

Abstract
Cells latently infected with HIV represent a currently insurmountable barrier to viral eradication in infected patients. Using the J-Lat human T-cell model of HIV latency, we have investigated the role of host factor binding to the κB enhancer elements of the HIV long terminal repeat (LTR) in the maintenance of viral latency. We show that NF-κB p50–HDAC1 complexes constitutively bind the latent HIV LTR and induce histone deacetylation and repressive changes in chromatin structure of the HIV LTR, changes that impair recruitment of RNA polymerase II and transcriptional initiation. Knockdown of p50 expression with specific small hairpin RNAs reduces HDAC1 binding to the latent HIV LTR and induces RNA polymerase II recruitment. Similarly, inhibition of histone deacetylase (HDAC) activity with trichostatin A promotes binding of RNA polymerase II to the latent HIV LTR. This bound polymerase complex, however, remains non-processive, generating only short viral transcripts. Synthesis of full-length viral transcripts can be rescued under these conditions by expression of Tat. The combination of HDAC inhibitors and Tat merits consideration as a new strategy for purging latent HIV proviruses from their cellular reservoirs.
Keywords: HDAC, HIV, latency, NF-κB, trichostatin A
Introduction
A small pool of memory CD4+ T cells latently infected with integrated but transcriptionally silent HIV proviruses has undermined all attempts at curative eradication of virus in HIV-infected patients. Although active replication of HIV can be reduced to undetectable levels by multidrug therapies, the long half-life of these latently infected memory CD4+ T cells predicts that treatment must be continued for more than 60 years for effective clearance of this reservoir (Finzi et al, 1997; Siliciano et al, 2003). Systemic reseeding of HIV from this latent reservoir appears to play an important role in the recrudescence of viral replication that routinely follows the withdrawal of antiviral therapy in fully suppressed patients. To advance therapeutic strategies beyond an expectation of life-long treatment, novel approaches for dealing with the latent HIV reservoir must be developed (Pomerantz, 2003). An important first step toward this goal is to understand at a molecular level how HIV latency is established and maintained.
HIV latency is associated with a lack of proviral gene expression. Thus, latently infected memory CD4+ T cells differ from their uninfected counterparts only by the presence of the integrated and transcriptionally silent HIV provirus. Expression of the viral genome is regulated by the enhancer and promoter elements contained within the HIV long terminal repeat (LTR) located at the 5′ end of the integrated HIV provirus (see Rohr et al, 2003 for review). In the prevailing model of HIV transcription, the RNA polymerase II (RNA Pol II) complex is constitutively bound to the HIV promoter and persistently initiates the synthesis of short RNA transcripts (Kao et al, 1987; Ratnasabapathy et al, 1990; Yankulov et al, 1994). However, in the absence of the virally encoded transactivating protein Tat or cellular activation signals, the processivity of this bound polymerase is sharply limited, leading to the production of abortive short viral transcripts (Laspia et al, 1989). Tat binds to an RNA stem–loop structure termed the transactivating responsive element (TAR), which is located at the 5′ end of all initiated viral transcripts (Kao et al, 1987), and also recruits cellular cyclin T1 and CDK9 proteins (the p-TEFb complex), which phosphorylate multiple serines in the C-terminal domain (CTD) of RNA Pol II (Zhu et al, 1997). These events culminate in an elongating RNA Pol II complex that effectively synthesizes full-length HIV transcripts (Marshall and Price, 1995; Zhou et al, 1998). Similarly, induction of transcriptionally active NF-κB strongly promotes recruitment of RelA to κB-binding sites within the LTR, driving the initiation and elongation of HIV transcripts (West et al, 2001). Of note, RelA also binds p-TEFb, providing a Tat-independent mechanism for initial RNA Pol II-dependent synthesis of HIV Tat mRNA (Barboric et al, 2001).
The prototypical NF-κB transcription factor is a positively acting heterodimeric complex composed of RelA and NF-κB1 p50 (see Hayden and Ghosh, 2004 for review). Both RelA and p50 bind DNA through a shared Rel homology domain; however, RelA also contains C-terminal transcriptional activation domains (TAD), whereas p50 does not. In the absence of NF-κB activating stimuli, RelA–p50 heterodimers are bound by a specific inhibitor, IκBα, which prevents this transcription factor from associating with cognate κB enhancers present in NF-κB-responsive target genes. In unstimulated cells, NF-κB1 p50–p50 homodimers are the predominant nuclear species of Rel proteins and bind a range of NF-κB-responsive genes in vivo. The ability of NF-κB1 p50 to bind DNA coupled with its lack of a TAD has suggested that p50–p50 homodimers may act as κB-specific transcriptional repressors. Consistent with this notion, p50 homodimers also bind to histone deacetylases (HDACs) and recruit these enzymes to various NF-κB target genes under basal conditions (Zhong et al, 2002), where they promote deacetylation of surrounding histones and reduced basal transcriptional activity.
Histones are subject to dynamic covalent modifications that modify gene expression, including acetylation, methylation, ubiquitylation, and ADP-ribosylation (Khorasanizadeh, 2004). Compaction of chromatin associated with HDAC-mediated deacetylation of histone tails can markedly reduce the binding of basal transcription factors to their DNA targets. In contrast, histone acetylation induced by histone acetyltransferases (HATs) is associated with relaxation of chromatin structure, a characteristic of transcriptionally active genes (Kurdistani and Grunstein, 2003). The integrated HIV provirus is assembled into an ordered chromatin structure altered by NF-κB-inducing stimuli or inhibitors of HDAC activity (Pazin et al, 1996; Van Lint et al, 1996). How this restructuring impacts HIV transcription is unclear.
The exceeding rarity of latently HIV-infected cells coupled with the lack of a distinctive surface marker makes purification and biochemical analysis of these cells impractical. As an experimentally tractable and relevant model of postintegration HIV latency, we have employed Jurkat CD4+ T-cell-based J-Lat clones to explore the molecular underpinnings of HIV latency (Jordan et al, 2003). J-Lat cells contain a single, full-length integrated HIV provirus in which GFP has been substituted for Nef. This substitution allows rapid assessment of HIV transcriptional activity by cytometric detection of GFP epifluorescence. Our recent studies have shown that NF-κB-inducing agents rapidly promote the binding of RelA to the latent HIV LTR, leading in turn to GFP expression (Williams et al, 2004). In the present study, we explored how HIV proviral latency is maintained in J-Lat cells, investigating the potential role of κB-specific transcriptional repressors. Using chromatin immunoprecipitation (ChIP) analyses, we found that the HIV LTR is actively repressed through local histone deacetylation and restriction of promoter access to RNA Pol II mediated by NF-κB1 p50–HDAC1 complex binding.
Results
RNA Pol II binding to the HIV LTR is greatly reduced in latently infected T cells
To examine the potential occupancy of the κB enhancers in the HIV LTR of latently infected human CD4+ T lymphocytes, we first assessed NF-κB1 p50 and RelA binding in ChIP assays using the human J-Lat T-cell model of postintegration HIV latency. J-Lat T cells were incubated with or without TNF-α (20 ng/ml) for 30 min, formaldehyde-crosslinked sheared chromatin extracts were prepared, and these extracts were immunoprecipitated with antibodies specific for RelA, NF-κB1 p50, or RNA Pol II. These immunoprecipitates were then interrogated for the presence of HIV-1 LTR sequences spanning the κB enhancers by PCR (Figure 1B, lanes 1 and 2). More distal HIV sequences (Figure 1B, lanes 3 and 4) and DNA 3 kb upstream (Figure 1B, lanes 5 and 6) or downstream of the β-actin promoter (Figure 1B, lanes 7 and 8) served as specificity controls (Figure 1B, lanes 5 and 6). RelA or RNA Pol II antibodies did not detectibly co-immunoprecipitate HIV LTR DNA in unstimulated samples, suggesting that these factors do not bind to the LTR in unstimulated J-Lat cells in vivo (Figure 1B, lane 1). In contrast, unstimulated samples immunoprecipitated with NF-κB1 p50 antibodies were strongly enriched in HIV LTR DNA, suggesting constitutive binding of NF-κB1 p50 to the latent LTR (Figure 1B, lane1). Immunoprecipitation of TNF-α-stimulated samples with anti-RelA and anti-RNA Pol II antibodies led to marked enrichment in HIV LTR DNA, indicating the recruitment of both RelA and RNA Pol II under these stimulated conditions. Binding of NF-κB1 p50 did not appreciably change with TNF-α stimulation (Figure 1B, lane 2).
Figure 1.
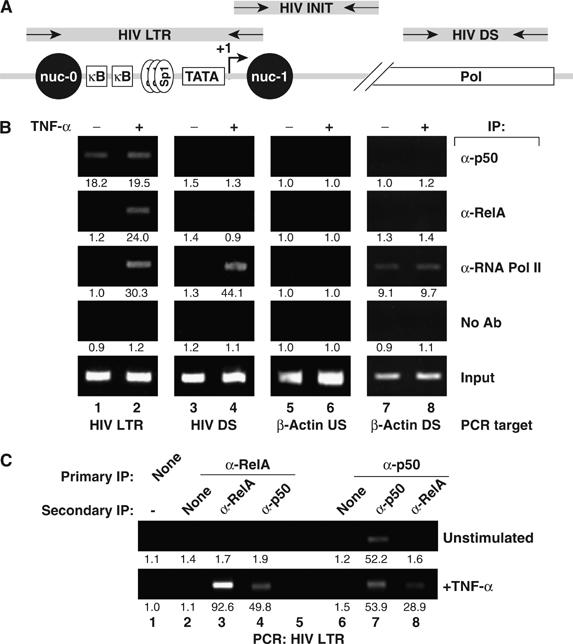
p50/RelA complexes displace NF-κB1 p50 from the latent HIV promoter. (A) Schematic of the HIV genome and location of oligonucleotide primers. ChIP primers for analysis of the HIV LTR were designed to span the nuc-0 to nuc-1 region, including the duplicated κB enhancers, the TATA box, and the transcriptional initiation site. Primers for analysis of initiated HIV transcripts were directed against TAR, and primers for downstream analysis were targeted at HIV tat. (B) NF-κB1 p50 is constitutively recruited to the HIV-1 promoter, whereas RelA and RNA Pol II are inducibly recruited. Fixed chromatin extracts from J-Lat 6.3 cells treated with 20 ng/ml TNF-α for 30 min or left unstimulated were immunoprecipitated with the indicated antibodies. Samples were assessed for enrichment in HIV LTR DNA, downstream HIV DNA (HIV DS), nonspecific control DNA (β-actin US), or transcriptionally active control DNA (β-actin DS) by UV visualization of PCR products in an ethidium bromide-stained agarose gel. Specific enrichment was quantitated by real-time PCR; mean of three measurements is indicated beneath each band image. Data are representative of three independent experiments. (C) A RelA–NF-κB1 p50-containing complex is recruited to the activated HIV LTR. Fixed chromatin extracts from J-Lat 6.3 cells treated as in panel B were immunoprecipitated with the indicated antibodies, and enriched complexes were subjected to a second round of immunoprecipitation with the indicated antibodies. Samples were assessed for enrichment of HIV LTR DNA. Nonspecific enrichment in β-actin DNA was not detected, supporting the specificity of these immunoprecipitations (data not shown). Data are representative of three independent experiments.
The absence of detectable binding of RNA Pol II to the latent HIV LTR by ChIP under unstimulated conditions was surprising in view of the prevailing model of a constitutively bound but non-processive polymerase in the absence of Tat or activating stimuli. To confirm that generalized immunoprecipitation of DNA-associated RNA Pol II was not TNF-α dependent in all situations, anti-RNA Pol II immunoprecipitation was performed and followed by interrogation of a constitutively transcribed region of the β-actin gene (β-ACT DS), expected to associate with RNA Pol II under basal conditions. β-ACT DS DNA was readily detected in both untreated and TNF-α-stimulated samples (Figure 1B, lanes 7 and 8, third panel), demonstrating stimulation-independent association of RNA Pol II association with a constitutively transcribed cellular gene. To assess whether RNA Pol II was similarly present on downstream regions of HIV DNA, a real-time indicator of effective polymerase elongation (Sandoval et al, 2004), immunoprecipitated samples were assessed for enrichment in HIV Pol DNA (DS HIV). RNA Pol II antibodies strongly enriched HIV DS DNA in TNF-α treated but not untreated samples, indicating that cellular activation was required for the appearance of an effective elongating Pol II complex (Figure 1B, lanes 3 and 4, third panel).
The detection of HIV LTR DNA with antibodies specifically reacting with NF-κB1 p50 and RelA after TNF-α induction is consistent with the recruitment of p50 and RelA as a heterodimeric NF-κB complex (p50/RelA). However, this analysis did not exclude the possibility that homodimers of p50 and RelA were recruited to separate HIV LTR DNA fragments. To distinguish between these possibilities, sequential ChIP (seqChIP) with antibodies specific for RelA or p50 was performed. TNF-α stimulated, but not unstimulated, samples incubated with anti-RelA antibodies in both the first and second rounds of immunoprecipitation were highly enriched in HIV LTR DNA (Figure 1C, lane 3). Immunoprecipitation first with anti-RelA and then with anti-p50 showed enrichment in HIV LTR DNA only after TNF-α induction (Figure 1C, lane 4), indicating coassociation of RelA and NF-κB1 p50 with a common fragment of HIV LTR DNA. To confirm this result, we performed seqChIP in the reverse manner, using anti-p50 antibodies in the first immunoprecipitation and anti-p50 or anti-RelA antibodies in the second. In unstimulated cells, HIV LTR binding of p50, but not RelA, was detected; however, in TNF-α-treated samples, RelA-bound HIV LTR DNA was detected in the anti-p50 immunoprecipitates (Figure 1C, lane 8), supporting the conclusion that the HIV LTR in TNF-α-activated cells is bound by p50/RelA heterodimers.
NF-κB1/p50 knockdown induces increased basal transcription of latent HIV
In view of the constitutive binding of NF-κB1 p50 to the latent HIV LTR, we investigated the possibility that p50, which lacks a transactivation domain, might function as a transcriptional repressor of the HIV LTR, thereby reinforcing HIV latency in these cells. To assess this possibility, a small hairpin RNA (shRNA) targeting NF-κB1 p50 was cloned into an expression vector coexpressing mouse MHC class I H2Kk, the latter permitting rapid identification of successfully transfected cells. J-Lat cells were transfected with various quantities of p50-shRNA vector, stained for H2Kk expression, and H2Kk-expressing cells were enriched by cell sorting. Whole-cell lysates were prepared and analyzed by immunoblotting for NF-κB1 p50 expression. Cells transfected with the p50-targeted shRNA vector exhibited an shRNA dose-dependent decrease in NF-κB1 p50 expression (Figure 2A).
Figure 2.
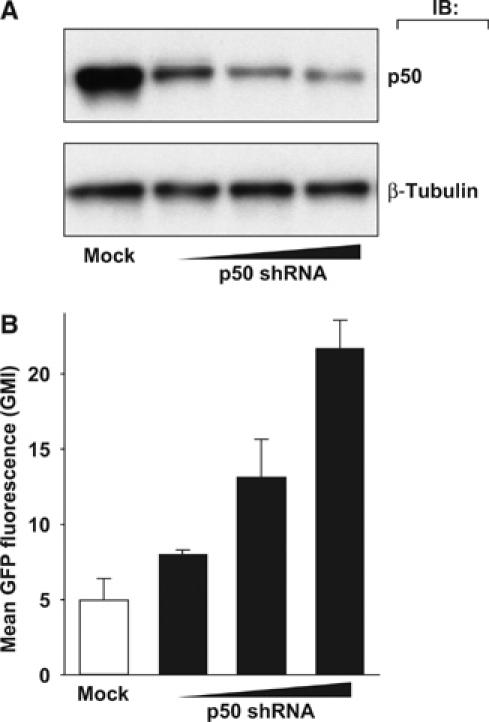
NF-κB1 p50 inhibits basal transcription of HIV in latently infected T cells. (A) Transient reduction of NF-κB1 p50 expression with anti-p50 shRNA vector. J-Lat 6.3 cells were cotransfected with an shRNA vector directed against NF-κB1 p50 and a plasmid expressing the cell-surface H-2Kk marker to identify transfected cells. H-2Kk-expressing cells were sorted and lysed 72 h after transfection, and samples were assessed for NF-κB p50 by Western blot. Data are representative of three independent experiments. (B) Reduction of NF-κB1 p50 expression is associated with increased basal HIV expression. Cells were treated as in panel A, and H-2Kk-expressing cells were assessed for HIV-LTR-driven expression of GFP by FACS. Experiments were conducted in triplicate; error bars represent standard deviation. Data are representative of three independent experiments.
HIV gene expression within these p50-shRNA-transfected cells was examined by flow cytometric analysis of LTR-driven GFP expression in J-Lat cells. In contrast to the induction achieved with TNF-α stimulation, p50-shRNA transfection was not sufficient to drive multi-log increases in GFP expression (data not shown); however, cells transfected with p50-shRNA consistently displayed higher GFP fluorescence as measured by increased mean geometric fluorescence (Figure 2B). Induction of p50-shRNA-transfected cells with TNF-α produced a GFP expression response in a similar percentage of the cells as observed with untransfected cells (data not shown). Taken together, these data suggest that a modest increase in HIV transcription occurs following ‘knockdown' of p50 expression in the absence of additional cellular stimulation.
HDAC1 is recruited to the latent HIV promoter and is inducibly removed
NF-κB1 p50 has been suggested to act as a transcriptional repressor not only because it lacks a TAD but also because it assembles with HDAC1. HDAC1 action is associated with repressive changes in chromatin structure, leading to diminished transcription (Zhong et al, 2002). To assess whether p50 effectively recruits HDAC1 to the latent HIV LTR, J-Lat chromatin extracts were immunoprecipitated with specific HDAC1 antibodies. The immunoprecipitates were enriched in HIV LTR DNA, indicating that HDAC1 is associated with the latent HIV LTR (Figure 3A). In contrast, immunoprecipitation of HDAC4 or HDAC7 did not yield similar enrichment in HIV LTR DNA (data not shown). To assess the effect of cellular activation and the recruitment of p50/RelA complexes to the HIV LTR on HDAC1 binding, chromatin extracts from TNF-α-treated cells were analyzed by ChIP. Levels of HIV LTR DNA detected with anti-HDAC1 antibodies were markedly decreased in TNF-α-treated samples (Figure 3A). This finding suggests that an HDAC1-containing complex is inducibly depleted from the HIV LTR upon TNF-α induction, consistent with the notion that HDAC1 is recruited to the HIV LTR by a transcriptionally repressive p50–p50 complex constitutively bound to the HIV LTR under basal conditions, which dissociates upon induction of p50/RelA binding. Similar results were observed with PMA stimulation, indicating that NF-κB-inducing stimuli with alternate upstream signaling intermediates correspondingly regulate transcription factor recruitment to the HIV LTR (Supplementary Figure 1).
Figure 3.
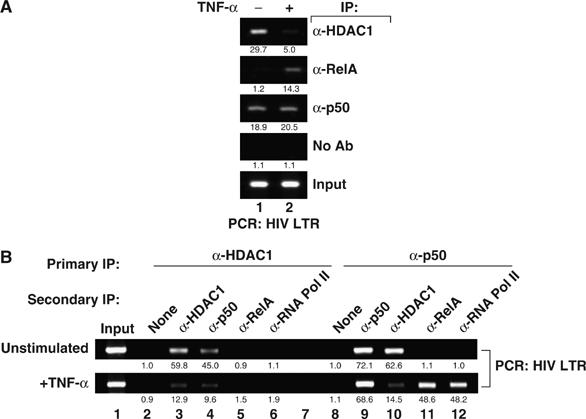
HDAC1 recruitment and loss from the latent HIV LTR RNA Pol II complexes. (A) HDAC1 is present on the latent HIV-1 promoter and is lost when T cells are activated. Fixed chromatin extracts from J-Lat 6.3 cells treated with 20 ng/ml TNF-α for 30 min or left untreated were immunoprecipitated with the indicated antibodies. Samples were analyzed for enrichment in HIV LTR DNA by UV visualization of PCR products on an ethidium bromide-stained agarose gel. Specific enrichment was quantitated by real-time PCR; mean of three measurements is indicated beneath each band image. (B) HDAC1 is excluded from RelA- or RNA Pol II-containing complexes on the HIV-1 LTR. Fixed chromatin extracts from J-Lat 6.3 cells treated as in panel A were immunoprecipitated with the indicated antibodies and enriched complexes were subjected to a second round of immunoprecipitation with the indicated antibodies. Data are representative of three independent experiments.
HDAC1 recruitment to the HIV LTR was strongly reduced by TNF-α treatment, but binding of the deacetylase was not entirely eliminated. To assess whether the residual HDAC1 is bound to the HIV LTR in concert with RelA, seqChIP assays were performed. Untreated chromatin extracts were first immunoprecipitated with anti-HDAC1 antibodies and then with anti-p50 antibodies. The immunoprecipitates were highly enriched in HIV LTR DNA, supporting the notion that NF-κB1/p50 and HDAC1 exist in an interacting complex on the latent HIV promoter (Figure 3B, lane 4, top panel). TNF-α stimulation reduced this enrichment (Figure 3B, lane 4, bottom panel), likely reflecting the replacement of p50 homodimers with p50/RelA heterodimers (Figure 3B, lanes 9 and 11). Secondary immunoprecipitation of anti-HDAC1-immunoprecipitated samples with anti-RelA or anti-RNA Pol II antibodies revealed no detectable enrichment in unstimulated or TNF-α-induced samples, suggesting that HDAC1 is excluded from RelA- or RNA Pol II-containing complexes (Figure 3B, lanes 5 and 6). In contrast, after TNF-α stimulation, sequential immunoprecipitation of p50 and RelA or p50 and RNA Pol II complexes was highly enriched in HIV LTR DNA (Figure 3B, lanes 11 and 12). Thus, the residual HDAC1 present in the TNF-α-treated samples likely reflects either continued binding in the subset of cells that did not respond to the stimulus or recruitment of HDAC1 to the promoter through its association with a different factor.
Inhibition of HDAC activity is associated with RNA Pol II recruitment to the latent HIV LTR and transcriptional initiation but not elongation
To further assess the role of p50-dependent recruitment of HDAC1 in repression of HIV transcription, J-Lat cells were treated with trichostatin A (TSA), a cell-permeable chemical inhibitor of class I/II HDACs. Analysis of GFP expression revealed a fourfold increase in GFP expression as measured by mean geometric fluorescence, a pattern similar to that observed with p50-shRNA (data not shown). One key function of HDACs is the deacetylation of local histone tails, which alters chromatin structure and is associated with impaired transcription of resident genes. To assess the acetylation status of the histones surrounding the latent HIV LTR before and after activation, chromatin extracts were prepared from J-Lat cells treated with TSA or TNF-α and immunoprecipitated with antibodies specific for acetyl-K14 histone H3. Samples treated with TNF-α or TSA, but not untreated controls, were strongly enriched in HIV LTR DNA (Figure 4A, top panel). K14 acetylation of histone H3 is associated with a less compacted chromatin structure, which correlates with increases in the access of basal transcription factors to DNA and transcriptional activation.
Figure 4.
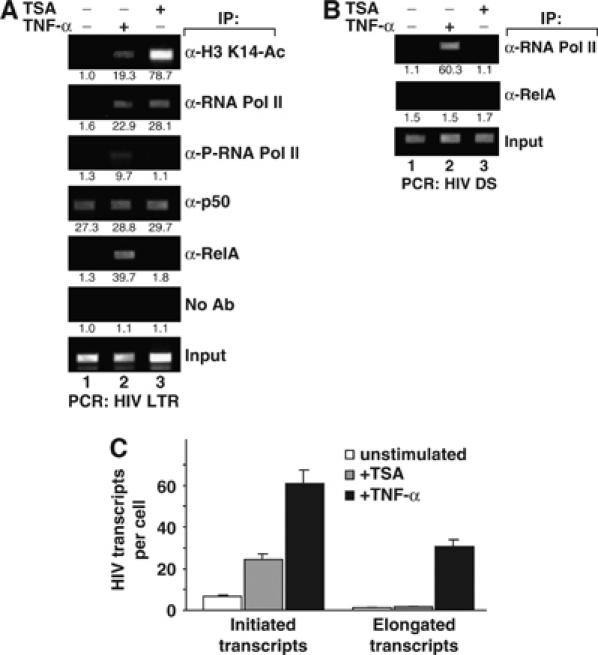
Recruitment of RNA Pol II to the HIV promoter is modulated by histone acetylation. (A) Both the activation of NF-κB and addition of the HDAC inhibitor TSA induce histone hyperacetylation and RNA Pol II recruitment to the latent HIV LTR. Fixed chromatin extracts from J-Lat 6.3 cells treated with 20 ng/ml TNF-α for 30 min, TSA for 4 h, or untreated cells were immunoprecipitated with the indicated antibodies. Samples were assessed for enrichment in HIV-LTR DNA by UV visualization of PCR products on an ethidium bromide-stained agarose gel. Specific enrichment was quantitated by real-time PCR; mean of three measurements is indicated beneath each band image. (B) TNF-α, but not TSA, induces a processive RNA Pol II complex. Fixed chromatin extracts prepared as in panel A were analyzed for enrichment in downstream HIV DNA (HIV DS). (C) TSA induces initiation but not elongation of HIV RNA transcripts. Total RNA was extracted from J-Lat 6.3 cells treated as in panel A, and initiated or elongated HIV RNA transcripts were quantitated by real-time RT–PCR. Bars represent the mean of triplicate samples; error bars represent standard deviation. Data are representative of three independent experiments.
To assess the effect of HDAC inhibition on the association of basal transcription factors with latent HIV LTR DNA, extracts were immunoprecipitated with RNA Pol II antibodies. Both TNF-α and TSA treatments were associated with enhanced binding of RNA Pol II to the HIV LTR (Figure 4A, second panel). Next, we investigated whether the CTD of RNA Pol II was phosphorylated, a change that signifies a fully active elongating polymerase complex. While both TSA and TNF-α treatment induced greater binding of RNA Pol II, only TNF-α induced phosphorylation of its CTD (Figure 4A, third panel). ChIP assays further showed that TSA did not interfere with the binding of RelA or NF-κB1 p50 to the HIV promoter (Figure 4A, fourth and fifth panels, and data not shown).
To test independently whether TSA induction was insufficient to induce RNA Pol II processivity, samples immunoprecipitated with anti-RNA Pol II antibodies were probed for enrichment of downstream HIV DNA. TNF-α stimulation induced strong enrichment of DS HIV, but TSA did not (Figure 4B, top panel).
To explore the functional relevance of the association of RNA Pol II with HIV LTR and DS DNA, initiated versus elongated HIV transcripts were measured by quantitative real-time RT–PCR with probe sets targeting HIV TAR in the 5′ 60 bp or the HIV Tat exon 1, roughly 5 kb downstream of the HIV LTR. The specificity of primers and the ability to isolate short transcripts were confirmed through analysis of samples treated with TNF-α alone or in combination with DRB, an inhibitor of CDK9-dependent RNA Pol II phosphorylation (data not shown). To assess the effect of TSA on transcription of HIV from the latent promoter, RNA was extracted from J-Lat cells treated with TNF-α, TSA, and untreated controls, and initiated and elongated transcripts were quantified. TSA induced a fourfold increase in initiated HIV transcripts, but failed to enhance elongated transcripts (Figure 4C, gray bars). In contrast, TNF-α increased initiated transcripts eightfold and elongated transcripts from a level below the detection threshold to over 30 copies per cell (Figure 4C, black bars). These observations coupled with the ChIP data highlight a key role for HDAC-mediated inhibition of RNA Pol II association with the latent HIV LTR in the basal state, and consequent reduction of transcriptional initiation. While inhibition of HDAC activity is associated with increased RNA Pol II binding, this polymerase remains unphosphorylated in its CTD and only capable of generating short viral transcripts.
NF-κB1/p50 regulates HDAC1 recruitment to the latent HIV promoter
To determine if recruitment of HDAC1 to the HIV LTR is dependent on NF-κB1 p50 expression, polyclonal populations of J-Lat cells stably expressing p50-shRNA or scrambled control shRNA were isolated. Immunoblotting of the p50-shRNA cells revealed marked reduction of NF-κB1 p50 expression compared with the control or parental cell lines (Figure 5A). Immunoprecipitation of chromatin extracts of these cells with anti-p50 antibodies revealed strong enrichment in HIV LTR DNA in control cells, but not in p50-shRNA knockdown cells (Figure 5B, top panel). Reduction of NF-κB1 p50 expression did not affect the binding of RelA to the HIV LTR following TNF-α stimulation, nor did it lead to spontaneous RelA binding to the HIV LTR in unstimulated cells (Figure 5B, second panel). In uninduced p50-shRNA cells, the association of HDAC1 with the latent HIV LTR was markedly reduced (Figure 5B, third panel), supporting a key role for p50 in the recruitment of HDAC1 to the latent HIV LTR. Consistent with the predicted effect of the removal of local HDAC activity, anti-acetyl-K14 histone H3 immunoprecipitates revealed marked enrichment in HIV LTR DNA in unstimulated p50-shRNA samples, but not in unstimulated controls (Figure 5B, fourth panel).
Figure 5.
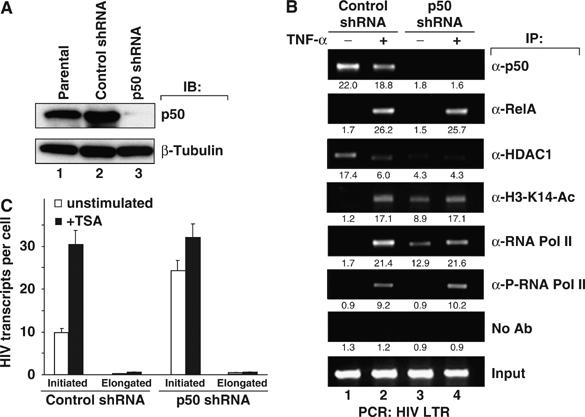
NF-κB1 p50 inhibits basal HIV expression in latently infected T cells by excluding RNA Pol II. (A) Stable shRNA knockdown of NF-κB1 p50 in latently infected T cells. Parental J-Lat 6.3 cells or stably transfected clones expressing an shRNA vector directed against a scrambled sequence or NF-κB1 p50 were selected and lysates were analyzed for p50 or control β-tubulin expression. (B) Fixed chromatin extracts from J-Lat 6.3 cells stably transfected with scrambled control shRNA or anti-NF-κB1 p50 shRNA treated with 20 ng/ml TNF-α for 30 min or left untreated were immunoprecipitated with the indicated antibodies. Samples were analyzed for enrichment of HIV LTR DNA by UV visualization of PCR products on an ethidium bromide-stained agarose gel. Specific enrichment was quantitated by real-time PCR; mean of three measurements is indicated beneath each band image. (C) Cells stably transfected with control- or NF-κB1 p50-shRNA were treated with 100 nM TSA for 2 h or left untreated. Total RNA was extracted, and initiated or elongated HIV RNA transcripts were quantitated by real-time RT–PCR. Bars represent the mean of triplicate samples; error bars represent standard deviation. Data are representative of three independent experiments.
The transcriptional consequences of reduced NF-κB1 p50 expression were further explored. Unstimulated p50-shRNA samples immunoprecipitated with anti-RNA Pol II antibodies revealed a strong enrichment in HIV LTR DNA, suggesting increased polymerase binding in the absence of p50. In contrast, no detectable enrichment was observed in the scrambled control shRNA samples. Immunoprecipitation with anti-phospho-RNA Pol II antibodies, however, showed little or no enrichment in HIV LTR DNA in either control- or p50-shRNA-treated samples in the absence of stimulation (Figure 5B, sixth panel).
The observed increase in HIV LTR RNA enrichment in RNA Pol II from cells expressing p50-shRNA and the absence of enrichment in phospho-RNA Pol II suggest that an unphosphorylated, non-elongating RNA Pol II complex is recruited to the latent HIV LTR in the absence of p50. These results were similar to those obtained with TSA-treated cells, suggesting a common mechanism of action. To further test this notion, control- and p50-shRNA stably transfected cells were treated with TSA or left unstimulated, and initiated or elongated HIV transcripts were quantified by real-time PCR. Initiated HIV transcripts were increased by TSA stimulation in control-shRNA cells. In contrast, and like TSA-treated cells, initiated HIV transcripts in untreated p50-shRNA cells were elevated relative to untreated controls (Figure 5C). Further, treatment of p50-shRNA cells with TSA induced only a modest additional increase in the level of initiated transcripts. Either TSA treatment or p50 knockdown was sufficient to promote the accumulation of initiated HIV transcripts but did not appreciably increase elongated transcripts. The fact that reduction of p50 expression was associated with hyperacetylation of histones surrounding the latent HIV promoter is consistent with the observed p50-dependent recruitment of HDAC1 in unstimulated cells. The failure of TSA to further increase initiated transcript formation in p50-shRNA samples adds additional support to the notion that these manipulations alter the same pathway leading to modulation of LTR transcription.
NF-κB1/p50 knockdown or TSA sensitizes latent HIV transcription to Tat
We next assessed whether increased RNA Pol II binding associated with TSA treatment or knockdown of p50 expression confers sensitivity of latently infected cells to the viral Tat transactivator protein. J-Lat cells were transfected with an empty H2Kk expression plasmid DNA or DNA encoding Tat or RelA. Ectopic expression of Tat induced a small subset of latently infected cells to express HIV, as measured by GFP expression. Expression of RelA, however, induced robust HIV gene expression, with ∼85% of the transfected cells expressing GFP (Figure 6A). Treatment of transfected cells with a 1 h pulse of TSA strongly enhanced GFP expression in Tat-transfected cell but had little effect on RelA-transfected cells, suggesting that induction of increased initiation of HIV transcripts by TSA sensitizes cells to the transactivating effects of Tat. Similar results were observed in J-Lat clones 8.4 and 15.2 (Supplementary Figure 2), suggesting that a similar mechanism might govern HIV latency in these clones. To examine the influence of TSA treatment on the inherent transactivating potential of Tat on a non-chromatinized substrate, J-Lat cells were transfected with an HIV-LTR luciferase reporter vector in conjunction with control, Tat, or RelA expression vectors, and treated or not with a 1 h pulse of TSA. Both Tat and RelA induced strong upregulation of HIV transcription, with minimal enhancement by TSA (Figure 6B), suggesting that these treatment conditions do not significantly alter the inherent transcriptional activity of Tat and RelA. Taken together, these results indicate that TSA treatment sensitizes latently infected J-Lat cells to Tat action through enhanced recruitment of RNA Pol II, leading to increased transcriptional initiation.
Figure 6.
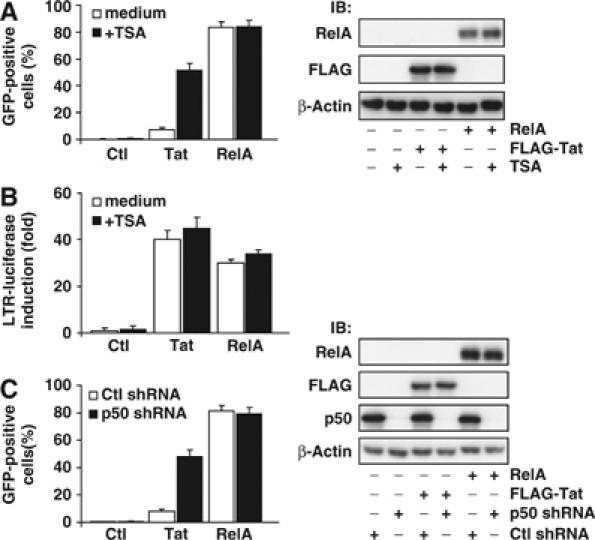
p50 and TSA-sensitive HDACs mediate desensitization of the latent HIV LTR to Tat. (A) TSA enhances Tat induction of latent HIV expression. J-Lat cells were transfected with control, Tat, or RelA expression vectors, pulse treated with TSA (400 nM) for 1 h or left untreated, and transfected cells were assessed for GFP expression (left panel). Lysates were prepared and probed for β-actin, RelA, and Tat expression as a control (right panel). Note the low basal sensitivity of J-Lat cells to Tat induction and the sensitization to Tat induced by TSA treatment. (B) TSA treatment does not alter inherent Tat transactivating potential. Jurkat cells were transfected with an HIV LTR firefly luciferase reporter vector and a control Renilla luciferase vector in conjunction with control, Tat, or RelA expression vectors, pulse treated with TSA (400 nM) for 1 h or left untreated, and relative increase in firefly luciferase activity was quantitated. (C) p50 shRNA enhances Tat induction of latent HIV expression. Scramble- or p50-shRNA stable cells were transfected with control, Tat, or RelA expression vectors, pulse treated with TSA (400 nM) for 1 h or left untreated, and transfected cells were assessed for GFP expression (left panel). Lysates were prepared and probed for β-actin, RelA, and Tat expression as a control (right panel). Note the sensitization to Tat expression in p50-shRNA cells and relative lack of additional TSA sensitivity.
To examine whether NF-κB1 p50 similarly limits the transactivating potential of Tat in latently infected cells, p50- or scramble-shRNA stable cell lines were transfected with control, Tat, or RelA expression vectors. Expression of Tat strongly promoted production of GFP in p50 knockdown cells, but not in the control p50 scramble-shRNA cells (Figure 6C). Further, treatment of Tat-transfected p50-shRNA cells with TSA did not appreciably enhance GFP expression above levels in untreated controls (data not shown), again suggesting that these manipulations likely exert their Tat-sensitizing effects through a common mechanism.
These results strongly favor a model where NF-κB1 p50–HDAC1 complexes promote the maintenance of HIV latency through changes in chromatin structure that impair effective recruitment of RNA Pol II. The absence of RNA Pol II binding to the latent HIV LTR and the consequent failure of transcriptional initiation render these cells unresponsive to Tat.
Discussion
In this study, we used the J-Lat model of postintegration HIV-1 latency to investigate the potential role of κB-specific transcriptional repressors in the maintenance of viral latency. We found that the latent HIV LTR in J-Lat cells is constitutively bound by p50, likely as a p50–p50 homodimer. Additionally, we found that p50 mediates recruitment of HDAC1 to the latent LTR, leading to deacetylation of local histone tails and thus altering the chromatin environment (Figure 7A). The resultant changes in chromatin structure limit the association of RNA Pol II with the latent proviral LTR, emphasizing how p50–HDAC1 repressor complex reinforces transcriptional latency of the integrated HIV provirus. These repressive effects are forfeited either when NF-κB1 p50 expression is knocked down by shRNA or when the enzymatic activity of HDAC1 is inhibited by the addition of TSA (Figure 7C and D). Under these conditions, chromatin structure is altered in a manner that favors increased RNA Pol II binding and enhanced initiation of HIV transcription. Yet, in the absence of transcriptional activators like RelA or the viral Tat protein, the bound polymerase does not effectively elongate, reflecting the absence of cyclin T1–CDK9-mediated phosphorylation of the CTD of the polymerase.
Figure 7.
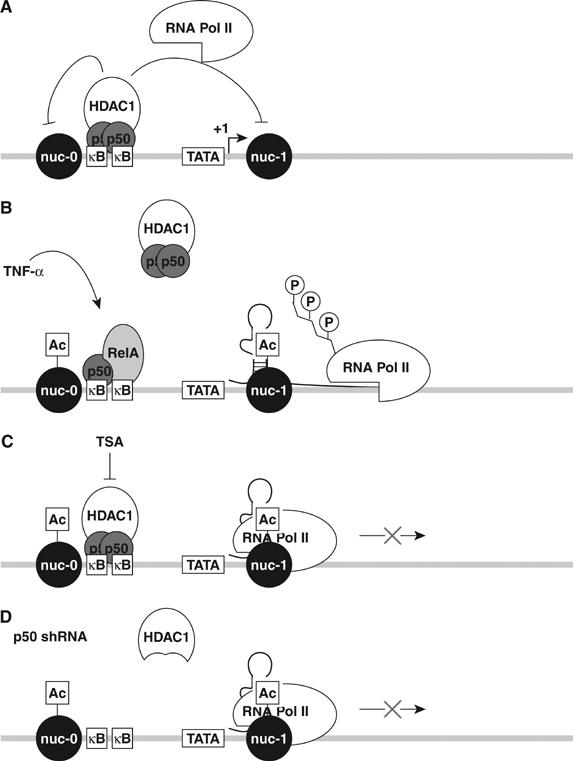
Model of p50-mediated repression of basal HIV expression in latently infected T cells. (A) NF-κB1 p50 homodimers bound to the latent HIV-1 promoter recruit HDAC1, which deacetylates regional histones and compacts local histone structure, thereby inhibiting the binding of RNA Pol II. (B) TNF-α liberates p50/RelA heterodimers, which displace constitutively bound p50/p50 homodimers present on the HIV LTR, thereby removing HDAC1. The regional shift in favor of HAT activity promotes increased acetylation of surrounding histones, relaxation of chromatin, and increased accessibility to RNA Pol II. Recruitment of CTD kinases by RelA induces transcriptional elongation. (C) TSA treatment inhibits HDAC1-mediated deacetylation of regional histones, inducing local histone acetylation, chromatin relaxation, and increased RNA Pol II binding. Under these conditions, RNA Pol II is non-processive owing to the absence of phosphorylation of its CTD. (D) shRNA knockdown of p50 displaces the p50–HDAC1 complexes from the latent LTR, promoting local histone acetylation and increased recruitment of RNA Pol II. Similarly, the lack of CTD kinases recruited to the LTR in this context produces a non-processive RNA polymerase complex.
Our findings thus support a model where p50-dependent changes in the chromatin structure of the latent HIV LTR importantly contribute to the maintenance of proviral latency. These results extend and modify a prevailing model of HIV latency, which proposes that constitutive association of RNA Pol II can initiate transcription, but cannot efficiently support elongation in the absence of Tat (Kao et al, 1987) or activated NF-κB (West et al, 2001). However, as noted earlier, HIV latency is likely multifactorial and blocks could occur at different levels in different latently infected cells (Lassen et al, 2004).
The function of p50 as a transcriptional repressor of the latent HIV LTR involving the recruitment of HDAC1 is very similar in many respects to the reported regulation of IL-8 gene expression (Zhong et al, 2002), a TSA-responsive host gene (Ashburner et al, 2001). The latent HIV and IL-8 promoters are both enriched in acetylated histone H3 after TSA treatment and are constitutively bound to repressive HDAC1/p50-containing complexes, supplanted by active p50/RelA heterodimers (Baek et al, 2002). However, the fact TSA is sufficient to drive IL-8 gene expression but is insufficient to activate the latent HIV LTR highlights an important difference in the regulation of these transcription units.
Studies in HeLa cells identified HDAC1 repression of an integrated HIV reporter in HeLa cells through its association with YY1 and LSF (Coull et al, 2000; He and Margolis, 2002). In our studies, shRNA knockdown of p50 expression reduced, but did not eliminate, the association of HDAC1 with the HIV LTR. The residual binding of HDAC1 might reflect LSF/YY1-mediated recruitment of the deacetylase to regions downstream of the κB-binding site. Studies of YY1 association with the latent HIV LTR in the J-Lat system did not show a reduction of YY1 binding to the promoter upon activation of gene expression (data not shown). It is possible that both YY1/LSF and p50–p50 homodimers are required to efficiently recruit HDAC1 and that the association of the deacetylase with the LTR involves coordinated recruitment by both factors. Such a scenario could explain the residual HDAC1 binding to the LTR after p50 shRNA knockdown.
The transcriptional activity of NF-κB is itself modulated by dynamic acetylation. For example, RelA acetylation by p300/CBP enhances its nuclear retention and transactivation potential (Chen et al, 2001). Additionally, NF-κB1 p50 is inducibly acetylated, promoting increased binding to and transcriptional activation of the COX-2 and iNOS promoters in the context of p50/RelA heterodimers (Deng and Wu, 2003; Deng et al, 2003). Of note, we found no change in the binding of NF-κB1 p50 to the latent HIV LTR in TSA-treated samples, nor was binding of RelA induced. In contrast, TSA induced strong acetylation of histone H3, supporting the notion that local HDAC activity promotes the deacetylation of histones arrayed around the HIV LTR. These findings suggest that the enrichment in RNA Pol II at the latent HIV promoter in TSA-treated cells is principally a consequence of histone modification rather than modulation of transcription factor binding.
Our observations are limited to the analysis of various J-Lat T-cell clones containing latent HIV proviruses. Unfortunately, it is not currently possible to identify and purify primary CD4 T cells latently infected with HIV in sufficient numbers to confirm these results in primary cells. However, in a prior study of viral propagation from cultures enriched in latently infected cells, the addition of the HDAC inhibitor valproic acid proved sufficient to rescue low-level viral recovery (Ylisastigui et al, 2004). Thus, the processes we have observed in the J-Lat system may reflect important biological events underlying HIV latency in vivo. Our observations suggest that HDAC inhibitors might be valuable adjuncts in future strategies aimed at eliminating the latent reservoir in infected patients. Indeed, our findings raise the interesting possibility that HDAC inhibitors in combination with Tat could prove a potent combination for activation of latent HIV proviruses. Such a strategy would be facilitated by the ‘protein transducing' properties of Tat, which allow its successful transit across cellular membranes.
Materials and methods
Cell lines and culture conditions
J-Lat 6.3 cells were maintained in RPMI with 10% fetal calf serum, penicillin, streptomycin, and L-glutamine. For stimulation, cells were treated with 20 ng/ml TNF-α (R&D Systems) or 100 nM TSA (Calbiochem), alone or in combination. For pulsed stimulation with TSA, cells were incubated with 400 nM TSA for 1 h, washed in medium, and suspended in complete medium.
Expression vectors and construction of shRNA vectors
pMACS Kk.II was obtained from Miltenyi Biotech. To ‘knock down' p50 expression in J-Lat cells, shRNA vectors were constructed by annealing synthetic DNA oligonucleotide primers with the sequences P50SH_S (5′-GATCCCCGGGGCTATAATCCTGGACTTTCAAG AGAAGTCCAGGATTATAGCCCCTTTTTA-3′) and P50SH_AS (5′-AGCTTAAAAAGGGGCTATAATCCTGGACTTCT CTTGAAAGTCCAGGATTATAGCCCCGGG-3′). A scrambled control version of the same sequence was prepared: SCRAMSH_S (5′-GATCCCCGTCTTACCCTCAGGTCAAATTCAAG AGATTTGACCTGAGGGTAAGACTTTTTA-3′) and SCRAMSH_AS (5′-AGCTTAAAAAGTCTTACCCTCAGGTCAAATCT CTTGAATTTGACCTGAGGGTAAGACGGG-3′). These DNAs were ligated into the BglII/HindIII sites in digested pTER shRNA cloning vector. The pMACS Kk.II SV40 promoter was eliminated by digestion with NheI/Kle and then autoligated to produce pMACSΔSV40. pTER.NF-κB1/p50 and pTER.Scramble were digested with EcoRI/HindIII, and the resulting fragment containing the modified H1 promoter and shRNA sequence was ligated into pMACSΔSV40 digested with EcoRI/HindIII to produce pMT.NF-κB1/p50 and pMT.Scramble vectors encoding both the targeted shRNA cassette and the H-2Kk marker of transfection driven by the PGK promoter.
Transfection and flow cytometric detection of transfected cells
Transfections were performed by electroporation as described (Williams et al, 2004). After 16 h of incubation, the cells were stained with biotin-anti-H-2Kk antibody, washed, and counterstained with streptavidin-APC (Pharmingen). Cells were analyzed on a FACSCalibur flow cytometer (Becton Dickinson) using FlowJo software (Treesoft). Cells were sorted with a FACS-DIVA (Becton Dickinson).
To produce stably transfected cell lines, cells were transfected with pMT.NF-κB1/p50 or pMT.Scramble vectors. At 7 days after transfection, H-2Kk-positive cells were enriched by fluorescence-based sorting. This process was repeated four times at 7-day intervals, resulting in >95% of cells expressing the H-2Kk marker of transfection.
Chromatin immunoprecipitation
J-Lat 6.3 T cells were adjusted to 1 × 107 cells/ml and incubated in medium or stimulated with TNF-α (20 ng/ml) for 30 min or TSA (100 nM) for 4 h. ChIP assays were performed as described (Williams et al, 2004).
For seqChIP experiments, following an initial round of ChIP and washing, the immunoprecipitated complexes were eluted in buffer containing 1.5% SDS and heated to 65°C for 15 min. Samples were then diluted 10-fold in elution buffer and 10 μl of the second antibody was added. Samples were incubated with agitation at 4°C for 2 h, followed by the addition of salmon sperm DNA/agarose protein A beads at 4°C for 2 h. Antibody–bead complexes were washed and eluted as described in the standard ChIP protocol.
For detection of specific HIV-1 LTR sequences in the ChIP eluates, DNA oligonucleotide primers LTRκBprimer5 (5′-AGGTTTGACAGCCGCCTA-3′) and LTRκBprimer3 (5′-AGAGACCCAGTACAGGCAAAA-3′) specific for a 203 bp region encompassing the κB binding sites in the HIV LTR were used for PCR amplification. To detect downstream HIV sequences, primers directed against the Pol gene with sequences 5HIVDS (5′-TGACTCAGATTGGCTGCAC-3′) and 3HIVDS (5′-AATTTCTACTAATGCTTTA-3′) were employed. To detect sequences in the coding region of the β-actin gene (β-actin DS), oligonucleotide primers with the sequences 5DSBACT (5′-GTCGACAACGGCTCCGGC-3′) and 3DSBACT (5′-GGTGTGGTGCCAGATTTTCT-3′) were used. To detect sequences in the 5′ flanking region of the β-actin gene, primers were targeted at a region 4 kb upstream of the β-actin gene with sequences 5USBACT (5′-GCCAGCTGCAAGCCTTGG-3′) and 3USBACT (5′-GCCACTGGGCCTCCATTC-3′). Amplification was performed using Taq polymerase (Qiagen) for 35–40 cycles and products were analyzed on 2.5% agarose gels. Images were acquired with an EagleEye II digital imaging system (Stratagene). Specific enrichment in HIV LTR DNA was quantitated by real-time PCR analysis of ChIP eluates, normalized to enrichment in nonspecific actin DNA. All values are reported in fold-enrichment relative to no-antibody control ChIP eluates.
RNA extraction and analysis of initiated and elongated HIV transcripts
J-Lat 6.3 cells (1 × 106 cells/ml) were treated with TSA (100 nM) or TNF-α (20 ng/ml) for 2 h at 37°C. For analysis of HIV mRNA synthesis in nucleofected primary T cells, RNA was extracted from 0.5 × 106 cells with an RNAWiz kit (Ambion). RNA transcripts were quantitated with the QuantiTect SYBR Green RT–PCR kit (Qiagen). To quantitate viral transcripts, serial dilutions of a quantitated RNA stock of full-length viral genome were used as a reference standard (gift of R Grant). Initiated transcripts were detected with primers HIVTAR5 (5′-GTTAGACCAGATCTGAGCCT-3′) and HIVTAR3 (5′-GTGGGTTCCCTAGTTAGCCA-3′). Elongated transcripts were detected with primers HIVTat5 (5′-ACTCGACAGAGGAGAGCAAG-3′) and HIVtat3 (5′-GAGTCTGACTGTTCTGATGA-3′). β-Actin mRNA copies were quantitated with primers β-actin5 (5′-GTCGACAACGGCTCCGGC-3′) and β-actin3 (5′-GGTGTGGTGCCAGATTTTCT-3′) specific for a 239 bp region in the β-actin mRNA and samples were normalized for β-actin copies. Fluorescence profiles were collected on an ABI 7700 real-time thermal cycler and analyzed with SDS v1.91 (Applied Biosystems). The absence of nonspecific bands in RT–PCR products was confirmed on 2% agarose gels.
Immunoblotting analysis
J-Lat 6.3 or stably transfected J-Lat 6.3 cells were pelleted and lysed on ice in egg lysis buffer (ELB) for 20 min and clarified by microcentrifugation. Protein concentration was quantitated using the Bradford protein assay (Bio-Rad), and 10 μg of each sample was added to an equal volume of 2 × Laemmli buffer and heated to 95°C for 5 min. Samples were separated on 10% acrylamide Tris–HCl-buffered SDS–PAGE gels (Bio-Rad), transferred to polyvinylidene difluoride membranes, and immunoblotted with anti-NF-κB p50, RelA (Santa Cruz Biotechnology), anti-FLAG, or anti-β-tubulin (Sigma) antibodies.
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
References
- Ashburner BP, Westerheide SD, Baldwin AS Jr (2001) The p65 (RelA) subunit of NF-kappaB interacts with the histone deacetylase (HDAC) corepressors HDAC1 and HDAC2 to negatively regulate gene expression. Mol Cell Biol 21: 7065–7077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek SH, Ohgi KA, Rose DW, Koo EH, Glass CK, Rosenfeld MG (2002) Exchange of N-CoR corepressor and Tip60 coactivator complexes links gene expression by NF-kappaB and beta-amyloid precursor protein. Cell 110: 55–67 [DOI] [PubMed] [Google Scholar]
- Barboric M, Nissen RM, Kanazawa S, Jabrane-Ferrat N, Peterlin BM (2001) NF-kappaB binds P-TEFb to stimulate transcriptional elongation by RNA polymerase II. Mol Cell 8: 327–337 [DOI] [PubMed] [Google Scholar]
- Chen L, Fischle W, Verdin E, Greene WC (2001) Duration of nuclear NF-kappaB action regulated by reversible acetylation. Science 293: 1653–1657 [DOI] [PubMed] [Google Scholar]
- Coull JJ, Romerio F, Sun JM, Volker JL, Galvin KM, Davie JR, Shi Y, Hansen U, Margolis DM (2000) The human factors YY1 and LSF repress the human immunodeficiency virus type 1 long terminal repeat via recruitment of histone deacetylase 1. J Virol 74: 6790–6799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng WG, Wu KK (2003) Regulation of inducible nitric oxide synthase expression by p300 and p50 acetylation. J Immunol 171: 6581–6588 [DOI] [PubMed] [Google Scholar]
- Deng WG, Zhu Y, Wu KK (2003) Up-regulation of p300 binding and p50 acetylation in tumor necrosis factor-alpha-induced cyclooxygenase-2 promoter activation. J Biol Chem 278: 4770–4777 [DOI] [PubMed] [Google Scholar]
- Finzi D, Hermankova M, Pierson T, Carruth LM, Buck C, Chaisson RE, Quinn TC, Chadwick K, Margolick J, Brookmeyer R, Gallant J, Markowitz M, Ho DD, Richman DD, Siliciano RF (1997) Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science 278: 1295–1300 [DOI] [PubMed] [Google Scholar]
- Hayden MS, Ghosh S (2004) Signaling to NF-kappaB. Genes Dev 18: 2195–2224 [DOI] [PubMed] [Google Scholar]
- He G, Margolis DM (2002) Counterregulation of chromatin deacetylation and histone deacetylase occupancy at the integrated promoter of human immunodeficiency virus type 1 (HIV-1) by the HIV-1 repressor YY1 and HIV-1 activator Tat. Mol Cell Biol 22: 2965–2973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan A, Bisgrove D, Verdin E (2003) HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. EMBO J 22: 1868–1877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao SY, Calman AF, Luciw PA, Peterlin BM (1987) Anti-termination of transcription within the long terminal repeat of HIV-1 by tat gene product. Nature 330: 489–493 [DOI] [PubMed] [Google Scholar]
- Khorasanizadeh S (2004) The nucleosome: from genomic organization to genomic regulation. Cell 116: 259–272 [DOI] [PubMed] [Google Scholar]
- Kurdistani SK, Grunstein M (2003) In vivo protein–protein and protein–DNA crosslinking for genomewide binding microarray. Methods 31: 90–95 [DOI] [PubMed] [Google Scholar]
- Laspia MF, Rice AP, Mathews MB (1989) HIV-1 Tat protein increases transcriptional initiation and stabilizes elongation. Cell 59: 283–292 [DOI] [PubMed] [Google Scholar]
- Lassen K, Han Y, Zhou Y, Siliciano J, Siliciano RF (2004) The multifactorial nature of HIV-1 latency. Trends Mol Med 10: 525–531 [DOI] [PubMed] [Google Scholar]
- Marshall NF, Price DH (1995) Purification of P-TEFb, a transcription factor required for the transition into productive elongation. J Biol Chem 270: 12335–12338 [DOI] [PubMed] [Google Scholar]
- Pazin MJ, Sheridan PL, Cannon K, Cao Z, Keck JG, Kadonaga JT, Jones KA (1996) NF-kappa B-mediated chromatin reconfiguration and transcriptional activation of the HIV-1 enhancer in vitro. Genes Dev 10: 37–49 [DOI] [PubMed] [Google Scholar]
- Pomerantz RJ (2003) Reservoirs, sanctuaries, and residual disease: the hiding spots of HIV-1. HIV Clin Trials 4: 137–143 [DOI] [PubMed] [Google Scholar]
- Ratnasabapathy R, Sheldon M, Johal L, Hernandez N (1990) The HIV-1 long terminal repeat contains an unusual element that induces the synthesis of short RNAs from various mRNA and snRNA promoters. Genes Dev 4: 2061–2074 [DOI] [PubMed] [Google Scholar]
- Rohr O, Marban C, Aunis D, Schaeffer E (2003) Regulation of HIV-1 gene transcription: from lymphocytes to microglial cells. J Leukoc Biol 74: 736–749 [DOI] [PubMed] [Google Scholar]
- Sandoval J, Rodriguez JL, Tur G, Serviddio G, Pereda J, Boukaba A, Sastre J, Torres L, Franco L, Lopez-Rodas G (2004) RNAPol-ChIP: a novel application of chromatin immunoprecipitation to the analysis of real-time gene transcription. Nucleic Acids Res 32: e88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siliciano JD, Kajdas J, Finzi D, Quinn TC, Chadwick K, Margolick JB, Kovacs C, Gange SJ, Siliciano RF (2003) Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat Med 9: 727–728 [DOI] [PubMed] [Google Scholar]
- Van Lint C, Emiliani S, Ott M, Verdin E (1996) Transcriptional activation and chromatin remodeling of the HIV-1 promoter in response to histone acetylation. EMBO J 15: 1112–1120 [PMC free article] [PubMed] [Google Scholar]
- West MJ, Lowe AD, Karn J (2001) Activation of human immunodeficiency virus transcription in T cells revisited: NF-kappaB p65 stimulates transcriptional elongation. J Virol 75: 8524–8537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams SA, Chen LF, Kwon H, Fenard D, Bisgrove D, Verdin E, Greene WC (2004) Prostratin antagonizes HIV latency by activating NF-kappaB. J Biol Chem 279: 42008–42017 [DOI] [PubMed] [Google Scholar]
- Yankulov K, Blau J, Purton T, Roberts S, Bentley DL (1994) Transcriptional elongation by RNA polymerase II is stimulated by transactivators. Cell 77: 749–759 [DOI] [PubMed] [Google Scholar]
- Ylisastigui L, Archin NM, Lehrman G, Bosch RJ, Margolis DM (2004) Coaxing HIV-1 from resting CD4T cells: histone deacetylase inhibition allows latent viral expression. Aids 18: 1101–1108 [DOI] [PubMed] [Google Scholar]
- Zhong H, May MJ, Jimi E, Ghosh S (2002) The phosphorylation status of nuclear NF-kappa B determines its association with CBP/p300 or HDAC-1. Mol Cell 9: 625–636 [DOI] [PubMed] [Google Scholar]
- Zhou Q, Chen D, Pierstorff E, Luo K (1998) Transcription elongation factor P-TEFb mediates Tat activation of HIV-1 transcription at multiple stages. EMBO J 17: 3681–3691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Pe'ery T, Peng J, Ramanathan Y, Marshall N, Marshall T, Amendt B, Mathews MB, Price DH (1997) Transcription elongation factor P-TEFb is required for HIV-1 tat transactivation in vitro. Genes Dev 11: 2622–2632 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2