

Abstract
ATM and ATR are key components of the DNA damage checkpoint. ATR primarily responds to UV damage and replication stress, yet may also function with ATM in the checkpoint response to DNA double-strand breaks (DSBs), although this is less clear. Here, we show that atl-1 (Caenorhabditis elegans ATR) and rad-5/clk-2 prevent mitotic catastrophe, function in the S-phase checkpoint and also cooperate with atm-1 in the checkpoint response to DSBs after ionizing radiation (IR) to induce cell cycle arrest or apoptosis via the cep-1(p53)/egl-1 pathway. ATL-1 is recruited to stalled replication forks by RPA-1 and functions upstream of rad-5/clk-2 in the S-phase checkpoint. In contrast, mre-11 and atm-1 are dispensable for ATL-1 recruitment to stalled replication forks. However, mre-11 is required for RPA-1 association and ATL-1 recruitment to DSBs. Thus, DNA processing controlled by mre-11 is important for ATL-1 activation at DSBs but not following replication fork stalling. We propose that atl-1 and rad-5/clk-2 respond to single-stranded DNA generated by replication stress and function with atm-1 following DSB resection.
Keywords: ATR, checkpoint, DSBs, replication stress
Introduction
In eukaryotic cells DNA-damage checkpoints sense the physical state of the genome and coordinate the orderly progression of the cell cycle with the completion of critical events such as DNA replication and repair (Shiloh, 2003; Kastan and Bartek, 2004). In response to DNA damage, checkpoint activation triggers a cascade of events that culminate in a transient cell cycle arrest, thus providing the necessary time for DNA repair to ensue. In metazoan organisms, checkpoint pathways can also induce apoptosis in the presence of irreparable or persistent DNA damage to eliminate compromised cells. The importance of DNA damage response (DDR) pathways in maintaining genomic integrity and preventing tumorigenesis is highlighted by the existence of inherited cancer predisposition syndromes, such as ataxia-telengiectasia (A–T) and Nijmegan breakage syndrome, which are caused by mutations in the checkpoint genes ATM and Nbs1, respectively (Carney et al, 1998; Shiloh, 2003).
ATM (ataxia-telangiectasia, mutated) and ATR (ATM and Rad3-related), members of the phosphatidylinositol 3-kinase related kinase (PIKK) family, are the primary sensors of DNA damage that are generally believed to function independently of each other in distinct DNA damage checkpoint signalling pathways (Abraham, 2001; Shiloh, 2003; Kastan and Bartek, 2004). Indeed, ATM and ATR primarily respond to different types of DNA damage and exhibit distinct differences in how they are recruited and activated by DNA lesions (Falck et al, 2005). ATM responds to the presence of DNA double-strand breaks (DSBs) generated following ionizing radiation (IR)-treatment and is activated by autophosphorylation on Ser-1981 (Bakkenist and Kastan, 2003) prior to its recruitment to DSBs through an interaction with the Mre11–Rad50–Nbs1 (MRN) complex (Costanzo et al, 2004; Falck et al, 2005; Lee and Paull, 2005). ATR on the other hand primarily responds to DNA lesions generated following replication fork stalling in S-phase or following UV damage (Kastan and Bartek, 2004). Accumulation of RPA bound to single-stranded DNA (ssDNA) generated during DNA lesion processing or following replication stress are believed to recruit and activate ATR via its interacting partner, ATRIP (Zou and Elledge, 2003).
Once activated ATM and ATR trigger checkpoint responses by phosphorylating distinct as well as overlapping downstream target proteins, such as Chk1, Chk2 and p53 to bring about cell cycle arrest, DNA repair or apoptosis (Durocher and Jackson, 2001; Kastan and Bartek, 2004). Although ATM and ATR are primarily activated by different DNA lesions, placing them into separate checkpoint pathways maybe an oversimplification as ATR can compensate for loss of ATM under certain conditions. For example, overexpression of ATR can partially restore radio-resistance to A–T cells, phosphorylation of p53 and Chk1 after IR-treatment is delayed rather than abolished in A–T cells, and overexpression of a dominant-negative kinase dead mutant of ATR confers defects in cell cycle checkpoint activation in response to DSBs generated following IR (Cliby et al, 1998; Wright et al, 1998; Kastan and Bartek, 2004). Collectively, these observations suggest that ATR may participate with ATM in the checkpoint response to DSBs. In the absence of ATR-deficient cells that survive in culture (Brown and Baltimore, 2000), it has been difficult to determine the importance of ATR in the checkpoint response to DSBs. However, it may be possible to analyze the role of ATR in the DSB response in cell lines carrying hypomorphic mutations in ATR that were derived from two related Seckel syndrome patients (O'Driscoll et al, 2003).
Several features of the Caenorhabditis elegans adult hermaphrodite germline, required for generating haploid gametes, make it a powerful model for dissecting the underlying mechanisms required for the DDR. Each germline is spatially polarized in a distal-to-proximal manner with respect to mitotic proliferation and progression through meiotic prophase I. Following the detection of DNA damage, conserved DNA damage checkpoint pathways transduce signals to the cell cycle machinery to induce arrest of mitotically dividing cells, or to the apoptosome to induce death of pachytene cells (Gartner et al, 2000; Boulton et al, 2002). Currently, rad-5/clk-2 is the only C. elegans gene shown to be essential for the S-phase checkpoint, but how rad-5/clk-2 performs its functions is not known (Ahmed et al, 2001). The C. elegans 9-1-1 complex components, mrt-2 (ScRad17/SpRad1) and hus-1, together with rad-5/clk-2, are required for IR-induced cell cycle arrest and germ cell death, yet are surprisingly dispensable for the S-phase checkpoint (Ahmed and Hodgkin, 2000; Ahmed et al, 2001; Boulton et al, 2002; Hofmann et al, 2002). It is currently unclear how checkpoint activation brings about cell cycle arrest in the mitotic compartment of the germline. However, like in mammals, checkpoint activation in response to IR triggers germ cell death by activating the C. elegans p53 homolog (CEP-1) that induces expression of the BH3 domain containing protein, EGL-1, that in turn regulates the general cell death regulators, CED-3 and CED-4 (Conradt and Horvitz, 1998; Derry et al, 2001; Hengartner, 2001; Schumacher et al, 2001, 2005).
Here we report the characterization of a mutant in the C. elegans homolog of ATR (atl-1). Although atl-1 is essential, maternal rescue permits analysis of atl-1 loss-of-function in adult tissues. Both atl-1 and rad-5/clk-2 mutants exhibit mitotic catastrophe and defects in the S-phase checkpoint. ATL-1 is recruited to stalled replication forks by RPA-1 bound to ssDNA and functions upstream of RAD-5/CLK-2 in the checkpoint pathway. Surprisingly, we find that atl-1, as well as atm-1 and rad-5/clk-2, are required for checkpoint dependent cell cycle arrest and apoptosis via the cep-1/egl-1 pathway in response to DSBs generated by IR. ATL-1 recruitment to DSBs also requires rpa-1, but unlike its recruitment to stalled replication forks, there is an additional requirement for mre-11. The failure to recruit ATL-1 to DSBs in the absence of mre-11 is due to a defect in processing DBSs to generate resected ssDNA ends bound by RPA-1. Thus, ATL-1 and RAD-5/CLK-2 function in S-phase checkpoint and ensure replication fork stability, and also cooperate with ATM-1 in the checkpoint response to DSBs.
Results
C. elegans ATR is essential for viability
The lack of ATR-deficient animals or cells has been an obstacle to functional studies of this gene as knockout mice are embryonic lethal (Brown and Baltimore, 2000; de Klein et al, 2000), cells generated from these mice fail to survive in culture and analysis of ATR-deficient cells generated by siRNA or cre-lox excision of ATR is restricted to one cell division (Cortez et al, 2001). We reasoned that analysis of ATR loss-of-function might be possible in C. elegans as many lethal mutations can be studied in homozygous progeny, if rescued into adulthood by maternal mRNA contribution.
atl-1, the C. elegans homolog of ATR, is located at the T06E4.3 locus on chromosome V. The atl-1 gene spans 10.47 kb, including 18 exons, and is predicted to encode two splice variants, T06E4.3a and T06E4.3b, that produce 2531 and 2514 amino-acid proteins, respectively (Figure 1A and B). The function of the two predicted isoform's is currently unknown but both possess extensive sequence conservation throughout all known functional domains in human ATR, displaying 20% identity and 36% similarity in the FAT domain, 39% identity and 57% similarity in the phosphatidylinositol 3- and 4-kinase catalytic motif (PI3Kc) and 36% identity and 50% similarity in the FATC domain compared with the corresponding regions of human ATR (Figure 1A). In C. elegans there is another member of the PI3Kase family called atm-1, with high homology to HsATM (Supplementary Figure S1A).
Figure 1.
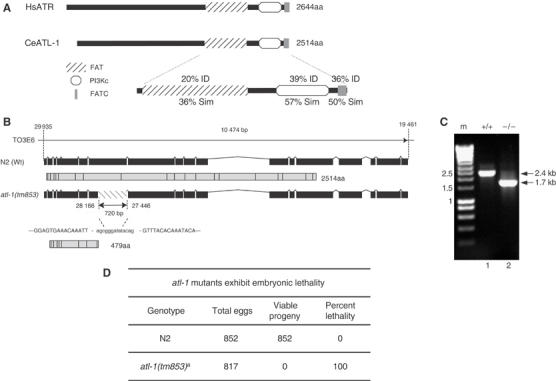
Mutation in atl-1, the C. elegans homolog of ATR, results in embryonic lethality. (A) Schematic representation of C. elegans atl-1 compared with H. sapiens ATR. The various conserved domains positions, including the FAT domain (diagonal stripes box), the PI3K domain (white box) and the FATC domain (gray box), and their respective identity (ID) and similarity (Sim) compared to HsATR are indicated. (B) Schematic of atl-1 gene structure and the predicted protein product for wild-type (Wt) N2 (2514aa protein) and the atl-1(tm853) deletion mutant (479aa predicted). atl-1(tm853) carries a 720 bp deletion in the 7th exon that generates a premature stop codon. The numbers indicates the nucleotide positions in the cosmid T03E6. (C) Nested PCR with atl-1 specific primers from a single wild-type N2 worm (lane1) and a single atl-1 (tm853) mutant worm (lane 2) showing the 720-bp deletion. (D) Embryonic lethality was scored by counting the number of eggs laid (Total Eggs), which was compared to the number of eggs that hatch to produce viable progeny. aThe hermphrodites whose progeny were scored in these experiments were homozygous atl-1 mutants derived from atl-1 −/+ parents.
We have characterized an atl-1 deletion mutant allele, atl-1(tm853), that was isolated by PCR using atl-1 specific primers from a UV-trimethylpsoralen mutagenized library. A 720 bp region within exon 7 of the T06E4.3 locus is deleted in atl-1(tm853) creating a frameshift in the atl-1 gene that results in a premature stop codon, which prevents expression of the last 12 exons, including the FAT, PI3Kc kinase and FATC domains (Figure 1B and C). The atl-1(tm853) allele is predicted to encode a 479 amino-acid truncated product. This product is undetectable by Western blotting and is unlikely to function as a dominant negative as atl-1(tm853)/+ heterozygotes are viable and exhibit wild-type responses to DNA damage (Supplementary Figure S2). The major phenotype associated with depletion of atl-1 using RNA-mediated interference (RNAi) is a Him (high incidence of males) phenotype, reflecting chromosome segregation defects, and partial lethality during early embryogenesis, explained in part, by a cell-cycle timing defect during early development (Aoki et al, 2000; Brauchle et al, 2003). In contrast, atl-1(tm853) confers complete embryonic lethality (Emb phenotype), suggesting that RNAi depletion of atl-1 does not necessarily reflect complete loss-of-function for this gene. Although the brood size produced by atl-1(tm853) mutant animals is similar to wild-type of 817 embryos analyzed from atl-1(tm853) animals none survived, whereas 852 embryos analyzed from wild-type (N2) worms all survive to adulthood (Figure 1D). These findings suggest that as in mice (Brown and Baltimore, 2000; de Klein et al, 2000), atl-1 is an essential gene in C. elegans and is required for completion of embryogenesis.
atl-1 and rad-5/clk-2 prevent mitotic catastrophe
Although atl-1 is an essential gene, atl-1(tm853)/+ heterozygous hermaphrodite parents produce atl-1(tm853) homozygous progeny by self-fertilization. These progeny are rescued through to adulthood by maternal mRNA contribution, thus permitting analysis of atl-1 loss-of-function within adult tissues. atl-1(tm853) adult hermaphrodites display normal locomotion, touch responses, pharyngeal pumping, egg laying and overall morphology, suggesting that atl-1 is dispensable for generating functional somatic tissues during development (data not shown). The C. elegans hermaphrodite germline is responsible for generating germ cells and is spatially polarized in a distal-to-proximal manner with respect to mitotic proliferation and progression through meiotic prophase (Figure 2A). The distal end of the germline comprises a stem cell compartment of mitotically proliferating nuclei that are followed, more proximally, by cells in progressive stages of meiosis I. It is possible to distinguish nuclei in the mitotic zone from those in the early stages of meiosis I by the characteristic transition to ‘crescent-shaped' nuclei that define the initial stages of homolog alignment and pairing in early meiosis.
Figure 2.
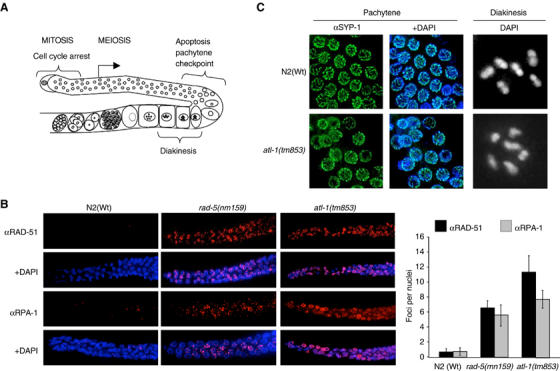
atl-1 inactivation leads to mitotic catastrophe but is dispensable for meiotic prophase I. (A) Diagram of the adult C. elegans hermaphrodite gonad. In wild-type animals, genotoxic stress leads to checkpoint-dependent cell cycle arrest of nuclei in the mitotic compartment and apoptosis of meiotic germ cells via the pachytene checkpoint (Gartner et al, 2000). (B) Representative images of fixed germlines of the indicated genotype immunostained with RPA-1 and RAD-51 antibodies and counterstained with DAPI. Wild type (N2) and atl-1 mutant worms were analyzed under normal growth at 20°C, rad-5(mn159) was analyzed after growth at the nonpermissive temperature of 25°C. Quantification of the number of RPA-1 and RAD-51 foci per nucleus for the indicated genotypes is shown in the graph on the right. Error bars indicate standard error of mean from at least 20 nuclei from 10 to 15 worms of each genotype. (C) Representative images of fixed pachytene nuclei from N2(Wt) and atl-1(tm853) mutants immunostained with the core SC component, SYP-1 (MacQueen et al, 2002). Representative images of a single oocyte nucleus arrested at diakinesis counterstained with DAPI from N2(Wt) and atl-1(tm853) mutants.
Cytological analysis of fixed germlines isolated from atl-1(tm853) mutants reveals a highly disorganized mitotic zone with aberrant nuclei and evidence of chromosomal aberrations resembling the mitotic catastrophe and nuclear fragmentation phenotype described for ATR−/− cells (Brown and Baltimore, 2000; Alderton et al, 2004) (Supplementary Figure S1B). Unlike wild-type (N2) worms, atl-1 mutants exhibit regions of ssDNA and multiple DSBs in mitotic germline nuclei, as indicated by the presence of multiple replication protein A (RPA-1) and RAD-51 nuclear foci, respectively (Figure 2B) (Martin et al, 2005). Early meiotic nuclei, distinguished from mitotic nuclei by their ‘crescent-shaped' appearance, are devoid of detectable RPA-1 and RAD-51 staining in atl-1 mutants, indicating that the genome instability resulting from the loss of ATL-1 arises in mitotic nuclei.
The mn159 mis-sense allele in the rad-5/clk-2 checkpoint gene is viable at 15°C but confers a temperature-sensitive embryonic lethal phenotype when grown at the restrictive temperature of 25°C (Ahmed et al, 2001). Although no obvious defects were observed in the first two embryonic cell division in rad-5(mn159) mutants following a shift to the nonpermissive temperature, it is possible that the lethality observed at 25°C could be caused by a defect during S-phase (Ahmed et al, 2001). Indeed, L4 stage rad-5(mn159) mutants shifted to the nonpermissive temperature of 25°C for 12 h exhibit a dramatic accumulation of single stranded DNA and DSBs that are restricted to cells undergoing S-phase in the mitotic compartment of the germline (Figure 2B). This phenotype is very similar to that observed in atl-1 mutants, implying that atl-1 and rad-5/clk-2 may function in a common pathway to prevent mitotic catastrophe and genome instability in S-phase cells. These observations are consistent with the proposed role for ATR in stabilizing stalled replication forks and also implicate RAD-5/CLK-2 in promoting fork stability (Lopes et al, 2001; Tercero and Diffley, 2001; Shechter et al, 2004).
atl-1 is dispensable for SC assembly and crossover recombination during meiotic prophase
Following exit from the mitotic cell cycle and completion of premeiotic S-phase, homologs align, ‘zip' together (synapsis) and are held in place along their entire length by a protein/DNA structure called the synaptonemal complex (SC). Although the mitotic zone is severely compromised in atl-1 mutants, meiotic prophase nuclei are cytologically normal and correctly assemble the SC at the interface between homolog pairs, as revealed by DAPI staining of DNA and immunostaining with antibodies against the core SC component, SYP-1 (MacQueen et al, 2002) (Figure 2C). Furthermore, six DAPI-stained bivalent chromosomes are detectable at diakinesis in atl-1(tm853) oocyte nuclei, indicative of successful crossover recombination (Figure 2C). Thus, at1-1 is dispensable for SC assembly and crossover recombination during meiotic prophase I.
atl-1 is required for the S-phase checkpoint
It has been proposed that the primary role of ATR in human cells is to respond to problems encountered during S-phase that culminate in replication fork stalling, whereas ATM primarily responds to DSBs (Shiloh, 2003; Kastan and Bartek, 2004). The mitotic defects in atl-1 and rad-5/clk-2 mutants are consistent with shared roles as checkpoint sensors required for stabilizing stalled replication forks during S-phase. Since rad-5/clk-2 is the only C. elegans gene so far shown to be essential for the S-phase checkpoint (Ahmed et al, 2001), we next determined the integrity of this response in atl-1 and atm-1 mutants by subjecting L4 staged animals to treatment with hydroxyurea (HU), an inhibitor of ribonucleotide reductase that leads to replication fork stalling. Consistent with previous reports (Ahmed et al, 2001), HU treatment of wild-type (N2) animals leads to S-phase arrest that manifests as enlarged nuclei with an overall reduction in the number of nuclei in the mitotic compartment of the germline (72.3±2.4 versus 12.6±0.8 after treatment, P<0.01). Cell cycle arrest is also evident in animals RNAi depleted for the C. elegans ATM homolog, atm-1 (67.4±3.6 versus 16.8±1.4 after treatment, P<0.01), whereas rad-5/clk-2 mutants fail to arrest cell cycle progression after HU-treatment (Figure 3A) (Ahmed et al, 2001). Similar to rad-5/clk-2 mutants, mitotic nuclei in atl-1 mutants also fail to arrest following HU-treatment as revealed by an absence of enlarged mitotic nuclei and no measurable decrease in the number of nuclei in the mitotic compartment when compared with untreated animals (Figure 3A) (49.7±2.6 versus 48.4±4.8 after treatment, P<0.5). The same results was also obtained with atl-1(RNAi) (Supplementary Figure S2B). These results indicate that both atl-1 and rad-5/clk-2 are components of the S-phase checkpoint required to induced cell cycle arrest in response to replication fork stalling. In contrast, atm-1 is dispensable for the S-phase checkpoint response.
Figure 3.
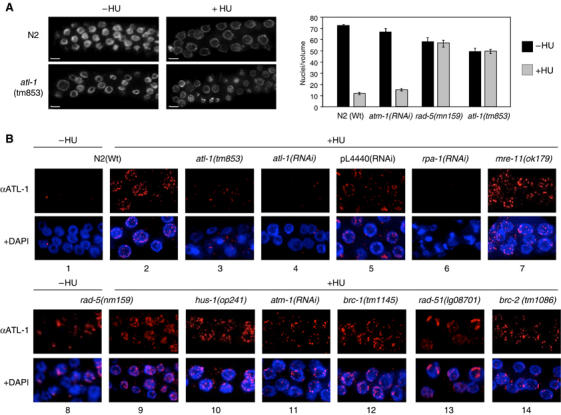
ATL-1 is required for cell cycle arrest after HU and is recruited to stalled replication forks in an rpa-1 dependent manner. (A) Representative images of a single focal plane through the mitotic region of the germline from animals of the indicated genotype counterstained with DAPI, with and without HU treatment (scale bar=5 μm). The graph on the right shows quantification of HU-induced cell cycle arrest of mitotic germline nuclei that was determined in animals of the indicated genotype by scoring the number of nuclei in a volume of 54 000 μm3 16 h after exposure of L4 stage animals to 40 mM hyrdroxyurea, as previously described (Ahmed et al, 2001). At least 10 germlines were scored for each experiment. (B) Representative images of ATL-1 staining of fixed mitotic nuclei for the indicated genotypes following 16 h of HU treatment (quantification of this data is shown in Supplementary Figure S3C and ATL-1 staining without treatment is shown in Supplementary Figure S3A).
ATL-1 is recruited to stalled replication forks by RPA-1/ssDNA
To further explore the role of ATL-1 in responding to S-phase insults, we raised antibodies to examine ATL-1 localization in cells arrested during DNA replication by HU-treatment. Under normal growth conditions, germline nuclei from wild-type (N2) animals are devoid of any detectable ATL-1 staining (Figure 3B1). In contrast, ATL-1 is detected in multiple nuclear foci in mitotic germline nuclei arrested in S-phase following HU-treatment, but is absent in nuclei at all stages of meiotic prophase (Figure 3B2; data not shown) and is dramatically reduced in atl-1(tm853) mutants or in animals subjected to atl-1 RNAi depletion (Figure 3B3 and 4). This result indicates that ATL-1 localizes to sites of replication fork stalling in mitotic cells after HU treatment.
In order to define the position and function of ATL-1 within the signalling cascade required to trigger the S-phase checkpoint in C. elegans, we analyzed ATL-1 focus formation in mutants defective for several DDRs genes, including rpa-1, mre-11, atm-1, rad-5/clk-2, hus-1, atm-1, brc-1, brd-1, rad-51 and brc-2 (Ahmed et al, 2001; Chin and Villeneuve, 2001; Hofmann et al, 2002; Boulton et al, 2004; Martin et al, 2005). RPA-1 and ATL-1 focus formation following HU-treatment is abolished in animals subjected to rpa-1(RNAi) depletion (Supplementary Figure S3B; Figure 3B6), indicating that RPA-1 is required for ATL-1 recruitment to stalled replication forks, a result in agreement with previous reports in mammalian cells (Zou and Elledge, 2003). In untreated rad-5/clk-2 mutants, ATL-1 foci accumulate in the majority of nuclei within the mitotic zone (Figure 3B8), consistent with the presence of ssDNA and DSBs within this region (Figure 2B). ATL-1 foci also increase in number in rad-5/clk-2 mutants after HU treatment (Figure 3B9). These data indicate that rad-5/clk-2 is dispensable for recruitment of ATL-1 to stalled replication forks, despite being defective for the S-phase checkpoint. Animals harboring mutations in mre-11 or hus-1 or animals depleted for atm-1 by RNAi are also competent for ATL-1 focus formation after HU (Figure 3B7, 10 and 11) (Chin and Villeneuve, 2001). Furthermore, ATL-1 foci form as normal after HU-treatment in mutants defective for DSB repair and recombinational restart of collapsed replication forks, including brc-1, brd-1, rad-51 and brc-2 (Boulton et al, 2004; Martin et al, 2005) (Figure 3B12–14; Supplementary Figure S3D). Together, these data indicate that ATL-1 recruitment to stalled replication forks does not require rad-5/clk-2, mre-11, atm-1, hus-1 or DSB repair genes, but does require rpa-1.
ATL-1 is recruited to processed DSBs in an rpa-1 and mre-11 dependent manner
It is currently unclear if ATR cooperates with ATM in the checkpoint response to DSBs generated following IR-treatment. To examine this possibility in C. elegans, we next assessed whether atl-1 has any role in the response to IR as has been previously suggested using atl-1 RNAi (Boulton et al, 2002). Following exposure to an intermediate dose of IR (75 Gy) for C. elegans (Ahmed et al, 2001; Chin and Villeneuve, 2001; Hofmann et al, 2002; Boulton et al, 2004; Martin et al, 2005), ATL-1 responds by accumulating at discrete nuclear foci at sites of DSBs (Figure 4A2). ATL-1 foci are first detected in wild-type N2 animals 30 min after IR-treatment and persist for more than 8 h (data not shown). This signal corresponds to ATL-1 protein as IR-induced foci are absent in atl-1(tm853) mutants (Figure 4A3).
Figure 4.
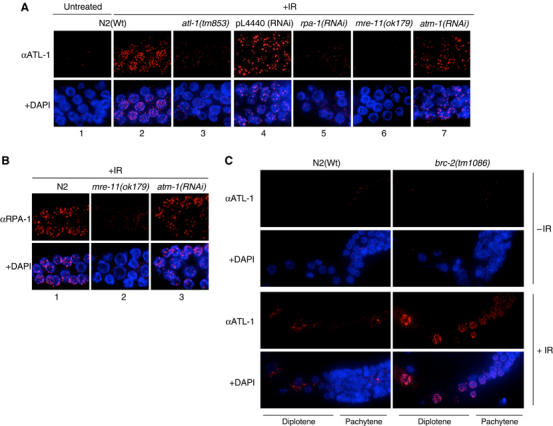
ATL-1 is recruited to processed DSBs following exposure to IR in an rpa-1 and mre-11 dependent manner. (A) ATL-1 staining of fixed germline nuclei from N2(Wt) animals before and after 1 h post-IR-treatment (75 Gy) and in animals of the indicated genotypes 1 h after IR-treatment (75 Gy). (B) RPA-1 staining of fixed mitotic nuclei for the indicated genotypes 1 h after IR-treatment (75 Gy). (C) ATL-1 staining of fixed diplotene nuclei during meiotic prophase for the indicated genotypes before and 1 h after IR-treatment (75 Gy). All images shown are representative of fixed germline staining with the respective antibodies.
To examine the genetic requirements for ATL-1 recruitment to IR-induced DSBs, we assessed ATL-1 focus formation in several mutants required for the DNA damage checkpoint (rpa-1, mre-11, atm-1, hus-1 and rad-5) (Ahmed et al, 2001; Chin and Villeneuve, 2001; Hofmann et al, 2002). RNAi depletion of rpa-1 abolishes IR-induced ATL-1 focus formation (Figure 4A5), indicating that ATL-1 recruitment to DSBs and to stalled replication forks are both dependent on the formation of RPA-ssDNA complexes. Moreover, mutants defective for the mre-11 component of the MRN complex are also severely defective for ATL-1 focus formation after IR-treatment (Figure 4A6), revealing a distinct difference in the genetic requirements for ATL-1 focus formation at stalled replication forks versus ATL-1 recruitment to IR-induced DSBs. One plausible interpretation of these data is that ATL-1 is recruited by RPA-1 to ssDNA generated by replication stress or following resection of DSBs following IR. The observations that nuclease deficient alleles of Mre11 are sensitive to IR (Moreau et al, 1999), confer moderate defects in processing meiosis specific DSBs (Tsubouchi and Ogawa, 1998; Moreau et al, 1999) and display reduced resection of HO DSBs (Tsubouchi and Ogawa, 1998; Moreau et al, 1999) raises the possibility that C. elegans mre-11 may regulate processing of IR-induced DSBs to generate ssDNA overhangs bound by RPA. Consistent with this hypothesis, Figure 4B2 shows that mre-11 mutants are severely impaired for RPA-1 focus formation at sites of DSBs following IR-treatment. The majority of mre-11 mutant germlines have no detectable RPA-1 or ATL-1 staining. However in a few germlines, we observed a small number of RPA-1 and ATL-1 foci in a subset of nuclei (data not shown). Recent studies have shown that the MRN complex recruits ATM to sites of DSBs (Falck et al, 2005; Lee and Paull, 2005). Given our finding that ATL-1 recruitment to DSBs also requires mre-11, we next assessed the role of the atm-1 in the recruitment of ATL-1 to IR-induced DSBs. Contrary to mre-11 mutants, loss of atm-1 does not affect ATL-1 focus formation or the formation of RPA–ssDNA complexes at DSBs following IR-treatment (Figure 4A7 and B3). Together, these results indicate that mre-11, but not atm-1, are required for RPA-1 and ATL-1 recruitment to DSBs and also suggests that the predominant mechanism for DSB resection relies on mre-11 function, although DSBs can be resected inefficiently in their absence.
ATL-1 is recruited to persistent DSBs in mitotic and meiotic cells
To analyze the response of ATL-1 to an alternative source of DSBs, we monitored recruitment of ATL-1 following phleomycin treatment. Although ATL-1 foci are detectable, the dose and conditions required to induce DSB formation in the C. elegans germline, as revealed by RAD-51 focus formation, are excessive (25 mg/ml for 4 days), indicating that phleomycin is not a satisfactory method for producing DSB in this system (data not shown). Since we have previously reported that C. elegans BRCA2 mutants (brc-2), defective for nuclear localization and recruitment of RAD-51 to DSBs, exhibit RPA-1 foci at persistent DSBs in late pachytene and diplotene stages of meiotic prophase (Martin et al, 2005), we also monitored ATL-1 recruitment to DSBs in meiotic cells. In contrast to wild-type (N2) animals, ATL-1 foci accumulate in brc-2 mutants in pachytene and diplotene nuclei similar to that previously reported for RPA-1 (Figure 4C; Martin et al, 2005). These data indicate that ATL-1 recruitment to DSBs is not restricted to mitotic cells, but can also respond to persistent DSBs in meiotic prophase nuclei.
In brc-1, brd-1, rad-51 and brc-2 mutants, IR-induced DSBs persist due to defects in DSB repair (Boulton et al, 2004; Martin et al, 2005). ATL-1 nuclear foci are elevated in number in the mitotic zone of these mutants following IR (Supplementary Figure S4A and B). ATL-1 foci also persist in rad-5 and hus-1 mutants (Ahmed et al, 2001; Hofmann et al, 2002), which are both compromised for the checkpoint response to IR (Supplementary Figure S4C). These data suggest that ATL-1 responds to persistent and/or irreparable DSBs.
atl-1, atm-1 and rad-5/clk-2 are required for the checkpoint response following IR
Our observation that ATL-1 is recruited to sites of DSBs following IR-treatment raises the possibility that like atm-1 and rad-5/clk-2 mutants (Ahmed et al, 2001), atl-1 may be required for checkpoint responses to DSBs, including the induction of cell cycle arrest in the mitotic compartment or triggering apoptosis of pachytene nuclei during meiotic prophase (Gartner et al, 2000). Similar to atm-1(RNAi) and the rad-5/clk-2 mutant, the atl-1(tm853) mutant is severely compromised for mitotic cell cycle arrest following IR (Figure 5A), with no measurable difference in the number or size of mitotic nuclei before or after treatment (49.7±2.6 versus 49.3±1.8, P<0.1). Following IR-treatment of wild-type (N2) animals, pachytene cells with persistent DNA damage undergo checkpoint dependent apoptosis (Figure 5B), showing a progressive increase in the number of apoptotic corpses over 12–36 h (P<0.001 versus nonirradiated worms). In contrast to wild type, but similar to atm-1(RNAi) and the rad-5/clk-2 mutant, after exposure to 75 Gy of IR, no measurable increase in the number of apoptotic corpses is detectable in the atl-1(tm853) mutant (at 12 h the apoptotic corpses in the wild type are 6±0.1 versus the atl-1 0.8±0.01; at 24 h 7.9±0.3 versus 0.73±0.1; at 36 h 4.9±0.5 versus 1.7±0.3. P<0.001 each time). The same results was also obtained with atl-1(RNAi) (Supplementary Figure S2C). These results indicate that atm-1, rad-5/clk-2 and atl-1 are required for checkpoint-dependent cell cycle arrest and apoptosis in response to IR.
Figure 5.
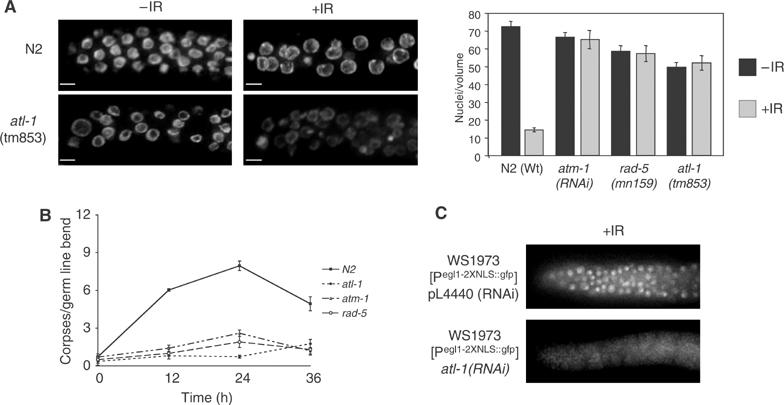
atl-1 is required for checkpoint-dependent cell cycle arrest and apoptosis following IR via the cep-1/egl-1 proapoptotic pathway. (A) Representative images of a single focal plane through the mitotic region of the germline from animals of the indicated genotype counterstained with DAPI following IR-treatment (scale bar=5 μm). The graph on the right shows quantification of IR-induced cell cycle arrest of mitotic germline nuclei in animals of the indicated genotype that was determined by scoring the number of nuclei in a volume of 54 000 μm3 12 h after exposure of L4 stage animals to 75 Gy IR, as previously described (Gartner et al, 2000). Error bars indicate standard error of mean from at least ten germlines for each experiment. (B) Germ cell apoptosis was measured by differential interference contrast (DIC) microscopy in animals of the indicated genotype at the indicated time points post IR-treatment. Error bars indicate standard error of mean from at least ten germlines for each time point. (C) Representative images of GFP expression from the Pegl-1::gfp transcriptional reporter (Hofmann et al, 2002) 30 h after IR treatment (120 Gy) in animals subjected to L4440 RNAi (control) and atl-1(RNAi).
atl-1 triggers apoptosis via cep-1/egl-1 in response to IR
In C. elegans, DNA-damage induced apoptosis of pachytene cells requires the core apoptotic machinery (CED-9/4/3) and the proapoptotic gene egl-1, whose expression is induced following IR-treatment in a cep-1 (C. elegans p53) dependent manner (Derry et al, 2001; Schumacher et al, 2001). To determine if atl-1 triggers apoptosis following IR-treatment through a pathway dependent upon cep-1, we monitored the transcriptional activation of a Pegl-1::gfp transcriptional reporter (Hofmann et al, 2002) in animals subjected to atl-1(RNAi) depletion. As previously reported (Hofmann et al, 2002), GFP expression is induced in mitotic nuclei of RNAi control animals following IR-treatment (Figure 5C), with 35/45 germlines showing GFP-expressing nuclei. In contrast, no detectable expression from the Pegl-1::gfp reporter is observed in mitotic nuclei in animals subjected to atl-1(RNAi) after IR-treatment (Figure 5C), with 1/45 germlines showing GFP-expressing nuclei. Collectively, these results indicate that ATL-1 triggers cep-1 and egl-1 dependent apoptosis of pachytene cells following IR-treatment.
Discussion
This study reveals that atl-1 and rad-5/clk-2 prevent genomic instability and mitotic catastrophe during normal cell cycle progression and function in the checkpoint response to replication stress to bring about cell cycle arrest during S-phase. It has been proposed that the essential function of the S-phase checkpoint is to prevent fork collapse during replication fork pausing (Lopes et al, 2001; Tercero and Diffley, 2001). The accumulation of ssDNA and DSBs in atl-1 and rad-5/clk-2 mutants, specifically in mitotically dividing cells, is consistent with the accumulation of collapsed replication forks in these mutants. A role for atl-1 in stabilizing stalled replication forks is further supported by our finding that ATL-1 is recruited to sites of replication forks pausing. While this role is predicted for ATL-1, based on analogy to Mec1p in yeast, this was not known for RAD-5/CLK-2. It is currently unclear how RAD-5/CLK-2 functions in the S-phase checkpoint and prevents replication fork collapse (Ahmed et al, 2001). However, it is intriguing to note that Tel2p, the yeast homolog of RAD-5/CLK-2, interacts with the Cdc7p kinase (Uetz et al, 2000), a component of the replisome required for late origin firing, for the normal checkpoint response to HU-treatment and for trans-lesion synthesis as part of the RAD6 epistasis group (Bousset and Diffley, 1998; Weinreich and Stillman, 1999; Pessoa-Brandao and Sclafani, 2004). While it remains to be seen if RAD-5/CLK-2 associates with CDC-7 kinase in C. elegans, defects in regulating CDC-7 could impact on fork stability. Further studies of RAD-5/CLK-2 are likely to provide important insight into the S-phase checkpoint and the mechanism through which it functions with ATL-1 to stabilize stalled replication forks.
It has been shown that ATRIP, the interacting partner of ATR, senses DNA damage by recognizing and binding to RPA-ssDNA complexes that arise during replication stress or following DNA damage (Zou and Elledge, 2003). Consistent with these observations, we find that ATL-1 is also recruited to stalled replication forks by RPA-1/ssDNA (Figure 3B), indicating that the mechanism of ATL-1 recruitment and its activation at stalled replication forks is conserved. A C. elegans homolog of ATRIP (Zou and Elledge, 2003; Ball et al, 2005; Falck et al, 2005) has not been identified to date so this remains to be formally shown in the nematode. Although rad-5/clk-2 mutants are compromised for the S-phase checkpoint and exhibit evidence of replication fork collapse, these mutants still remain competent for ATL-1 recruitment to stalled replication forks (Figure 3B). This result suggests that recruitment of ATL-1 to stalled replication forks is not sufficient to prevent collapse of stalled forks and also indicates that ATL-1 functions upstream of RAD-5/CLK-2 in the S-phase checkpoint. Rather, we propose that ATL-1 and RAD-5/CLK-2 function together or in nonredundant parallel pathways to stabilize stalled replication forks (Figure 6A).
Figure 6.
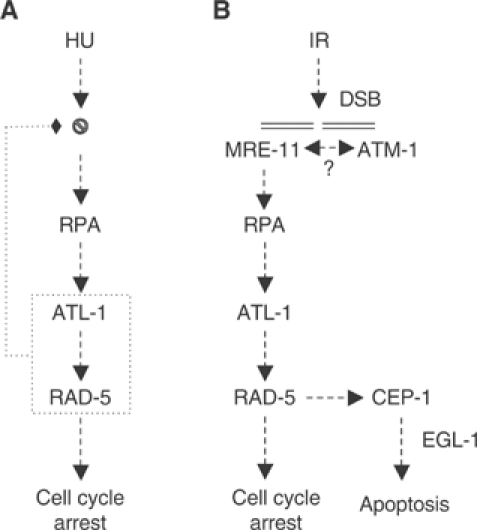
A model for ATL-1 activation in response to replication fork stalling and following processing of DSBs. (A) RPA-ssDNA complexes generated following replication fork pausing/stalling following hydroxyurea (HU) treatment recruit ATL-1. ATL-1 and RAD-5/CLK-2 are both required to stabilize stalled replication forks and to activate the S-phase checkpoint to transiently arrest the cell cycle to provide time to restart DNA synthesis. (B) MRE-11 is recruited to DSBs generated following IR-treatment. Once recruited, MRE-11 is believed to recruit ATM-1 based on studies in mammalian cells and also regulates DSB processing. RPA-ssDNA complexes generated by DSB resection recruit ATL-1. Together, ATL-1, RAD-5/CLK-2 and ATM-1 cooperate to induce cell cycle arrest or apoptosis via the cep-1/egl-1 pathway.
We also show that ATL-1 is recruited to IR-induced DSBs in both mitotic and meiotic cells, suggesting that ATL-1 also responds to DSBs even in cells outside of S-phase. Our analyses highlight a distinct difference in the mechanistic requirements for ATL-1 focus formation at stalled replication forks versus ATL-1 recruitment to IR-induced DSBs. Specifically, we have shown that ATL-1 recruitment to IR-induced DSBs is dependent upon mre-11 and rpa-1 (Figure 4A). In contrast, the generation or exposure of ssDNA bound by RPA that signals ATL-1 recruitment following replication stress does not require the action of either atm-1 or mre-11 (Figure 3B). We propose that DSBs generated following IR-treatment require prior processing to generate ssDNA overhangs that become bound by RPA and recruit and activate ATL-1. What is particularly surprising is that processing of DSBs required to recruit and activate ATL-1 is genetically dependent on mre-11, but not on atm-1. How MRE-11 regulates DSB processing is not clear. One possible explanation is that MRE-11 recruits an exonuclease that mediates DSB processing to generate a resected ssDNA end bound by RPA. Alternatively, MRE-11 possesses exonuclease activity in vitro and could itself be responsible for DSB processing (Moreau et al, 1999; Paull and Gellert, 1999).
The observation that ATR is recruited to DSBs has led to speculation that it may function with ATM in the checkpoint response to DSBs. However, the essential nature of ATR has precluded the direct analysis of its role in this response. Maternal rescue of the atl-1 mutant circumvents this problem and has allowed us to assess the role of atl-1 in the checkpoint response to DSBs generated following IR-treatment. Our data show that atl-1, atm-1 and rad-5/clk-2 are all required for checkpoint dependent cell cycle arrest and cep-1(p53)/egl-1 mediated apoptosis in response to IR (Figures 5 and 6B). Moreover, our phenotypic comparison of atm-1, atl-1 and atm-1; atl-1 double mutants strongly suggest that ATM-1 and ATL-1 function in a common pathway, rather than in parallel or partially parallel pathways (Supplementary Figure S5). Our data suggest that the conventional model in which ATM mediates the checkpoint response to DSBs, whereas ATR responds to UV damage or replication stress may be an oversimplification. Taken together with recent findings, we hypothesize that in response to DSBs, MRE-11 (MRN complex) is targeted to DSBs where it facilitates recruitment of ATM-1 (ATM; Falck et al, 2005; Lee and Paull, 2005). While the role of the MRN complex in ATM recruitment to DSBs is widely accepted in mammalian cells this has not been formally shown in C. elegans and requires further study. Our findings indicate that MRE-11 also regulates DSB processing to generate RPA-ssDNA complexes that target ATL-1 recruitment to DSBs. Once recruited, ATL-1 (ATR), together with RAD-5/CLK-2 and ATM-1, are required to activate the checkpoint response to induce cell cycle arrest or apoptosis (Figure 6B).
Materials and methods
Strains and culture conditions
C. elegans strains were cultured and maintained using standard procedures. The following strains were kindly provided by the Caenorhabditis Genetics Centre (University of Minnesota, St Paul, MN), including wild-type Bristol N2, mre-11(ok179) (Chin and Villeneuve, 2001), rad-51(lg08701), brc-1(tm1145), rad5(mn159) (Ahmed et al, 2001), hus-1(op241) (Hofmann et al, 2002), brd-1(dw1), brc-2(tm1086) (Martin et al, 2005) and WS1973 [Pegl-1_2nls::gfp] (Hofmann et al, 2002) were described previously. atl-1(tm853) was generated and kindly provided by Shoehi Mitani of the National Bioresource Project for the nematode, Japan. Deletions were outcrossed six times with the wild-type Bristol N2 and then balanced with JK2663 [dpy-11 (e224) mes-4 (bn67) V/nT1(qIs50) IV;V]. The phenotypes we describe for atl-1 cosegregate with the tm853 mutation. tm853/+ hets are wild type for all of the assays we have described, whereas atl-1(RNAi) to a large extent recapitulates the tm853 homozygous mutant phenotype (Supplementary Figure S2). RNA interference (RNAi) depletion of rpa-1, atm-1 and atl-1 was performed by the feeding method described previously (Boulton et al, 2002). Hydroxyurea (HU) treatment was performed as previously described (Ahmed et al, 2001). L4 larvae were transferred to plates with 40 mM final HU for 16 and 24 h prior to analysis. To assay radiation-induced germ-cell death, apoptotic cell corpses were measured 0, 12, 24 and 36 h after irradiation using Nomarski optics, as previously described (Gartner et al, 2000; Boulton et al, 2002). RNAi experiments were performed in the RNAi sensitive C. elegans strain, rrf-3(pk1426) as previously described (Simmer et al, 2002). atm-1, atl-1 and rpa-1 RNAi was found to be more penetrant in rrf-3 strains compared with previous studies using wild-type N2 strains (Boulton et al, 2002).
Gateway recombinational cloning
ORF specific primers compatible with the Gateway® system (http://www.invitrogen.com) were designed to full-length rpa-1 (F18A1.5) and atm-1 (Y48G1BL.2) genes, PCR amplified and cloned into the p221 Entry vector as previously described (Boulton et al, 2002). The cloned open reading frames were transferred by LR destination cloning into L4440-dest for RNAi by feeding.
ATL-1 antibodies
An N-terminal synthetic peptide corresponding to the 20 first amino acids (CTAKKYVTQKEFNRQKMKFL) of ATL-1 was generated by the Peptide Synthesis Laboratory, Cancer Research UK, and used to generate rabbit anti-ATL-1 polyclonal antibodies. Animals were immunized (Harlan Sera Labs, Loughborough, UK) with 1 mg of each peptide coupled to activated keyhole limpet hemocyanin (Pierce, Rockford, III). Affinity purified antibodies were tested by Western blotting against recombinant proteins expressed in Escherichia coli and subsequently by immunofluorescence in N2(Wt) and atl-1(tm853) to test for specificity. The signal observed following HU and IR-treatment is abolished by atl-1(RNAi), by incubation with a competitor peptide corresponding to the epitope recognized by the ATL-1 antibody, and is dramatically reduced in atl-1(tm853).
Cytological preparation and immunostaining
Gravid hermaphrodites were transferred to 30 μl PBS on a poly-L lysine coated slide (slides were washed in 70% ethanol, then given 2 coats of 100% poly-L-lysine, air drying between each coat). The worms were washed in PBS before transferring to 50 μl 10 mM levamisole. Germlines were extruded by removing the head and tail using a fine gauge needle (27 G). Levamisole was replaced with 1% para-formaldehyde (PFA) in PBS for 10 min and germlines were permeabilized for 5 min in TBSBT (TBS+0.5% BSA+0.1% Triton X-100), then washed in TBSB for at least 2 × 5 min, followed by blocking for 30 min. Primary antibodies were diluted in TBSB (1:200 for RAD-51, 1:200 SYP-1, 1:500 RPA-1, 1:500 ATL-1) and incubated overnight at 4°C in a humid chamber. Germlines were subsequently washed at least 3 × 5 min in TBSB before incubation with the secondary antibody for 1–2 h at room temperature (anti-rabbit Cy3 1:10 000; anti-guinea FITC 1:5000 (Sigma)). Finally, germlines were washed at least 3 × 5 min in TBSB before mounting with a coverslip on Vectashield containing DAPI (Vector Laboratories).
Fluorescence microscopy
Deltavision microscopy was used to examine germlines with × 40 or × 63, 1.4 NA Planapochromat lens on an Olympus inverted microscope (IX71), and images captured using SoftWorx computer software (Applied Precision). Three-dimensional data sets were computationally deconvolved, and regions of interest then projected into one dimension. Merged or single color images were recorded using GIMP software.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Legends
Acknowledgments
We thank the Bioresource Project for the nematode (Shohei Mitani) for kindly providing atl-1(tm853), E. Randel Hofmann and Michael Hengartner for the WS1973 [Pegl-2nls::gfp] strain and the Caenorhabditis Genetic Centre and the Sanger Centre for providing C. elegans strains and cosmids. We also thank members of the Boulton lab for helpful discussions and to Vincenzo Costanzo, Spencer Collis, Louise Barber, Monica Segurado and Julie Martin for comments on the manuscript. This work was funded by Breast Cancer Campaign (GA3221) and Cancer Research UK.
Competing interests statement The authors declare that they have no competing financial interests.
References
- Abraham RT (2001) Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev 15: 2177–2196 [DOI] [PubMed] [Google Scholar]
- Ahmed S, Alpi A, Hengartner MO, Gartner A (2001) C. elegans RAD-5/CLK-2 defines a new DNA damage checkpoint protein. Curr Biol 11: 1934–1944 [DOI] [PubMed] [Google Scholar]
- Ahmed S, Hodgkin J (2000) MRT-2 checkpoint protein is required for germline immortality and telomere replication in C. elegans. Nature 403: 159–164 [DOI] [PubMed] [Google Scholar]
- Alderton GK, Joenje H, Varon R, Borglum AD, Jeggo PA, O'Driscoll M (2004) Seckel syndrome exhibits cellular features demonstrating defects in the ATR-signalling pathway. Hum Mol Genet 13: 3127–3138 [DOI] [PubMed] [Google Scholar]
- Aoki H, Sato S, Takanami T, Ishihara T, Katsura I, Takahashi H, Higashitani A (2000) Characterization of Ce-atl-1, an ATM-like gene from Caenorhabditis elegans. Mol Gen Genet 264: 119–126 [DOI] [PubMed] [Google Scholar]
- Bakkenist CJ, Kastan MB (2003) DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 421: 499–506 [DOI] [PubMed] [Google Scholar]
- Ball HL, Myers JS, Cortez D (2005) ATRIP binding to replication protein A-single-stranded DNA promotes ATR-ATRIP localization but is dispensable for Chk1 phosphorylation. Mol Biol Cell 16: 2372–2381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulton SJ, Gartner A, Reboul J, Vaglio P, Dyson N, Hill DE, Vidal M (2002) Combined functional genomic maps of the C. elegans DNA damage response. Science 295: 127–131 [DOI] [PubMed] [Google Scholar]
- Boulton SJ, Martin JS, Polanowska J, Hill DE, Gartner A, Vidal M (2004) BRCA1/BARD1 orthologs required for DNA repair in Caenorhabditis elegans. Curr Biol 14: 33–39 [DOI] [PubMed] [Google Scholar]
- Bousset K, Diffley JF (1998) The Cdc7 protein kinase is required for origin firing during S phase. Genes Dev 12: 480–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brauchle M, Baumer K, Gonczy P (2003) Differential activation of the DNA replication checkpoint contributes to asynchrony of cell division in C. elegans embryos. Curr Biol 13: 819–827 [DOI] [PubMed] [Google Scholar]
- Brown EJ, Baltimore D (2000) ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev 14: 397–402 [PMC free article] [PubMed] [Google Scholar]
- Carney JP, Maser RS, Olivares H, Davis EM, Le Beau M, Yates JR III, Hays L, Morgan WF, Petrini JH (1998) The hMre11/hRad50 protein complex and Nijmegen breakage syndrome: linkage of double-strand break repair to the cellular DNA damage response. Cell 93: 477–486 [DOI] [PubMed] [Google Scholar]
- Chin GM, Villeneuve AM (2001) C. elegans mre-11 is required for meiotic recombination and DNA repair but is dispensable for the meiotic G(2) DNA damage checkpoint. Genes Dev 15: 522–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cliby WA, Roberts CJ, Cimprich KA, Stringer CM, Lamb JR, Schreiber SL, Friend SH (1998) Overexpression of a kinase-inactive ATR protein causes sensitivity to DNA-damaging agents and defects in cell cycle checkpoints. EMBO J 17: 159–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conradt B, Horvitz HR (1998) The C. elegans protein EGL-1 is required for programmed cell death and interacts with the Bcl-2-like protein CED-9. Cell 93: 519–529 [DOI] [PubMed] [Google Scholar]
- Cortez D, Guntuku S, Qin J, Elledge SJ (2001) ATR and ATRIP: partners in checkpoint signaling. Science 294: 1713–1716 [DOI] [PubMed] [Google Scholar]
- Costanzo V, Paull T, Gottesman M, Gautier J (2004) Mre11 assembles linear DNA fragments into DNA damage signaling complexes. PLoS Biol 2: E110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Klein A, Muijtjens M, van Os R, Verhoeven Y, Smit B, Carr AM, Lehmann AR, Hoeijmakers JH (2000) Targeted disruption of the cell-cycle checkpoint gene ATR leads to early embryonic lethality in mice. Curr Biol 10: 479–482 [DOI] [PubMed] [Google Scholar]
- Derry WB, Putzke AP, Rothman JH (2001) Caenorhabditis elegans p53: role in apoptosis, meiosis, and stress resistance. Science 294: 591–595 [DOI] [PubMed] [Google Scholar]
- Durocher D, Jackson SP (2001) DNA-PK, ATM and ATR as sensors of DNA damage: variations on a theme? Curr Opin Cell Biol 13: 225–231 [DOI] [PubMed] [Google Scholar]
- Falck J, Coates J, Jackson SP (2005) Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 434: 605–611 [DOI] [PubMed] [Google Scholar]
- Gartner A, Milstein S, Ahmed S, Hodgkin J, Hengartner MO (2000) A conserved checkpoint pathway mediates DNA damage--induced apoptosis and cell cycle arrest in C elegans. Mol Cell 5: 435–443 [DOI] [PubMed] [Google Scholar]
- Hengartner MO (2001) Apoptosis: corralling the corpses. Cell 104: 325–328 [DOI] [PubMed] [Google Scholar]
- Hofmann ER, Milstein S, Boulton SJ, Ye M, Hofmann JJ, Stergiou L, Gartner A, Vidal M, Hengartner MO (2002) Caenorhabditis elegans HUS-1 is a DNA damage checkpoint protein required for genome stability and EGL-1-mediated apoptosis. Curr Biol 12: 1908–1918 [DOI] [PubMed] [Google Scholar]
- Kastan MB, Bartek J (2004) Cell-cycle checkpoints and cancer. Nature 432: 316–323 [DOI] [PubMed] [Google Scholar]
- Lee JH, Paull TT (2005) ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science 308: 551–554 [DOI] [PubMed] [Google Scholar]
- Lopes M, Cotta-Ramusino C, Pellicioli A, Liberi G, Plevani P, Muzi-Falconi M, Newlon CS, Foiani M (2001) The DNA replication checkpoint response stabilizes stalled replication forks. Nature 412: 557–561 [DOI] [PubMed] [Google Scholar]
- MacQueen AJ, Colaiacovo MP, McDonald K, Villeneuve AM (2002) Synapsis-dependent and -independent mechanisms stabilize homolog pairing during meiotic prophase in C. elegans. Genes Dev 16: 2428–2442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin JS, Winkelmann N, Petalcorin MI, McIlwraith MJ, Boulton SJ (2005) RAD-51-dependent and -independent roles of a Caenorhabditis elegans BRCA2-related protein during dna double-strand break repair. Mol Cell Biol 25: 3127–3139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau S, Ferguson JR, Symington LS (1999) The nuclease activity of Mre11 is required for meiosis but not for mating type switching, end joining, or telomere maintenance. Mol Cell Biol 19: 556–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Driscoll M, Ruiz-Perez VL, Woods CG, Jeggo PA, Goodship JA (2003) A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nat Genet 33: 497–501 [DOI] [PubMed] [Google Scholar]
- Paull TT, Gellert M (1999) Nbs1 potentiates ATP-driven DNA unwinding and endonuclease cleavage by the Mre11/Rad50 complex. Genes Dev 13: 1276–1288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pessoa-Brandao L, Sclafani RA (2004) CDC7/DBF4 functions in the translesion synthesis branch of the RAD6 epistasis group in Saccharomyces cerevisiae. Genetics 167: 1597–1610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher B, Hanazawa M, Lee MH, Nayak S, Volkmann K, Hofmann R, Hengartner M, Schedl T, Gartner A (2005) Translational repression of C. elegans p53 by GLD-1 regulates DNA damage-induced apoptosis. Cell 120: 357–368 [DOI] [PubMed] [Google Scholar]
- Schumacher B, Hofmann K, Boulton S, Gartner A (2001) The C. elegans homolog of the p53 tumor suppressor is required for DNA damage-induced apoptosis. Curr Biol 11: 1722–1727 [DOI] [PubMed] [Google Scholar]
- Shechter D, Costanzo V, Gautier J (2004) ATR and ATM regulate the timing of DNA replication origin firing. Nat Cell Biol 6: 648–655 [DOI] [PubMed] [Google Scholar]
- Shiloh Y (2003) ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer 3: 155–168 [DOI] [PubMed] [Google Scholar]
- Simmer F, Tijsterman M, Parrish S, Koushika SP, Nonet ML, Fire A, Ahringer J, Plasterk RH (2002) Loss of the putative RNA-directed RNA polymerase RRF-3 makes C. elegans hypersensitive to RNAi. Curr Biol 12: 1317–1319 [DOI] [PubMed] [Google Scholar]
- Tercero JA, Diffley JF (2001) Regulation of DNA replication fork progression through damaged DNA by the Mec1/Rad53 checkpoint. Nature 412: 553–557 [DOI] [PubMed] [Google Scholar]
- Tsubouchi H, Ogawa H (1998) A novel mre11 mutation impairs processing of double-strand breaks of DNA during both mitosis and meiosis. Mol Cell Biol 18: 260–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uetz P, Giot L, Cagney G, Mansfield TA, Judson RS, Knight JR, Lockshon D, Narayan V, Srinivasan M, Pochart P, Qureshi-Emili A, Li Y, Godwin B, Conover D, Kalbfleisch T, Vijayadamodar G, Yang M, Johnston M, Fields S, Rothberg JM (2000) A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature 403: 623–627 [DOI] [PubMed] [Google Scholar]
- Weinreich M, Stillman B (1999) Cdc7p-Dbf4p kinase binds to chromatin during S phase and is regulated by both the APC and the RAD53 checkpoint pathway. EMBO J 18: 5334–5346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright JA, Keegan KS, Herendeen DR, Bentley NJ, Carr AM, Hoekstra MF, Concannon P (1998) Protein kinase mutants of human ATR increase sensitivity to UV and ionizing radiation and abrogate cell cycle checkpoint control. Proc Natl Acad Sci USA 95: 7445–7450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou L, Elledge SJ (2003) Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 300: 1542–1548 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Legends