

Abstract
Individual ubiquitin (Ub)–protein ligases (E3s) cooperate with specific Ub-conjugating enzymes (E2s) to modify cognate substrates with polyubiquitin chains. E3s belonging to the Really Interesting New Gene (RING) and Homologous to E6-Associated Protein (E6AP) C-Terminus (HECT) domain families utilize distinct molecular mechanisms. In particular, HECT E3s, but not RING E3s, form a thiol ester with Ub before transferring Ub to the substrate lysine. Here we report that different HECT domain E3s can employ distinct mechanisms of polyubiquitin chain synthesis. We show that E6AP builds up a K48-linked chain on its HECT cysteine residue, while KIAA10 builds up K48- and K29-linked chains as free entities. A small region near the N-terminus of the conserved HECT domain helps to bring about this functional distinction. Thus, a given HECT domain can specify both the linkage of a polyubiquitin chain and the mechanism of its assembly.
Keywords: conjugation, HECT domain, polyubiquitin, ubiquitin, ubiquitin ligase
Introduction
The covalent modification of cellular proteins with ubiquitin (Ub) modulates a host of biological processes. Ubiquitylation signals substrate degradation by 26S proteasomes, thus regulating cell cycle progression, antigen presentation, inflammatory responses, and many other pathways (Pickart, 2004). Ub is also a nonproteolytic signal, especially in protein trafficking (Hicke and Dunn, 2003). At least three factors contribute to Ub's ability to act as a multifunctional signal: the exquisite specificity of Ub conjugation, the existence of structurally distinct Ub modifications, and the presence of recognition factors that can translate specific Ub modifications into appropriate downstream consequences.
Ub conjugation occurs through the sequential actions of three enzymes (Pickart and Eddins, 2004). An activating enzyme (E1) uses ATP to drive the formation of a thiol ester with Ub's C-terminal Gly residue (G76). Ub is then passed to the Cys residue of a Ub conjugating enzyme (E2). Finally, a Ub-protein ligase (E3) catalyzes the transfer of Ub from the E2 to a substrate Lys residue. The conjugation cascade is hierarchically organized, usually consisting of a single E1, a limited number of E2s, and many E3s. The E3 plays the key role in target protein selection through its ability to recognize a structural motif presented by its cognate substrates (Pickart and Eddins, 2004; Petroski and Deshaies, 2005).
Most known E3 enzymes belong to one of two families. Members of the first family, whose founding member is E6-Associated Protein (E6AP), share a conserved ∼350-residue region called the Homologous to E6AP C-Terminus (HECT) domain, which harbors an essential Cys residue (Huibregtse et al, 1993, 1995; Scheffner et al, 1995). Members of the second family, called Really Interesting New Gene (RING) E3s, are defined by the presence of multiple Cys and His residues that coordinate two zinc ions to form a globular E2-binding domain (Petroski and Deshaies, 2005). Although HECT and RING E3s interact with similar surfaces of their cognate E2s (Huang et al, 1999; Zheng et al, 2000), the two types of E3s lack sequence or structural similarity and employ divergent catalytic mechanisms. RING E3s are scaffolds that dock the charged E2 and the substrate so as to facilitate direct attack of the substrate Lys on the E2-linked Ub (Zheng et al, 2000, 2002). In HECT E3 catalysis, the E2-bound Ub is transferred to the E3 Cys residue prior to attack of the substrate Lys residue (Scheffner et al, 1995).
Although E3s play a well-established role in substrate selection, E3 catalysis is poorly understood (Wu et al, 2003; Pickart and Eddins, 2004). Large distances separate the E2 active site and the substrate Lys residue in several RING E3 complexes (Zheng et al, 2000, 2002; Orlicky et al, 2003), and the first HECT E3/E2 structure revealed a separation of 41 Å between the E2 and E3 active sites (Huang et al, 1999). While the latter distance can be reduced through a conformational change in the HECT domain (Verdecia et al, 2003), the molecular details of Ub transfer between E2 and HECT E3 remain unknown.
Ub modifications take several structural forms, which can confer different functional readouts. Modification by a single Ub frequently leads to altered protein trafficking (Hicke and Dunn, 2003). In other cases, substrates are modified by multiple Ubs linked to one another in the form of a polyUb chain. Chains linked through Ub-K48 and K63 are the best-characterized, signaling proteasome proteolysis in the first case and (nonproteolytic) DNA damage tolerance or kinase activation in the second (Pickart and Fushman, 2004). Despite the demonstrated importance of polyUb signals, the mechanisms used by E3 enzymes in linkage site selection and catalysis are poorly understood.
Here we report that two different HECT E3s employ distinct mechanisms of chain synthesis. The mechanism observed with E6AP, in which the chain is built up at the E3 active site, resembles that proposed from recent structural studies (Verdecia et al, 2003). In contrast, KIAA10 synthesizes the chain as a free entity. Thus, HECT E3s display an unexpected level of mechanistic diversity.
Results
In vitro chain assembly by KIAA10–C-terminal domain (CD) and E6AP–HECT domains
The HECT E3 KIAA10 rapidly synthesizes unanchored polyubiquitin chains linked through K29 or K48; the related yeast E3 Hul5 shows a similar dual linkage preference (Mastrandrea et al, 1999; You and Pickart, 2001). The CD of KIAA10, which includes the canonical HECT domain plus 60 upstream residues, is necessary and sufficient for chain synthesis (You and Pickart, 2001). These properties of KIAA10 suggested that chain synthesis could be a conserved biochemical activity of the HECT domain.
We tested this hypothesis using E6AP, a HECT domain E3 that cooperates with the viral E6 protein to target p53 for proteasome degradation in cells infected with oncogenic human papilloma viruses (HPVs) (Scheffner et al, 1990, 1993; Huibregtse et al, 1991). E6AP also targets the DNA repair protein HHR23A and other substrates for degradation in uninfected cells (Kuhne and Banks, 1998; Kumar et al, 1999; Oda et al, 1999). Mutations in E6AP cause Angelman syndrome (Kishino et al, 1997; Matsuura et al, 1997; Cooper et al, 2004).
The E6AP–HECT and KIAA10–CD proteins were expressed and purified as fusions to glutathione-S-transferase (GST) and released from GST by proteolytic cleavage (Figure 1A). Proper folding of the E6AP–HECT domain was shown by its ability to form a Ub thiol ester in the presence of UbcH5A or UbcH7, which are cognate E2s (Scheffner et al, 1994; Hatakeyama et al, 1997; Schwarz et al, 1998) (see below). In an earlier work, we observed Ub thiol ester formation with full-length KIAA10, but we were unable to detect this intermediate with KIAA10–CD; evidently, it is too rapidly discharged to permit detection in our assays (You and Pickart, 2001). However, we found that mutating the HECT Cys residue of KIAA10–CD (to Ala) ablated chain synthesis, confirming that polyubiquitylation proceeds through a thiol ester intermediate (Supplementary Figure 1). As expected, mutating the E6AP–HECT active-site Cys (to Ala) eliminated the conjugation activities described below (data not shown).
Figure 1.
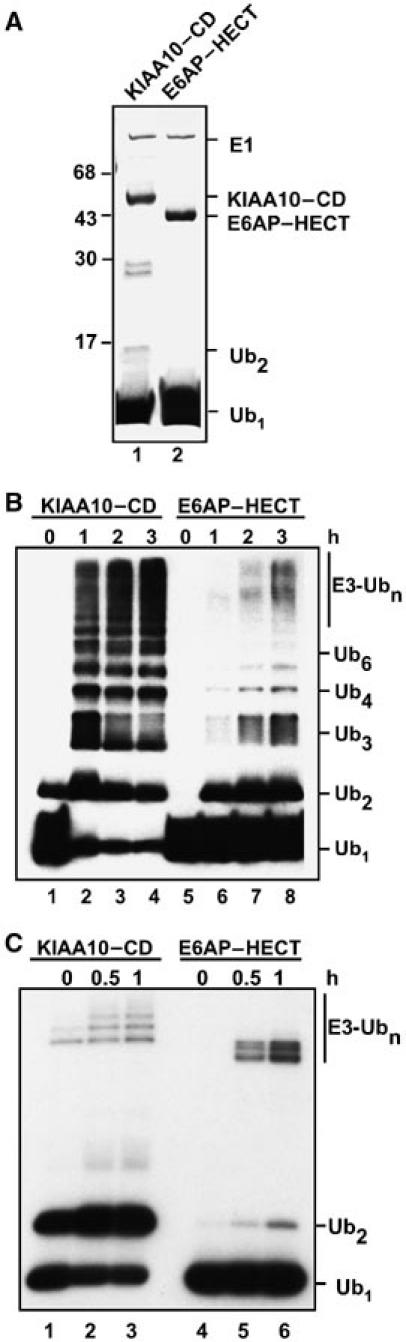
PolyUb chain synthesis activity of KIAA10–CD and E6AP–HECT proteins. Assays were conducted as described in Materials and methods. (A) Coomassie-stained gel of zero-time points from panel B confirms E3 purity and concentration. Lane 1, KIAA10–CD; lane 2, E6AP–HECT. (B) Time courses of steady-state chain synthesis assays with Ub76 as substrate (Ub Western blot; ECL detection). Lanes 1–4, KIAA10–CD; lanes 5–8, E6AP–HECT. (C) Steady-state Ub2 synthesis assay with Ub74 as acceptor (autoradiograph). Lanes 1–3, KIAA10–CD; lanes 4–6, E6AP–HECT.
Both the KIAA10–CD and E6AP–HECT proteins synthesized unanchored polyUb chains from wild-type Ub, hereafter called Ub76 (Figure 1B). The activity of each E3 enzyme depended strictly on the presence of E1, E2, and the respective HECT domain protein (data not shown.) However, there was a marked difference in the spectrum of chains produced by the two E3s. The KIAA10–CD protein produced substantial amounts of long chains (up to n∼9). In contrast, Ub2 was the principal chain product produced by the E6AP–HECT domain; longer chains, although detectable, were of lower abundance (Figure 1B). It will be shown below that these properties also apply to full-length E6AP, with the full-length E3 displaying a more robust chain synthesis activity than the isolated HECT domain.
As discussed above, the KIAA10–CD protein uses Ub-K29 or K48 as the linkage site during chain synthesis (You and Pickart, 2001) (Supplementary Figure 2A). To determine the linkage specificity of chain synthesis catalyzed by the E6AP–HECT domain, we tested a series of mutant Ubs carrying K29, K48, or K63 (the major known sites of chain synthesis) as the sole Lys residue (Arnason and Ellison, 1994; You et al, 1999). The E6AP–HECT domain strongly prefers to make K48-linked chains when using free Ub as the substrate (Supplementary Figure 2B). This specificity is consistent with the structure of chains produced during E6AP autoubiquitylation and p53 polyubiquitylation, and with the ability of E6AP to target substrates for proteasome degradation (Scheffner et al, 1993; Kumar et al, 1999; Oda et al, 1999; Glockzin et al, 2003).
Competence of free Ub as an acceptor distinguishes KIAA10–CD from E6AP–HECT
Previously, we developed a quantitative Ub2 synthesis assay in which one of the two Ub substrates is truncated at its C-terminus. Ub74 lacks the GlyGly dipeptide and therefore cannot be activated by E1. This variant binds with Km ∼10–50 μM to KIAA10–CD, serving as a functional acceptor for the transfer of 125I–Ub76 (Mastrandrea et al, 1999; You and Pickart, 2001). 125I–Ub2 is formed as the sole product because the high concentration of Ub74 prevents labeled Ub2 or Ub76 from occupying the acceptor-binding site and nucleating the formation of longer chains. With KIAA10–CD, this reaction is so rapid that significant Ub2 synthesis occurred within the mixing time in Figure 1C, left. Surprisingly, the E6AP–HECT domain displayed negligible activity in this assay even though it was active in Ub2 synthesis when Ub76 was the sole substrate (Figure 1C versus 1B, right sides).
The KIAA10–CD protein has a 60-residue N-terminal extension that is absent in the E6AP–HECT domain. To determine if the absence of this region explained the failure of Ub74 to be used as a substrate by the E6AP–HECT domain, we made a new construct that included the 60 upstream residues of E6AP. The E6AP–CD and E6AP–HECT proteins behaved identically in chain synthesis assays with Ub76 as substrate and displayed the same K48 linkage specificity (data not shown). However, neither protein showed significant activity in chain synthesis with Ub74 as acceptor (data not shown). Therefore, the absence of the N-terminal extension in E6AP–HECT cannot explain the functional distinction revealed in Figure 1C.
Mechanism of chain synthesis by E6AP–HECT domain
Figure 2 outlines three possible mechanisms for polyUb chain synthesis catalyzed by a HECT domain E3. In model A, a free chain is synthesized by the sequential addition of mono-Ub moieties. Here, a specific Lys residue of a noncovalently bound acceptor Ub attacks G76 of the E3-linked Ub. (In substrate polyubiquitylation, this acceptor Ub could be conjugated to the noncovalently bound substrate.) In model B, the acceptor Ub is covalently bound to the HECT Cys; its Lys residue attacks G76 in a second molecule of HECT–Ub thiol ester. In the simplest case, this mechanism would occur through an E3 homodimer (Figure 2B). In model C, the acceptor Ub is again bound covalently at the HECT active site, but it attacks Ub-G76 in the E2–Ub thiol ester intermediate, that is, model C involves an E2/E3 heterodimer. Model C corresponds to the mechanism proposed from recent structural studies of the WWP1–HECT domain (Verdecia et al, 2003). In a different version of model C, not shown, the polarity of bond synthesis is reversed. In models B and C, transfer of the pre-made chain to the substrate occurs subsequent to chain assembly at the E3 active site.
Figure 2.
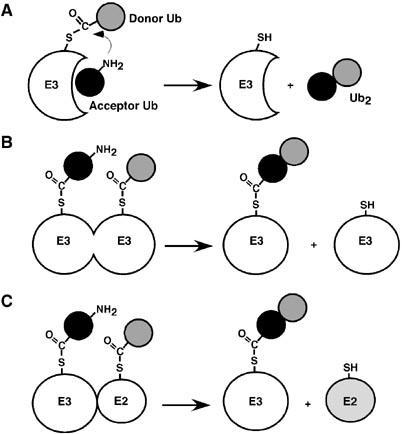
Potential mechanisms for polyUb chain synthesis catalyzed by a HECT domain E3. (A) Sequential model: because the acceptor Ub binds noncovalently, the chain is built up as a free entity. (The acceptor Ub molecule contributes the lysine residue to the isopeptide bond.) (B, C) As the acceptor Ub is covalently linked to the HECT cysteine, the chain is built up at the E3 active site. In these models, G76 must be present in the acceptor Ub so that this molecule can be linked to the HECT (or E2) active site. (B) A specific lysine side chain of this acceptor Ub attacks Ub-G76 in a different molecule of HECT–Ub thiol ester (in an E3 homodimer). (C) Alternatively, the lysine of the acceptor attacks Ub-G76 in the E2–Ub thiol ester (in an E2/E3 heterodimer).
The E6AP–HECT domain catalyzes chain synthesis only when Ub-G76 is present (Figure 1B versus C). Therefore, if model A applies, then G76 must be required for noncovalent binding of the acceptor Ub. So far, such a mode of interaction is known only for the deubiquitylating enzyme isopeptidase T, which discriminates strongly between Ub76 and Ub74 (Wilkinson et al, 1995). To address if the E6AP–HECT domain shows such selectivity, we tested the competence of Ub76 as an acceptor in a one-turnover pulse-chase assay. This method has been used previously to study chain synthesis by E2 enzymes and certain E2 complexes (Chen and Pickart, 1990; Hofmann and Pickart, 2001). In the present work, E6AP–HECT was first incubated briefly with a low concentration of 125I–Ub. During this pulse, most of the labeled Ub is transferred to the HECT Cys, as shown by depletion of free Ub and the appearance of a labile Ub adduct migrating slightly above the position of E6AP–HECT (Figure 3A, top, lanes 1, 3 versus 5). Following addition of EDTA to prevent further Ub activation, a high concentration of unlabeled Ub76 or Ub74 was added to serve as acceptor and samples were quenched in the absence of mercaptoethanol to preserve thiol ester intermediates. There was no transfer of the E3-bound, labeled Ub to either acceptor (Figure 3A, bottom), contrary to prediction from model A (Figure 2A). In contrast, there is robust Ub2 synthesis in similar experiments with E2–25K and the Mms2/Ubc13 complex (Chen and Pickart, 1990; Hofmann and Pickart, 2001). In the present case, most of the E6AP–HECT thiol ester hydrolyzed (Figure 3A, top, lanes 1, 3 versus 2, 4), while a fraction of the thiol-bound Ub was transferred to a lysine residue of E6AP–HECT, as indicated by the appearance of a mercaptoethanol-resistant adduct (lane 6). The failure to synthesize Ub2 in this experiment (Figure 3A) also argues against model B (Figure 2B), since there is no obvious reason why E6AP would have dimerized in Figure 1B but not in Figure 3A. Further evidence against a dimerization-dependent mechanism of chain synthesis comes from gel filtration data, which suggest that the E6AP–HECT domain is monomeric (Supplementary Figure 3A). Although we cannot exclude that E6AP–HECT undergoes transient dimerization under certain conditions, other results, presented below, argue against the relevance of such dimerization in free-chain synthesis.
Figure 3.
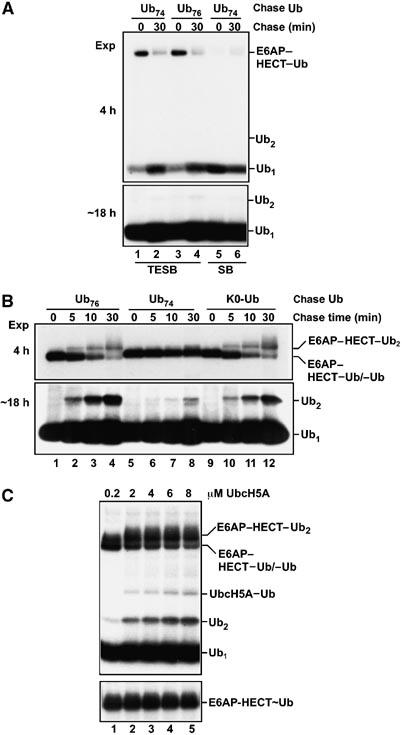
Requirements for Ub2 synthesis catalyzed by E6AP–HECT (pulse-chase experiments; autoradiographs). (A) One-turnover pulse-chase assays. The HECT–Ub thiol ester was formed in a pulse with a low concentration of 125I–Ub. Further ATP utilization was blocked with EDTA and the chase was initiated by adding a 50-fold excess of unlabeled Ub76 or Ub74. Reactions were quenched at the indicated times in buffer lacking (TESB) or containing (SB) mercaptoethanol. Top, short exposure focusing on HECT–Ub adducts. Bottom, long exposure confirming the absence of Ub2 synthesis. (B) Ub2 synthesis requires continued Ub activation and the presence of G76 in the acceptor Ub. ATP was maintained throughout chases with the indicated unlabeled Ub variants. Top, short exposure focusing on E3 adducts; bottom, long exposure showing Ub2 production. Reactions were quenched with TESB, but quenching with SB gave identical results (not shown), confirming that the HECT–Ub adducts are conjugates. (C) Distinct (E2) dependence of HECT–Ub thiol ester formation versus Ub2 production. Top, ATP-replete pulse-chase assays (20-min chase with K0–Ub) contained the indicated concentrations of UbcH5A and were quenched with TESB. Bottom, E6AP–HECT–Ub thiol ester band from a simultaneous experiment in which the chase contained Ub74; under this condition, the thiol ester is maintained during the chase (see A).
The negative results shown in Figure 3A suggest that E6AP–HECT-catalyzed chain synthesis could proceed through model C (Figure 2C). To gain positive evidence in favor of this model, we carried out conventional pulse-chase assays. Here, EDTA was omitted from the chase. Therefore, the continued presence of Mg·ATP supports activation of the (unlabeled) Ub added during the chase, which re-populates the E2–Ub thiol ester pool. Although multiple turnovers occur during the chase, only the first turnover is observed since we detect only the labeled Ub activated during the pulse. As shown in Figure 3B, addition of Ub76 under this condition resulted in the synthesis of labeled Ub2 (bottom, lanes 1–4). Note that only one of the two constituent Ubs is labeled; therefore, the intensity of this band underestimates, by two-fold, the Ub2 concentration. As expected from Figure 3A, minimal Ub2 was produced in chases with Ub74 (Figure 3B, bottom, lanes 5–9). Thus, the Ub2 product derives from the Ub76 added in the chase and not from free labeled Ub carried over from the pulse. To exclude the possibility that labeled Ub produced by hydrolysis during the chase (cf. Figure 3A) contributed significantly to labeled Ub2 synthesis, we showed that delaying the addition of 125I–Ub until the chase caused a severe inhibition of 125I–Ub2 synthesis, as expected given the strong reduction in specific radioactivity caused by the addition of a 50-fold excess of unlabeled Ub (Supplementary Figure 4). Taken together, these results show that continued Ub activation and the presence of G76 in the acceptor Ub are both needed for E6AP–HECT-dependent synthesis of Ub2.
These requirements can be explained if Ub2 comes from the reaction of two thiol-linked Ubs—one bound at the E2 active site and the other at the E3 active site. Although data presented so far do not indicate which Ub contributes the Lys residue to the isopeptide bond, we found that Ub2 synthesis was largely unimpeded when we chased with Lys-less Ub (K0–Ub; Figure 3B, bottom, lanes 9–12). This outcome is only possible if a Lys residue of the E3-linked Ub attacks G76 of the E2-linked Ub, as shown in Figure 2C. The initial product of this reaction will be an E3–Ub2 thiol ester, but we could not detect this intermediate. An E6AP–HECT–Ub2 adduct was formed during chases with full-length Ub (Figure 3B, top), but it was a conjugate rather than a thiol ester as indicated by resistance to mercaptoethanol (data not shown). The data shown in Figure 3A and B suggest that the initial HECT–Ub2 thiol ester is rapidly discharged through competing processes of hydrolysis (yielding free Ub2) and transfer to a Lys residue of the E6AP–HECT domain (yielding the HECT–Ub2 conjugate). Evidence presented below suggests that Ub2 can also be discharged to a substrate protein.
Model C (Figure 2) predicts that a heterodimeric HECT–Ub/E2–Ub thiol ester complex, rather than a simple HECT–Ub thiol ester, is the key intermediate in chain synthesis catalyzed by the E6AP–HECT domain. In accordance with this prediction, the yield of free Ub2 (and HECT–Ub2 conjugate) produced in these pulse-chase assays depended strongly on the concentration of UbcH5A (Figure 3C, top) even though the level of HECT–Ub thiol ester was already maximal at the lowest E2 concentration used in this experiment (Figure 3C, bottom). Thus, the E2–Ub thiol ester plays a critical role in E6AP–HECT-catalyzed chain synthesis that is distinct from its function in delivering activated Ub to the E3 active site. This result also argues that Ub2 synthesis does not occur through the reaction of two E3-linked Ub molecules (Figure 2B) in a stable or transient dimer.
Mechanism of chain synthesis catalyzed by KIAA10–CD domain
As we could not detect the KIAA10–CD–Ub thiol ester, we were unable to perform pulse-chase experiments with this protein. However, the competence of Ub74 as an acceptor in steady-state assays (Figure 1C) rigorously excludes any model in which the acceptor Ub must be covalently linked to the E2 (or E3) active site. We conclude that the KIAA10–CD protein employs a simple sequential mechanism by which the acceptor Ub binds noncovalently to the HECT–Ub thiol ester (Figure 2A).
Mechanistic distinctions between full-length HECT E3s
We next addressed if the above-described properties of the E6AP–HECT domain extend to full-length E6AP. Proper folding of the purified, bacterially expressed enzyme was confirmed by its ability to ubiquitylate a cognate substrate, HHR23A (Figure 4A and B, lane 4), and its robust activity in self-ubiquitylation (Figure 4A and C, lanes 2, 4). Importantly, full-length E6AP produced K48-linked Ub2 as the principal chain product in steady-state chain synthesis assays with Ub76, as detected by Coomassie staining and Western blotting (Figure 4A and C, lanes 2, 4; Supplementary Figure 2C), while Ub3 and Ub4 were detectable by Western blotting (Figure 4C). The chain synthesis results are very similar to those obtained with the E6AP–HECT domain (Figure 1B), although the chain synthesis activity of the full-length E3, like its autoubiquitylation activity, is more robust. Our finding that UbcH5A and E6AP can produce strong ubiquitylation of HHR23A contrasts with an earlier report that UbcH5A is an inefficient partner of E6AP (Kumar et al, 1997). We speculate that the earlier result reflected the use of in vitro-translated UbcH5A, since wheat germ lysate contains endogenous Ubc5-family E2s that could have competed for access to E6AP (Scheffner et al, 1994). The ability of submicromolar concentrations of UbcH5A to support the ubiquitylation of HHR23A suggests that UbcH5A binds efficiently to E6AP. However, the stability of the resulting complex may be lower than that of the E6AP/UbcH7 complex.
Figure 4.
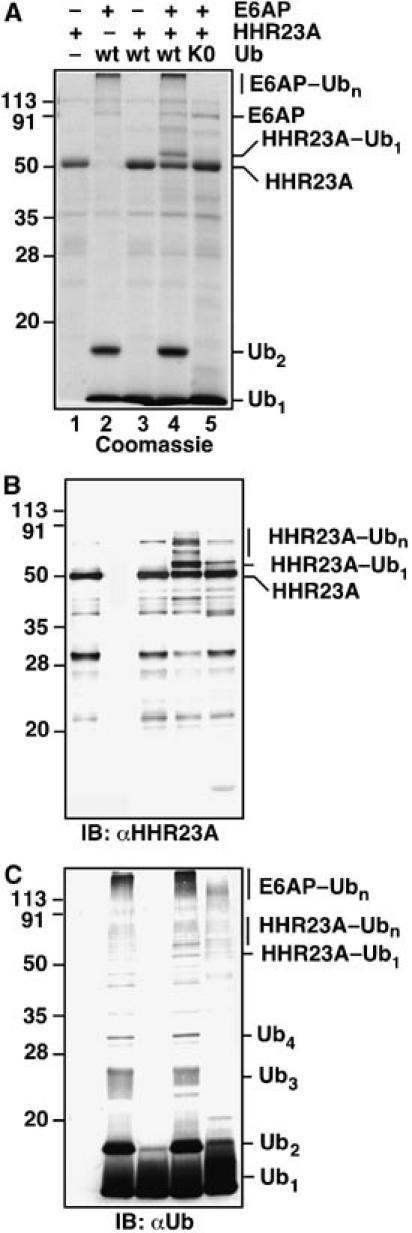
Full-length E6AP catalyzes Ub2 synthesis, self-ubiquitylation, and HHR23A ubiquitylation. (A) Coomassie-stained gel. The incubations contained 0.1 μM E1, 0.2 μM UbcH5A, 0.4 μM E6AP, and 234 μM wt Ub or K0–Ub. Where indicated, 2 μM HHR23A was added. The same samples were analyzed by Western blotting against HHR23A (B) and Ub (C), with colorimetric detection.
To characterize the mechanism of chain synthesis by full-length E6AP more fully, we again turned to pulse-chase assays. We could not use precisely the same assay procedure as in Figure 3, however, because of unexpected discrimination by full-length E6AP against K0–Ub and 125I–Ub. We found that E6AP was virtually unable to transfer K0–Ub either to itself or to HHR23A (Figure 4A–C, lane 5). Similar findings have been reported previously (Glockzin et al, 2003; E Cooper and P Howley, personal communication). 125I–Ub supported the self-ubiquitylation of E6AP, but it was a poor substrate for transfer to HHR23A (data not shown). To circumvent these idiosyncrasies, we replaced 125I–Ub with unlabeled Ub76 in the pulse, and replaced K0–Ub with Ub-K48R in the chase. These procedural modifications were feasible because E6AP uses only K48 in chain synthesis, making Ub-K48R functionally equivalent to K0–Ub (Supplementary Figure 2C).
Free-chain synthesis data from these pulse-chase assays are shown in Figure 5A. When the pulse contained wild-type Ub76, addition of Ub-K48R in the chase was permissive for Ub2 production (lanes 2–4). No Ub2 was observed when Ub-K48R was substituted for wild-type Ub in the pulse (Figure 5A, lanes 8, 9), consistent with the isopeptide Lys (K48) deriving from the E3-linked Ub. Moreover, Ub2 was virtually absent when Ub74 was substituted for Ub-K48R during the chase (lanes 5–7), indicating that Ub2 production depends on de novo activation of Ub-K48R in the chase, rather than carryover of Ub76 or E2–Ub from the pulse. The results shown in Figures 4 and 5A suggest that full-length E6AP, like its isolated HECT domain, synthesizes Ub2 by the mechanism shown in Figure 2C. We showed in an earlier work that the chain synthesis activity of full-length KIAA10 closely mimics the activity of the isolated C-domain. The important point for the present study is that the full-length enzyme efficiently uses Ub74 as an acceptor in chain synthesis (You and Pickart, 2001). Therefore, full-length KIAA10 and E6AP, like their respective CD and HECT domains, employ distinct mechanisms in the synthesis of polyUb chains (Figure 2A and C, respectively).
Figure 5.
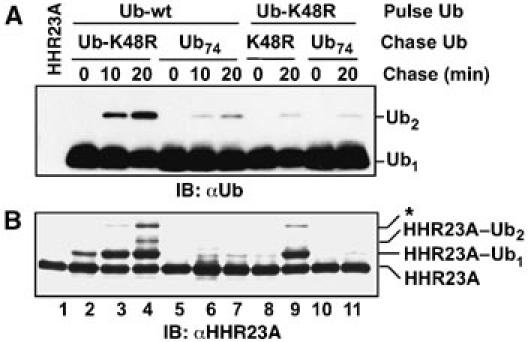
Molecular mechanism of chain synthesis by full-length E6AP (Western blots). (A) The pulse (3 min) contained 0.2 μM E1, 5 μM UbcH5A, 5 μM E6AP, and 5 μM of wild-type Ub or Ub-K48R (as indicated). The chase was initiated by adding Ub-K48R or Ub74 (117 μM, as indicated) together with HHR23A (1 μM). ATP was maintained during the chase. Samples were quenched at the indicated times with SB and analyzed by blotting against Ub (ECL detection). (B) The procedure was identical to (A), except that the E2 was UbcH7 and blotting was against HHR23A (colorimetric staining). *See the text. Detectable reaction occurred in the mixing time in the experiment shown in lanes 2–4.
We also monitored the ubiquitylation of HHR23A by E6AP during a second set of pulse-chase studies. Here we replaced UbcH5A with UbcH7, because preliminary experiments indicated that UbcH7 was a more efficient partner of E6AP in HHR23A conjugation assays. As a control, we first showed that synthesis of free Ub2 by E6AP–HECT was readily observable with UbcH7 (Supplementary Figure 5), as expected from results obtained with UbcH5A (Figure 3C). As shown in Figure 5B, mono- and diubiquitylated HHR23A were produced as major and secondary products, respectively, by full-length E6AP/UbcH7 when the chase contained Ub-K48R (Figure 5B, lanes 2–4). Although the level of HHR23A–Ub2 was modest, it is important to note that it is limited by the low concentration of wt Ub in the experiment (5% of total Ub). The HECT–Ub thiol ester formed in the pulse with full-length E6AP is also discharged through rapid self-ubiquitylation of the E3 (Figure 4C), further limiting the yield of HHR23A products. The formation of HHR23A–Ub2 was strongly diminished when Ub-K48R was substituted for wild-type Ub in the pulse (lane 9). This result suggests that the HHR23A–Ub2 product originates through the mechanism discussed above (Figure 2C). The low yield of HHR23A–Ub2 in lane 9 also shows that HHR23A–Ub2 (lane 4) is principally modified with a Ub2 chain and not with two Ub-K48R molecules linked to different lysines of HHR23A. A higher-mass band (asterisk; Figure 5B), whose intensity was significantly diminished when the pulse contained Ub-K48R, may represent a combination of HHR23A carrying three mono-Ub moieties and HHR23A carrying one mono-Ub and a Ub2 chain conjugated to a distinct lysine of HHR23A. That almost no HHR23A-derived products were produced when Ub74 was used in the chase (Figure 5B, lanes 5–7) indicates that most of the HHR23A–Ub1 seen in lanes 3 and 4 arose from Ub-K48R activation and conjugation during the chase. This is not unexpected given the 20-fold excess of Ub-K48R (chase) over Ub76 (pulse). Taken together, the results shown in Figure 5 suggest that full-length E6AP employs the same mechanism of chain synthesis as the E6AP–HECT domain and that this mechanism is relevant in substrate polyubiquitylation.
The N-terminus of KIAA10–CD contributes to acceptor Ub binding
Our results indicate that KIAA10 possesses a noncovalent Ub binding site that is competent for free-chain synthesis, while E6AP lacks such a site. We noticed that the N-terminal 60 amino acids of KIAA10–CD and E6AP–CD are poorly conserved compared to their HECT domains (no significant similarity and 54% similarity, respectively). To test if the N-terminal part of the KIAA10–CD protein contributes to the mechanistic distinction revealed in this work, we fused the first 60 residues of KIAA10–CD to the N-terminus of the E6AP–HECT domain (Figure 6A). Remarkably, the chimeric protein displayed catalytic properties that were intermediate between E6AP–HECT and KIAA10–CD. (As discussed above, the E6AP–HECT and E6AP–CD proteins behaved identically.) Unlike the E6AP–HECT domain, which produced Ub2 as the main product in steady-state assays with Ub76, the chimera produced a ladder of chains similar to that seen using KIAA10–CD (compare Figure 6B to Figure 1B). In pulse-chase assays (plus ATP), the chimera produced Ub2 when the chase contained K0–Ub (Figure 6C, lanes 1–4 versus 9–12); this is diagnostic for a pathway involving tethered chain synthesis, as seen with the E6AP–HECT domain (Figure 2C). However, the chimera also produced Ub2 when the chase contained Ub74 (Figure 6C, lanes 5–8). Competence of Ub74 as an acceptor is diagnostic for a KIAA10-like mechanism of chain synthesis (Figure 2A). Therefore, the chimera possesses a noncovalent Ub-binding site, which is either contained within the 60 KIAA10-derived residues or forms through interactions between this region and the E6AP–HECT domain. Our results show that this fusion protein is chimeric from both structural and mechanistic perspectives—that is, it can build up the chain either as a free or a tethered entity (Figure 2A and C). However, despite having acquired several KIAA10-like properties, the chimera was unable to use Ub–K29 as a chain linkage site (data not shown).
Figure 6.
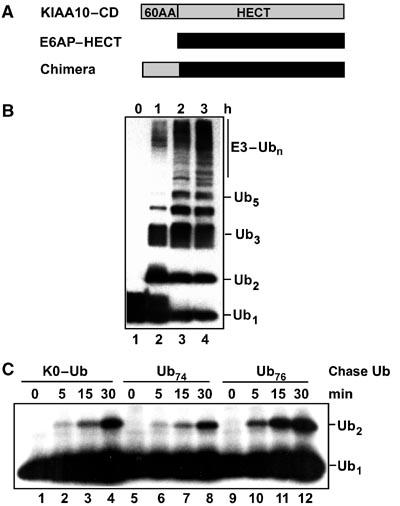
The N-terminus of KIAA10–CD contributes to acceptor Ub binding. (A) Schematic structure of KIAA10/E6AP chimera. (B) Steady-state chain synthesis activity of chimera (Ub76 substrate; Western blot). Samples were quenched in SB after the indicated incubation times and analyzed by blotting against Ub (ECL detection). (C) ATP-supplemented pulse-chase assay (autoradiograph). The HECT–Ub thiol ester was formed in a short pulse with 125I–Ub prior to initiating the chase with a high concentration of the indicated (unlabeled) Ub variant (as in Figure 3C).
Discussion
Substrate selection by E3 enzymes is now understood in considerable molecular detail and increasingly, at atomic resolution. Substrate binding is mediated by complementary interaction motifs presented by the E3 and its cognate substrates. For example, PPXY motifs in the amiloride-sensitive sodium channel are bound by WW domains in the N-terminal region of the HECT E3 Nedd4 (Kanelis et al, 2001; Ingham et al, 2004). By comparison, the molecular events that transpire during E3 catalysis remain poorly characterized. This is true even though the vast majority of E3s utilize one of just two catalytic modules and in spite of the availability of high-resolution structures of RING/E2 and HECT/E2 complexes (Huang et al, 1999; Zheng et al, 2000; Brzovic et al, 2003; Dominguez et al, 2004).
A major challenge in understanding E3 catalysis lies in the apparent necessity for large conformational transitions to achieve catalytic competence. Although the E2 is known to transfer Ub directly to the HECT Cys (Scheffner et al, 1995), the structures of the E6AP–HECT/UbcH7 complex and a Smurf–HECT/UbcH7 model show separations of 41–50 Å between the E2 and E3 active-site Cys residues (Huang et al, 1999; Ogunjimi et al, 2005). A partial resolution of this puzzle was provided by the crystal structure of the WWP1–HECT domain, which showed that, relative to E6AP, rotation about a hinge region had occurred, resulting in a predicted separation of only 16 Å between the E2 and E3 Cys residues (Verdecia et al, 2003). Continued rotation along the same pathway could bring these two residues to within 6 Å of each other—a distance that might reasonably be bridged by local reorganization of the active sites. Verdecia and co-workers proposed that these motions could be coupled to the assembly of a polyUb chain during repeated catalytic cycles. They suggested that the chain is built up at the E3 active site through successive transfers of E2-bound Ub to the E3-bound Ub/chain. With each successive catalytic cycle, the distal Ub's target lysine is more distant from the HECT Cys, such that less displacement of the E2–Ub thiol ester is needed to reach the Lys. At some point, the growing chain is released through the attack of a substrate Lys side chain on the HECT active site.
To our knowledge, we have provided the first direct evidence supporting this model. We found that synthesis of K48-linked Ub2 catalyzed by the E6AP–HECT domain occurs through the reaction of two covalently bound Ubs: one linked to the E2 and the other linked to the E3. This key conclusion derives from several lines of evidence. First, the pre-formed E6AP–HECT–Ub thiol ester is unable to discharge its covalently bound Ub to an acceptor Ub that cannot form a thiol ester intermediate. This is shown by blockade of Ub2 synthesis in the absence of ATP hydrolysis (Figure 3A) or when the acceptor Ub lacks its C-terminal GlyGly moiety (Figures 1C, and 3A and B). Moreover, HECT–Ub thiol ester formation and HECT-catalyzed Ub2 synthesis depend differently on E2 concentration (Figure 3C). E3 charging is maximal at highly substoichiometric E2 levels, whereas optimal rates of chain synthesis require higher E2 concentrations. This difference shows that the function of the E2 in chain synthesis is not limited to loading the HECT active site with Ub; it is consistent with chain synthesis occurring through an E2/E3 heterodimer. That Ub2 synthesis still occurs when the E2-bound Ub lacks lysines (Figure 3B and Supplementary Figure 5) reveals the directionality of the chemical step: K48 of the E3-bound Ub attacks G76 of the E2-bound Ub. These results are in precise agreement with prediction from the model proposed by Verdecia et al (2003). Importantly, full-length E6AP appears to catalyze the synthesis of K48-linked Ub2 by a similar mechanism (Figures 4A and C, and 5A).
Free-chain synthesis by E6AP occurs exclusively through this mechanism. However, it is unlikely that free-chain synthesis is a major activity of E6AP because this reaction is slow and falls off steeply above n=2 in reactions with wt Ub (Figures 1B, 3B, and 4A). Since it would be wasteful to allow unregulated chain assembly, it is possible that elongation and discharge of the active-site-linked chain are normally held in check, pending the availability of a bound substrate. This could help to explain our unexpected failure to observe the E6AP–HECT–Ub2 thiol ester directly (Figure 3B). Interestingly, autoubiquitylation of E6AP–HECT occurred more readily when Ub2, versus Ub1, was bound at the active site (Figure 3A versus B and C), suggesting a possible regulatory relationship between chain length and self-ubiquitylation. The greater lability of the Ub2 thiol ester likely contributed to the difficulty of its detection.
We investigated the relevance of tethered chain assembly for substrate polyubiquitylation using the E6AP substrate HHR23A. The production of HHR23A–Ub2 correlated with the production of free Ub2 according to the mechanism described above, consistent with the idea that HHR23A modification relies on the transfer of Ub2. (Due to the design of this experiment, HHR23A–Ub2 was the most extensively modified product that could have been formed; so the failure to observe extensive polyubiquitylation sheds no light on the potential regulatory mechanism discussed above.) Our finding that Ub2 can be transferred from the E6AP active site to HHR23A (Figure 5B) argues that tethered chains are competent in substrate polyubiquitylation, but more work is needed to determine if this is the major mechanism. It will be critical, in the future, to show that tethered chains can yield extensively polyubiquitylated substrates. HHR23A is probably not the best substrate for detecting long-chain transfer from E6AP because its Ub-associated domains could bind the chain linked to the E3 active site and impede elongation or transfer (Ortolan et al, 2000; Raasi and Pickart, 2003; E Cooper and P Howley, personal communication). Consistent with this idea, the principal products of HHR23A conjugation are oligoubiquitylated, rather than polyubiquitylated (Figure 4B) (Kumar et al, 1999). Attempts to circumvent this problem by using p53 as a substrate (in conjunction with HPV-E6) were defeated by the presence of multiple target lysines in p53 (Glockzin et al, 2003), which severely complicated the interpretation of pulse-chase experiments. In summary, further work is needed to establish the generality and significance of the tethered chain synthesis mechanism.
Remarkably, KIAA10 uses a different mechanism of chain synthesis than E6AP, even though both E3s belong to the HECT domain family. In the case of KIAA10, a noncovalently bound acceptor Ub (or chain) is the substrate for sequential Ub transfer events. The key feature that enables this mechanism to operate with KIAA10, but not with E6AP, is the existence of a noncovalent Ub-binding site that gives the target lysine access to the active site. Fusing the first 60 residues of KIAA10–CD to E6AP–HECT generated a chimeric protein able to synthesize chains (in part) by a KIAA10-like mechanism (Figure 6B and C), indicating that this region of KIAA10 harbors a cis-determinant of acceptor Ub binding. Studies are in progress to address whether this region, which lacks any known Ub-binding motif, binds Ub directly. Interestingly, while E6AP–HECT behaved as a monomer (Supplementary Figure 3A), the chimeric protein displayed an apparent molecular mass of ∼80 kDa, suggestive of dimerization or a highly asymmetric conformation (Supplementary Figure 3B). KIAA10–CD shares this property (You and Pickart, 2001).
The mechanism of chain synthesis used by KIAA10 resembles that used by two other well-characterized chain synthesis factors, both of which are E2 enzymes. In vitro, E2–25K and the Mms2/Ubc13 complex both employ Ub74 as an acceptor for Ub transfer, producing unanchored K48- and K63-linked chains, respectively (Chen and Pickart, 1990; Hofmann and Pickart, 2001). This is the only possible mechanism for these factors since each of them has just one covalent binding site for Ub. In contrast, KIAA10 binds UbcH5A strongly (You and Pickart, 2001). This interaction has the potential to provide a second covalent binding site for Ub, which could enable KIAA10 to use an E6AP-like mechanism under conditions not yet identified. KIAA10 is highly expressed in skeletal muscle and has at least one (non-Ub) cognate substrate that binds to its N-terminal domain, in accordance with expectation for a HECT E3 (You et al, 2003). One interesting possibility is that KIAA10 uses an E6AP-like mechanism when acting on a non-Ub substrate, but a different mechanism when using Ub as the sole substrate. The physiological relevance of free-chain synthesis by KIAA10 is unknown. Interestingly, KIAA10 and yeast Hul5 can bind to the proteasome's regulatory complex, suggesting that they could function in an E4-like manner to modulate the length or topology of chains linked to proteasome-targeted substrates (Koegl et al, 1999; You and Pickart, 2001; Leggett et al, 2002; You et al, 2003). As such a role is inherently nonspecific with respect to the target protein, it requires the E3 to engage in a productive noncovalent interaction with the acceptor Ub (as observed), which in this case would be linked to a target protein.
HECT E3 enzymes are defined by the presence of diverse N-terminal regions and conserved HECT domains. This conjunction of a unique substrate-interacting domain and a conserved catalytic domain, which is mirrored in the much larger RING E3 family, has led to a modular model of E3 action. A key feature of this model is that it assigns the molecular determinants of functional specificity to the substrate-interacting domain. While the general features of the modular model are supported by overwhelming evidence, including the ability to design E3s with novel specificities (Zhou, 2005), it is likely that the individual domains within a given E3 family have also evolved to perform specific functions. For example, the N-domain of Smurf binds an accessory factor that modulates E2 binding to this E3's HECT domain (Ogunjimi et al, 2005). In addition, domain-swapping studies showed that one HECT domain could not be substituted for another (Schwarz et al, 1998). This functional nonequivalence can be explained if the HECT domain plays a role in substrate binding or if there are functionally significant interactions between the N-terminal and HECT domains (Huibregtse et al, 1993; Kao et al, 2000). The results reported here offer another possibility—that the HECT catalytic module is a site of functional specialization that specifies both the topology of the chain product and the mode of its assembly.
Materials and methods
Plasmids and cloning
Plasmids specifying GST–E6AP–HECT (residues 495–852 of human E6AP) and GST–E6AP were from A Weissman, NIH (Hatakeyama et al, 1997). We amplified the E6AP–CD ORF (residues 437–852) with flanking sites for ligation into the pGEX* vector (Haldeman et al, 1997). A cDNA specifying the chimeric KIAA10/E6AP–CD protein (residues 656–714 of human KIAA10 fused to the N-terminus of E6AP–HECT) was created by overlap extension PCR.
Protein expression and purification
Ub proteins were expressed in Escherichia coli strain BL21(DE3)pJY2, and Ub76/Ub74 were purified, as described (Haldeman et al, 1997; You et al, 1999). K0–Ub (Lys-less) and the single-Lys mutants were purified avoiding strong acid precipitation (You et al, 1999). Ub76 was radioiodinated using chloramine T. UbcH7 was from BostonBiochem. Affinity-purified polyclonal anti-Ub antibodies were produced in rabbits (Haas and Bright, 1985). Anti-HHR23A serum was a gift of P Howley, Harvard Med. Sch. Immune complexes were detected using enhanced chemiluminescence (ECL) or colorimetric (alkaline phospatase) staining.
E1 was produced as described (Raasi and Pickart, 2003). GST–HHR23A, GST–UbcH5A, GST–E6AP–HECT, GST–E6AP–CD, and GST–chimera were expressed as soluble proteins in E. coli strain BL21(DE3)pJY2 at 30°C according to published procedures (You and Pickart, 2001). A soluble full-length GST–E6AP fusion protein was expressed using a heat-shock/low-temperature procedure (Russell and Wilkinson, 2004). In general, cell pellets were frozen overnight at −80°C, thawed, suspended in 2 ml/g of lysis buffer, and used to make a soluble extract (Haldeman et al, 1997). GST fusion proteins were absorbed onto GSH beads (Sigma) prior to releasing the protein by incubating the beads with 20 mM Tris–HCl (pH 7.6), 0.1 mM EDTA, 0.2 mM DTT, and 100 U/ml thrombin (Amersham-Pharmacia Biotech) using 1 ml buffer/mg fusion protein. Thrombin was sometimes removed by cation exchange at pH 7.6. KIAA10–CD was produced from refolded GST–KIAA10–CD as described (You and Pickart, 2001).
Steady-state chain synthesis assay (Ub76 substrate)
Assays (37°C) employed 0.1 μM E1, ∼0.2 μM UbcH5A, and ∼4 μM of the specified E3. Assays were initiated by adding a reaction cocktail contributing (final concentrations): 50 mM Tris–HCl (pH 7.6), 5 mM MgCl2, 2 mM ATP (plus regenerating system), 0.3 U/ml inorganic pyrophosphatase, 0.5 mM DTT, and 117 μM Ub76 (1 mg/ml, wild type or single lysine). Reactions were incubated for up to 4 h, quenched with mercaptoethanol-containing sample buffer (SB), and resolved by SDS–PAGE. Products were detected by Coomassie staining or Western blot.
Steady-state Ub2 synthesis assay (Ub74 acceptor)
Ub2 synthesis was assayed as described in the preceding paragraph, except that (E3) was 1 μM and Ub76 was replaced with 117 μM Ub74 and 2 μM 125I–Ub. Products were resolved by SDS–PAGE and visualized by autoradiography.
Pulse-chase assays
Single-turnover pulse-chase assays combined 0.1 μM E1 with 2 μM UbcH5A (or UbcH7) and ∼4 μM E3 (see figure legends). Assays were initiated by adding reaction cocktail (above, except lacking Ub) together with 2 μM 125I–Ub. After 3 min at 37°C, EDTA (0.1 M) was added to quench Ub activation. Ub74 or Ub76 (1 mg/ml) was added and incubation was continued. Aliquots removed at increasing times were usually quenched with a modified sample buffer lacking mercaptoethanol (TESB) to preserve enzyme–Ub thiol esters. Products were detected by SDS–PAGE/autoradiography. ATP-replete pulse-chase assays were conducted in the same manner, except without the EDTA quench (where indicated, K0–Ub was employed in the chase). In some cases, HHR23A (1 μM) was also added at the start of the chase.
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Acknowledgments
This research was funded by a grant from the NIH (DK46984). We thank P Howley for a generous gift of anti-HHR23A antibodies, A Weissman for plasmid reagents, J Huibregste for help with experiments involving p53, and E Cooper and P Howley for communicating unpublished findings. We thank E Cooper and R Cohen for comments on the manuscript.
References
- Arnason T, Ellison MJ (1994) Stress resistance in Saccharomyces cerevisiae is strongly associated with assembly of a novel type of multiubiquitin chain. Mol Cell Biol 14: 7876–7883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brzovic PS, Keeffe JR, Nishikawa H, Mayamoto K, Fox D, Fukuda M, Ohta T, Klevit R (2003) Binding and recognition in the asembly of an active BRCA1–BARD1 ubiquitin ligase complex. Proc Natl Acad Sci USA 100: 5646–5651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Pickart CM (1990) A 25-kilodalton ubiquitin carrier protein (E2) catalyzes multi-ubiquitin chain synthesis via lysine-48 of ubiquitin. J Biol Chem 265: 21835–21842 [PubMed] [Google Scholar]
- Cooper EM, Hudson AW, Amos J, Wagstaff J, Howley PM (2004) Biochemical analysis of Angelman syndrome-associated mutations in the E3 ubiquitin ligase E6-associated protein. J Biol Chem 279: 41208–41217 [DOI] [PubMed] [Google Scholar]
- Dominguez C, Bonvin AM, Winkler GS, van Schaik FM, Timmers HT, Boelens R (2004) Structural model of the UbcH5B/CNOT4 complex revealed by combining NMR, mutagenesis, and docking approaches. Structure 12: 633–644 [DOI] [PubMed] [Google Scholar]
- Glockzin S, Ogi F-X, Hengstermann A, Scheffner M, Blattner C (2003) Involvement of the DNA repair protein hHR23 in p53 degradation. Mol Cell Biol 23: 8960–8969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas AL, Bright PM (1985) The immunochemical detection and quantitation of intracellular ubiquitin–protein conjugates. J Biol Chem 260: 12464–12473 [PubMed] [Google Scholar]
- Haldeman MT, Xia G, Kasperek EM, Pickart CM (1997) Structure and function of ubiquitin conjugating enzyme E2–25K: the tail is a core-dependent activity element. Biochemistry 36: 10526–10537 [DOI] [PubMed] [Google Scholar]
- Hatakeyama S, Jensen JP, Weissman AM (1997) Subcellular localization and ubiquitin-conjugating enzyme (E2) interactions of mammalian HECT family ubiquitin ligases. J Biol Chem 272: 15085–15092 [DOI] [PubMed] [Google Scholar]
- Hicke L, Dunn R (2003) Regulation of membrane protein transport by ubiquitin and ubiquitin-binding proteins. Annu Rev Cell Dev Biol 19: 141–172 [DOI] [PubMed] [Google Scholar]
- Hofmann RM, Pickart CM (2001) In vitro assembly and recognition of K63 polyubiquitin chains. J Biol Chem 276: 27936–27943 [DOI] [PubMed] [Google Scholar]
- Huang L, Kinnucan E, Wang G, Beaudenon S, Howley PM, Huibregtse JM, Pavletich NP (1999) Structure of an E6AP–UbcH7 complex: insights into ubiquitination by the E2–E3 enzyme cascade. Science 286: 1321–1326 [DOI] [PubMed] [Google Scholar]
- Huibregtse JM, Scheffner M, Beaudenon S, Howley PM (1995) A family of proteins structurally and functionally related to the E6-AP ubiquitin–protein ligase (published erratum appears in Proc Natl Acad Sci USA 1995; 92: 5249). Proc Natl Acad Sci USA 92: 2563–2567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huibregtse JM, Scheffner M, Howley PM (1991) A cellular protein mediates association of p53 with the E6 oncoprotein of human papillomavirus types 16 or 18. EMBO J 10: 4129–4135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huibregtse JM, Scheffner M, Howley PM (1993) Cloning and expression of the cDNA for E6-AP, a protein that mediates the interaction of the human papillomavirus E6 oncoprotein with p53. Mol Cell Biol 13: 775–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingham RJ, Gish G, Pawson T (2004) The Nedd4 family of E3 ubiquitin ligases: functional diversity within a common modular architecture. Oncogene 23: 1972–1984 [DOI] [PubMed] [Google Scholar]
- Kanelis V, Rotin D, Forman-Kay JD (2001) Solution structure of a Nedd4 WW domain-ENaC peptide complex. Nat Struct Biol 8: 407–412 [DOI] [PubMed] [Google Scholar]
- Kao WH, Beaudenon SL, Talis AL, Huibregtse JM, Howley PM (2000) Human papillomavirus type 16 E6 induces self-ubiquitination of the E6AP ubiquitin–protein ligase. J Virol 74: 6408–6417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishino T, Lalande M, Wagstaff J (1997) UBE3A/E6-AP mutations cause Angelman syndrome. Nat Genet 15: 70–73 [DOI] [PubMed] [Google Scholar]
- Koegl M, Hoppe T, Schlenker S, Ulrich HD, Mayer TU, Jentsch S (1999) A novel ubiquitination factor, E4, is involved in multiubiquitin chain assembly. Cell 96: 635–644 [DOI] [PubMed] [Google Scholar]
- Kuhne C, Banks L (1998) E3-ubiquitin ligase/E6-AP links multicopy maintenance protein 7 to the ubiquitination pathway by a novel motif, the L2G box. J Biol Chem 273: 34302–34309 [DOI] [PubMed] [Google Scholar]
- Kumar S, Kao WH, Howley PM (1997) Physical interaction between specific E2 and Hect E3 enzymes determines functional cooperativity. J Biol Chem 272: 13548–13554 [DOI] [PubMed] [Google Scholar]
- Kumar S, Talis AL, Howley PM (1999) Identification of HHR23A as a substrate for E6-associated protein-mediated ubiquitination. J Biol Chem 274: 18785–18792 [DOI] [PubMed] [Google Scholar]
- Leggett DS, Hanna J, Borodovsky A, Crosas B, Schmidt M, Baker RT, Walz T, Plough H, Finley D (2002) Multiple associated proteins regulate proteasome structure and function. Mol Cell 10: 495–507 [DOI] [PubMed] [Google Scholar]
- Mastrandrea LD, You J, Niles EG, Pickart CM (1999) E2/E3-mediated assembly of lysine 29-linked polyubiquitin chains. J Biol Chem 274: 27299–27306 [DOI] [PubMed] [Google Scholar]
- Matsuura T, Sutcliffe JS, Fang P, Galjaard R-J, Jiang Y-h, Benton CS, Rommens JM, Beaudet AL (1997) De novo truncating mutations in E6-AP ubiquitin–protein ligase gene (UBE3A) in Angelman syndrome. Nat Genet 15: 74–77 [DOI] [PubMed] [Google Scholar]
- Oda H, Kumar S, Howley PM (1999) Regulation of the Src family tyrosine kinase Blk through E6AP-mediated ubiquitination. Proc Natl Acad Sci USA 96: 9557–9562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogunjimi AA, Briant DJ, Pece-Barbara N, Le Roy C, Di Guglielmo GM, Kavsak P, Rasmussen RK, Seet BT, Sicheri F, Wrana JL (2005) Regulation of Smurf2 ubiquitin ligase activity by anchoring the E2 to the HECT domain. Mol Cell 19: 297–308 [DOI] [PubMed] [Google Scholar]
- Orlicky S, Tang X, Willems A, Tyers M, Sicheri F (2003) Structural basis for phosphodependent substrate selection and orientation by the SDFCdc4 ubiquitin ligase. Cell 112: 243–256 [DOI] [PubMed] [Google Scholar]
- Ortolan TG, Tongaonkar P, Lambertson D, Chen L, Schauber C, Mardura K (2000) The DNA repair protein Rad23 is a negative regulator of multi-ubiquitin chain assembly. Nat Cell Biol 2: 601–608 [DOI] [PubMed] [Google Scholar]
- Petroski MD, Deshaies RJ (2005) Function and regulation of culling–RING ubiquitin ligases. Nat Rev Mol Cell Biol 6: 9–20 [DOI] [PubMed] [Google Scholar]
- Pickart CM (2004) Back to the future with ubiquitin. Cell 116: 181–190 [DOI] [PubMed] [Google Scholar]
- Pickart CM, Eddins MJ (2004) Ubiquitin: structures, function, mechanisms. Biochem Biophys Acta 1695: 55–72 [DOI] [PubMed] [Google Scholar]
- Pickart CM, Fushman D (2004) Polyubiquitin chains: polymeric protein signals. Curr Opin Chem Biol 8: 610–616 [DOI] [PubMed] [Google Scholar]
- Raasi S, Pickart CM (2003) Rad23 ubiquitin-associated domains (UBA) inhibit 26S proteasome-catalyzed proteolysis by sequestering lysine 48-linked polyubiquitin chains. J Biol Chem 278: 8951–8959 [DOI] [PubMed] [Google Scholar]
- Russell NS, Wilkinson KD (2004) Identification of a novel 29-linked polyubiquitin binding protein, Ufd3, using polyubiquitin chain analogues. Biochemistry 43: 4844–4854 [DOI] [PubMed] [Google Scholar]
- Scheffner M, Huibregtse JM, Howley PM (1994) Identification of a human ubiquitin-conjugating enzyme that mediates the E6-AP-dependent ubiquitination of p53. Proc Natl Acad Sci USA 91: 8797–8801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheffner M, Huibregtse JM, Vierstra RD, Howley PM (1993) The HPV-16 E6 and E6-AP complex functions as a ubiquitin–protein ligase in the ubiquitination of p53. Cell 75: 495–505 [DOI] [PubMed] [Google Scholar]
- Scheffner M, Nuber U, Huibregtse JM (1995) Protein ubiquitination involving an E1–E2–E3 enzyme ubiquitin thioester cascade. Nature 373: 81–83 [DOI] [PubMed] [Google Scholar]
- Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM (1990) The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 63: 1129–1136 [DOI] [PubMed] [Google Scholar]
- Schwarz SE, Rosa JL, Scheffner M (1998) Characterization of human hect domain family members and their interaction with UbcH5 and UbcH7. J Biol Chem 273: 12148–12154 [DOI] [PubMed] [Google Scholar]
- Verdecia MA, Joazeiro CAP, Wells NJ, Ferrer J-L, Bowman ME, Hunter T, Noel JP (2003) Conformational flexibility underlies ubiquitin ligation mediated by the WWP1 HECT domain E3 ligase. Mol Cell 11: 249–259 [DOI] [PubMed] [Google Scholar]
- Wilkinson KD, Tashayev VL, O'Connor LB, Larsen CN, Kasperek E, Pickart CM (1995) Metabolism of the polyubiquitin degradation signal: structure, mechanism and role of isopeptidase T. Biochemistry 34: 14535–14546 [DOI] [PubMed] [Google Scholar]
- Wu P-Y, Hanlon M, Eddins M, Tsui C, Rogers R, Jensen JP, Matunis MJ, Weissman AM, Wolberger C, Pickart CM (2003) A conserved catalytic residue in the E2 enzyme family. EMBO J 22: 5241–5250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- You J, Cohen RE, Pickart CM (1999) Construct for high-level expression and low misincorporation of lysine for arginine during expression of pET-encoded eukaryotic proteins in Escherichia coli. BioTechniques 27: 950–954 [DOI] [PubMed] [Google Scholar]
- You J, Pickart CM (2001) A hect domain E3 enzyme assembles novel polyubiquitin chains. J Biol Chem 276: 19871–19878 [DOI] [PubMed] [Google Scholar]
- You J, Wang M, Aoki T, Tamura T, Pickart CM (2003) Proteolytic targeting of transcriptional regulator TIP120B by a HECT domain E3 ligase. J Biol Chem 278: 23369–23375 [DOI] [PubMed] [Google Scholar]
- Zheng N, Schulman BA, Song L, Miller JJ, Jeffrey PD, Wang P, Chu C, Koepp DM, Elledge SJ, Pagano M, Conaway RC, Conaway JW, Harper JW, Pavletich NP (2002) Structure of the Cul1–Rbx1–Skp1-F boxSkp2 SCF ubiquitin ligase complex. Nature 416: 703–709 [DOI] [PubMed] [Google Scholar]
- Zheng N, Wang P, Jeffrey PD, Pavletich NP (2000) Structure of a c-Cbl–UbcH7 complex: RING domain function in ubiquitin–protein ligases. Cell 102: 533–539 [DOI] [PubMed] [Google Scholar]
- Zhou P (2005) Targeted protein degradation. Curr Opin Chem Biol 9: 51–55 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5