

Abstract
Integrons play a major role in the dissemination of antibiotic resistance genes among Gram-negative pathogens. Integron gene cassettes form circular intermediates carrying a recombination site, attC, and insert into an integron platform at a second site, attI, in a reaction catalyzed by an integron-specific integrase IntI. The IntI1 integron integrase preferentially binds to the ‘bottom strand' of single-stranded attC. We have addressed the insertion mechanism in vivo using a recombination assay exploiting plasmid conjugation to exclusively deliver either the top or bottom strand of different integrase recombination substrates. Recombination of a single-stranded attC site with an attI site was 1000-fold higher for one strand than for the other. Conversely, following conjugative transfer of either attI strand, recombination with attC is highly unfavorable. These results and those obtained using mutations within a putative attC stem-and-loop strongly support a novel integron cassette insertion model in which the single bottom attC strand adopts a folded structure generating a double strand recombination site. Thus, recombination would insert a single strand cassette, which must be subsequently processed.
Keywords: antibiotic resistance, DNA, evolution, lateral gene transfer, tyrosine recombinase
Introduction
Integron cassettes are small DNA units that carry open reading frames generally without promoters. They integrate into an integron platform, consisting of a site-specific recombinase, an associated primary recombination target called the attI site and two appropriately orientated (divergent) promoters, one driving an integrase gene, intI, and the other driving expression of the cassette-associated gene. The integron is the generic name for the integron platform–cassette ensemble. Integrons are key players in the capture and dissemination of antibiotic resistance genes among Gram-negative bacteria (see Hall and Collis, 1998). Their importance has recently been underlined by the discovery of large integrons in the chromosomes of a wide range of bacterial species (Rowe-Magnus et al, 2001). These superintegrons (SI) contain arrays of hundreds of genes for various adaptive functions. The corresponding recombinases, IntI integrases, belong to the phage λ integrase family of tyrosine (Y) recombinases (see Azaro and Landy, 2002). The IntI integrases mediate recombination between their specific attI site and a second type of recombination site carried by a gene cassette, called the attC site (or 59-base element), and which is formed following circularization of the integron cassette. IntI integrases can also catalyze recombination between two attC sites. Although related to λ int, several lines of evidence imply that Int1-mediated recombination may be quite different from that of phage λ.
A unique feature of the integron recombination system is the structure of the attI and attC recombination sites. These differ significantly from the canonical Y-recombinase core sites, which are composed of a pair of highly conserved 9- to 13-bp inverted binding sites separated by a 6- to 8-bp central spacer region (see Figure 1A). One of the putative IntI binding sites, within the core of attI, is extremely degenerate and the spacer region differs widely from that of the partner attC sites (see Figure 1A and B). IntI1 recombinase binds to four regions of double-stranded (ds) attI in vitro. Two correspond to the core repeats and two to direct repeats located upstream of the core (Figure 1A) (Collis et al, 1998; Gravel et al, 1998a). The role of the two direct repeats of the attI1 site for the recombination reaction is still unclear (Hansson et al, 1997). The structure of attC is more complex. It consists of two potential core sites, R″–L″ and L′–R′ (also called 1L–2L and 2R–1R (Stokes et al, 1997)), separated by a region that is variable in sequence and length (Figure 1C). A number of these have been demonstrated to be efficiently recombined by IntI1 (Collis et al, 2001; Biskri et al, 2005). While recombination occurs at L′-R′, directed mutagenesis showed that R″-L″ is also essential. All attC sites exhibit extensive potential cruciform structures (Hall et al, 1991; Stokes et al, 1997; Rowe-Magnus et al, 2003). Purified IntI1 binds specifically to the ‘bottom' strand (bs) of single-stranded (ss) attC site, attCaadA1, DNA but not to a ds attCaadA1 site (Francia et al, 1999). This seminal observation was confirmed, and several key elements that act as recognition determinants for in vitro IntI1 binding were identified in the attCaadA1 sequence. Some appear to play important roles in the potential secondary structure of the attC site (Johansson et al, 2004).
Figure 1.
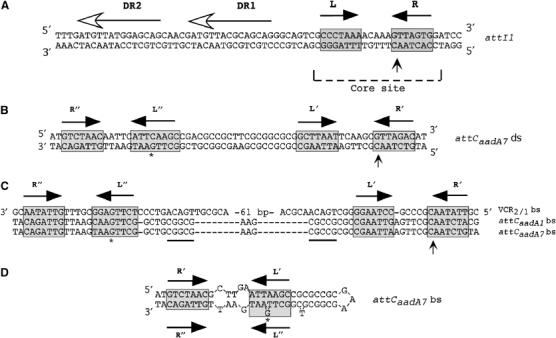
Integron recombination sites. (A) Sequence of the ds attI1 site. (B) Sequence of the ds attCaadA7 site. (C) Multiple sequence alignment of the attC sites bs studied in this work. (D) Proposed secondary structure for the attCaadA7 bs. The inverted repeats L, L′ and L″, R, R′ and R″ are indicated with black arrow; the asterisk (*) shows the position of the protruding G present in L″ relative to L′. The attI1 direct repeats bound by InI1 are indicated by horizontal lines with an empty arrowhead (Collis et al, 1998; Gravel et al, 1998a). The putative IntI1 binding domains, as defined by Stokes et al (1997), are marked with gray boxes. Vertical arrows indicate crossover position. The secondary structure was determined using the MFOLD (Walter et al, 1994) online interface at the Pasteur Institute.
Integron cassettes are thought to move using an excised circular intermediate (Collis and Hall, 1992). These would have the capacity to form extensive secondary structures if produced as a single strand. For most cassettes, self-pairing on the same single strand can be extended up to the R′ and R″ sequences, which usually show a stretch of 9–11 consecutive complementary nucleotides (Figure 1; Hansson et al, 1997; Rowe-Magnus et al, 2003). Such a self-paired stem could be seen as an almost canonical core site consisting of the L″–L′ duplex and an unpaired central region followed by an R″–R′ duplex (Figure 1D).
In the present study, we have addressed the mechanism of integron cassette transfer. We have extended previous observations in vitro (Francia et al, 1999; Johansson et al, 2004) demonstrating specific binding of IntI to the bs of attCaadA1. We show that IntI1 has a similar single strand preference for two additional and structurally distinct attC sites. This demonstrates that strand choice is a general phenomenon and is not associated specifically with attCaadA1. More importantly, we also present evidence strongly suggesting that integration occurs via a single strand intermediate and that a specific single strand of the cassette (that which is bound by IntI) is used. This conclusion is based on recombination frequencies obtained following delivery of one or other single strand by conjugation to a suitable recipient Escherichia coli strain carrying the integron platform and expressing the appropriate integrase. While the attC sites recombine in single strand form, our results suggest that attI must be present in a double strand configuration. However, although the attC recombination intermediate may be single stranded, recombination appears to occur using a ds attC region generated by the secondary structure within the single strand cassette. Thus, while mutations disrupting the potential pairing of non-conserved positions in a putative stem-and-loop structure of the attC bs decreased the recombination frequency, restoration of the complementarity by mutation of the partner sequence restored a high frequency of recombination.
We propose an unusual recombination model to explain the insertion of integron cassettes at the attI site. In this model, a first strand exchange occurs using the attC bs folded into a stem-and-loop structure to generate a Holliday junction (HJ), which is then resolved by replication of the recipient replicon.
Results
IntI1 in vitro binding properties for single- and double-stranded forms of the attI1 site and two attC sites
To determine whether bs-specific binding of IntI1 was specific to attCaadA1 (Francia et al, 1999) or is a general feature of attC sites, we tested two additional unrelated sites: attCaadA7 site, which differs only in two positions from the attCaadA1 site, and VCR2/1, the attC site from a Vibrio cholerae SI cassette, which is larger and unrelated to these two sites (Figure 1C). We also repeated IntI1 binding experiments (Francia et al, 1999) using attI1 (68 bp). The attCaadA7 site was carried on a 76 bp DNA fragment and the VCR2/1 on a 149 bp fragment. We used an MBP-IntI1 fusion protein in our in vitro binding experiments, as previous studies had shown that addition of an MBP tag did not disturb IntI1 function in vitro or in vivo (Gravel et al, 1998a, 1998b). As previously observed (Francia et al, 1999), we found that 48 pmol of IntI1 specifically retarded 0.5 pmol of ds attI1 site, but not the corresponding top strands (ts) or bs (Figure 2A and B). In the case of attCaadA7 ds, there are traces of retarded complex visible in Figure 2A. This might be explained by sufficient instability of this 76 bp duplex to leave a fraction of non-paired ss material, which could be bound by IntI1. These complexes likely correspond to attCaadA7 bs–IntI1 complexes, as nuclease S1 treatment led to their elimination (not shown). It is noteworthy that incubation with the larger (149 bp and likely more stable) VCR2/1 ds did not lead to such complexes. Under the same conditions, IntI1 did not alter the mobility of either of the ds attCaadA7 or VCR2/1 sites (Figure 2B). Conversely, we observed specific retardation when 0.5 pmol of either attCaadA7 bs or VCR2/1 bs was incubated with 4.8 pmol of IntI1, while incubation with the ts of either of these attC sites did not lead to any retardation (Figure 2A).
Figure 2.
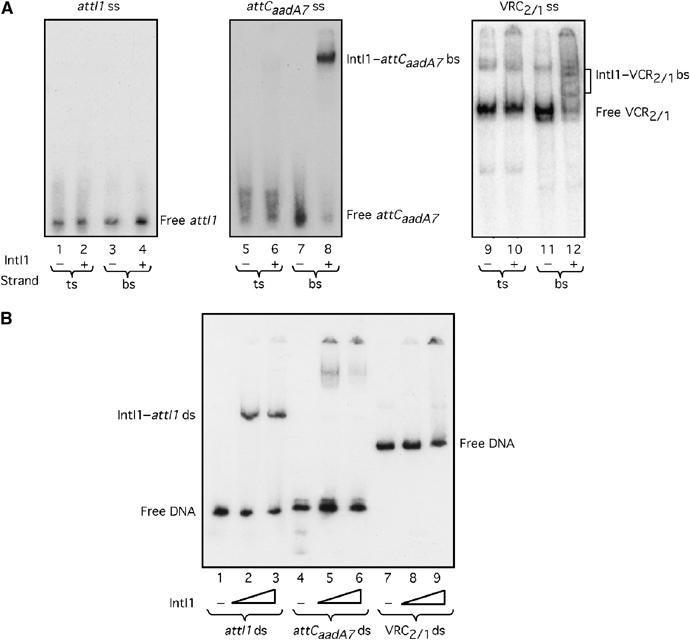
Gel retardation of ss or ds attI1, attCaadA7 and VCR2/1 by IntI1. (A) Single strand substrates. A 4.8 pmol portion of IntI1 was incubated with 0.5 pmol of ssDNA containing the ts or the bs of attI1, attCaadA7 or VCR2/1. Lanes 1–4 show the attI1 ts or bs binding study; lanes 5–8 correspond to the attCaadA7 ts or attCaadA7 bs binding study; the last four lanes (9–12) show the VCR2/1 ts or VCR2/1 bs binding study. (B) Double strand substrates. Lanes 1, 2 and 3 correspond to incubation of 0, 24 or 48 pmol of IntI1 with ds attI1, respectively; lanes 4, 5 and 6 correspond to incubation of 0, 24 or 48 pmol of IntI1 with ds attCaadA7, respectively; lanes 7, 8 and 9 correspond to incubation of 0, 24 or 48 pmol of IntI1 with ds VCR2/1, respectively.
Recombination of attC sites after conjugative transfer
To assess whether an ss structure could be the substrate for recombination in vivo, we used a recombination assay that we developed to compare attC × attI site recombination, which mimics the cassette integration process (Biskri et al, 2005). This assay used conjugation to deliver one of the recombination substrates into a recipient cell expressing the IntI1 integrase and carrying a second recombination target on a pSU38 plasmid derivative (see Figure 3). Conjugative transfer of plasmids occurs by transfer of a single DNA strand (rather than duplex DNA) from donor to recipient. In addition, the orientation of the oriT sequence determines which of the two strands is transferred. The integron recombination site provided by conjugation was carried on an R6K-derived plasmid of the pSW family. Replication of these plasmids relies on the Π protein, provided by a pir gene inserted in the donor genome (Demarre et al, 2005). Transfer functions are also provided by the appropriate plasmid genes inserted into the donor chromosome. Following conjugation, re-circularization of the single transferred strand is catalyzed by the conjugative relaxase enzyme (Pansegrau et al, 1993; Pansegrau and Lanka, 1996). Complete ss transfer and re-circularization precede the complete second strand synthesis. Since the recipient does not supply the Π protein, the transferred plasmid is unable to replicate (Figure 3). This procedure has been called suicide conjugation. Insertion of attC in one orientation or the other in a given pSW derivative would lead to transfer of either attC ts or attC bs. If recombination uses a strand-specific ss substrate, a difference in the recombination rate measured after transfer of either attC ts or attC bs would be expected. On the other hand, if recombination involves a ds substrate, and thus requires second strand synthesis to be effective, no difference in the recombination rate is expected.
Figure 3.
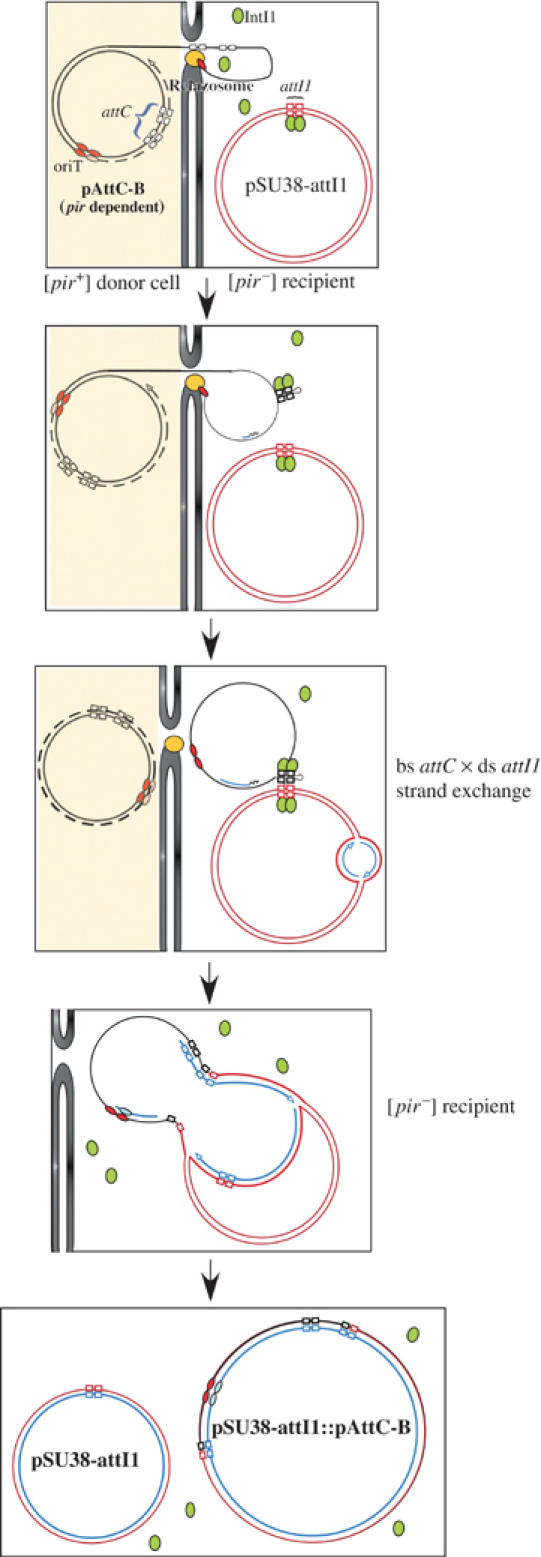
Schematic representation of the conjugation–recombination assay used for the integron cassette integration reaction. Briefly, the donor cell expresses the Π protein, encoded by pir and required for pAttC replication. This strain also provides the transfer functions necessary for its conjugation. The recipient is devoid of a pir gene and therefore cannot sustain pAttC replication. The recipient also contains a plasmid carrying the attI1 site (pSU38-attI1) and expresses IntI1 (symbolized by green ovals). Core site sequences in the attC and attI1 sites are represented as empty boxes, and correspond to those of Figure 1; red and pink ovals indicate the oriT; de novo synthesized strands are shown in blue. The relaxosome, which cleaves and pumps DNA into the recipient, is shown in yellow, and the donor and recipient cell walls and membranes are shown as gray vertical lines. The donor is represented with a pale yellow background.
In a first set of experiments, we compared the recombination of the VCR2/1 bs and VCR2/1 ts after transfer using plasmids pVCR-B and pVCR-T (Table II), which transfer respectively the VCR2/1 bs and VCR2/1 ts in the recipient. pVCR-B and pVCR-T are identical except for the VCR2/1 fragments, which are carried in opposite orientations. We established that both plasmids were transferred at similar rates (2 × 10−1) using strain UB5201-Pi (a UB5201 derivative able to sustain pSW replication) as a recipient. We also determined their IntI1-mediated recombination frequencies with the target attI1 site carried on the compatible plasmid, pSU38-attI1, in the same pSW replication permissive context. This was about 1–5 × 10−2 (Figure 4). We then tested their recombination frequencies following suicide conjugation to a UB5201 recipient (i.e. without Π) carrying the target pSU38-attI1 plasmid and expressing IntI1. An overall rate of 5.5 × 10−3 was obtained for pVCR-B while pVCR-T recombined at a rate of 1 × 10−6. In order to establish that this difference was not due to an unknown contextual difference between plasmids, we inverted the oriT orientation in the two plasmids, leading to plasmids pVCR-TINV (starting from the pVCR-T) and pVCR-BINV (starting from the pVCR-B) (Table II). Again, these two plasmids were transferred at similar rates, 2 × 10−1, yet a 2 × 104-fold higher recombination rate was still obtained when the transferred strand contained the VCR2/1 bs (pVCR-TINV; Figure 4). When recombination of the same constructs was tested in UB5201-Pi, a [pir+] host permitting replication, the ratio of recombination frequencies of the two plasmids obtained with the UB5201 recipient dropped from 2 × 104 to 68. This 68-fold discrepancy may be due to the specific plasmid constructions in some way and is under investigation.
Table 2.
Plasmids used and constructed in this study
| Plasmids | Description |
|---|---|
| pTRC99A::intI1 | oriColE1 [Ap]R (Rowe-Magnus et al, 2001) |
| pSU38Δ | orip15A [Km]R (Biskri et al, 2005) |
| pSU38-attI1 | pSU38Δ::attI1 (Biskri et al, 2005) |
| pSU38-attCaadA7 | 76 bp PstI/BamHI attCaadA7 fragment (annealing between attC-GD1 and attC-GD2) in pSU38Δ digested by PstII/BamHI |
| pSU38Δ-attP | 420 bp SalI λattP fragment from pG-attP in pSU38Δ digested by SalI |
| pTSA29-CXI-AK | oriPSC101ts; intλ (Valens et al, 2004) |
| pSU711ΔoriT::aac(3)-IV | oriVR388 (IncW) [Km Gm]R (Demarre et al, 2005) |
| pG-λattP | oriColE1; λattP lacZ::λattB; [Ap]R (Valens et al, 2004) |
| pMalC-2X::intI1 | 1023 bp SmaI/BamHI intI1 PCR fragment (Fmal2 and Eibam2) amplified from pTRC99A::intI1 in pMalC-2X (New England Biolab) digested by XmnI/BamHI |
| pSW23T | pSW23T::oriTRP4; oriVR6K [Cm]R (Demarre et al, 2005) |
| pSW23TISS | 1772 bp inverse PCR fragment (Isal/sac-1 and Isal/sac-2) amplified from pSW23T, digested by BamHI and religated |
| pVCR-B | pSW23T::VCR2/1B (Biskri et al, 2005) |
| pVCR-T | 207 bp SalI/SacI VCR2/1 fragment from pVCR-B in pSW23Tiss digested by SalI/SacI |
| pAttI1-B | 155 bp EcoRI/BamHI attI1 fragment from pSU38-attI1 in pSW23T digested by EcoRI/BamHI |
| pAttI1-T | 155 bp EcoRI/BamHI attI1 fragment from pSU38-attI1 in pSW23Tiss digested by EcoRI/BamHI |
| pVCR-BIKSL | 1723 bp inverse PCR fragment (Ikpn/sal-1 and Ikpn/sal-2) amplified from pVCR-B, digested by SmaI and religated |
| pVCR-TIKSC | 1723 bp inverse PCR fragment (Ikpn/sac-1 and Ikpn/sac-2) amplified from pVCR-T, digested by SmaI and religated |
| pVCR-BINV | 260 bp KpnI/SalI oriTRP4 fragment from pVCR-B in pVCR-BIKSL digested by KpnI/SalI |
| pVCR-TINV | 260 bp KpnI/SacI oriTRP4 fragment from pVCR-T in pVCR-TIKSC digested by KpnI/SacI |
| pAttC-B | 76 bp MfeI/BamHI attCaadA7 fragment (annealing between attC-GD3 and attC-GD4) in pSW23TISS digested by EcoRI/BamHI |
| pAttC-T | 76 bp MfeI/BamHI attCaadA7 fragment (annealing between attC-GD3 and attC-GD4) in pSW23T digested by EcoRI/BamHI |
| pAttC-B-Mut1 | 70 bp EcoRI/BamHI attCaadA7-Mut1 fragment (annealing between attC-Mut1 UP and DW) in pSW23T digested by EcoRI/BamHI |
| pAttC-B-Mut2 | 70 bp EcoRI/BamHI attCaadA7-Mut2 fragment (annealing between attC-Mut2 UP and DW) in pSW23T digested by EcoRI/BamHI |
| pAttC-B-Mut3 | 70 bp EcoRI/BamHI attCaadA7-Mut3 fragment (annealing between attC-Mut1 DW and attC-Mut3) in pSW23T digested by EcoRI/BamHI |
| pAttC-B-Mut4 | 70 bp EcoRI/BamHI attCaadA7-Mut4 fragment (annealing between attC-Mut4 UP and DW) in pSW23T digested by EcoRI/BamHI |
| pλattB-1 | 34 bp EcoRI/BamHI λattB fragment (annealing between attB-1 and attB-2) in pSW23T digested by EcoRI/BamHI |
| pλattB-2 | 34 bp EcoRI/BamHI λattB fragment (annealing between attB-1 and attB-2) in pSW23TISS digested by EcoRI/BamHI |
| pSW27 | pSW23T::oriTR388; oriT orientation −; oriVR6K [Cm]R (Demarre et al, 2005) |
| pSW27ISS | 1844 bp inverse PCR fragment (Isal/sac-3 and Isal/sac-4) amplified from pSW27, digested by KpnI and religated |
| p388VCR-B | 203 bp SalI/SacI VCR2/1 fragment from pVCR-B in pSW27ISS digested by SalI/SacI |
| p388VCR-T | 184 bp EcoRI/SacI VCR2/1 fragment from pVCR-B in pSW27 digested by EcoRI/SacI |
| pΔ388VCR-B | 1678 bp inverse PCR fragment (Imfe/sac-1 and Imfe/sac-2) amplified from p388VCR-B, digested by KpnI and religated |
| pSW26 | pSW23T::oriTR388; oriT orientation +; oriVR6K [Cm]R (Demarre et al, 2005) |
| p388VCR-TINV | 184 bp EcoRI/SacI VCR2/1 fragment from p388VCR-T in pSW26 digested by EcoRI/SacI |
| p388VCR-BINV | 373 bp MfeI/SacI oriTR388 fragment from p388VCR-B in pΔ388VCR-B digested by MfeI/SacI |
Figure 4.
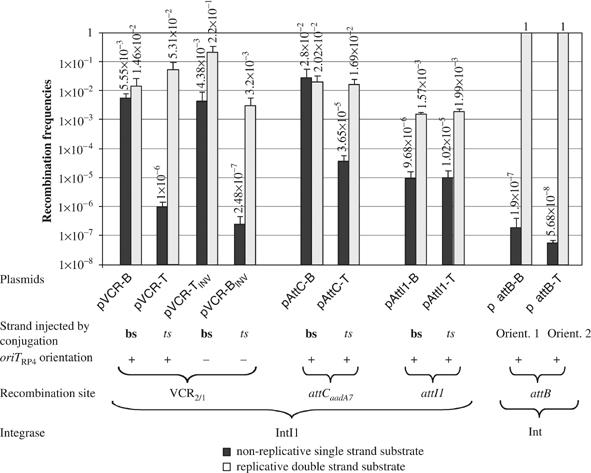
Recombination frequencies of the different recombination sites and substrates. For a given substrate, the black bar indicates the recombination frequencies established in the in vivo recombination assay with non-replicative ss substrate and the light gray bar the corresponding value in the recombination control assay in replication permissive conditions, as described in Materials and methods. Recombination frequencies (vertical axis of histogram) correspond to the average of three independent trials. Error bars show standard deviations. For clarity, the recombination site, the strand–bs (bottom) or ts (top)—injected by conjugation, the orientation (+) and (−) of the oriT and the integrase used are indicated below plasmid names.
We extended our strand recombination analysis to the attCaadA7 site used in the in vitro electrophoretic mobility shift assay (EMSA) (Figure 2). Two plasmids, pAttC-B and -T, were constructed (Table II), allowing the conjugative transfer of either attCaadA7 bs or attCaadA7 ts, respectively. As in the case of pVCR-B and -T, we found that in a [pir+] host, the B and T derivatives were recombined at similar rates (2 × 10−2), whereas in the suicide conjugation assay, attCaadA7 bs recombined at a rate (7.6 × 102) higher than attCaadA7 ts (Figure 4).
To eliminate the possibility that these results were specifically linked to the RP4 transfer machinery, we repeated several of these experiments using plasmid R388, which specifies a different transfer system. Using plasmids pSW26 and pSW27, which carry the R388 oriT in opposite orientations (Demarre et al, 2005), we constructed two derivatives of each containing VCR2/1 in either orientation, leading to plasmids p388VCR-B and -T, and p388VCR-BINV and -TINV (Table II). These four plasmids were found to recombine with attI1 at similar rates when tested in UB5201-Pi, a [pir+] host expressing IntI1 (2.7–13 × 10−2; Figure 5). When measured after suicide conjugation from strain Π1977, which expresses the R388 transfer machinery, recombination of VCR2/1 bs occurred at rates 3.4 × 103 (p388VCR-B) and 6.2 × 103 (p388VCR-TINV) higher than VCR2/1 ts, after conjugation from p388VCR-T and p388VCR-BINV, respectively (Figure 5).
Figure 5.
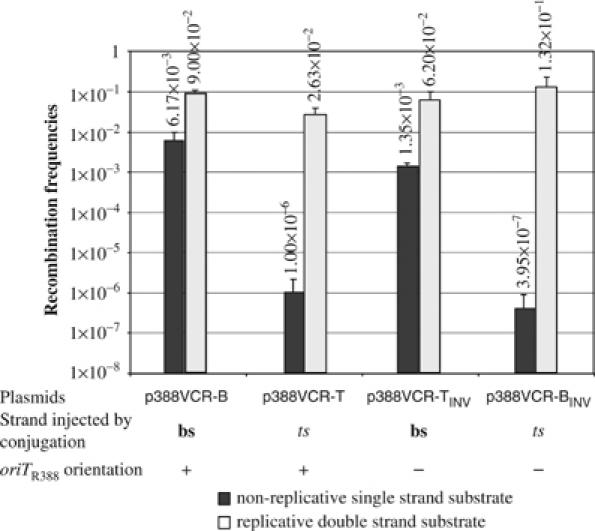
Recombination frequencies of the VCR2/1 site obtained with the R388-based suicide conjugation assay. For a given substrate, the black bar indicates the recombination frequencies established in the in vivo recombination assay with non-replicative ss substrate and the light gray bar the corresponding value in the recombination control assay in replication permissive conditions, as described in Materials and methods. Recombination frequencies (vertical axis of histogram) correspond to the average of three independent trials. Error bars show standard deviations. For clarity, the recombination site, the strand—bs (bottom) or ts (top)—injected by conjugation and the type and orientation (+) and (−) of the oriT are indicated below plasmid names.
Effect of mutations in the potential stem sequence
To test our model of recombination involving the attC bs folding into a stem-and-loop structure, we introduced mutations that would disrupt the potential base pairing, and measured their effect on the recombination frequency. As the last positions involved in the potential stem formed by the various ss attC sites are not conserved and cannot be involved directly in the chemistry of the reaction (positions underlined in Figure 1C), we substituted the last 5 nucleotides of the attCaadA7 stem (attCaadA7Mut1; Figure 6). These mutations lead to a 10-fold decrease of the recombination frequency of the attCaadA7 bs, as established after suicide conjugation of plasmid pAttC-B-Mut1 (Figure 6). We then increased the destabilization of the potential secondary structure by the introduction of three additional substitutions further down in the stem structure and covering the last two positions in the L′/L″ potential hybrid (attCaadA7Mut3; Figure 6). These mutations lead to a 90-fold decrease of the recombination frequency of the attCaadA7. We then tested for both mutants the effect of restoration of complementarity, which would stabilize the attCaadA7Mut1 and attCaadA7Mut3 bs folding, on the recombination frequency. In both cases, these secondary mutations restored a level of recombination similar to the one obtained with the wild-type (WT) attCaadA7 site (attCaadA7Mut2 and attCaadA7Mut4; Figure 6), after suicide conjugation of the corresponding bs from pAttC-B-Mut2 and pAttC-B-Mut4.
Figure 6.
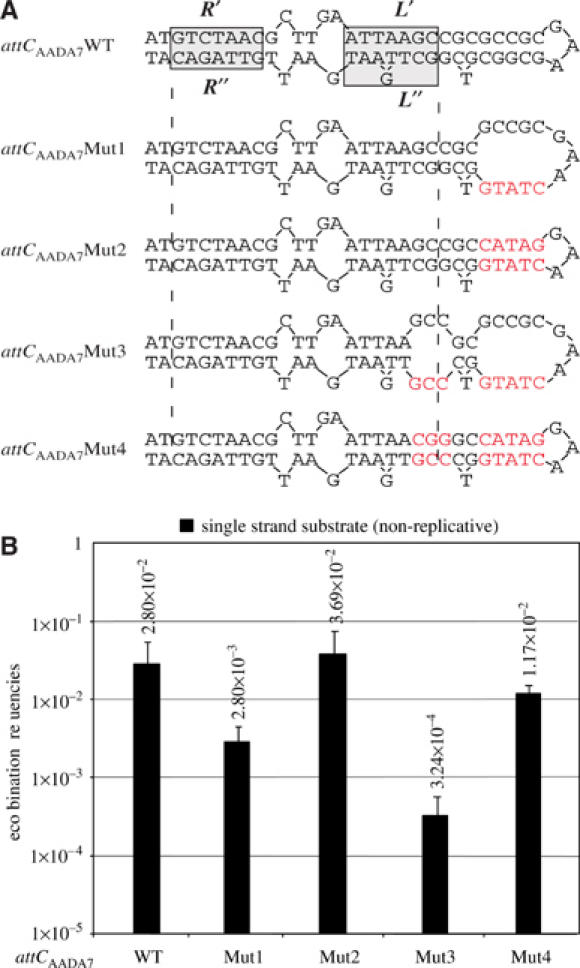
Proposed secondary structure for the attCaadA7 mutants bottom strand (A) and their recombination frequencies (B), as established in the suicide conjugation assay. Red letters indicate mutations introduced in the attCaadA7. Symbols are as in Figure 1.
λ phage attB × attP recombination using the suicide conjugative transfer assay
To confirm that these results truly reflect a single strand preference, we investigated the related phage λ recombination system that is known to require two strands. Here, orientation should have no effect on recombination. We used the λ phage integration, since its mechanism is known in detail (reviewed by Azaro and Landy, 2002). In this reaction, recombination between the phage attP and chromosomal attB sites requires ds substrates and is catalyzed by the λ integrase, Intλ. The attP site was cloned into pSU38 and introduced into the recipient, which supplied the accessory and necessary host protein IHF. The attB site was cloned in both orientations into pSW23T to create pλattB-1 and pλattB-2 (Table II). When tested in a [pir+] host containing pSU38-λattP and a plasmid expressing IntIλ, a 90 min induction was sufficient to obtain 100% attP × attB cointegrate formation with pλattB-1 and pλattB-2. Recombination of each of the λattB strands was then tested following suicide conjugation of either pλattB-1 or pλattB-2 into MG1657-PIλ, a ΔattB∷aadA E. coli that contained pSU38-λattP as recombination target and expressed Intλ. Conjugation was for 2 h and transconjugants were selected for the pλattB marker. This resulted in cointegrate formation (integration) frequencies of 2 × 10−7 and 0.6 × 10−7 for λattB-1 and λattB-2, respectively (Figure 4). Interestingly, increasing the conjugation time up to 3 h resulted in an ≈103 increase of cointegrate formation for both λattB ss substrates. This suggested that the increased time allowed for an increase in the amount of complementary strand synthesis in the recipient, generating the ds sequences necessary for an efficient attB × attP recombination to be catalyzed.
Recombination properties of the attI1 site after suicide conjugative transfer
From EMSA assays, neither the ts nor bs DNA of the other recombination partner attI1 appeared to be bound by IntI1, although ds attI1 site was clearly recognized (Figure 2). To determine whether this is also reflected in recombination, we tested recombination proficiency after suicide conjugative transfer. The attI1 site was cloned in both orientations in pSW23T, leading to pAttI1-B and pAttI1-T, and the attCaadA7 was inserted into pSU38, leading to pSU38-attCaadA7 (Table II). When tested in a [pir+] host expressing IntI1, pAttI1-B and pAttI1-T were found to recombine with the target pSU38-attCaadA7 at similar rates of 1.6–2 × 10−3 (Figure 4). These results, which do not significantly differ from those obtained when recombination sites and vector plasmids were reciprocally reversed, showed that under conditions that permit autonomous replication of all plasmids, the properties of the different recombination sites were independent of the backbone plasmid. Conversely, recombination following suicide conjugative transfer of either attI1 strands with the attCaadA7 site on pSU38 in the recipient was found to occur at identical low rates, about 1 × 10−5 (Figure 4), strongly suggesting that ss attI1 are not bona fide substrates.
Recombination of attC and λattB sites after transformation using double-stranded non-replicative plasmids
To determine whether the ds attC can be used as a recombination substrate, for example by adopting the necessary structure recognized by IntI1, without single strand passage, we transformed the ds circular plasmids pVCR-B and pVCR-T into the [pir−] strain UB5201-I1. This strain expresses IntI1 and carries the target plasmid pSU38-attI1. Competent cells were then transformed with 1, 10 and 50 μg of each of the pVCR derivatives and selected for CmR transformants, as cointegration between the VCR2/1 and the attI1 site carried on the pSU38 would lead to viable CmR transformants. No transformants were obtained for either of the tested plasmids, although the frequency of transformation for a compatible control plasmid, pSC101, was 1.1 × 105 transformants/μg. The maximal recombination frequencies for the ds test plasmids were therefore lower than 5.5 × 10−6. We performed the same type of experiment using plasmids pλattB-1 and pλattB-2, and transforming the [pir−] strain MG1657-PIλ expressing Intλ and containing the target plasmid pSU38-λattP. We obtained CmR transformants at frequencies of 2.1 × 104transformants/μg (pλattB-1) and 3.4 × 104 transformants/μg (pλattB-2), compared to 7.2 × 104 transformants/μg of the control plasmid pSC101. This gives recombination frequencies of 2.9 × 10−1 and 4.7 × 10−1, respectively, for these ds substrates.
Discussion
We have analyzed the mechanism of IntI1-mediated recombination that occurs during integron cassette acquisition and provide evidence that cassette integration occurs by recombination between ss attC and ds attI. Johansson et al (2004) reported covalent complex formation between IntI1 and the attCaadA1 bs, demonstrating that IntI1 not only recognized the bs (Francia et al, 1999), but was also able to catalytically cleave this substrate without the necessity of a complex cruciform structure formed from both attC strands. We have extended these studies to the related attCaadA7 and the poorly related VCR2/1 sites. Like attCaadA1, IntI1 bound only to the bs of ss attCaadA7 and ss VCR2/1. These observations led us to consider a model in which recombination would only involve a structured attC bs and a canonical ds attI site. The attC bs can potentially adopt a ds DNA-like structure by annealing of L″ to L′ and R″ to R′, which has almost all the structural features of a canonical recombination site (Figure 1D). These regions would be separated by an unpaired central segment. In most circularized cassettes, self-pairing of the same single strand could cover almost the entire attC site and even extend slightly further. Indeed, in most cases, the 7 bp R′ and R″ sequence complementarity is extended on the external part, to form a stretch of 9–11 consecutive complementary nucleotides (Hansson et al, 1997; Rowe-Magnus et al, 2003). In addition, comparison of the secondary structure adopted by the different attC sites, which are efficiently recombined by IntI1, shows that apart from the conserved AAC and GTT in the R′ and R″ boxes, and the flipped out G present in all L′/L″ stem, all the other positions show no conservation (Supplementary Figure 1).
This hypothesis was tested in vivo using suicide conjugative transfer of one of the recombination sites, in order to provide an ss substrate (Figure 3). Conjugative transfer of mobile plasmids, such as RP4, occurs by transfer of a single DNA strand from donor to recipient. In addition, the orientation of the oriT sequence determines which of the two strands is transferred. Furthermore, re-circularization of the transferred RP4 strand, catalyzed by the relaxase enzyme, occurs between ss substrates (Pansegrau et al, 1993; Pansegrau and Lanka, 1996). Thus, complete ss transfer and re-circularization precede complete second strand synthesis. In E. coli, second strand synthesis had been shown to be initiated by either a specific primase (TraC) cotransferred with the DNA, or by DnaG (Lanka and Barth, 1981). In our case, specific priming is unlikely, as the transferred plasmid carries only a small piece of the original RP4, a 256 bp fragment centered on the nick site defining the origin of transfer and sufficient to ensure optimal transfer of the carrier plasmid (Demarre et al, 2005). Using this assay, in which the transferred plasmid is unable to replicate in the recipient (suicide conjugation), we can potentially deliver largely ss circularized DNA as a substrate for recombination by IntI1. Our in vivo recombination assays showed that suicide transfer of the attC bs, be it attCaadA7 or a VCR (the attC site specific of the V. cholerae super-integron cassettes), led to cointegrate formation at rates similar to those obtained in the classical assay, which involves recombination between replicative plasmids (Figure 4). In contrast, recombination was three orders of magnitude lower with attC ts and the VCR1/2 ts. It is noteworthy that these in vivo results correlate with the EMSA results.
In addition, we showed that mutations potentially disrupting the putative attC bs folding in a stem-and-loop structure lead to a significant decrease of the recombination frequency of the corresponding bs site (Figure 6). Furthermore, we showed that there was a correlation between the potential destabilization of the secondary structure and the recombination rates (Figure 6). We also showed that compensatory mutations introduced in order to re-establish a stem-and-loop structure similar to the one folded from the WT bs, completely restored the recombination properties of the mutated sites. These observations clearly support the model we propose, in which, the global folding of the bs structure is essential for the recombination to proceed.
If the recombination substrate was not the bs but any other structure involving both strands of the site, we would expect to obtain identical low rates of recombination irrespective of the transferred strand since the limiting step would be synthesis of the strand complementary to that injected by conjugation. Results of this type were observed for λattB × λattP recombination following suicide transfer, for which recombination rates after injection of either strand were found to be 6 orders of magnitude lower than recombination between replicative plasmids in identical conditions (Figure 4). It is noteworthy that a similar but less extreme result was obtained after suicide transfer of either attI1 strand (Figure 4). These last results are in complete agreement with those obtained by EMSA, showing that IntI1 recognizes only the ds attI1.
Finally, inversion of oriT in each plasmid resulted in a reciprocal exchange of recombination rates, and identical recombination biases for ts or bs following suicide transfer were also observed using the R388 conjugative system, strengthening the observations we made using the RP4 conjugative machinery (Figure 5).
Although DNA is not generally maintained in an ss form in vivo, ss DNA is formed during both replication and transcription. It is possible that binding of IntI1 to attC bs somehow stabilizes this form. This model would explain the lack of attC site recombination when sites are delivered on non-replicative ds plasmids by transformation into the recipient cell, in comparison to the high rate of recombination obtained after delivery of the λattB site in similar tests.
Recognition by IntI1 of the folded structure that can be adopted by the bs of attC, and recombination of this structure with a canonical ds attI site would lead to an HJ intermediate that could be productively resolved through an additional replication step (Figure 7). Indeed, once the first strand exchange occurred, resolution of the HJ by a second pair of strand exchanges would lead to the formation of dead-end, linear, covalently closed molecules (axis B in Figure 7). However, if this HJ intermediate was replicated, this would generate the original attI1 site together with the integrated product. Such a model can also be applied to cassette deletion through attC × attC site recombination, which would also only involve the attC bs. The production of circular cassettes at a very low but undetermined frequency has been demonstrated from an array of three complete cassettes driven by IntI1 (Collis and Hall, 1992). These circular cassettes were mainly ds, covalently closed molecules. At first sight, these results are in contradiction with the recombination model we propose. However, the lifetime of ss, non-replicating circular molecules could be short; the synthesis rate inside the cell of the complementary strand is unknown. In addition, open circular molecules were also detected but were attributed to the plasmid preparation protocol used. These could also correspond to single strand molecules in which complementary strand synthesis was incomplete. The same authors also claimed that cassette integration occurred through recombination of ds substrates (Collis et al, 1993). This was based on the recovery of cointegrates following transformation with ds circular cassettes produced in vitro. This was carried out in a similar manner to the experiment described here, in which we were unable to detect cointegrates following transformation using a ds cassette. However, a careful analysis of their data reveals that the events observed occurred at a very low frequency. Indeed, in their assay, recombination proficiency was measured through the number of cointegrates obtained after electroporation of 0.1 μg of cassette into a recipient strain expressing IntI1. This varied from 3 to 28 using different cassettes. Although the competence of the cells was not determined, it is reasonable to estimate a transformation frequency of about 107 transformants/μg for these electrocompetent DH5α cells. This would give frequencies of cointegrate formation of between 3 × 10−6 and 3 × 10−5. Such low rates are of the same order as the background recombination rates we obtained after transfer of the attC ts and might correspond to either the erratic production of the attC bs or reflect a poor IntI1 recombination activity on the ds attC substrate, compared to attC bs substrate.
Figure 7.
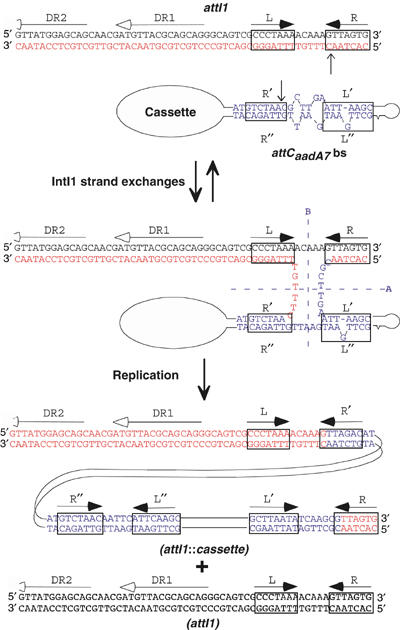
Model of the integron recombination molecular mechanism using an ss attC substrate folded through pairing of the imperfect palindromic sequences. Steps are identical to classical site-specific recombination steps catalyzed by other Y-recombinases, up to the HJ intermediate. Classical resolution through the A axis reverses the recombination to the original substrates, while resolution through the B axis, giving rise to covalently closed linear molecules, is abortive. The non-abortive productive resolution necessitates a replication step. Putative IntI1 binding domains and crossover positions are indicated by boxes and vertical arrows, respectively. The extent of the attI1-protected regions in methylation interference assays (Gravel et al, 1998a), corresponding to repeats DR1 and DR2, is shown by horizontal lines with empty arrowhead.
Our model involving the attC bs as a substrate for recombination may also in part explain the characteristic low recombination rate of the integron machinery. Indeed for canonical Y-recombinase catalyzed reactions in vivo, such as λ phage integration or the yeast 2 μm FRT × FRT inversion catalyzed by Flp, recombination yields are almost 100%. In integron reactions, even in conditions of IntI1 overexpression, the recombination yield never exceeds a few percent (e.g. Collis et al, 2001). This might directly reflect the relative abundance of ss substrate accessible to IntI1 that is generated through replication and/or transcription processes during the cell cycle. Indeed, the recombination rates measured after delivery of attC bs was identical to that obtained in the classical in vivo assay that employs ds attC and ds attI sites, that is, carried on compatible replicative plasmids. It is likely that the IntI1 binding on the attC bs stem-and-loop could stabilize this otherwise transient structure, rendering it prone for recombination. The rest of the cassette sequence can be maintained in a canonical ds DNA form; this will not interfere with the attC bs × attI ds recombination reaction or with its resolution through a replication step.
These results suggest a novel site-specific recombination mechanism that uses a non-canonical substrate. They also explain why the overall complementarity is more conserved than the primary sequence in the different attC sites found in the integron cassettes. This model may be more general and not limited to the integron recombination system. Indeed, we have recently obtained evidence that integration of the single strand V. cholerae CTX phage genome into the dimer resolution site of chromosome 1 by the XerCD recombinases depends on the formation of a stem-and-loop structure (Val et al, 2005). The selective advantages that have led to the development of these ss recombination sites and processes are still elusive. However, this might be linked to the phenomenon of gene dissemination by horizontal transfer, which in many cases goes through an ss stage, as demonstrated for filamentous phages, for conjugation and for natural transformation in bacteria.
Materials and methods
Bacterial strains, plasmids and media
Bacterial strains and plasmids are described in Tables I and II. E. coli strains were grown in Luria-Bertani or, when specified, in Mueller-Hinton (MH) broth at 37°C, or 30°C for the Intλ experiments. Antibiotics were used at the following concentrations: ampicillin (Ap), 100 μg/ml; chloramphenicol (Cm), 25 μg/ml; erythromycin (Em), 200 μg/ml; kanamycin (Km), 25 μg/ml; nalidixic acid (Nal), 30 μg/ml. Thymidine (Thy) and diaminopimelic acid (DAP) were supplemented when necessary to a final concentration of 0.3 mM. Isopropyl-β-D-thiogalactopyranoside (IPTG) was added at 0.5 mM final concentration. Chemicals were from Sigma.
Table 1.
Bacterial strains used in this study
| E. coli strains | Description/relevant characteristics | Reference |
|---|---|---|
| DH5α | supE44 ΔlacU169 (Φ80lacZ′ΔM15) ΔargF hsdR17 recA1 endA1 gyrA96 thi-1 relA1 | Laboratory collection |
| Π1 | DH5α ΔthyA::(erm-pir116) [ErmR] | Demarre et al (2005) |
| Π1977 | Π1 pSU711ΔoriT::aac(3)-IV [GmR KmR ErmR] | Demarre et al (2005) |
| ED9 | F-ΔlacU169 araD139 rpsL relA flbB ΔmalE444 srl::Tn10 recA1 [TcR] | E Dassa (unpublished) |
| β2163 | MG1655::ΔdapA::(erm-pir)RP4-2-Tc::Mu [KmR] | Demarre et al (2005) |
| UB5201 | F-pro met recA56 gyrA [NalR] | Martinez and de la Cruz (1990) |
| UB5201-I1 | UB5201 pTRC99A::intI1 pSU38-attI1 | This study |
| UB5201-Pi | UB5201ΔthyA::(erm-pir116) [NalRErmR] | This study |
| MG1655 | E. coli K12 | Laboratory collection |
| MG1657 | MG1655 ΔλattB::aadA ΔlacZ recA [SpecR] | F Boccard (unpublished) |
| MG1657-PIλ | MG1657 pTSA29-CXI-AK pSU38Δ-attP | This study |
Polymerase chain reaction procedures
Polymerase chain reaction (PCR) for plasmid assembly used the Pfu DNA polymerase (Promega). Other PCR reactions used the PCR Reddy mix (Abgene, UK). Both were used according to the manufacturer's instructions. PCR primers listed in Table III were obtained from Proligo (France).
Table 3.
Oligonucleotides
| Oligonucleotides | Sequences |
|---|---|
| EMSA experiments | |
| aadA7-TB1 | GATCCTGCCTAACAATTCATTCA |
| aadA7-TB2 | TGCAGCAATTGCCTAACGCTTG |
| aadA7bot | TGCAGCAATTGCCTAACGCTTGAATTAAGCCGCGC CGCGAAGCGGCGTCGGCTTGAATGAATTGTTAGGC AGGATC |
| aadA7top | GATCCTGCCTAACAATTCATTCAAGCCGACGCCGC TTCGCGGCGCGGCTTAATTCAAGCGTTAGGCAATT GCTGCA |
| attI1-TB1 | GCGGGATCCGCACTAACTTTG |
| attI1-TB2 | GGAATTCAGCAGCAACGATG |
| attI1bot | GCGGGATCCGCACTAACTTTGTTTTAGGGCGACTG CCCTGCTGCGTAACATCGTTGCTGCTGAATTCCGG |
| attI1top | CCGGAATTCAGCAGCAACGATGTTACGCAGCAGGG CAGTCGCCCTAAAACAAAGTTAGTGCGGATCCCGC |
| VCR-TB1 | GCGGGATCCGTTATAACGCC |
| VCR-TB2 | CCGGAATTCGTTATAACAAACGC |
| VCRbot | GCGGGATCCGTTATAACGCCCGCCTAAGGGGCTGA CAACGCACTACCACTAAACTCAAACACAACAACAG CAACCAC |
| CGCGGCTCAATGGGACTGGAAACGCCACGCGTTGA CAGTCCCTCTTGAGGCGTTTGTTATAACGAATTGG | |
| VCRtop | CCGGAATTCGTTATAACAAACGCCTCAAGAGGGAC TGTCAACGCGTGGCGTTTCCAGTCCCATTGAGCCG CGGTGGT |
| TGCTGTTGTTGTGTTTGAGTTTAGTGGTAGTGCGT TGTCAGCCCCTTAGGCGGGCGTTATAACGGATCGC | |
| Plasmid constructions | |
| Fmal2 | CCGGAATTCTAATAGGAGACCCGGGATGAAAACCG CCACTGCGCC |
| Eibam2 | CGCGGATCCTTACCTCTCACTAGTGAGGGGC |
| Isal/sac1 | CGCGGATCCCATATGCTCGAGGAGCTCGCCGGCCA GCCTCGCAGAGCAGGATTCCCG |
| Isal/sac2 | CGCGGATCCGCTAGCGAATTCGTCGACCAGCTTTT GTTCCCTTTAGTGAGGGTTAATTGCG |
| Ikpn/sal-1 | TCCCCCGGGCATATGCTCGAGGGTACCGGTATCGA TAAGCTTGATATCGAATTCAGATCTG |
| Ikpn/sal-2 | TCCCCCGGGGCTAGCGAATTCGTCGACCAATTCGC CCTATAGTGAGTCGTATTACGCGCGC |
| Ikpn/sac-1 | TCCCCCGGGCATATGCTCGAGGGTACCCACCGCGG TGGCGGCCGCTCTAGAACTAGTGGAT |
| Ikpn/sac-2 | TCCCCCGGGGCTAGCGAATTCGAGCTCCAATTCGC CCTATAGTGAGTCGTATTACGCGCGC |
| Isal/sac-3 | CGGGGTACCCATATGCTCGAGGAGCTCCCGCTCTA GAACTAGTGGATCCCCCGGGCTGCAG |
| Isal/sac-4 | CGGGGTACCGCTAGCGAATTCGTCGACCAGCTTTT GTTCCCTTTAGTGAGGGTTAATTGCG |
| Imfe/sac-1 | CGGGGTACCCATATGCTCGAGCAATTGCACCGCGG TGGCGGCCGCTCTAGAACTAGTGGAT |
| Imfe/sac-2 | CGGGGTACCGCTAGCGAATTCGAGCTCCGCCGAAT AAATACCTGTGACGGAAGATCACTTC |
| attB-1 | AATTCAGCCTGCTTTTTTATACTAACTTGG |
| attB-2 | GATCCCAAGTTAGTATAAAAAAGCAGGCTG |
| attC-GD1 | GATCCTGCCTAACAATTCATTCAAGCCGACGCCGC TTCGCGGCGCGGCTTAATTCAAGCGTTAGGCAATT GCTGCA |
| attC-GD2 | GCAATTGCCTAACGCTTGAATTAAGCCGCGCCGCG AAGCGGCGTCGGCTTGAATGAATTGTTAGGCAG |
| attC-GD3 | GATCCTGCCTAACAATTCATTCAAGCCGACGCCGC TTCGCGGCGCGGCTTAATTCAAGCGTTAGGC |
| attC-GD4 | AATTGCCTAACGCTTGAATTAAGCCGCGCCGCGAA GCGGCGTCGGCTTGAATGAATTGTTAGGCAG |
| attC-Mut1 UP | CTAGAATTCGGTTATAACAATTCATTCAAGCCGAC CATAGTTCGCGGCGC |
| attC-Mut1 DW | CGCGGATCCGGTTATAACGCTTGAATTAAGCCGCG CCGCGAACTATGGTC |
| attC-Mut2 UP | CTAGAATTCGGTTATAACAATTCATTCAAGCCGAC CATAGTTCCTATGGCGGC |
| attC-Mut2 DW | CGCGGATCCGGTTATAACGCTTGAATTAAGCCGCC ATAGGAACTATGGTCGGC |
| attC-Mut3 | CTAGAATTCGGTTATAACAATTCATTCAACGGGAC CATAGTTCGCGGCGC |
| attC-Mut4 UP | CTAGAATTCGGTTATAACAATTCATTCAACGGGAC CATAGTTCCTATGGCCCG |
| attC-Mut4 DW | CGCGGATCCGGTTATAACGCTTGAATTAACGGGCC ATAGGAACTATGGTCCCG |
| MRV | AGCGGATAACAATTTCACACAGGA |
| SW23begin | CCGTCACAGGTATTTATTCGGCG |
| SW23end | CCTCACTAAAGGGAACAAAAGCTG |
In vitro binding assay
In vitro binding assays used an MBP-IntI1 fusion protein, which retains full IntI1 functionality in vitro and in vivo (Gravel et al, 1998a, 1998b).
Purification of MBP-IntI1 integrase. IntI1 was amplified by PCR with primers Fmal2 and Eibam2 (Table III), digested with SmaI and BamHI and cloned into pMalC-2X (New England Biolabs, USA) that had been digested with XmnI and BamHI. MBP-IntI1 fusion protein was purified according to the manufacturer's instructions after expression in strain ED9 (Table I). Concentration and purity of the purified MBP-IntI1 protein preparation were determined on an SDS–PAGE gel.
Double strand substrate. Double strand DNA fragments containing attI1 (68 bp), attCaadA7 (76 bp) and VCR2/1 (149 bp) were generated by PCR with primers attI-TB1 and attI-TB2, aadA7-TB1 and aadA7-TB2 and VCR-TB1 and VCR-TB2 (Table III), respectively, using pSU38-attI1, pSU38-attCaadA7 and pVCR-BIKSL (Table II) as templates. One primer in each pair used (attI-TB1, aadA7-TB2 and VCR-TB1) was previously labeled at its 5′ terminus with radioactive phosphate transferred from [γ-32P]ATP (Amersham) by T4 polynucleotide kinase. PCR products were purified with the QIAquick PCR purification Kit (Qiagen).
Single strand substrate. Oligonucleotides corresponding to the attI1 ts (attI1top) and bs (attI1bot), attCaadA7 ts (aadA7top) and bs (aadA7bot) and VCR2/1 ts (VCRtop) and VCR2/1 bs (VCRbot) (Table III) were obtained from Proligo (France) and MWG Biotech AG (Germany), 5′ labeled with radioactive phosphate and purified as stated above.
Electrophoretic mobility shift assay. Purified labeled DNA fragments (20 000 c.p.m., 0.5 pmol) were incubated with MBP-IntI1 for 15 min at 30°C in a 20 μl final volume containing 50 mM Tris (pH 7.5), 100 mM NaCl, 1 mM CHAPS, 0.2 mM EDTA, 5% glycerol, 1 mM dithiothreitol, 1.5 μg of poly(dI-dC) DNA and 0.7 μg of bovine serum albumin. Following this incubation, the binding reaction mixtures were electrophoresed at room temperature in 6% native polyacrylamide gels (50 mM Tris, 400 mM glycine, 1.73 mM EDTA) (Derre et al, 1999).
In vivo recombination assay
Recombination with non-replicative single strand substrate. The in vivo recombination assay was based on that of Biskri et al (2005). It used conjugation to deliver one of the recombination substrates into a recipient cell expressing the IntI1 integrase and carrying a second recombination substrate on a pSU38 plasmid (Bartolome et al, 1991) derivative. The recombination sites provided by conjugation were carried on suicide vectors from the R6K-based pSW family (Demarre et al, 2005). Plasmids are described in Table II. IntI1 integrase was expressed under the control of LacI from pTRC99A∷intI1 (p112 in Rowe-Magnus et al, 2002). Plasmids carrying different recombination sites are listed in Table II. Briefly, the RP4 (IncPα) conjugation system used the donor strain, β2163 [dapA−, pir+] (Demarre et al, 2005) and the recipient, UB5201-I1, which does not carry a pir gene copy. β2163 carries an RP4 integrated into its chromosome, requires DAP to grow in rich medium and can sustain pSW replication through the expression of a chromosomally integrated pir gene. UB520-I1 is a UB5201 derivative, which contains pTRC99A∷intI1 [AmpR] and the pSU38 plasmid derivative [KmR] carrying the targeted recombination site (Table I). As pSW replication absolutely requires the Π protein, the number of recipients expressing the pSW marker directly reflects the frequency of cointegrate formation between the conjugated pSW plasmid and the target replicon in the recipient cell. Conjugations were performed as previously described (Biskri et al, 2005). The integration activity using this assay was calculated as the ratio of transconjugants expressing the pSW marker CmR to the total number of recipient AmpR, KmR clones. attC × attI cointegrate formation was checked by PCR with appropriate primers (MRV and SW23begin or MRV and SW23end; Table III) on eight randomly chosen clones per experiment. Backgrounds were established using recipient strains containing an empty pTRC99A in place of the pTRC99A∷intI1, and were found to be <8 × 10−7.
Similar experiments were performed using the IncW R388 conjugation system. In this case, the donor strain was Π1977, a [thyA−pir+] Π1 derivative containing the plasmid pSU711ΔoriT∷aac(3)-IV. This plasmid is deleted for its oriT but expresses all R388 transfer functions and has been shown to support the efficient transfer of the oriTR388 carrying pSW derivatives, pSW26 and pSW27 (Demarre et al, 2005). This strain requires Thy for growth in MH medium and sustain pSW replication through the expression of a chromosomally integrated pir gene. The recipient strains were identical to those used in the RP4-based assay, and the integration activity was determined using the same calculation.
Recombination control: recombination with a replicative double-stranded substrate
The three plasmids, pTRC99A∷intI1, pSU38-attI1 and pSW∷attC, harboring the different attC site derivatives were transformed into UB5201-Pi, a UB5201 derivative rendered [pir+], by thyA allelic replacement, with allele ΔthyA∷(erm-pir116) as described (Demarre et al, 2005). This [pir+] strain allows pSW∷attC replication. After overnight growth in the presence of appropriate antibiotics and 0.5 mM IPTG to allow intI1 expression, cells were harvested and total plasmid DNA extracted. This was then introduced by transformation into DH5α, a [pir−] strain, and transformants were selected for CmR (the pSW∷attC marker) or KmR (the pSU38-attI1 marker). As pSW∷attC cannot replicate in DH5α, CmR clones should correspond to cointegration of the plasmid with pSU38-attI1 through intI1-mediated recombination between the attI1 and attC sites. Recombination activity is calculated as the ratio of CmR to KmR transformants.
Conjugation control. Conjugation was performed using the counterselectable donor strains β2163 or Π1977, respectively DAP and Thy auxotrophs, carrying the different pSW∷attC derivatives, and the [pir+] recipient strain UB520-Pi, carrying pTRC99A∷intI1 and pSU38-attI1. Control conjugations were performed in the same conditions as the suicide conjugative transfer assay. Frequencies were measured as the fraction of CmR to AmpR KmR clones in the recipient population.
Phage λ attP × attB recombination assay
The same suicide conjugative transfer assays were applied to the well-known reaction of phage λ integration (attP × attB recombination).
Two pSW plasmids carrying the attB site in both orientations, pλattB-1 and pλattB-2, to permit suicide transfer of either strand, were constructed. The attP site was cloned in pSU38Δ, leading to pSU38-λattP, and the expression of intλ was controlled by temperature shift from plasmid pTSA29-CX1-AK. Recombination was measured after 2 h of conjugation using MG1657-PIλ, an MG1657 derivative carrying pTSA29-CX1-AK and pSU38-λattP as a recipient. Recombination activity is established as the ratio of the CmR transconjugants to the AmpR, KmR recipients.
We determined that under these conditions, the frequency of conjugation of both pλattB-1 and pλattB-2 plasmids to a [pir+] recipient strain was about 2–5 × 10−2. Recombination controls with ds substrates were established in a [pir−] host as for the integron reaction, but the induction of the intλ expression was limited to 90 min, as we found that it was sufficient to obtain 100% recombination.
Supplementary Material
Supplementary Figure 1
Acknowledgments
We thank Dr T Msadek for helpful advices in the EMSA experiments. We thank Dr F Boccard and E Dassa for kindly providing strains MG1657 and ED9. We thank Drs M Chandler, F-X Barre, F Cornet and D Rowe-Magnus for helpful comments on the manuscript. GD and MB are doctoral fellows from the MENESR. This work was supported by the Institut Pasteur, the CNRS (URA 2171 and GDR 2157 on transposable elements) and the Programme de ‘Microbiologie fondamentale et appliquée, maladies infectieuses, environnement et bioterrorisme' from the MENESR.
References
- Azaro MA, Landy A (2002) Chapter 7-λ integrase and the λ Int family. In Mobile DNA II, Craig NL, Craigie R, Gellert M, Lambowitz AM (eds) pp 118–148. Washington, DC: ASM Press [Google Scholar]
- Bartolome B, Jubete Y, Martinez E, de la Cruz F (1991) Construction and properties of a family of pACYC184-derived cloning vectors compatible with pBR322 and its derivatives. Gene 102: 75–78 [DOI] [PubMed] [Google Scholar]
- Biskri L, Bouvier M, Guerout AM, Boisnard S, Mazel D (2005) Comparative study of class 1 integron and Vibrio cholerae superintegron integrase activities. J Bacteriol 187: 1740–1750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collis CM, Grammaticopoulos G, Briton J, Stokes HW, Hall RM (1993) Site-specific insertion of gene cassettes into integrons. Mol Microbiol 9: 41–52 [DOI] [PubMed] [Google Scholar]
- Collis CM, Hall RM (1992) Gene cassettes from the insert region of integrons are excised as covalently closed circles. Mol Microbiol 6: 2875–2885 [DOI] [PubMed] [Google Scholar]
- Collis CM, Kim MJ, Stokes HW, Hall RM (1998) Binding of the purified integron DNA integrase Intl1 to integron- and cassette-associated recombination sites. Mol Microbiol 29: 477–490 [DOI] [PubMed] [Google Scholar]
- Collis CM, Recchia GD, Kim MJ, Stokes HW, Hall RM (2001) Efficiency of recombination reactions catalyzed by class 1 integron integrase IntI1. J Bacteriol 183: 2535–2542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demarre G, Guerout AM, Matsumoto-Mashimo C, Rowe-Magnus DA, Marlière P, Mazel D (2005) A new family of mobilizable suicide plasmids based on the broad host range R388 plasmid (IncW) or RP4 plasmid (IncPα) conjugative machineries and their cognate E. coli host strains. Res Microbiol 156: 245–255 [DOI] [PubMed] [Google Scholar]
- Derre I, Rapoport G, Msadek T (1999) CtsR, a novel regulator of stress and heat shock response, controls clp and molecular chaperone gene expression in gram-positive bacteria. Mol Microbiol 31: 117–131 [DOI] [PubMed] [Google Scholar]
- Francia MV, Zabala JC, de la Cruz F, Garcia-Lobo JM (1999) The IntI1 integron integrase preferentially binds single-stranded DNA of the attC site. J Bacteriol 181: 6844–6849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gravel A, Fournier B, Roy PH (1998a) DNA complexes obtained with the integron integrase IntI1 at the attI1 site. Nucleic Acids Res 26: 4347–4355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gravel A, Messier N, Roy PH (1998b) Point mutations in the integron integrase IntI1 that affect recombination and/or substrate recognition. J Bacteriol 180: 5437–5442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall RM, Brookes DE, Stokes HW (1991) Site-specific insertion of genes into integrons: role of the 59-base element and determination of the recombination cross-over point. Mol Microbiol 5: 1941–1959 [DOI] [PubMed] [Google Scholar]
- Hall RM, Collis CM (1998) Antibiotic resistance in gram-negative bacteria: the role of gene cassettes and integrons. Drug Resist Update 1: 109–119 [DOI] [PubMed] [Google Scholar]
- Hansson K, Skold O, Sundstrom L (1997) Non-palindromic attl sites of integrons are capable of site-specific recombination with one another and with secondary targets. Mol Microbiol 26: 441–453 [DOI] [PubMed] [Google Scholar]
- Johansson C, Kamali-Moghaddam M, Sundstrom L (2004) Integron integrase binds to bulged hairpin DNA. Nucleic Acids Res 32: 4033–4043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanka E, Barth PT (1981) Plasmid RP4 specifies a deoxyribonucleic acid primase involved in its conjugal transfer and maintenance. J Bacteriol 148: 769–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez E, de la Cruz F (1990) Genetic elements involved in Tn21 site-specific integration, a novel mechanism for the dissemination of antibiotic resistance genes. EMBO J 9: 1275–1281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pansegrau W, Lanka E (1996) Mechanisms of initiation and termination reactions in conjugative DNA processing. Independence of tight substrate binding and catalytic activity of relaxase (TraI) of IncPalpha plasmid RP4. J Biol Chem 271: 13068–13076 [DOI] [PubMed] [Google Scholar]
- Pansegrau W, Schroder W, Lanka E (1993) Relaxase (TraI) of IncP alpha plasmid RP4 catalyzes a site-specific cleaving–joining reaction of single-stranded DNA. Proc Natl Acad Sci USA 90: 2925–2929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe-Magnus DA, Guerout A-M, Ploncard P, Dychinco B, Davies J, Mazel D (2001) The evolutionary history of chromosomal super-integrons provides an ancestry for multi-resistant integrons. Proc Natl Acad Sci USA 98: 652–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe-Magnus DA, Guerout AM, Biskri L, Bouige P, Mazel D (2003) Comparative analysis of superintegrons: engineering extensive genetic diversity in the vibrionaceae. Genome Res 13: 428–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe-Magnus DA, Guerout AM, Mazel D (2002) Bacterial resistance evolution by recruitment of super-integron gene cassettes. Mol Microbiol 43: 1657–1669 [DOI] [PubMed] [Google Scholar]
- Stokes HW, O'Gorman DB, Recchia GD, Parsekhian M, Hall RM (1997) Structure and function of 59-base element recombination sites associated with mobile gene cassettes. Mol Microbiol 26: 731–745 [DOI] [PubMed] [Google Scholar]
- Val ME, Bouvier M, Campos J, Sherratt D, Cornet F, Mazel D, Barre FX (2005) The single-stranded genome of phage CTX is the form used for integration into the genome of Vibrio cholerae. Mol Cell 19: 559–566 [DOI] [PubMed] [Google Scholar]
- Valens M, Penaud S, Rossignol M, Cornet F, Boccard F (2004) Macrodomain organization of the Escherichia coli chromosome. EMBO J 23: 4330–4341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter AE, Turner DH, Kim J, Lyttle MH, Muller P, Mathews DH, Zuker M (1994) Coaxial stacking of helixes enhances binding of oligoribonucleotides and improves predictions of RNA folding. Proc Natl Acad Sci USA 91: 9218–9222 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1