

Abstract
Glucocorticoids dramatically inhibit cytokine and chemokine production. They act through the glucocorticoid receptor (GR), a ligand-dependent transcription factor that binds to and represses activities of other DNA-bound regulators, activator protein 1 and nuclear factor κB, utilizing a p160 GRIP1 as a corepressor. A yeast two-hybrid screen with the GRIP1 corepression domain (RD) yielded interferon (IFN) regulatory factor (IRF)3—a downstream effector of Toll-like receptors (TLR) 3/4 and an essential activator of several IFN and chemokine genes. We defined the GRIP1:IRF3 interface and showed that endogenous GRIP1 and IRF3 interact in mammalian cells. Interestingly, GR and IRF3 competed for GRIP1 binding; GR activation or GRIP1 knockdown in macrophages blocked whereas GRIP1 overexpression rescued IRF3-dependent gene expression. GR interference persisted in MyD88- and IFNA receptor-deficient mice, suggesting a specific disruption of TLR3–IRF3 pathway, not of autocrine IFN signaling. Finally, IRF3-stimulated response elements were necessary and sufficient for TLR3-dependent induction and glucocorticoid inhibition. Thus, GRIP1 plays a cofactor role in innate immunity. Competition with GR for GRIP1 antagonizes IRF3-mediated transcription, identifying the GRIP1:IRF3 interaction as a novel target for glucocorticoid immunosuppression.
Keywords: glucocorticoid receptor, GRIP1, immunosuppression, interferon regulatory factor-3, transcriptional regulation
Introduction
Glucocorticoid hormones (GCs) control numerous physiological processes including metabolism, development and homeostasis, particularly under conditions of stress. At a cellular level, GCs regulate the balance between proliferation, differentiation and apoptosis in a tissue-, cell type- and developmental stage-specific manner. The immune system is under particularly tight control by GCs, which mediate proper development of the T-cell compartment (Ashwell et al, 2000; Pazirandeh et al, 2005). In addition to extensively studied T-cell apoptosis, GCs affect viability and function of many other immune cell types including B cells, monocytes, macrophages and granulocytes (Caramori and Adcock, 2005). Specifically, GCs block the production of many mediators of immune and inflammatory response, such as cytokines, chemokines and cell adhesion molecules (De Bosscher et al, 2003; Elenkov, 2004). As a consequence, exposure to pharmacologic quantities of GCs leads to dramatic immunosuppression, making GCs indispensable for managing autoimmune, inflammatory and lymphoproliferative diseases.
GCs act through the intracellular glucocorticoid receptor (GR), a transcription factor that transduces hormonal signals into changes in gene expression. Upon hormone binding and nuclear translocation, GR binds to glucocorticoid response elements (GREs) adjacent to target promoters and activates or represses gene transcription (Tsai and O'Malley, 1994). GREs have been subdivided into three classes (Lefstin and Yamamoto, 1998) depending on whether GR binds directly to DNA alone (‘simple'), along with another transcription factor (‘composite'), or is recruited to DNA indirectly through protein:protein interactions with other DNA-bound regulators (‘tethering'). The resultant transcriptional output (activation or repression) depends on the sequence and architecture of a given GRE, the identity of other regulators and GR cofactors bound at the site, and the surrounding chromatin structure; however, at tethering GREs, GR typically acts as a transcriptional repressor. Factors susceptible to such ‘transrepression' are activator protein (AP)1 and nuclear factor (NF)κB—critical regulators of cell proliferation, inflammation and immune response (Li and Verma, 2002; Shaulian and Karin, 2002). Not surprisingly, GR-mediated repression of AP1 and NFκB activities is believed to be the basis for GC-induced immunosuppression. Although profound and diverse effects of GR on the immune system suggest that other factors could potentially mediate these broad activities, their identity is unknown.
The molecular mechanisms of GR-mediated repression at tethering GREs are not well understood. There is no evidence that ‘conventional' nuclear receptor corepressors, NCoR or SMRT, confer agonist-dependent repression by GR, and using histone deacetylase (HDAC) inhibitors to test for the requirement for HDACs in repression yielded conflicting results (Ito et al, 2000; Nissen and Yamamoto, 2000). Although the question of whether and how HDACs contribute to GR agonist-dependent repression awaits resolution, other mechanisms of repression have been described: for example, at the interleukin (IL)8 gene, GR inhibits phosphorylation of serine 2 in the C-terminal domain (CTD) of RNA polymerase II owing to competition with a CTD kinase, pTEFb, for promoter binding (Nissen and Yamamoto, 2000; Luecke and Yamamoto, 2005), and under some conditions, displaces Pol II from the promoter (Rogatsky and Yamamoto, unpublished). It is likely that no unifying mechanism exists and that numerous pathways confer repression depending on the GRE, promoter, local chromatin structure and availability or activity of interacting cofactors.
One approach to dissecting the mechanisms of repression is to identify the components recruited into GR regulatory complexes. Using this strategy, TIF2/GRIP1 (Hong et al, 1996; Voegel et al, 1996), a member of the p160 family of nuclear receptor coactivators, was shown to function as a GR corepressor at the AP1 tethering GRE (Rogatsky et al, 2001). The GRIP1 corepression domain (RD) engaged in this context did not overlap with any established GRIP1 domain or resemble any known protein including other p160 family members; indeed, other p160s lacked this corepressor activity. Subsequent analysis of functional interactions between GR and GRIP1 at a series of GREs revealed that the requirement for RD was specific to AP1 and NFκB GR tethering sites (Rogatsky et al, 2002), at which GC actions are therapeutically desirable suggesting that GRIP1 RD may be a critical mediator of the immunosuppressive and anti-inflammatory actions of GCs. In the absence of any function suggested by the RD primary amino-acid sequence, we used this domain in a yeast two-hybrid screen for interacting factors. Two types of candidate interactors were envisioned: (1) known or novel proteins mediating GR repression—bona fide GR corepressors; (2) proteins known to be involved in other transcriptional networks, especially those in the immune system; even if unrelated to GR repression at tethering GREs, their interaction with GRIP1 will imply a broader role for GRIP1 in mammalian physiology and potential points of crosstalk between GR and diverse regulatory pathways.
One two-hybrid isolate encoded interferon regulatory factor (IRF)3, a key mediator of innate immune responses downstream of Toll-like receptors (TLR) 3 and 4 (activated by viral double-stranded RNA (dsRNA) and bacterial lipopolysaccharide (LPS), respectively) and an essential transcription factor for type I interferons (IFNα/β), chemokines IP10, RANTES and several other genes (Geiss et al, 2001; Doyle et al, 2002; Sharma et al, 2003). IFNα/β, in addition to their role in host responses to invading pathogens, are major contributors to the initiation and progression of systemic autoimmunity, rheumatic disease and diabetes (Hooks et al, 1979; Ronnblom et al, 1990; Alba et al, 2004); furthermore, accumulating evidence directly links TLRs and IRF3 to the pathogenesis of autoimmune diseases (Leadbetter et al, 2002; Eriksson et al, 2003). Here, we dissect the GRIP1:IRF3 interaction and examine its functional consequences with respect to IRF3-mediated transcription. Our studies identify GRIP1 as a novel link between GR signaling and the innate immune system and establish its interaction with IRF3 as a critical target of the anti-inflammatory and immunosuppressive activities of GCs.
Results
Isolation of IRF3 in a yeast two-hybrid screen
An LNCaP prostate cell cDNA library fused to the LexA DNA binding domain (DBD) was screened with the nuclear receptor interaction domain (NID)-RD region of GRIP1 (aa 631–1007; Figure 1A) fused to B42 acidic activation domain (AD) as ‘bait' (Supplementary Figure 1). A ‘reverse' orientation of the screen was used because the NID-RD slightly activates transcription in yeast when tethered to DNA; the modified screen enables isolation of factors interacting with transcriptional activators. The NID was included in the bait for two reasons: (1) In principle, the presence of NR boxes should permit the isolation of nuclear receptors; indeed, estrogen receptor-α, estrogen receptor-like receptor-α and retinoic acid receptor-γ were isolated further validating the screen. (2) Secondary structure prediction by the JPRED algorithm revealed that RD (GRIP1 aa 765–1007; Figure 1A) may contain two amphiphatic α-helices in the N-terminal portion (aa 804–814 and 837–849) followed by a large unstructured region typical of intrinsically disordered proteins that assume a defined structure upon protein:protein interaction (Obradovic et al, 2003). Hence, NID was retained to avoid potential destabilization of the RD.
Figure 1.
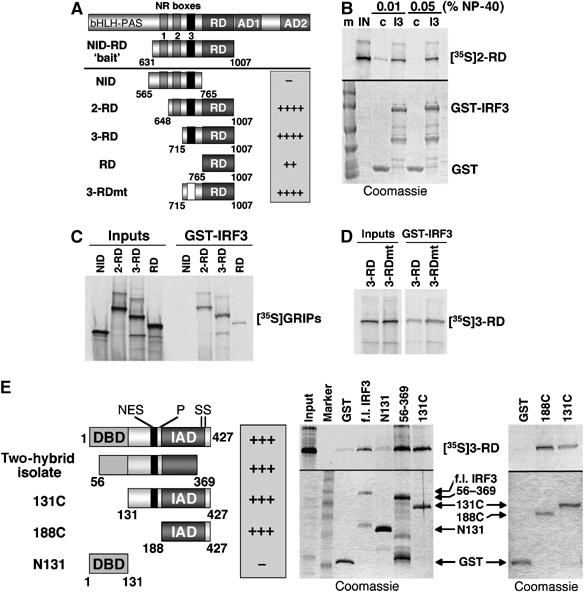
GRIP1 and IRF3 interact in vitro. (A) A diagram of GRIP1 derivatives tested for binding to GST-IRF3, with the relative strength of binding indicated as + or −. (B) GST-IRF3 interacts with in vitro-produced GRIP1 2-RD (648–1007). Interaction was tested in the presence of increasing amounts of NP-40. At 0.05% NP-40, nonspecific interaction with GST (control, c) is abolished, whereas interaction with GST-IRF3 (I3) is unaffected. (C) Mapping the interacting surface on GRIP1. In vitro-produced GRIP1 derivatives from panel A were tested for their ability to interact with GST-IRF3. (D) GRIP1 NR box-3 is dispensable for interaction with IRF3. The GRIP1 3-RD and 3-RDmt were assayed for the IRF3 interaction. (E) Mapping the GRIP1-interacting domain of IRF3. Functional domains of IRF3 (left) include N-terminal DBD, nuclear export signal (NES), a proline-rich region (P) and IRF-association domain (IAD) followed by a cluster of serine residues (SS). The two-hybrid clone (aa 56–369) lacks 55 aa of the DBD and the C-terminal CBP/p300-interacting region including the phosphorylation site cluster. Full-length IRF3, 56–369 isolate, 131C (lacking the DBD), 188C (the IAD) and N131 (DBD alone) were produced as GST-fusion proteins (Coomassie blue staining, right bottom) and tested for binding to in vitro-produced GRIP1 3-RD (right top). In all panels, 20% of ‘inputs' and ∼40% of the binding reaction was loaded on SDS–PAGE.
One library-encoded clone strongly activated LacZ and Leu2 reporters on galactose (when the GRIP1 bait was expressed) and weakly on glucose, consistent with some bait-independent transcriptional activity. It contained a single open reading frame corresponding to LexA DBD fused to aa 56–369 of the 427-aa human IRF3 protein (Figure 1E) lacking the 55 N-terminal residues of the DBD and the C-terminal phosphorylation site cluster known to regulate CBP binding and autoinhibition (Lin et al, 1999; Qin et al, 2003). A lack of autoinhibitory sequences may account for the mild bait-independent activation of the LacZ reporter by the IRF3 isolate.
The GRIP1:IRF3 interaction in vitro
A GST pull-down assay was used to independently confirm the GRIP1:IRF3 interaction. Full-length IRF3 fused to GST was tested with the in vitro-produced 2-RD fragment of GRIP1 (aa 648–1007; Figure 1A) containing the last two NR boxes and the corepression domain. Figure 1B shows that 2-RD bound IRF3, and that increasing the amount of NP-40 abolished nonspecific binding to GST-loaded beads but had no effect on the GRIP1:GST-IRF3 interaction; further increasing NP-40 concentration to 0.5% did not affect binding (not shown). In contrast, a well-known hydrophobic interaction between GR and GRIP1 was mostly disrupted by shifting NP-40 concentration from 0.05 to 0.1% (not shown). These data may suggest that interaction of GRIP1 with IRF3 is mediated by polar and/or charged rather than hydrophobic residues.
To better define the IRF3-interacting region of GRIP1, we tested a series of in vitro-produced GRIP1 derivatives, including NID, known to bind GR (Darimont et al, 1998), 2-RD, 3-RD and RD (Figure 1A). All but the NID interacted with IRF3 (Figure 1C); 2-RD and 3-RD interacted with IRF3 equally well, whereas RD fragment bound IRF3 less efficiently possibly owing to the altered conformation or stability of isolated RD. One well-defined feature distinguishing 3-RD from RD is NR box-3, the LxxLL motif responsible for GRIP1 interaction with nuclear receptors such as GR. To determine whether NR box-3 also mediates the interaction of GRIP1 with IRF3, we mutated the LxxLL motif to LxxAA and found that 3-RDmt bound IRF3 as well as wild type (wt) (Figure 1D). We conclude that 3-RD is the minimal GRIP1 fragment that binds well to IRF3 and that NR box-3 is dispensable for this interaction. Given secondary structure predictions for isolated RD discussed above and the fact that the N-terminal 50 aa upstream of RD (which differentiate it from 3-RD) do not confer binding to IRF3 in the context of NID, our results are consistent with the idea that RD is the major surface of GRIP1 interacting with IRF3 and that the N-terminal 50 aa extension serves to stabilize the RD conformation.
To map the GRIP1-interacting region of IRF3, we deleted its N-terminal 130 (IRF3 DBD) or 187 residues. The resultant IRF3 131C and 188C (IAD) interacted with GRIP1 3-RD as well as the full-length IRF3, suggesting that the DBD is not required for binding (Figure 1E). Consistent with this, the isolated DBD (N131) failed to interact with GRIP1. As expected, the original yeast two-hybrid isolate (aa 56–369) interacted with 3-RD similarly to the full-length, 131C or 188C IRF3 derivatives. Hence, the GRIP1-interacting region encompasses IRF3 residues 188–369 (IAD) and excludes the C-terminal CBP-binding and autoinhibitory domain.
Endogenous GRIP1 and IRF3 interact in mammalian cells
To determine the biological relevance of the observed two-hybrid and in vitro GRIP1:IRF3 interaction, we asked whether these proteins associate in mammalian cells. As we were interested in visualizing endogenous factors expressed at physiological levels, we selected antibodies suitable for co-immunoprecipitations and immunoblotting of GRIP1 and IRF3 in the same species and assessed their expression in several cell lines. GRIP1 is ubiquitously expressed, although its level varies between cell types (Rogatsky, unpublished). IRF3 is abundantly expressed as a latent transcriptionally inactive protein; its phosphorylation and nuclear translocation rely on signaling through TLRs 3/4 (Lin et al, 1998; Yoneyama et al, 1998; Kumar et al, 2000). The dsRNA–TLR3–IRF3 pathway controls innate immune responses to viral infection in macrophages; hence, murine RAW264.7 macrophage-like cells in which this pathway is functional (Doyle et al, 2002) were well suited for our experiment. As shown in Figure 2, treatment with a synthetic TLR3 agonist polyI:C (pIC) resulted in the formation of GRIP1:IRF3 complexes detectable by immunoblotting. Similar results were obtained in monkey CV1 cells (not shown), in which IRF3 pathway is also intact (see Figure 5C). Thus, endogenous IRF3 and GRIP1 interact in mammalian cells upon IRF3 activation.
Figure 2.
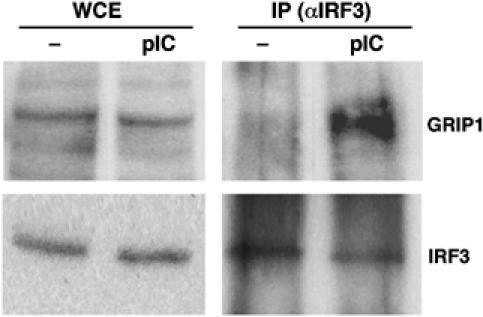
The GRIP1:IRF3 interaction in mammalian cells. RAW264.7 cells were treated for 2 h with 10 μg/ml pIC, as indicated, and lysates were prepared. A 20% of each lysate was boiled in sample buffer to generate whole-cell extracts (WCE), whereas the rest was precipitated with anti-IRF3 antibody (IP). Protein complexes were adsorbed on protein A/G agarose beads, boiled in sample buffer and separated by SDS–PAGE along with WCE. GRIP1 and IRF3 were detected by immunoblotting.
Figure 5.
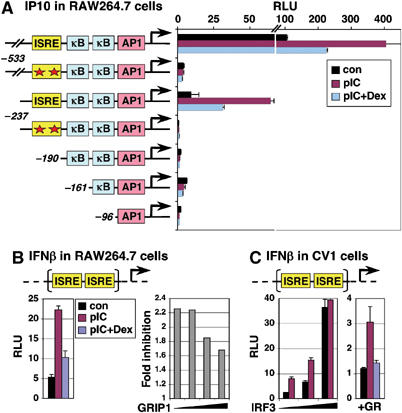
pIC induction and Dex inhibition are mediated by ISREs. (A) RAW264.7 cells were transfected with a series of IP10-Luc reporters, with wt or mutated (asterisks) ISREs, treated for 6 h as indicated, and luciferase activity was expressed as relative luminescence units (RLU). The Y-axis is broken to show the maximal activity of −533-Luc. (B) RAW264.7 cells were transfected with IFNβ promoter-derived p31x2-Luc reporter (left), with an increasing amount of GRIP1 (right), treated as indicated and luciferase activity was measured. ‘Fold inhibition' is a ratio of reporter activity in the presence of pIC versus pIC+Dex. (C) CV1 cells were transfected with p31x2-Luc reporter and (left) an increasing amount of IRF3 or (right) GR, treated as in panel B, and luciferase activity was measured.
GR and IRF3 compete for GRIP1 in vitro
GR interacts with GRIP1 NID (aa 563–765) through NR box-3 whereas the IRF3-interacting region of GRIP1 encompassed 3-RD (aa 715–1007), although the NR box-3 itself was not required. Thus, GR and IRF3 bind adjacent or overlapping regions of GRIP1, perhaps each invoking a distinct GRIP1 conformation incompatible with binding of the other factor. To test this possibility, we immobilized His-tagged 3-RD on cobalt affinity resin and performed a binding assay with in vitro-produced GR in the absence or presence of purified GST or GST-IRF3 131C. As shown in Figure 3A, GC dexamethasone (Dex)-activated GR bound GRIP1 and adding IRF3 significantly reduced binding (top; lane 3 versus lanes 7 and 8). At the same time, IRF3 bound 3-RD on its own (recapitulating the GRIP1:IRF3 interaction in a different system), and this interaction was greatly diminished by GR (bottom; lane 9 versus lanes 7 and 8). In contrast, GST did not bind 3-RD and did not interfere with GR binding (top; lane 3 versus lanes 4 and 5). Thus, GR and IRF3 compete with each other for binding to GRIP1 in vitro.
Figure 3.
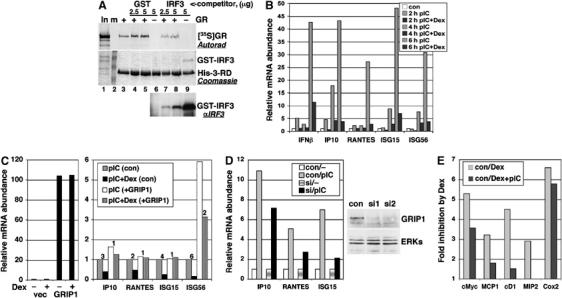
Physical and functional competition between GR and IRF3 for GRIP1. (A) GR and IRF3 compete for GRIP1 in vitro. The binding of in vitro-produced GR (top, autoradiogram) to immobilized His-tagged 3-RD (middle, Coomassie) was tested in the absence or presence of increasing amounts (in μg) of GST or GST-IRF3 131C (bottom, IRF3 immunoblot). (B) Inhibition of IRF3-mediated transcription by GCs. RAW264.7 cells were treated with pIC−/+Dex for indicated times; the expression of IRF3-responsive mRNAs was assessed by qPCR with Rpl19 as normalization control and expressed relative to untreated cells (con=1). (C) Overexpression of GRIP1 rescues Dex-dependent inhibition of IRF3 target gene expression. RAW264.7 cells were transfected with pcDNA3 vector or pcDNA3-GRIP1. The following day, cells were treated for 6 h as indicated and harvested for RNA isolation. qPCR was performed as in panel A with actinβ as normalization control. Fold inhibition by Dex is shown. (D) GRIP1 knockdown decreases IRF3 target gene expression. Scrambled siRNA duplexes (con) or siGRIP1 (si1 or si2) were nucleofected into RAW264.7 cells; 24 h later, one set of cells were harvested and GRIP1 expression in whole-cell extracts (WCE) was assessed by immunoblotting using re-probing with ERK antibodies as control for equal loading. A second set of cells were treated −/+pIC for 4 h and levels of IP10, RANTES and ISG15 RNAs were assessed by qPCR relative to untreated cells. (E) Activation of IRF3 in RAW264.7 cells relieves GR-mediated repression of AP1 and NFκB targets. Relative mRNA abundance of GR-repressible genes was assessed by qPCR and fold inhibition by Dex with and without pIC is shown after 6 h treatments.
GCs antagonize IRF3 primary target gene expression and GRIP1 relieves this inhibition
GRIP1 is an established coactivator for nuclear receptors (Hong et al, 1996; Voegel et al, 1996, 1998) and for the muscle-specific transcription factor Mef2C (Chen et al, 2000; Liu et al, 2004). It also serves as a GR corepressor at the AP1 and NFκB tethering GREs (Rogatsky et al, 2001). Given that GRIP1 and IRF3 interact, GRIP1 may function as a coregulator for IRF3 in which case its sequestration by hormone-activated GR may antagonize IRF3-dependent transcription. We therefore examined the effect of Dex on the expression of five primary IRF3 targets (IFNβ, IP10, RANTES, ISG15 and ISG56) in RAW264.7 cells, in which these genes are IRF3-inducible (Doyle et al, 2002). All five genes were induced by the TLR3 agonist pIC and strongly inhibited by Dex at all time points tested (Figure 3B). To focus on immediate IRF3-dependent transcriptional events, subsequent studies were performed at the earliest time points at which the responses were apparent.
Because competition for GRIP1 between GR and IRF3 should be at least partially relieved by ectopically provided GRIP1, we transiently overexpressed GRIP1 in RAW264.7 cells (Figure 3C, left) and measured the abundance of IRF3-regulated mRNAs. Although introduction of any plasmid DNA into RAW264.7 macrophages somewhat decreased the responsiveness of IRF3 targets (not shown), their Dex-dependent downregulation was nearly (IP10, ISG56) or completely (RANTES, ISG15) abrogated by overexpressed GRIP1 (Figure 3C, right), consistent with a model in which GRIP1 becomes a limiting component of IRF3 transcription complexes upon GR activation. In addition, ISG56 mRNA was significantly increased by ectopically expressed GRIP1, implying that GRIP1 can serve as an IRF3 coactivator for a subset of IRF3 targets. To examine this directly, we silenced GRIP1 expression in RAW264.7 cells at RNA (90–95%, not shown) and protein (Figure 3D, right) levels using small interfering (si)RNA and examined pIC-dependent induction of IRF3 targets unresponsive to GRIP1 overexpression. siGRIP1 reduced the induction of IP10, RANTES and ISG15 by 35, 50 and 70%, respectively (Figure 3D, left), further suggesting that GRIP1 is a component of IRF3 activation complexes.
The competition model predicts that in a reverse scenario, microbial TLR–IRF3 activators will limit the amount of GRIP1 available to GR, thereby inhibiting its transcriptional regulatory properties. As GR:GRIP1 complexes can either activate or repress transcription, in principle, both activities can be affected by IRF3. Yet, GR activation is mediated by all three p160 family members, and GR can rely on SRC1 or RAC3 if GRIP1 is no longer available. In contrast, GRIP1 is the only p160 corepressor at tethering GREs, suggesting that loss of GRIP1 may selectively impair GC repression. We therefore examined the effect of IRF3 activation on GR-mediated repression of AP1 and NFκB target genes (cMyc, cyclin D1, MCP1, MIP2 and COX2) not regulated by IRF3 directly. As expected, the expression of all five genes was repressed by Dex; remarkably, repression of all but COX2 gene was partially or completely relieved in the presence of pIC (Figure 3E), consistent with depletion of GRIP1 from GR repression complexes by activated IRF3.
GCs block IRF3-mediated transcription in primary macrophages in a MyD88-independent manner
Although RAW264.7 cells retain many macrophage characteristics, they are an A-MuLV-transformed tumor-derived line that does not require macrophage-colony stimulating factor (M-CSF) for growth. To test whether our model applies to primary cells, we examined the responses to pIC and Dex of bone marrow-derived macrophages from C57BL/6 mice. As shown in Figure 4A, pIC induced a dramatic accumulation of IRF3 target mRNAs in a strictly Dex-sensitive manner. To confirm these trends in primary macrophages, which do not require differentiation ex vivo, we injected mice with sterile thioglycollate and collected peritoneal macrophages for pIC−/+Dex treatment. As seen in Figure 4B, induction of IRF3 target genes by pIC in peritoneal macrophages was similarly antagonized by Dex. Importantly, the responses in peritoneal macrophages from Balb/c and C57BL/6 mice were nearly identical (Figure 4C), indicating that they were not specific to a particular genetic background. As pIC-activated TLR3 signals to IRF3 independent of the MyD88 adapter used by other TLRs, we examined the response to Dex in macrophages isolated from the MyD88-deficient mice (Adachi et al, 1998). IRF3 target gene expression was similarly affected by Dex in wt and MyD88−/− macrophages (Figure 4D), further implicating the TLR3–IRF3 pathway as a target for GC-mediated inhibition.
Figure 4.
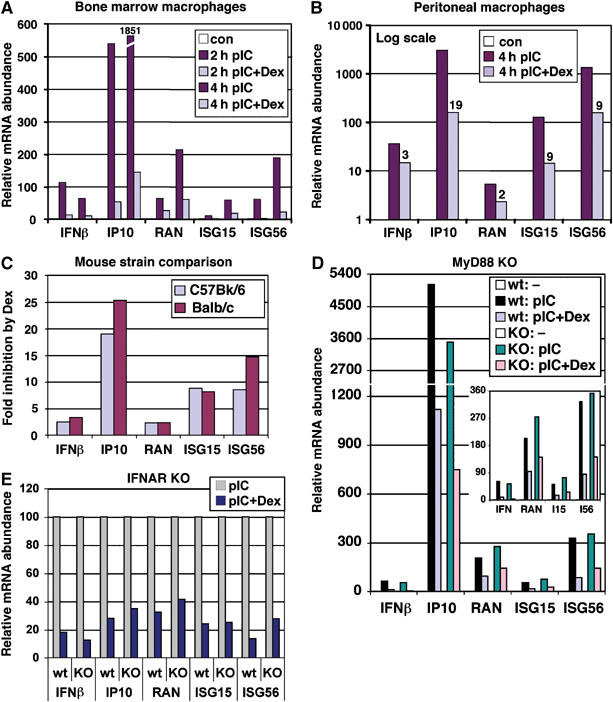
GR inhibits IRF3 target gene expression in primary macrophages independent of MyD88 and type I IFN signaling. (A) Bone marrow macrophages derived from C57BL/6 mice were treated as shown, total RNA was isolated and mRNA abundance of IRF3 target genes was determined by qPCR relative to untreated cells (con=1). (B) Peritoneal macrophages obtained from C57BL/6 mice were treated as shown, and the expression of IRF3 target genes was analyzed by qPCR. Fold inhibition by Dex is shown. (C) GC inhibition of IRF3 target gene expression is mouse strain-independent. Peritoneal macrophages were isolated from C57BL/6 or Balb/c mice and processed as in panel B. ‘Fold inhibition' is defined as the ratio of the level of each mRNA in pIC- versus pIC+Dex-treated cells. (D) Dex inhibits MyD88-independent pathway. Bone marrow macrophages derived from wt or MyD88 KO mice were treated as shown for 3 h and the expression of IRF3 target genes was assessed as in panel A. The Y-axis is broken to display maximal induction of IP10; the inset shows regulation of other mRNAs. (E) GC inhibition of IRF3 target gene expression does not require IFN signaling. Bone marrow-derived macrophages from wt or IFNAR KO mice were treated with pIC−/+Dex for 4 h and the expression of IRF3 target genes was analyzed by qPCR (the level of each mRNA in pIC-treated cells is set as 100%).
GC inhibition of IRF3-dependent genes does not require IFNβ signaling
IFNβ, one of the primary IRF3 targets, is also a critical component of the positive feedback loop whereby newly synthesized IFNβ binds to the IFNAR cell surface receptor and triggers the JAK/STAT signaling pathway, ultimately inducing many of the targets originally activated by IRF3 directly (Sato et al, 2000). As IFNβ expression might be downregulated by GR through unrelated mechanisms (e.g., direct repression through tethering to AP1 or NFκB), the observed GC suppression of numerous IRF3 target genes could reflect simply their impaired secondary induction due to reduced levels of secreted IFNβ. To uncouple primary effects of pIC from those of IFN signaling, we used IFNα/β-unresponsive IFNAR knockout (KO) mice (Muller et al, 1994). As shown in Figure 4E, Dex similarly downregulated pIC-induced genes in bone marrow-derived macrophages from IFNAR KO and wt controls, indicating that it acts independently of autocrine and paracrine signaling by IFNβ.
pIC-mediated induction and GC inhibition of IRF3 targets is mediated by their IFN-stimulated response elements
Although GC inhibition of IRF3-dependent genes was consistent with our hypothesis, most of them are regulated by multiple transcription factors including AP1 and NFκB: indeed, IFNβ and IP10 promoters contain both AP1 and NFκB binding sites, RANTES has two and ISG15 has one NFκB element (Doyle et al, 2002). Thus, downregulation by Dex could potentially result from GR-mediated repression at AP1 or NFκB tethering GREs. To establish whether IFN-stimulated response elements (ISREs) in the regulatory regions of IRF3 target genes were responsible for GC inhibition, a set of IP10 promoter-luciferase constructs (Figure 5A, left) were transfected into RAW264.7 cells, and luciferase activity was measured following a 6 h treatment with vehicle, pIC or pIC+Dex. As shown in Figure 5A, only constructs with an intact ISRE (−533-Luc and −237-Luc) were either activated by pIC or repressed by Dex, implicating IRF3 in both processes. Although the induction and inhibition of luciferase production in transient reporter assays is typically more apparent upon longer treatment, we focused on the same early 6 h time point at which the levels of endogenous IRF3-responsive mRNAs in RAW264.7 cells were examined (Figure 3). Our data indicate that under these conditions (IRF3 activation by pIC, rather than by LPS, and short duration of treatment), AP1 and NFκB elements do not appreciably contribute to the observed regulation.
To test whether the ISRE was sufficient to confer regulation by pIC and Dex, we used the p31x2-Luc reporter controlled by a dimerized IFNβ-derived ISRE (Qing et al, 2004), which serves as a specific readout for the IRF3-dependent transcriptional activity. As shown in Figure 5B, both positive and negative regulation was recapitulated by this simple ISRE reporter and, as seen with endogenous IRF3 targets (Figure 3C), ectopically introduced GRIP1 partially alleviated Dex-mediated inhibition. Furthermore, ISRE sufficiency was not a unique feature of macrophages. The p31x2-Luc reporter was also activated by pIC in CV1 monkey kidney cells (Figure 5C, left); the effect was further potentiated by transfected IRF3, indicating that both endogenous and transfected protein is inducible. Cotransfection of GR into these otherwise GR-negative cells conferred Dex-dependent inhibition of reporter activity (Figure 5C, right). Thus, GR interfered with IRF3-mediated transcription in two cell lines with reporters derived from two different IRF3-responsive genes, and in both cases ISREs mediated the effect.
Discussion
GCs are potent natural immunosuppressors that interfere with immune function at multiple levels. Apoptosis is one mechanism whereby GR eliminates immature T cells and transformed lymphoid and myeloid cells (Frankfurt and Rosen, 2004). The second GR property, not restricted to hematopoietic cells, is its ability to dramatically inhibit the production of cytokines, chemokines and their receptors, thereby disrupting cellular communication and migration. Furthermore, because many cytokines (e.g., IL2, IL8, GM-CSF, M-CSF) serve as proliferative, differentiation and trophic signals, suppression of cytokine production is often antiproliferative and may lead to apoptosis indirectly (Schimpl et al, 2002; Barreda et al, 2004). Consequently, inhibition of cytokine and chemokine synthesis is a desirable therapeutic goal in many inflammatory and autoimmune diseases. In addition, chemokines are increasingly recognized as players in tumorigenesis (Teruya-Feldstein et al, 2000; Moller et al, 2003), making them a potential therapeutic target in cancer.
The molecular dissection of the inhibitory effects of GCs on cytokine and chemokine gene expression identified AP1 and NFκB as key transcription factors susceptible to GC repression. Several models for transcriptional antagonism between these factors and GR have been proposed (reviewed in De Bosscher et al, 2003), ranging from mutual inhibition of DNA binding or competition for a common cofactor, to upregulation of an inhibitor (e.g., IκB or MKP phosphatase) or direct transcriptional repression by GR via protein:protein interaction. Although in principle, other transcriptional regulators and mechanisms may play critical roles in GR-mediated immunosuppression, their identity is obscure.
Through yeast two-hybrid and in vitro assays, we identified IRF3 as a GRIP1-binding protein (Figure 1). Endogenous GRIP1 and IRF3 interacted in RAW264.7 and CV1 cells upon IRF3 activation (Figure 2). The ability of GRIP1 to interact with both GR and IRF3 suggests that it is a shared and, perhaps, limiting subunit in their respective transcription complexes. Consequently, activated GR, in addition to directly repressing AP1 and NFκB, may sequester GRIP1 away from IRF3, thereby antagonizing production of IRF3-dependent mRNAs (Figure 6). Indeed, we found that GR and IRF3 competed for GRIP1 binding, and that GR activation nearly blocked pIC-induced expression of IRF3-responsive mRNAs and reporter constructs in RAW264.7 cells, in primary bone marrow-derived and peritoneal macrophages including those from MyD88 KO mice and in non-myeloid CV1 kidney cells in which GR expression was reconstituted (Figures 3, 4 and 5). Thus, Dex affects the MyD88-independent pathway inhibiting multiple IRF3-regulated genes in several cell types. Importantly, overexpression of GRIP1 alleviated GC inhibition of IRF3-dependent gene expression, whereas GR-mediated repression of AP1 and NFκB targets was relieved by activation of IRF3 (Figure 3C and D), consistent with the notion that GRIP1 is a limiting component in IRF3 and GR regulatory complexes. We considered the possibility that GC inhibition of IRF3 target genes reflected merely their impaired secondary induction by IFNβ, whose transcription could be affected by GR via unrelated mechanisms. Yet, GC interference persisted in IFNAR KO mice (Figure 4E), implicating IRF3 as its primary target. Finally, ISREs at target promoters were necessary and sufficient for Dex inhibition, whereas adjacent AP1 or NFκB sites were dispensable; in fact, these regulators appeared not to contribute to pIC-dependent induction of IP10 promoter activity (Figure 5). It is likely, however, that in other contexts, several mechanisms cooperate to effect GC-dependent downregulation of transcription. Indeed, promoter regions of most cytokines contain enhancers for multiple regulators close to each other, often forming composite elements. Our model predicts that GR can displace GRIP1 from the IRF3 activation complex and recruit it as a corepressor to an AP1 or NFκB tethering GRE at the same promoter (Figure 6). Such local ‘redistribution' of GRIP1 could be an important mechanism to coordinate functions of several response elements and ensure inhibition of transcription. Interestingly, while this manuscript was in review, C Glass's group defined through genomic and computational approaches an entirely different pathway linking GCs to IRF3-regulated genes. Specifically, in response to LPS/TLR4 signaling, NFκB subunit p65 was recruited to ISREs, which correlated with transcriptional activation, whereas GC treatment led to the loss of p65 and inhibition of transcription (Ogawa et al, 2005). Interestingly, semiquantitative microarray data in that study show that while Dex similarly inhibits the induction of RANTES by either pIC/TLR3 or LPS/TLR4, IP10 and ISG56 are more GC-sensitive when the inducer is LPS. It should be noted that a five-fold higher (50 μg/ml) dose of pIC in their study not only induces the above genes more potently than LPS, but could also account for a comparatively modest GC sensitivity and even invoke distinct mechanisms of repression as has been seen with other GR-repressible regulators (Nissen and Yamamoto, 2000; Rogatsky and Yamamoto, unpublished).
Figure 6.
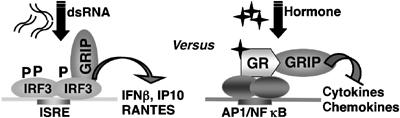
GR and IRF3 compete for GRIP1 cofactor. dsRNA promotes IRF3 phosphorylation (P) and ISRE binding. GRIP1 is recruited to ISRE as part of the IRF3 activation complex (left). Hormone-activated GR is tethered to AP1 or NFκB to repress transcription of cytokines or chemokines using GRIP1 as a corepressor (right). Activation of either pathway will deplete the other pathway from GRIP1.
Although the molecular consequences of the GRIP1:IRF3 interaction are unclear, we speculate that GRIP1 serves as an IRF3 coregulator. IRF3 was identified by homology cloning (Au et al, 1995) and was considered transcriptionally inert, until subsequent studies uncovered its phosphorylation-dependent interaction with CBP/p300 (Suhara et al, 2002; Fitzgerald et al, 2003), which remain its only well-established coactivators. Here, we identified GRIP1 as an IRF3-interacting protein. Although p160s are best known for their coactivator function for nuclear receptors, GRIP1 also regulates myoblast differentiation through interactions with Mef2C, and other p160s were recently shown to have unique functions unrelated to nuclear receptor signaling (Louie et al, 2004; Wu et al, 2005). Hence, GRIP1 interaction with IRF3 supports the emerging idea that p160s are pleiotropic transcriptional cofactors with non-redundant roles in mammalian physiology. In overexpression studies, GRIP1 not only restored IRF3 activity in the presence of Dex, but also potentiated the induction of ISG56 mRNAs by pIC, whereas knocking down endogenous GRIP1 reduced the expression of IP10, RANTES and ISG15 (Figure 3C and D). We envision several mechanisms whereby GRIP1 could facilitate IRF3-mediated transcription including (1) engagement of GRIP1 AD1/2, which can recruit CBP/p300 and CARM1 to target ISREs; (2) stabilization of IRF3 on the DNA, perhaps preventing its export or degradation; and (3) synergy with CBP/p300 recruited by the IRF3 C-terminus independent of GRIP1.
Functional interactions with IRF3 suggest unexpected roles for GRIP1 in the innate immune system. First, GRIP1 could enhance transcription of cytokines and chemokines required for a productive host response to microbial TLR agonists. Second, contribution of viral infections to autoimmune diseases (Gianani and Sarvetnick, 1996; Horwitz et al, 1998) and identification of endogenous TLR3 ligands (Kariko et al, 2004; Sen and Sarkar, 2005) implicate IRF3 and consequently GRIP1 in autoimmunity. Indeed, dysregulation of this low-level signaling could trigger massive IRF3 activation and transcription of IFNs whose key role in rheumatic disease and diabetes is established (Hooks et al, 1979; Preble et al, 1982; Alba et al, 2004). Third, GRIP1 depletion from IRF3 transcription complexes by liganded GR may promote immunosuppression and increased susceptibility to infections, a known detrimental consequence of long-term GC therapy. It will be informative to assess IRF3 function and innate immune responses in mice bearing a targeted disruption of the GRIP1 gene (Gehin et al, 2002).
Finally, an intriguing possibility exists that GRIP1 interacts with more than one IRF. The GRIP1-interacting fragment of IRF3 (IAD; Figure 1E) shares 25% identity (41% similarity) with IRF7 (Supplementary Figure 2), the closest ‘functional sibling' of IRF3 with an essential role in IFN production in response to numerous TLR agonists (Honda et al, 2005). Remarkably, in a preliminary experiment, the IRF7 IAD indeed strongly bound GRIP1 in vitro (Supplementary Figure 3). In addition to suggesting a broad role of GRIP1 in innate immunity, such interaction would explain why GCs are potent inhibitors of IFN production in plasmacytoid dendritic cells—a central cell type in the pathogenesis of autoimmune diseases (Blanco et al, 2001; Farkas et al, 2001; Shodell et al, 2003)—in which IRF7 rather than IRF3 is largely responsible for the IFNα/β synthesis (Honda et al, 2005).
It is becoming apparent that components of the innate immune system first described in the context of host defense against infection are critically involved in the pathogenesis of autoimmunity and can therefore be targeted therapeutically. Interestingly, ‘selective GR modulators' that display GC-like actions in the immune system but lack adverse side effects of conventional GCs permit differential interactions of GR with cofactors; one of these ligands specifically promotes GR binding to GRIP1 and not to other coactivators (Coghlan et al, 2003). It is tempting to speculate that one indirect target of such a compound is the GRIP1:IRF3 interaction, which will be disrupted or ‘remodeled' by the ligand-bound GR. Thus, the identification of novel molecular surfaces and mechanisms whereby GR affects the immune system should facilitate the design of more selective therapies.
Materials and methods
Yeast two-hybrid screen
The modified yeast two-hybrid screen was performed as described in Hittelman et al (1999) and legend to Supplementary Figure 1.
Plasmids
The generation of pJG4-5- and pGex4T1-GRIP1 NID-RD, pCDNA6-GRIP1, pET-GRIP1 3-RD, pGex4T1-IRF3 131C, 188C, N131 and 56–369 is described in Supplementary data. pRK5-Myc-IRF3, pGex-IRF3, pGex-IRF7 260C and p31x2-Luc (Qing et al, 2004) were kindly provided by R Derynck, UCSF. A set of IP10 promoter-luciferase constructs (−533-Luc, −237-Luc, −190-Luc, −161-Luc and −96-Luc) was a gift of D Muruve, University of Calgary (Borgland et al, 2000); −533-Luc.mt and −237-Luc.mt were generated as described in Supplementary data. βactin-LacZ and pCDNA3-GRIP1 were described previously (Rogatsky et al, 2001).
In vitro binding and competition
GST- or His-tagged fusion proteins were expressed in Escherichia coli and purified as described (Krstic et al, 1997). GRIP1 derivatives and GR were produced using the coupled in vitro transcription/translation system (Promega) in the presence of 35S-methionine. Detailed protocols are provided in Supplementary data.
Immunoprecipitation and immunoblotting
Immunoprecipitations and immunoblotting are described in Supplementary data. Antibodies used were IRF3 rabbit polyclonal (Santa Cruz) for co-immunoprecipitations, and IRF3 goat and ERK rabbit polyclonal (Santa Cruz) and GRIP1 mouse monoclonal (BD Transduction Laboratories) for immunoblotting.
RNA isolation and real-time PCR
Total RNA isolation, random-primed cDNA synthesis, qPCR and ∂∂Ct analysis were as described (Rogatsky et al, 2003). Rpl19 or actinβ was used as a normalization control. Primer pairs for target genes are listed in Supplementary Table 1.
Cell culture, transfections and siRNA
Mouse RAW264.7 macrophages and monkey CV1 kidney cells were maintained in DMEM (Invitrogen) supplemented with 10% FBS (HyClone). Lipofectamine-PLUS (Invitrogen) transfections were performed in 24-well plates in FBS-free media for 3 h, using 1 μl lipofectamine and 1.6 μl PLUS (RAW264.7) or 2 μl lipofectamine and 4 μl PLUS (CV1) per well. The following day, cells were treated with 10 μg/ml pIC and 100 nM Dex as described in figure legends and harvested for luciferase and β-gal assays as described (Rogatsky et al, 2001). GRIP1 siRNA in RAW264.7 cells was performed by AMAXA nucleofection (see Supplementary data).
Mouse primary macrophage isolation and culture
Bone-marrow derived or peritoneal macrophages were prepared from 10-week old C57BL/6 (wt or MyD88 KO (Adachi et al, 1998)), Balb/c, or SV129xC57BL/6 (wt or IFNAR KO (Muller et al, 1994)) mice as described (Hu et al, 2005). Detailed protocols are included in Supplementary data.
Supplementary Material
Supplementary Material
Supplementary Table 1
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Acknowledgments
We thank Drs M Garabedian, L Ivashkiv, K Yamamoto and K Zarember for comments on the manuscript, R Derynck and D Muruve for providing plasmids, M Garabedian for reagents and advice for the yeast two-hybrid screen, B Darimont for advice on in vitro competition assays, S Akira for MyD88 and L Ivashkiv for IFNAR KO mice. The initial stages of this work were supported by the NIH grant CA20535 to K Yamamoto and the Leukemia and Lymphoma Society Special Fellowship to IR.
References
- Adachi O, Kawai T, Takeda K, Matsumoto M, Tsutsui H, Sakagami M, Nakanishi K, Akira S (1998) Targeted disruption of the MyD88 gene results in loss of IL1- and IL18-mediated function. Immunity 9: 143–150 [DOI] [PubMed] [Google Scholar]
- Alba A, Puertas MC, Carrillo J, Planas R, Ampudia R, Pastor X, Bosch F, Pujol-Borrell R, Verdaguer J, Vives-Pi M (2004) IFNβ accelerates autoimmune type 1 diabetes in nonobese diabetic mice and breaks the tolerance to β cells in nondiabetes-prone mice. J Immunol 173: 6667–6675 [DOI] [PubMed] [Google Scholar]
- Ashwell JD, Lu FW, Vacchio MS (2000) Glucocorticoids in T cell development and function. Annu Rev Immunol 18: 309–345 [DOI] [PubMed] [Google Scholar]
- Au WC, Moore PA, Lowther W, Juang YT, Pitha PM (1995) Identification of a member of the interferon regulatory factor family that binds to the interferon-stimulated response element and activates expression of interferon-induced genes. Proc Natl Acad Sci USA 92: 11657–11661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barreda DR, Hanington PC, Belosevic M (2004) Regulation of myeloid development and function by colony stimulating factors. Dev Comp Immunol 28: 509–554 [DOI] [PubMed] [Google Scholar]
- Blanco P, Palucka AK, Gill M, Pascual V, Banchereau J (2001) Induction of dendritic cell differentiation by IFNα in systemic lupus erythematosus. Science 294: 1540–1543 [DOI] [PubMed] [Google Scholar]
- Borgland SL, Bowen GP, Wong NC, Libermann TA, Muruve DA (2000) Adenovirus vector-induced expression of the C-X-C chemokine IP10 is mediated through capsid-dependent activation of NFκB. J Virol 74: 3941–3947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caramori G, Adcock I (2005) Anti-inflammatory mechanisms of glucocorticoids targeting granulocytes. Curr Drug Targets Inflamm Allergy 4: 455–463 [DOI] [PubMed] [Google Scholar]
- Chen SL, Dowhan DH, Hosking BM, Muscat GE (2000) The steroid receptor coactivator, GRIP1, is necessary for MEF2C-dependent gene expression and skeletal muscle differentiation. Genes Dev 14: 1209–1228 [PMC free article] [PubMed] [Google Scholar]
- Coghlan MJ, Jacobson PB, Lane B, Nakane M, Lin CW, Elmore SW, Kym PR, Luly JR, Carter GW, Turner R, Tyree CM, Hu J, Elgort M, Rosen J, Miner JN (2003) A novel antiinflammatory maintains glucocorticoid efficacy with reduced side effects. Mol Endocrinol 17: 860–869 [DOI] [PubMed] [Google Scholar]
- Darimont BD, Wagner RL, Apriletti JW, Stallcup MR, Kushner PJ, Baxter JD, Fletterick RJ, Yamamoto KR (1998) Structure and specificity of nuclear receptor–coactivator interactions. Genes Dev 12: 3343–3356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bosscher K, Vanden Berghe W, Haegeman G (2003) The interplay between the glucocorticoid receptor and nuclear factor-κB or activator protein-1: molecular mechanisms for gene repression. Endocr Rev 24: 488–522 [DOI] [PubMed] [Google Scholar]
- Doyle S, Vaidya S, O'Connell R, Dadgostar H, Dempsey P, Wu T, Rao G, Sun R, Haberland M, Modlin R, Cheng G (2002) IRF3 mediates a TLR3/TLR4-specific antiviral gene program. Immunity 17: 251–263 [DOI] [PubMed] [Google Scholar]
- Elenkov IJ (2004) Glucocorticoids and the Th1/Th2 balance. Ann NY Acad Sci 1024: 138–146 [DOI] [PubMed] [Google Scholar]
- Eriksson U, Ricci R, Hunziker L, Kurrer MO, Oudit GY, Watts TH, Sonderegger I, Bachmaier K, Kopf M, Penninger JM (2003) Dendritic cell-induced autoimmune heart failure requires cooperation between adaptive and innate immunity. Nat Med 9: 1484–1490 [DOI] [PubMed] [Google Scholar]
- Farkas L, Beiske K, Lund-Johansen F, Brandtzaeg P, Jahnsen FL (2001) Plasmacytoid dendritic cells (natural interferon-α/β-producing cells) accumulate in cutaneous lupus erythematosus lesions. Am J Pathol 159: 237–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, Coyle AJ, Liao SM, Maniatis T (2003) IKKɛ and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol 4: 491–496 [DOI] [PubMed] [Google Scholar]
- Frankfurt O, Rosen ST (2004) Mechanisms of glucocorticoid-induced apoptosis in hematologic malignancies: updates. Curr Opin Oncol 16: 553–563 [DOI] [PubMed] [Google Scholar]
- Gehin M, Mark M, Dennefeld C, Dierich A, Gronemeyer H, Chambon P (2002) The function of TIF2/GRIP1 in mouse reproduction is distinct from those of SRC1 and p/CIP. Mol Cell Biol 22: 5923–5937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiss G, Jin G, Guo J, Bumgarner R, Katze MG, Sen GC (2001) A comprehensive view of regulation of gene expression by double-stranded RNA-mediated cell signaling. J Biol Chem 276: 30178–30182 [DOI] [PubMed] [Google Scholar]
- Gianani R, Sarvetnick N (1996) Viruses, cytokines, antigens, and autoimmunity. Proc Natl Acad Sci USA 93: 2257–2259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hittelman AB, Burakov D, Iniguez-Lluhi JA, Freedman LP, Garabedian MJ (1999) Differential regulation of glucocorticoid receptor transcriptional activation via AF1-associated proteins. EMBO J 18: 5380–5388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, Shimada N, Ohba Y, Takaoka A, Yoshida N, Taniguchi T (2005) IRF7 is the master regulator of type-I interferon-dependent immune responses. Nature 434: 772–777 [DOI] [PubMed] [Google Scholar]
- Hong H, Kohli K, Trivedi A, Johnson DL, Stallcup MR (1996) GRIP1, a novel mouse protein that serves as a transcriptional coactivator in yeast for the hormone binding domains of steroid receptors. Proc Natl Acad Sci USA 93: 4948–4952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooks JJ, Moutsopoulos HM, Geis SA, Stahl NI, Decker JL, Notkins AL (1979) Immune interferon in the circulation of patients with autoimmune disease. N Engl J Med 301: 5–8 [DOI] [PubMed] [Google Scholar]
- Horwitz MS, Bradley LM, Harbertson J, Krahl T, Lee J, Sarvetnick N (1998) Diabetes induced by Coxsackie virus: initiation by bystander damage and not molecular mimicry. Nat Med 4: 781–785 [DOI] [PubMed] [Google Scholar]
- Hu X, Ho HH, Lou O, Hidaka C, Ivashkiv LB (2005) Homeostatic role of interferons conferred by inhibition of IL1-mediated inflammation and tissue destruction. J Immunol 175: 131–138 [DOI] [PubMed] [Google Scholar]
- Ito K, Barnes PJ, Adcock IM (2000) Glucocorticoid receptor recruitment of histone deacetylase 2 inhibits IL1β-induced histone H4 acetylation on lysines 8 and 12. Mol Cell Biol 20: 6891–6903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kariko K, Ni H, Capodici J, Lamphier M, Weissman D (2004) mRNA is an endogenous ligand for Toll-like receptor 3. J Biol Chem 279: 12542–12550 [DOI] [PubMed] [Google Scholar]
- Krstic MD, Rogatsky I, Yamamoto KR, Garabedian MJ (1997) Mitogen-activated and cyclin-dependent protein kinases selectively and differentially modulate transcriptional enhancement by the glucocorticoid receptor. Mol Cell Biol 17: 3947–3954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar KP, McBride KM, Weaver BK, Dingwall C, Reich NC (2000) Regulated nuclear–cytoplasmic localization of interferon regulatory factor 3, a subunit of double-stranded RNA-activated factor 1. Mol Cell Biol 20: 4159–4168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ, Marshak-Rothstein A (2002) Chromatin–IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature 416: 603–607 [DOI] [PubMed] [Google Scholar]
- Lefstin JA, Yamamoto KR (1998) Allosteric effects of DNA on transcriptional regulators. Nature 392: 885–888 [DOI] [PubMed] [Google Scholar]
- Li Q, Verma IM (2002) NFκB regulation in the immune system. Nat Rev Immunol 2: 725–734 [DOI] [PubMed] [Google Scholar]
- Lin R, Heylbroeck C, Pitha PM, Hiscott J (1998) Virus-dependent phosphorylation of the IRF3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome-mediated degradation. Mol Cell Biol 18: 2986–2996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin R, Mamane Y, Hiscott J (1999) Structural and functional analysis of IRF3: localization of the transactivation and autoinhibitory domains. Mol Cell Biol 19: 2465–2474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Kang JS, Derynck R (2004) TGFβ-activated Smad3 represses MEF2-dependent transcription in myogenic differentiation. EMBO J 23: 1557–1566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louie MC, Zou JX, Rabinovich A, Chen HW (2004) ACTR/AIB1 functions as an E2F1 coactivator to promote breast cancer cell proliferation and antiestrogen resistance. Mol Cell Biol 24: 5157–5171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luecke HF, Yamamoto KR (2005) The glucocorticoid receptor blocks PTEFb recruitment by NFκB to effect promoter-specific transcriptional repression. Genes Dev 19: 1116–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moller C, Stromberg T, Juremalm M, Nilsson K, Nilsson G (2003) Expression and function of chemokine receptors in human multiple myeloma. Leukemia 17: 203–210 [DOI] [PubMed] [Google Scholar]
- Muller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, Aguet M (1994) Functional role of type I and type II interferons in antiviral defense. Science 264: 1918–1921 [DOI] [PubMed] [Google Scholar]
- Nissen RM, Yamamoto KR (2000) The glucocorticoid receptor inhibits NFκB by interfering with serine-2 phosphorylation of the RNA polymerase II carboxy-terminal domain. Genes Dev 14: 2314–2329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obradovic Z, Peng K, Vucetic S, Radivojac P, Brown CJ, Dunker AK (2003) Predicting intrinsic disorder from amino acid sequence. Proteins 53: 566–572 [DOI] [PubMed] [Google Scholar]
- Ogawa S, Lozach J, Benner C, Pascual G, Tangirala RK, Westin S, Hoffmann A, Subramaniam S, David M, Rosenfeld MG, Glass CK (2005) Molecular determinants of crosstalk between nuclear receptors and Toll-like receptors. Cell 122: 707–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pazirandeh A, Jondal M, Okret S (2005) Conditional expression of a glucocorticoid receptor transgene in thymocytes reveals a role for thymic-derived glucocorticoids in thymopoiesis in vivo. Endocrinology 146: 2501–2507 [DOI] [PubMed] [Google Scholar]
- Preble OT, Black RJ, Friedman RM, Klippel JH, Vilcek J (1982) Systemic lupus erythematosus: presence in human serum of an unusual acid-labile leukocyte interferon. Science 216: 429–431 [DOI] [PubMed] [Google Scholar]
- Qin BY, Liu C, Lam SS, Srinath H, Delston R, Correia JJ, Derynck R, Lin K (2003) Crystal structure of IRF3 reveals mechanism of autoinhibition and virus-induced phosphoactivation. Nat Struct Biol 10: 913–921 [DOI] [PubMed] [Google Scholar]
- Qing J, Liu C, Choy L, Wu RY, Pagano JS, Derynck R (2004) TGFβ/Smad3 signaling regulates IRF7 function and transcriptional activation of the β interferon promoter. Mol Cell Biol 24: 1411–1425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogatsky I, Luecke HF, Leitman DC, Yamamoto KR (2002) Alternate surfaces of transcriptional coregulator GRIP1 function in different glucocorticoid receptor activation and repression contexts. Proc Natl Acad Sci USA 99: 16701–16706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogatsky I, Wang JC, Derynck MK, Nonaka DF, Khodabakhsh DB, Haqq CM, Darimont BD, Garabedian MJ, Yamamoto KR (2003) Target-specific utilization of transcriptional regulatory surfaces by the glucocorticoid receptor. Proc Natl Acad Sci USA 100: 13845–13850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogatsky I, Zarember KA, Yamamoto KR (2001) Factor recruitment and TIF2/GRIP1 corepressor activity at a collagenase-3 response element that mediates regulation by phorbol esters and hormones. EMBO J 20: 6071–6083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronnblom LE, Alm GV, Oberg KE (1990) Possible induction of systemic lupus erythematosus by interferon-α treatment in a patient with a malignant carcinoid tumour. J Intern Med 227: 207–210 [DOI] [PubMed] [Google Scholar]
- Sato M, Suemori H, Hata N, Asagiri M, Ogasawara K, Nakao K, Nakaya T, Katsuki M, Noguchi S, Tanaka N, Taniguchi T (2000) Distinct and essential roles of transcription factors IRF3 and IRF7 in response to viruses for IFNα/β gene induction. Immunity 13: 539–548 [DOI] [PubMed] [Google Scholar]
- Schimpl A, Berberich I, Kneitz B, Kramer S, Santner-Nanan B, Wagner S, Wolf M, Hunig T (2002) IL2 and autoimmune disease. Cytokine Growth Factor Rev 13: 369–378 [DOI] [PubMed] [Google Scholar]
- Sen GC, Sarkar SN (2005) Transcriptional signaling by double-stranded RNA: role of TLR3. Cytokine Growth Factor Rev 16: 1–14 [DOI] [PubMed] [Google Scholar]
- Sharma S, tenOever BR, Grandvaux N, Zhou GP, Lin R, Hiscott J (2003) Triggering the interferon antiviral response through an IKK-related pathway. Science 300: 1148–1151 [DOI] [PubMed] [Google Scholar]
- Shaulian E, Karin M (2002) AP1 as a regulator of cell life and death. Nat Cell Biol 4: E131–136 [DOI] [PubMed] [Google Scholar]
- Shodell M, Shah K, Siegal FP (2003) Circulating human plasmacytoid dendritic cells are highly sensitive to corticosteroid administration. Lupus 12: 222–230 [DOI] [PubMed] [Google Scholar]
- Suhara W, Yoneyama M, Kitabayashi I, Fujita T (2002) Direct involvement of CREB-binding protein/p300 in sequence-specific DNA binding of virus-activated interferon regulatory factor-3 holocomplex. J Biol Chem 277: 22304–22313 [DOI] [PubMed] [Google Scholar]
- Teruya-Feldstein J, Tosato G, Jaffe ES (2000) The role of chemokines in Hodgkin's disease. Leuk Lymphoma 38: 363–371 [DOI] [PubMed] [Google Scholar]
- Tsai M-J, O'Malley BW (1994) Molecular mechanisms of action of steroid/thyroid receptor superfamily members. Annu Rev Biochem 63: 45–86 [DOI] [PubMed] [Google Scholar]
- Voegel JJ, Heine MJ, Tini M, Vivat V, Chambon P, Gronemeyer H (1998) The coactivator TIF2 contains three nuclear receptor-binding motifs and mediates transactivation through CBP binding-dependent and -independent pathways. EMBO J 17: 507–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voegel JJ, Heine MJ, Zechel C, Chambon P, Gronemeyer H (1996) TIF2, a 160 kDa transcriptional mediator for the ligand-dependent activation function AF2 of nuclear receptors. EMBO J 15: 3667–3675 [PMC free article] [PubMed] [Google Scholar]
- Wu HY, Hamamori Y, Xu J, Chang SC, Saluna T, Chang MF, O'Malley BW, Kedes L (2005) Nuclear hormone receptor coregulator GRIP1 suppresses, whereas SRC1A and p/CIP coactivate, by domain-specific binding of MyoD. J Biol Chem 280: 3129–3137 [DOI] [PubMed] [Google Scholar]
- Yoneyama M, Suhara W, Fukuhara Y, Fukuda M, Nishida E, Fujita T (1998) Direct triggering of the type I interferon system by virus infection: activation of a transcription factor complex containing IRF3 and CBP/p300. EMBO J 17: 1087–1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Supplementary Table 1
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3