPV1 by c-Src, TRPV1 protein was expressed in HEK293 cells and immunoprecipitated with anti-V5 antibody, then 20 U of recombinant c-Src (Upstate) and 100 μM ATP were added to the reaction tube to perform the Src kinase assay as described above.
Sodium pervanadate
Sodium pervanadate was freshly prepared as described (Zhao et al, 1996) by mixing 100 mM sodium orthovanadate with 100 mM hydrogen peroxide solution for 20 min at room temperature. The reaction was terminated by the addition of catalase (Sigma) to quench residual hydrogen peroxide, after which the pervanadate solution was kept on ice until use.
GST fusion protein expression and pulldown assay
GST fusion protein constructs encoding the TRPV1 N-terminal region (1–433, GST-N-TRPV1), TRPV1 C-terminal region (684–839, GST-C-TRPV1), Src SH3 domain (83–144, GST-Src-SH3), and Src SH2 domain (150–247, GST-Src-SH2) were PCR amplified. PCR products were subcloned into the pGEX-2T expression vector (Amersham Biosciences) with BamHI and EcoRI. For fusion protein expression, IPTG was added to BL-21 cells with different DNA constructs for 2 h. Cells were then solubilized, the supernatants were incubated with glutathione-agarose (Sigma) at 4°C with constant shaking. For the pulldown assay, bead-bound GST fusion proteins were incubated with lysates of HEK293 cells overexpressing TRPV1 or Src at 4°C. Protein-bound beads were thoroughly washed and resolved on 12% SDS–PAGE. Bound proteins were detected with anti-Src or anti-V5 antibody as above.
Biotinylation of cell surface protein
HEK293 cells transfected with both TrkA and TRPV1 were biotinylated with 1 mg/ml sulfo-NHS-LC-biotin (Pierce) for 30 min at 4°C and washed with PBS containing 100 mM glycine. Biotinylated cells were solubilized in RIPA buffer (20 mM HEPES, pH 7.4, 150 mM NaCl, 1 mM EDTA, 2 mM EGTA, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS plus protease inhibitors). Supernatant fractions were incubated with streptavidin-agarose (Sigma) overnight at 4°C, beads washed with RIPA buffer, and adsorbed proteins boiled in sample buffer. Proteins were separated, transferred to the blot, and surface TRPV1 was detected with anti-V5 tag antibody as described above.
Electrophysiology
HEK293-TrkA stable cells transfected with TRPV1 were whole-cell patch clamped as described by Vellani et al (2001). Cells were maintained at −60 mV and were pulsed with 10 μM capsaicin for 1 s at 30 s intervals until the response stabilized. NGF was then applied for 10 min and the current evoked by further applications of capsaicin was tested at the end of this period. [Na]o was reduced to 30 mM (substituted with choline chloride) in order to reduce the membrane current amplitude.
Immunocytochemistry
Cultured DRG neurons exposed to 100 ng/ml NGF for 30 min were fixed with 4% paraformaldehyde. Primary antibody was rabbit anti-TRPV1 antibody (Alomone Labs), followed by Alexa-488-conjugated anti-rabbit secondary antibody (Molecular Probes). Coverslips were examined using confocal microscopy (Bio-Rad). Fluorescence intensity was measured along a line across the cell and cells in which surface fluorescence intensity was greater than twice the mean cytoplasmic intensity were scored as positive. At least 700 cells were scored for each condition in five separate experiments.
Supplementary Material
Supplementary Fig. 1
Supplementary Fig. 2
Supplementary Fig. 3
Acknowledgments
This work was supported by the Biotechnology and Biological Sciences Research Council, UK. We thank E Smith and A Momin for help with patch clamp experiments, K Hinchliffe for suggesting the use of ProQ Diamond, H Cadiou, D Cooper, R Hardie, R Irvine and E Smith for helpful comments on the manuscript and M Young for ERK antibody.
References
- Barik S, Galinski MS (1991) ‘Megaprimer' method of PCR: increased template concentration improves yield. Biotechniques 10: 489–490 [PubMed] [Google Scholar]
- Besson JM, Chaouch A (1987) Peripheral and spinal mechanisms of nociception. Physiol Rev 67: 67–155 [DOI] [PubMed] [Google Scholar]
- Bessou P, Perl ER (1969) Response of cutaneous sensory units with unmyelinated fibers to noxious stimuli. J Neurophysiol 32: 1025–1043 [DOI] [PubMed] [Google Scholar]
- Bonnington JK, McNaughton PA (2003) Signalling pathways involved in the sensitisation of mouse nociceptive neurones by nerve growth factor. J Physiol 551: 433–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caterina MJ, Leffler A, Malmberg AB, Martin WJ, Trafton J, Petersen-Zeitz KR, Koltzenburg M, Basbaum AI, Julius D (2000) Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science 288: 306–313 [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Rosen TA, Tominaga M, Brake AJ, Julius D (1999) A capsaicin-receptor homologue with a high threshold for noxious heat. Nature 398: 436–441 [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D (1997) The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature 389: 816–824 [DOI] [PubMed] [Google Scholar]
- Cesare P, Dekker LV, Sardini A, Parker PJ, McNaughton PA (1999) Specific involvement of PKC-epsilon in sensitization of the neuronal response to painful heat. Neuron 23: 617–624 [DOI] [PubMed] [Google Scholar]
- Cesare P, McNaughton PA (1996) A novel heat-activated current in nociceptive neurons, and its sensitization by bradykinin. Proc Natl Acad Sci USA 93: 15435–15439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho CH, Song W, Leitzell K, Teo E, Meleth AD, Quick MW, Lester RA (2005) Rapid upregulation of alpha7 nicotinic acetylcholine receptors by tyrosine dephosphorylation. J Neurosci 25: 3712–3723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang HH, Prescott ED, Kong H, Shields S, Jordt SE, Basbaum AI, Chao MV, Julius D (2001) Bradykinin and nerve growth factor release the capsaicin receptor from PtdIns(4,5)P2-mediated inhibition. Nature 411: 957–962 [DOI] [PubMed] [Google Scholar]
- Davis JB, Gray J, Gunthorpe MJ, Hatcher JP, Davey PT, Overend P, Harries MH, Latcham J, Clapham C, Atkinson K, Hughes SA, Rance K, Grau E, Harper AJ, Pugh PL, Rogers DC, Bingham S, Randall A, Sheardown SA (2000) Vanilloid receptor-1 is essential for inflammatory thermal hyperalgesia. Nature 405: 183–187 [DOI] [PubMed] [Google Scholar]
- Frank C, Burkhardt C, Imhof D, Ringel J, Zschornig O, Wieligmann K, Zacharias M, Bohmer FD (2004) Effective dephosphorylation of Src substrates by SHP-1. J Biol Chem 279: 11375–11383 [DOI] [PubMed] [Google Scholar]
- Galoyan SM, Petruska JC, Mendell LM (2003) Mechanisms of sensitization of the response of single dorsal root ganglion cells from adult rat to noxious heat. Eur J Neurosci 18: 535–541 [DOI] [PubMed] [Google Scholar]
- Gamper N, Stockand JD, Shapiro MS (2003) Subunit-specific modulation of KCNQ potassium channels by Src tyrosine kinase. J Neurosci 23: 84–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T, Huganir RL (2004) Tyrosine phosphorylation and regulation of the AMPA receptor by SRC family tyrosine kinases. J Neurosci 24: 6152–6160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahnel R, Dreger M, Gillen C, Bender O, Kurreck J, Hucho F (2001) Biochemical characterization of the vanilloid receptor 1 expressed in a dorsal root ganglia derived cell line. Eur J Biochem 268: 5489–5496 [DOI] [PubMed] [Google Scholar]
- Jin X, Morsy N, Winston J, Pasricha PJ, Garrett K, Akbarali HI (2004) Modulation of TRPV1 by nonreceptor tyrosine kinase, c-Src kinase. Am J Physiol Cell Physiol 287: C558–C563 [DOI] [PubMed] [Google Scholar]
- Jordt S-E, Tominaga M, Julius D (2000) Acid potentiation of the capsaicin receptor determined by a key extracellular site. Proc Natl Acad Sci USA 97: 8134–8139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan DR, Miller FD (2000) Neurotrophin signal transduction in the nervous system. Curr Opin Neurobiol 10: 381–391 [DOI] [PubMed] [Google Scholar]
- Martin K, Steinberg TH, Cooley LA, Gee KR, Beechem JM, Patton WF (2003) Quantitative analysis of protein phosphorylation status and protein kinase activity on microarrays using a novel fluorescent phosphorylation sensor dye. Proteomics 3: 1244–1255 [DOI] [PubMed] [Google Scholar]
- Morenilla-Palao C, Planells-Cases R, Garcia-Sanz N, Ferrer-Montiel A (2004) Regulated exocytosis contributes to protein kinase C potentiation of vanilloid receptor activity. J Biol Chem 279: 25665–25672 [DOI] [PubMed] [Google Scholar]
- Numazaki M, Tominaga T, Toyooka H, Tominaga M (2002) Direct phosphorylation of capsaicin receptor VR1 by protein kinase Cepsilon and identification of two target serine residues. J Biol Chem 277: 13375–13378 [DOI] [PubMed] [Google Scholar]
- Obermeier A, Lammers R, Wiesmuller KH, Jung G, Schlessinger J, Ullrich A (1993) Identification of Trk binding sites for SHC and phosphatidylinositol 3′-kinase and formation of a multimeric signaling complex. J Biol Chem 268: 22963–22966 [PubMed] [Google Scholar]
- Pawson T, Gish GD, Nash P (2001) SH2 domains, interaction modules and cellular wiring. Trends Cell Biol 11: 504–511 [DOI] [PubMed] [Google Scholar]
- Premkumar LS, Ahern GP (2000) Induction of vanilloid receptor channel activity by protein kinase C. Nature 408: 985–990 [DOI] [PubMed] [Google Scholar]
- Prescott ED, Julius D (2003) A modular PIP2 binding site as a determinant of capsaicin receptor sensitivity. Science 300: 1284–1288 [DOI] [PubMed] [Google Scholar]
- Shah BH, Catt KJ (2002) Calcium-independent activation of extracellularly regulated kinases 1 and 2 by angiotensin II in hepatic C9 cells: roles of protein kinase Cdelta, Src/proline-rich tyrosine kinase 2, and epidermal growth receptor trans-activation. Mol Pharmacol 61: 343–351 [DOI] [PubMed] [Google Scholar]
- Shiratori T, Miyatake S, Ohno H, Nakaseko C, Isono K, Bonifacino JS, Saito T (1997) Tyrosine phosphorylation controls internalization of CTLA-4 by regulating its interaction with clathrin-associated adaptor complex AP-2. Immunity 6: 583–589 [DOI] [PubMed] [Google Scholar]
- Shu X, Mendell LM (1999) Nerve growth factor acutely sensitizes the response of adult rat sensory neurons to capsaicin. Neurosci Lett 274: 159–162 [DOI] [PubMed] [Google Scholar]
- Shu X, Mendell LM (2001) Acute sensitization by NGF of the response of small-diameter sensory neurons to capsaicin. J Neurophysiol 86: 2931–2938 [DOI] [PubMed] [Google Scholar]
- Tominaga M, Wada M, Masu M (2001) Potentiation of capsaicin receptor activity by metabotropic ATP receptors as a possible mechanism for ATP-evoked pain and hyperalgesia. Proc Natl Acad Sci USA 98: 6951–6956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Buren JJ, Bhat S, Rotello R, Pauza ME, Premkumar LS (2005) Sensitization and translocation of TRPV1 by insulin and IGF-I. Mol Pain 1: 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vellani V, Mapplebeck S, Moriondo A, Davis JB, McNaughton PA (2001) Protein kinase C activation potentiates gating of the vanilloid receptor VR1 by capsaicin, protons, heat and anandamide. J Physiol 534: 813–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vriens J, Watanabe H, Janssens A, Droogmans G, Voets T, Nilius B (2004) Cell swelling, heat, and chemical agonists use distinct pathways for the activation of the cation channel TRPV4. Proc Natl Acad Sci USA 101: 396–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang WH, Lin DH, Sterling H (2002) Regulation of ROMK channels by protein tyrosine kinase and tyrosine phosphatase. Trends Cardiovasc Med 12: 138–142 [DOI] [PubMed] [Google Scholar]
- Wennstrom S, Downward J (1999) Role of phosphoinositide 3-kinase in activation of ras and mitogen-activated protein kinase by epidermal growth factor. Mol Cell Biol 19: 4279–4288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodbury CJ, Zwick M, Wang S, Lawson JJ, Caterina MJ, Koltzenburg M, Albers KM, Koerber HR, Davis BM (2004) Nociceptors lacking TRPV1 and TRPV2 have normal heat responses. J Neurosci 24: 6410–6415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Zhao H, Tian W, Yoshida K, Roullet JB, Cohen DM (2003) Regulation of a transient receptor potential (TRP) channel by tyrosine phosphorylation. SRC family kinase-dependent tyrosine phosphorylation of TRPV4 on TYR-253 mediates its response to hypotonic stress. J Biol Chem 278: 11520–11527 [DOI] [PubMed] [Google Scholar]
- Zhao Z, Tan Z, Diltz CD, You M, Fischer EH (1996) Activation of mitogen-activated protein (MAP) kinase pathway by pervanadate, a potent inhibitor of tyrosine phosphatases. J Biol Chem 271: 22251–22255 [DOI] [PubMed] [Google Scholar]
- Zhuang ZY, Xu H, Clapham DE, Ji RR (2004) Phosphatidylinositol 3-kinase activates ERK in primary sensory neurons and mediates inflammatory heat hyperalgesia through TRPV1 sensitization. J Neurosci 24: 8300–8309 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Fig. 1
Supplementary Fig. 2
Supplementary Fig. 3