

Abstract
During Drosophila melanogaster eye development, signaling through receptor tyrosine kinases (RTKs) leads to activation of a mitogen activated protein tyrosine kinase, called Rolled. Key nuclear targets of Rolled are two antagonistic transcription factors: Yan, a repressor, and Pointed-P2 (Pnt-P2), an activator. A critical regulator of this process, Mae, can interact with both Yan and Pnt-P2 through their SAM domains. Although earlier work showed that Mae derepresses Yan-regulated transcription by depolymerizing the Yan polymer, the mechanism of Pnt-P2 regulation by Mae remained undefined. We find that efficient phosphorylation and consequent activation of Pnt-P2 requires a three-dimensional docking surface on its SAM domain for the MAP kinase, Rolled. Mae binding to Pnt-P2 occludes this docking surface, thereby acting to downregulate Pnt-P2 activity. Docking site blocking provides a new mechanism whereby the cell can precisely modulate kinase signaling at specific targets, providing another layer of regulation beyond the more global changes effected by alterations in the activity of the kinase itself.
Keywords: ETS-family transcription factors, Mae, MAPK, Pnt-P2, SAM domain, transcriptional regulation
Introduction
Receptor tyrosine kinase (RTK) activation at the cell surface is translated, via kinase cascades, into nuclear events that lead to changes in gene expression. To respond appropriately, the activation signal must be received and interpreted properly by many systems in the cell (Tan and Kim, 1999). In Drosophila melanogaster, eye development requires signaling through epidermal growth factor (EGF) and Sevenless RTKs. RTK activity leads to activation of Ras, which in turn leads to activation of the mitogen activated protein tyrosine kinase (MAPK) called Rolled (Rebay, 2002). The well-known nuclear targets of RTK stimulation include two ETS-family proteins (Sharrocks, 2001): the transcriptional repressor Yan (Lai and Rubin, 1992) and the transcriptional activator Pointed-P2 (Pnt-P2) (Klambt, 1993). In undifferentiated cells, Yan resides in the nucleus and represses transcription of its target genes. In response to RTK-initiated signaling, the activated Rolled MAPK phosphorylates a critical residue, Ser127, of Yan (Rebay and Rubin, 1995), which promotes CRM1-mediated nuclear export of Yan (Tootle et al, 2003). Concomitantly, Pnt-P2 is also phosphorylated by Rolled, becoming a functional transcriptional activator that stimulates genes previously repressed by Yan (Brunner et al, 1994; O'Neill et al, 1994).
More recently, a key regulator of this process, a protein called Mae, was identified (Baker et al, 2001; Yamada et al, 2003). Mae, Yan, and Pnt-P2 share a common protein–protein interaction motif—the SAM domain (Kim and Bowie, 2003; Qiao and Bowie, 2005). Mae can bind with both Yan and Pnt-P2 via SAM domain interactions. The SAM domain of Yan forms a head-to-tail polymer involving two surfaces on Yan-SAM called the Mid-Loop (ML) and End-Helix (EH) surfaces (Qiao et al, 2004). Polymerization is essential for Yan repression and Mae was found to act, in part, by efficiently depolymerizing the Yan polymer. Depolymerization of Yan by Mae may prevent the Yan polymer from spreading on chromatin, inhibit DNA binding (at least in part by loss of subunit cooperativity) and expose the site of phosphorylation, thereby facilitating the recruitment of the MAPK to Yan (Qiao et al, 2004). Phosphorylation enables displacement of Mae by CRM1, leading to nuclear export of Yan (Tootle et al, 2003; Song et al, 2005).
In contrast to our more extensive understanding of Mae action on Yan, the molecular mechanism of Mae regulation of Pnt-P2 has remained uncharacterized. The SAM domain of Pnt-P2 is monomeric in solution (Mackereth et al, 2004), and it is therefore unlikely that Mae acts to depolymerize Pnt-P2-SAM through their SAM domain interactions. While Mae was originally proposed to positively influence the transcriptional activity of Pnt-P2 by facilitating its phosphorylation by Rolled (Baker et al, 2001), transcriptional reporter assays in cultured S2 cells (Tootle et al, 2003; Yamada et al, 2003) and genetic studies in flies (Yamada et al, 2003; Vivekanand et al, 2004) indicate that Mae actually down-regulates Pnt-P2. The Rolled kinase phosphoacceptor site in Pnt-P2, Thr151, is crucial for its transcriptional activator function. The T151A mutant of Pnt-P2, which cannot be phosphorylated in vitro, is unable to rescue a pnt eye phenotype, and even in a wild-type background this mutant protein prevents R7 cell determination (O'Neill et al, 1994).
MAPKs can target their substrates through docking sites distal to the phosphoacceptors (Holland and Cooper, 1999; Sharrocks et al, 2000). Studies on the mammalian orthologs of Pnt-P2, ETS-1 and ETS-2 found that an LX(L/I)XXXF sequence motif in their SAM domains, which lies C-terminal to the Thr phosphoacceptor, acts as a docking site for the MAP kinase, ERK2 (Figure 1A and B) (Seidel and Graves, 2002). This kinase docking site increases the efficiency of ETS-1 and ETS-2 phosphorylation in vitro and is required for RTK-mediated transactivation activity of ETS-1 and ETS-2 in cultured cell assays. We suspected that such a docking site could also exist in their fly relative, Pnt-P2. Moreover, since Mae-SAM binds Pnt-P2-SAM (Baker et al, 2001), Mae might regulate Pnt-P2 activity by obstructing the kinase docking site, thereby inhibiting its phosphorylation.
Figure 1.
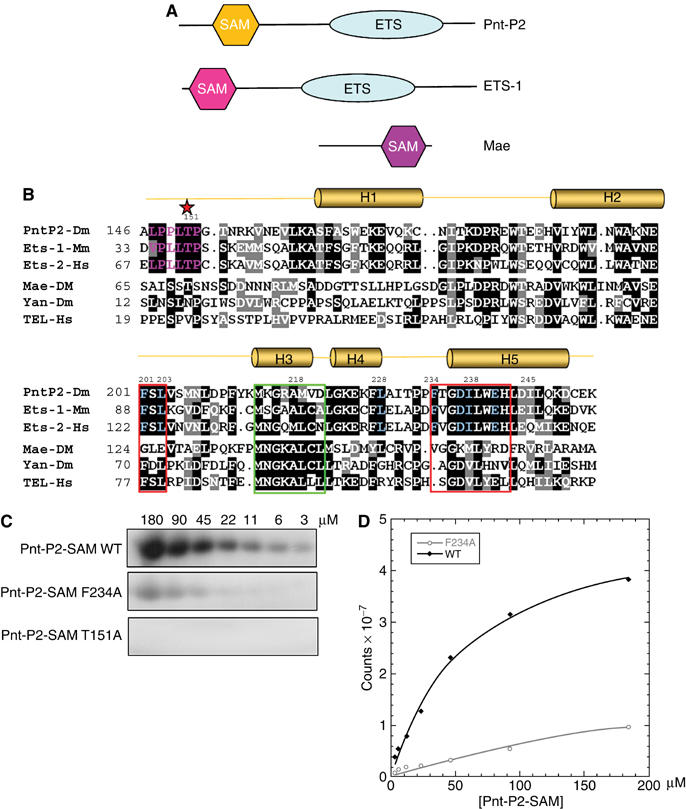
A MAPK docking site on the Pnt-P2 SAM domain. (A) Domain structure of Pnt-P2, ETS-1 and Mae. The SAM domain is often called the Pointed domain. ETS domain is a DNA-binding domain recognizing a 5′-GGA(A/T)-3′ sequence motif (Sharrocks, 2001). (B) Multiple sequence alignment of Pnt-P2 SAM, ETS-1 SAM, ETS-2 SAM, Mae-SAM, Yan-SAM and TEL-SAM. The sequence regions that comprise the ML and EH surfaces of the SAM domains are framed in green and red, respectively. The consensus MAPK phosphorylation sequences are colored in purple and the phosphoacceptor residues are marked by a star. The MAPK docking site residues are color in blue. (C) Kinase assays with wild type or the indicated mutant versions of Pnt-P2 SAM. Different concentrations of protein substrates were phosphorylated by an activated MAPK/Rolled with 32P-labeled ATP under steady-state conditions as described in Materials and methods. Phosphorylated proteins were detected after SDS–PAGE using a phosphorimager. (D) Initial reaction velocities were fit into Michaelis–Menten equation. Values are an average from two independent experiments.
We find that efficient phosphorylation of Pnt-P2 depends on a three-dimensional surface on Pnt-P2-SAM, which acts as a docking site for the MAPK/Rolled. In contrast to previously characterized docking sites that involve a small sequence motif, this surface covers about one-third of the surface area of the SAM domain. Furthermore, we also find that Mae-SAM specifically binds this kinase docking site on Pnt-P2-SAM with high affinity, which prevents the MAPK from docking onto Pnt-P2. These findings expand the concept of kinase docking site from a short primary sequence motif to a defined three-dimensional architecture. Regulation of kinase action by blocking the docking site on its substrate represents a new mechanism for specifically inhibiting the phosphorylation of certain substrates while maintaining phosphorylation competence of the kinase for other substrates.
Results
The EH surface of Pnt-P2-SAM forms a MAPK/Rolled docking site
To test the docking site hypothesis, we first established an in vitro kinase assay. Rolled (residues 18–376) was produced by coexpression with a constitutively active mutant of mammalian MEK1 (MEK1R4F) in Escherichia coli (Khokhlatchev et al, 1997). As shown in Figure 1C and D, Pnt-P2-SAM could be efficiently phosphorylated by Rolled in vitro. Mutation of Thr151 to Ala in Pnt-P2-SAM completely eliminated phosphorylation (Figure 1C), and quadrupole/time-of-flight tandem mass spectrometry (MS/MS) experiments further demonstrated that Thr151 is the phosphorylation site for Rolled (data not shown). By fitting the initial phosphorylation rates to the Michaelis–Menten equation, we obtained a Km value of 54 μM (Figure 1D), about 10-fold higher than the Km (Seidel and Graves, 2002) and Kd (Rainey et al, 2005) values reported for the ETS-1/ERK2 pair of ∼5 μM.
To test the importance of the putative LXIXXXF Pnt-P2-SAM docking site, we created docking site mutants L228R, I230R and F234A. The L228R and F234A mutations on Pnt-P2-SAM both resulted in significantly reduced phosphorylation efficiency (Figures 1C and 2A). The I230R mutation did not affect phosphorylation, however, indicating that it is not part of the docking surface (Figure 2A). These three mutants produced CD spectra almost identical to the wild-type Pnt-P2-SAM, implying that the amino-acid substitutions did not disrupt the SAM-fold (Figure 2B). Moreover, a Pnt-P2 peptide (residues 96–164), which contains the phosphoacceptor T151 but lacks the SAM domain, was also a poor substrate for the kinase (Figure 2A). Estimated Km values for Pnt-P2-SAM L228R, F234A and Pnt-P2 (96–164) (>350 μM) are comparable to the Km of the reactions of peptide substrates of ERK2 that do not contain docking sites (∼300–500 μM) (Gonzalez et al, 1991; Robinson et al, 1996).
Figure 2.
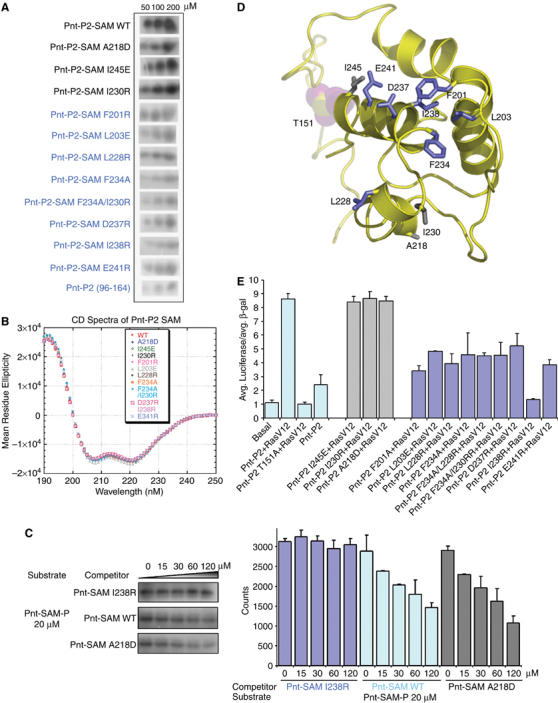
The EH surface of Pnt-P2 SAM acts as MAPK docking surface. (A) Kinase assays with the wild-type Pnt-P2 SAM or the indicated mutants. Equal amounts of each protein were used in the assays. Three concentrations of each protein were assayed. (B) Far-UV CD spectra of wild type or different mutant versions of Pnt-P2-SAM. Similar spectra characterizing α-helical structure were observed for all the proteins tested. (C) The SAM domain competes for substrate phosphorylation. Phosphorylation of the Pnt substrate was monitored in the presence of increasing concentrations of Pnt-P2-SAM domain variants that did not possess the phosphoacceptor site (see Materials and methods). Both the wild-type SAM domain and a variant bearing a mutation outside the docking site (A281D) compete with the substrate, but a variant with a mutation in the docking surface (I238R) does not compete. The bar graph shows the average and range of three independent experiments. (D) A structural model of Pnt-P2 SAM based on the NMR structure of its mammalian homolog ETS-1-SAM (see Materials and methods). Identified MAPK docking site residues were colored in blue. The phosphoacceptor T151, colored in purple, is in a flexible region N-terminal to the H1 of Pnt-P2-SAM. Several residues outside the kinase docking surface are colored in gray. (E) Pnt-P2 MAPK docking site mutants affect Ras induced transactivation. Luciferase assays were performed by using Drosophila S2 cells transfected with the pEBS-luciferase reporter, constitutively active form of Ras, RasV12, and either wild type or mutant forms of full-length Pnt-P2 constructs. A pAC5.1-lacZ was used as an internal control. The normalized units are shown based on experiments performed in triplicate.
The presence of a docking site on the SAM domain is also supported by a competition assay shown in Figure 2C. Adding increasing concentrations of a Pnt-P2-SAM domain competitor, with the phosphorylation site deleted, diminishes phosphorylation of the Pnt-P2 substrate. Moreover, a Pnt-P2-SAM competitor bearing a mutation outside the putative docking site (A281D) remains effective, but the I238R mutation in the putative docking surface fails to compete.
The SAM domain of Pnt-P2 and the SAM domain of its mammalian homolog ETS-1 share about 60% sequence identity (Figure 1B), so it was possible to build a reasonable structural model of Pnt-P2-SAM (residue 142–249) based on the NMR structure of ETS-1-SAM (PDB code:1BQV) (Slupsky et al, 1998) (Figure 2D). Examination of the Pnt-P2-SAM structural model indicated that docking site residue F234 is in a surface of the SAM domain that is commonly used in protein–protein interactions, called the EH surface (framed in red in Figure 1B) (Kim et al, 2001). Moreover, the sequence of the EH surface region of Pnt-P2 is almost identical to that of both ETS-1- and ETS-2-SAM, implying some functional conservation in this region among these three proteins. These observations led us to speculate that more of the EH surface of Pnt-P2-SAM might be part of the docking interface with Rolled.
To test this hypothesis, we decided to determine the effect of additional EH surface mutations on the phosphorylation of Pnt-P2-SAM by Rolled. The following mutants were made (colored in blue in Figures 1B and 2D): F201A, L203E, D237R, I238R, and E241R. A mutation on the ML surface of Pnt-P2-SAM (A218D) and a mutation near the EH surface (I245E) were also made as controls (colored gray in the Figure 2D). Purified mutants were then analyzed for their capacity to be phosphorylated by Rolled. Indeed, all the Pnt-P2-SAM EH surface mutants were phosphorylated with reduced efficiency (Figure 2A). In contrast, the ML surface mutation, A218D, and another mutation outside the EH surface, I245E, had only modest affects on Pnt-P2-SAM phosphorylation (Figure 2A). None of the mutants exhibited altered CD spectra, indicating that they remain properly folded (Figure 2B). These experiments indicate that the EH surface of Pnt-P2-SAM, comprising at least residues F201, L203, L228, F234, D237, I238, E241, acts as a docking interface that contacts the Rolled MAPK (Figure 2D).
We next examined the biological consequences of introducing the docking site mutations into full-length Pnt-P2, by testing the ability of the mutant proteins to activate Ras-dependent gene transcription. We utilized a luciferase transcriptional reporter assay in Drosophila S2 cells (Tootle et al, 2003; Qiao et al, 2004). Luciferase expression was brought under Pnt-P2 control by placing high-affinity ETS binding sites upstream of a basal promoter (EBS-luciferase). As shown in Figure 2E, wild-type Pnt-P2 activated transcription of the reporter gene in a Ras-dependent manner, while the mutation of its phosphoacceptor (Thr151) completely abolished its transactivation ability (O'Neill et al, 1994). The Pnt-P2 proteins that bear mutations in their SAM domain kinase docking surface (F201A, L203E, L228R, F234A, D237R, I238R, and E241R) failed to activate transcription as effectively as the wild-type protein. Notably, the mutation at the center of the EH surface of the SAM domain, I238R, reduced the activation to around the level of the phosphorylation site deletion (T151A). On the other hand, mutation of a residue on the ML surface (A218D) and of a residue with its side-chain pointing away from the EH surface (I230R) can activate the transcription as well as the wild-type Pnt-P2. Western blots showed that all these Pnt-P2 variants were equivalently expressed in S2 cells (not shown).
Collectively, these in vitro and in vivo data demonstrate that the EH surface of the Pnt-P2 SAM domain is the docking surface for MAPK/Rolled and this docking surface is essential for Pnt-P2-mediated enhancement of transactivation in response to Ras signaling. This suggests that the Rolled MAPK recognizes its Pnt-P2-SAM docking site in the context of the tertiary structure. Our results expand the concept of kinase docking site from a one-dimensional sequence motif to a three-dimensional structural motif.
The ML surface of Mae-SAM specifically interacts with the EH surface of Pnt-P2-SAM
We next examined how Mae interacts with Pnt-P2 (Tootle et al, 2003). SAM domains often bind to one another utilizing their ML or EH binding surfaces (Kim and Bowie, 2003). Indeed, Mae-SAM utilizes its ML surface to bind to Yan-SAM (Qiao et al, 2004). We therefore speculated that Mae-SAM and Pnt-P2-SAM could interact via their ML and EH surfaces. We tested this possibility by using surface plasmon resonance binding experiments. An N-terminal fusion of Maltose Binding Protein (MBP) to Mae-SAM (MBP-Mae-SAM) was used in these assays (Qiao et al, 2004). As shown in Figure 3A, when a Pnt-P2-SAM ML surface mutant, A218D, was attached to the chip, strong binding of the MBP-Mae-SAM was observed. Kinetic fits to the data yield a Kd of 11±3 nM (Supplementary Figure S1). The affinity is comparable to the Mae-SAM/Yan-SAM interaction, which has a Kd of 11 nM (Qiao et al, 2004), suggesting that Mae can regulate both proteins in a similar concentration range. When a Pnt-P2-SAM EH surface mutant, I238R, was attached to the chip, however, no binding was detected to the MBP-Mae-SAM fusion at a concentration as high as 3.45 μM. These experiments suggest that the EH surface of Pnt-P2-SAM is required for its interaction with Mae-SAM. We then prepared two mutants of MBP-Mae-SAM to test their binding to Pnt-P2-SAM. One Mae-SAM mutant, A141D, is in the ML surface and the other mutant, M160E, is in the EH surface. As summarized in Figure 3A, the MBP-Mae-SAM M160E mutant binds to Pnt-P2-SAM with approximately wild-type affinity (Kd=20±4 nM). However, no binding was detected with MBP-Mae-SAM A141D at concentrations as high as 4.2 μM. These results indicate that the Mae-SAM binds to both Pnt-P2-SAM and Yan-SAM in a similar fashion, namely the ML surface of Mae binds to the EH surface of both proteins (Qiao et al, 2004).
Figure 3.
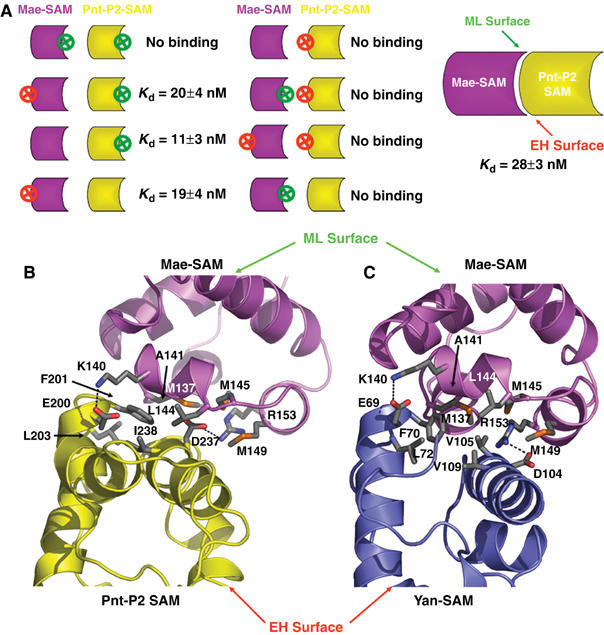
The EH surface of Pnt-P2 SAM interacts with the ML surface of Mae-SAM. (A) Evaluation of interactions between different combinations of MBP-Mae-SAM and Pnt-P2 SAM variants using surface plasmon resonance (SPR). The symbols represent the SAM domains of Mae (purple) and Pnt-P2 (yellow). The convex side is used to depict the ML-surface and the concave side the EH surface. A red circled X represents a mutation on the EH surface and a green circled X represents a mutation on the ML surface. The dissociation constants were obtained from kinetic parameters calculated from the sensograms (see Supplementary Figure S1 for details). No binding indicates that we could see no interaction at the highest MBP-Mae concentration used of 3.45 μM. (B, C) Detailed view comparing the Mae-SAM/Pnt-P2-SAM and the Mae-SAM/Yan-SAM interfaces. The structural model of Mae-SAM/Pnt-P2-SAM complex was built by superimposing the Pnt-P2-SAM structure on Yan-SAM structure in the Mae-SAM/Yan-SAM crystal structure. Mae-SAM, Pnt-P2-SAM and Yan-SAM are colored in purple, yellow, and blue, respectively. Hydrophobic residues that make up the core of the interface are colored by atom type. They are C—gray, S—orange, N—blue, and O—red. Potential hydrogen bonds are indicated by dotted lines. The core of the interface in the Mae-SAM/Pnt-P2-SAM model is composed of hydrophobic residues M137, A141, L144, M145, M149 on the ML surface of Mae-SAM and F201, L203, I238, I245 on the EH surface of Pnt-P2-SAM. Mutations in each of these hydrophobic interface residues lead to the disruption of Mae-SAM/Pnt-P2-SAM interaction as assessed by SPR experiments (data not shown).
Given the biochemical data discussed above, the high degree of sequence similarity between Pnt-P2-SAM and Yan-SAM, and the accumulated knowledge of hetero-SAM domain interactions (Ramachander and Bowie, 2004; Kim et al, 2005), it seems reasonable to build a Mae-SAM/Pnt-P2-SAM model based on the high-resolution crystal structure of the Mae-SAM/Yan-SAM complex (Qiao et al, 2004). We therefore built a model of the Mae-SAM/Pnt-P2-SAM heterodimer by superimposing the structure of Pnt-P2-SAM on the Yan-SAM in Mae-SAM/Yan-SAM complex (Figure 3B and C). The core of the interface in the Mae-SAM/Pnt-P2-SAM model is composed of hydrophobic residues M137, A141, L144, M145, M149 on the ML surface of Mae-SAM and F201, L203, I238, I245 on the EH surface of Pnt-P2-SAM (Figure 3B). Mutations in each of these hydrophobic interface residues lead to the disruption of Mae-SAM/Pnt-P2-SAM interaction as assessed by SPR experiments (data not shown).
Our results demonstrate that both Mae and Rolled bind to the EH surface of Pnt-P2-SAM suggesting that Mae binding would block MAPK/Rolled docking. Moreover, the binding affinity between Mae-SAM and Pnt-P2-SAM is approximately 3000 times higher than the Rolled/Pnt-P2 enzyme–substrate complex as judged by the difference between the Km of the kinase reaction and the binding affinity of Mae-SAM to Pnt-P2-SAM, implying that Mae could compete effectively for Pnt-P2 binding (Figure 4A and B).
Figure 4.
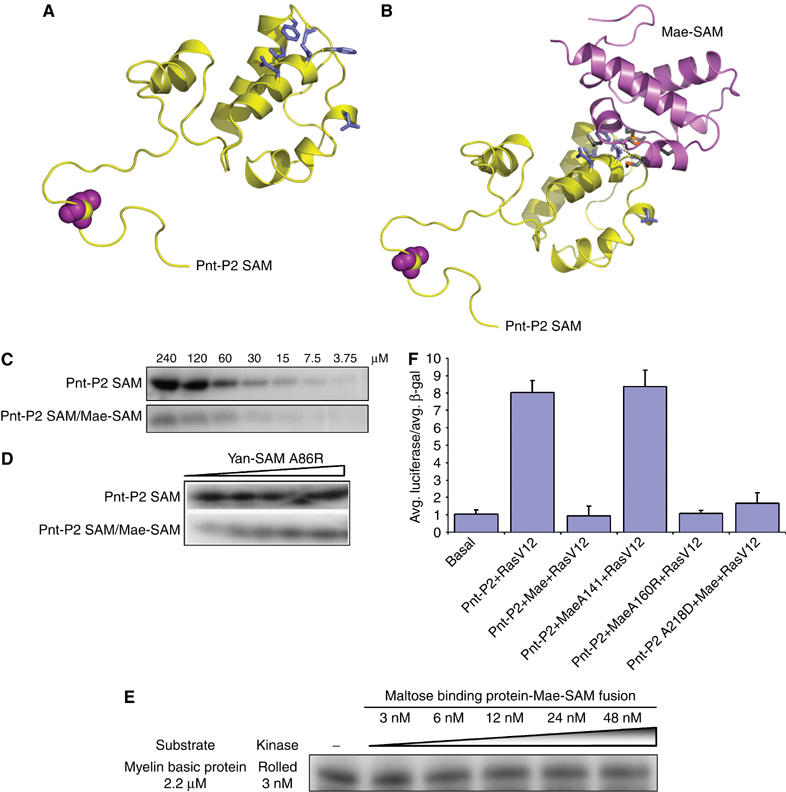
Mae blocks the MAPK docking site on Pnt-P2. (A, B) Comparison of the structural models between Pnt-P2-SAM and Pnt-P2-SAM/Mae-SAM complex. On binding of Mae-SAM to Pnt-P2-SAM, the kinase docking surface on Pnt-P2-SAM is fully blocked, which results in much less efficient phosphorylation of Pnt-P2 by MAPK/Rolled, as shown by kinase assays in (C). (D) The addition of Yan-SAM-A86R, which can compete with Pnt-P2-SAM for Mae-SAM, led to the increasing phosphorylation of Pnt-P2-SAM by Rolled. Yan-SAM-A86R has no effect on Pnt-P2-SAM alone. (E) Mae-SAM has little effect on the phosphorylation of a heterologous substrate. Myelin basic protein was incubated with Rolled kinase and increasing concentrations of MBP-Mae-SAM. (F) Luciferase reporter assays showing that the specific interaction between Mae-SAM and Pnt-P2-SAM inhibits Ras induced transactivation.
Mae inhibits the phosphorylation of Pnt-P2 by blocking its docking surface for MAPK/Rolled
To evaluate the effect of Mae-SAM on the phosphorylation of Pnt-P2-SAM by Rolled, we performed in vitro kinase assays comparing the phosphorylation efficiency of Pnt-P2-SAM in the presence and absence of stoichiometric Mae-SAM. The Pnt-P2-SAM/Mae-SAM complex was obtained by coexpression in E. coli, followed by purification through a gel filtration column as a 1:1 complex. The proper folding of the complex was further validated by CD spectroscopy (Supplementary Figure S2). The kinase assays unambiguously showed that Mae-SAM significantly diminished the phosphorylation of Pnt-P2-SAM by Rolled (Figure 4C). Moreover, when we added increasing amounts of Yan-SAM-A86R, which should compete for the binding to the ML surface of Mae-SAM and release Pnt-P2-SAM monomer for phosphorylation, increasing phosphorylation of Pnt-P2-SAM was observed (Figure 4D). This result indicates that Pnt-P2-SAM is competent for phosphorylation in these preparations. As an additional control, the addition of Yan-SAM-A86R did not affect Pnt-P2-SAM phosphorylation in the absence of Mae-SAM (Figure 4D). We also tested whether Mae-SAM could act by directly inhibiting the kinase by examining the effects of Mae-SAM on the phosphorylation of a heterologous substrate, myelin basic protein. At concentrations more than 10-fold higher than the kinase and nearly five-fold over the Kd for Mae binding to Pnt-P2, we see little effect on kinase activity (Figure 4E). These experiments strongly support the conclusion that the EH surface of Pnt-P2-SAM acts as docking surface for the Rolled/MAPK and the specific interaction between the ML surface of Mae-SAM and the EH surface of Pnt-P2-SAM interferes with the phosphorylation of Pnt-P2-SAM by Rolled.
Negative regulation of Pnt-P2 transcription activity by Mae
To study the functional importance of the Mae-SAM/Pnt-P2-SAM interaction in vivo, and the consequences of Mae-SAM mediated blocking of the MAPK/Rolled docking site on Pnt-P2, we examined the abilities of the wild type and the mutant Mae proteins to regulate Ras-dependent Pnt-P2 activated transcription of the EBS-luciferase reporter construct. We found that in Drosophila S2 cells, expression of myc-tagged wild-type Mae or Mae with a point mutation on the EH surface of its SAM domain (Mae M160E) inhibited Pnt-P2 mediated transactivation (Figure 4F). Mae A141D, which bears a mutation rendering it incapable of interacting with Pnt-P2-SAM, had no effect on Pnt-P2 mediated transactivation (Figure 4F). These three versions of myc-tagged Mae were all well expressed as shown by Western blots (data not shown).
Discussion
Regulation of Pnt-P2 by Mae
Overexpression of Mae in the fly visual system results in loss of eye tissue, mimicking the loss of Pnt-P2 phenotype (Vivekanand et al, 2004). This phenotype, however, could be fully suppressed by coexpression of Pnt-P2, suggesting that Mae negatively regulates Pnt-P2 function (Vivekanand et al, 2004). Our work reveals the biochemical mechanism underlying the genetic interactions between Mae and Pnt-P2: the binding of Mae to Pnt-P2 specifically blocks the Rolled docking surface on Pnt-P2 and consequently inhibits Pnt-P2 phosphorylation. We cannot rule out the possibility that Mae binding to Pnt-P2 has additional consequences for Pnt-P2 function in the cell, however, perhaps by blocking interactions with other proteins.
A unified model for Mae, Yan and Pnt-P2
Our findings in this work, combined with prior results, suggest how Yan, Pnt-P2 and Mae work together to determine cell fate in response to the activation of the RTK pathways (Voas and Rebay, 2004; Tootle and Rebay, 2005). As illustrated in Figure 5A, in the absence of MAPK activation, unphosphorylated Yan polymers outcompete Pnt-P2 for access to ETS-binding sites, creating a repressed state of the target genes. Upon RTK activation (Figure 5B), activated phospho-Rolled MAPK enters the nucleus (Khokhlatchev et al, 1998) and phosphorylates a small amount of the monomeric Yan. By binding to Yan and blocking polymer interactions, basal levels of Mae likely help to maintain an appropriate concentration of free Yan in the nucleus (Song et al, 2005). Phosphorylation of Yan triggers its cytoplasmic export with the help of CRM1 (Tootle et al, 2003). The decrease in free Yan then drives the equilibrium away from the DNA-bound polymer. Meanwhile, the antagonist of Yan, Pnt-P2, becomes activated by Rolled MAPK phosphorylation, presumably through enhanced binding to transcriptional coactivators CREB binding protein (CBP) and p300, which act to bridge the DNA-bound transcription factors and the basal transcription complex (Foulds et al, 2004). As Mae is regulated by Yan and Pnt-P2 (Vivekanand et al, 2004), inactivation of Yan and activation of Pnt-P2 leads to increasing amounts of Mae and further removal of Yan repression. These processes could easily lead to runaway expression of differentiation genes. As revealed in this study, however, another job of Mae is to block the MAPK/Rolled docking site on Pnt-P2, inhibiting Pnt-P2 phosphorylation, which in turn attenuates transcriptional activity. This negative feedback loop ensures a level of transcription appropriate for normal development.
Figure 5.
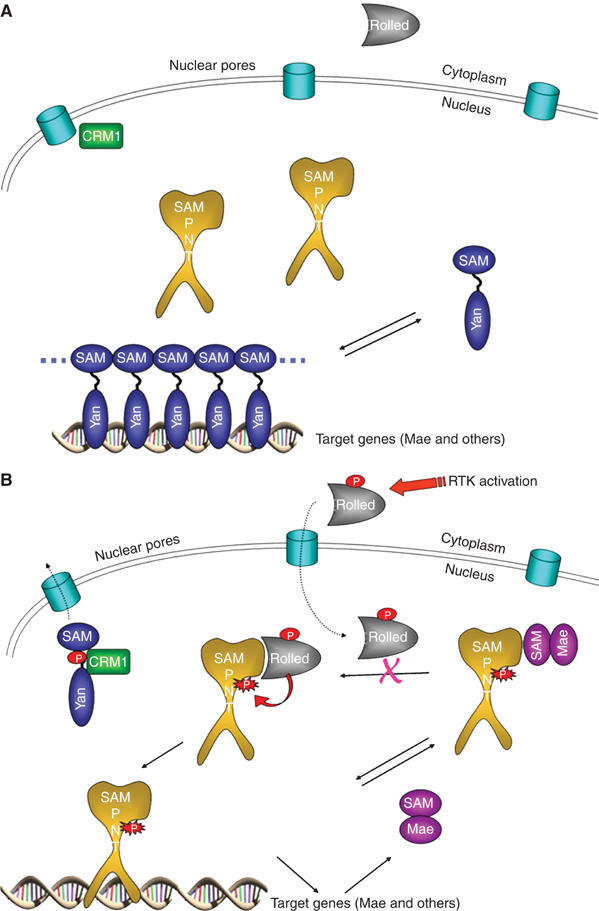
A unified model for Mae, Yan, and Pnt-P2 regulation of gene transcription. (A) The state prior to RTK stimulation. (B) The state after RTK stimulation. See text for details.
The kinase docking surface in Pnt-P2-SAM
In the last decade, several short amino-acid sequence motifs have been found to be MAPK docking sites based on studies of different transcription factors (Holland and Cooper, 1999; Jacobs et al, 1999; Biondi and Nebreda, 2003; Yang et al, 2003). Most of these docking sites belong to two main groups: ‘LXL' and ‘FXF' motifs (Lee et al, 2004). The characteristic hydrophobic residues in these motifs were found to be indispensable for the interaction between the kinases and their substrate proteins. These sequence signatures have been used to identify MAP kinase substrates. In this study, we showed that the EH surface of Pnt-P2 SAM domain greatly enhances the phosphorylation of a distal Thr residue by MAPK/Rolled in vitro and therefore is essential for Pnt-P2 mediated gene activation in response to Ras signaling in vivo. The docking surface on Pnt-P2-SAM is noteworthy because it appears to be constructed by residues that are distant in sequence but in close proximity in the folded protein structure. Residues spanning almost 50 amino acids can form a three-dimensional kinase docking surface covering about 1/3 of the surface area of a protein domain.
Fine tuning MAPK signaling
To our knowledge, this is the first example of a transcriptional regulator functioning by blocking a kinase docking site on a substrate. A different form of docking site blocking has been reported, in which the PKR kinase can be inhibited by a virally encoded protein that binds to the substrate recognition surface of the kinase itself (Dar and Sicheri, 2002; Dey et al, 2005). This mechanism will inhibit phosphorylation of all substrates that interact with that surface of the kinase, however. Blocking the docking surface on the substrate, as reported here, provides a highly targeted regulatory mechanism. Specific interaction between the ML surface of Mae-SAM with the EH surface of Pnt-P2-SAM blocks the MAPK/Rolled docking site on Pnt-P2, which inhibits phosphorylation and therefore the transcriptional activation activity of Pnt-P2. This mechanism adds another layer of regulation to the MAPK pathway. In addition to controlling the overall level of kinase activity, and the targeting of the kinase to specific sites, cells also have a mechanism to adjust the phosphorylation activity at specific proteins in the context of a particular level of MAPK activity. The profound loss of Pnt-P2 activity that occurs when the kinase docking site is occluded, combined with the relatively modest affinity of the kinase-docking site interactions, suggests that it may be possible to develop small molecules that bind to the docking site and block phosphorylation. These inhibitors could serve as highly specific drugs because they would not affect the normal, global functions of the kinase.
Materials and methods
Sequence alignment and homology modeling
Multiple sequence alignments were performed using PILEUP in the GCG software package. For homology modeling, the NMR structure of ETS-1 SAM domain (Slupsky et al, 1998) was used as a template to generate a Pointed-P2-SAM coordinate model using SWISS-MODEL (Schwede et al, 2003). Structure alignment and superposition were carried out using the incremental combinatorial extension (CE) algorithm (Shindyalov and Bourne, 1998). Ribbon and surface representations were generated using Pymol (www.pymol.org).
Plasmid constructs and protein preparation
The coding sequences for the wild-type Pnt-P2 SAM domain (residues 142–249) and Mae SAM domain (residues 94–173) were PCR-amplified from a D. melanogaster cDNA library and then cloned into a modified pET-3c vector (Novagen). The expressed protein sequences include an additional N-terminal ME leader sequence and C-terminal His6-tag. All mutants of the Pointed-P2 SAM domain and the Mae SAM domain were prepared using the QuikChange Site-Directed Mutagenesis Kit (Stratagene) and verified by DNA sequencing. For in vitro biochemical analysis, the wild type and mutant, Pointed-P2-SAM or Mae-SAM were expressed as GST-fusion proteins using pGEX-3T (Pharmacia) or MBP-fusion proteins using pETM-40 (gift from Dr Jeanne Perry, UCLA). Both GST and MBP fusions also have C-terminal His6-tags.
All the proteins were expressed in BL21-CodonPlus(DE3)-RP cells. The SAM domain proteins were purified from the soluble fraction of cell lysates using Ni-NTA (Qiagen) affinity column followed by cation exchange chromatography on a HiTrap SP column (Pharmacia) as described for other SAM domain proteins (Kim et al, 2001, 2002). Electrospray ionization mass spectrometry was used to verify the identity of each purified protein. MBP-Mae-SAM wild type and its mutants were prepared as described previously (Qiao et al, 2004).
To express the activated form of Rolled, a coexpression system for MEK1R4F, a constitutively active mutant of human MEK1, and ERK2 (Khokhlatchev et al, 1997) was modified by replacing ERK2 gene with the Rolled (residue 18–376) gene. The Rolled kinase was expressed and purified according to the method described for ERK2 (Canagarajah et al, 1997). Dual-phosphorylation of Rolled was confirmed by Western blot analysis using an anti-dual-phosphorylated ERK2 antibody (Cell Signaling Technology).
In vitro kinase assays
The kinase assays were carried out in 20 μl reaction volumes with 1.5 nM Rolled in kinase buffer (50 mM Tris–HCl pH 7.5, 10 mM MgCl2, 1 mM EGTA, 2 mM DTT and 0.01% Brij 35). Reactions were initiated by the addition of [32P]ATP to a final concentration of 2 mM. After 15 min at 30°C, the reactions were terminated by the addition of 4 × SDS sample buffer and boiled at 95°C for 3 min. The phosphorylation of substrate proteins was examined by using a Phosphorimager (Molecular Dynamics) following SDS–PAGE, and quantified using ImageQuant software.
For the competition assays, 20 μM of Pnt-P2-SAM domain with an intact phospho-acceptor site were incubated for 30 min in kinase buffer with the Rolled kinase as well as 0, 15, 30, 60 and 120 μM of competing Pnt-P2-SAM domains variants with the phospho-acceptor region deleted. After the incubation, the reactions were initiated by adding [32P]ATP to a final concentration of 2 mM.
Far-UV circular dichroism
CD spectra were recorded using a JASCO-J720 spectrapolarimeter using a 1 mm cuvette at room temperature. All the protein samples were diluted to the same concentration (0.25 mg/ml) with water. Spectra were acquired by averaging five scans from 250 to 190 nm in 0.5 nm steps.
Surface plasmon resonance
The surface plasmon resonance experiments for the Pnt-P2-SAM or Mae-SAM mutants were performed using a Biacore × at 20°C in 10 mM HEPES, pH 7.0, 150 mM NaCl, 3 mM EDTA and 0.005% Surfactant P20. Proteins were immobilized on a Biacore Pioneer CM5 sensor chip and various concentrations of the mobile phase proteins were applied to the chip. The resulting binding data were analyzed with the BIAevaluation 3.0 software.
Luciferase reporter assays
Drosophila S2 cells were transfected with an EBS-luciferase reporter, which contains six tandem copies of the ETS-binding site upstream of the luciferase coding sequence. CuSO4 inducible pRMHa-3 vectors encoding wild type or mutated forms of Pnt-P2 or Mae were cotransfected in various combinations. A pAC5.1-lacZ reporter was cotransfected as an internal control. Cells were harvested two days after 0.5 mM CuSO4 induction. The luciferase and the β-galactosidase activities were measured as previously described (Tootle et al, 2003).
Supplementary Material
Supplementary Figures
Acknowledgments
We thank I Rebay for reagents for the luciferase assays, MH Cobb and BJ Graves for the MEK1/ERK2 coexpression plasmid, MH Nie for help with cell culture, and Mary Jane Budny for comments on the manuscript. JUB is a Leukemia and Lymphoma Society Scholar. This work was supported by NIH Grants RO1-CA081000 to JUB and RO1-GM44522 to AJC.
References
- Baker DA, Mille-Baker B, Wainwright SM, Ish-Horowicz D, Dibb NJ (2001) Mae mediates MAP kinase phosphorylation of Ets transcription factors in Drosophila. Nature 411: 330–334 [DOI] [PubMed] [Google Scholar]
- Biondi RM, Nebreda AR (2003) Signalling specificity of Ser/Thr protein kinases through docking-site-mediated interactions. Biochem J 372: 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunner D, Ducker K, Oellers N, Hafen E, Scholz H, Klambt C (1994) The ETS domain protein pointed-P2 is a target of MAP kinase in the sevenless signal transduction pathway. Nature 370: 386–389 [DOI] [PubMed] [Google Scholar]
- Canagarajah BJ, Khokhlatchev A, Cobb MH, Goldsmith EJ (1997) Activation mechanism of the MAP kinase ERK2 by dual phosphorylation. Cell 90: 859–869 [DOI] [PubMed] [Google Scholar]
- Dar AC, Sicheri F (2002) X-ray crystal structure and functional analysis of vaccinia virus K3L reveals molecular determinants for PKR subversion and substrate recognition. Mol Cell 10: 295–305 [DOI] [PubMed] [Google Scholar]
- Dey M, Trieselmann B, Locke EG, Lu J, Cao C, Dar AC, Krishnamoorthy T, Dong J, Sicheri F, Dever TE (2005) PKR and GCN2 kinases and guanine nucleotide exchange factor eukaryotic translation initiation factor 2B (eIF2B) recognize overlapping surfaces on eIF2alpha. Mol Cell Biol 25: 3063–3075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foulds CE, Nelson ML, Blaszczak AG, Graves BJ (2004) Ras/mitogen-activated protein kinase signaling activates Ets-1 and Ets-2 by CBP/p300 recruitment. Mol Cell Biol 24: 10954–10964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez FA, Raden DL, Davis RJ (1991) Identification of substrate recognition determinants for human ERK1 and ERK2 protein kinases. J Biol Chem 266: 22159–22163 [PubMed] [Google Scholar]
- Holland PM, Cooper JA (1999) Protein modification: docking sites for kinases. Curr Biol 9: R329–R331 [DOI] [PubMed] [Google Scholar]
- Jacobs D, Glossip D, Xing H, Muslin AJ, Kornfeld K (1999) Multiple docking sites on substrate proteins form a modular system that mediates recognition by ERK MAP kinase. Genes Dev 13: 163–175 [PMC free article] [PubMed] [Google Scholar]
- Khokhlatchev A, Xu S, English J, Wu P, Schaefer E, Cobb MH (1997) Reconstitution of mitogen-activated protein kinase phosphorylation cascades in bacteria. Efficient synthesis of active protein kinases. J Biol Chem 272: 11057–11062 [DOI] [PubMed] [Google Scholar]
- Khokhlatchev AV, Canagarajah B, Wilsbacher J, Robinson M, Atkinson M, Goldsmith E, Cobb MH (1998) Phosphorylation of the MAP kinase ERK2 promotes its homodimerization and nuclear translocation. Cell 93: 605–615 [DOI] [PubMed] [Google Scholar]
- Kim CA, Bowie JU (2003) SAM domains: uniform structure, diversity of function. Trends Biochem Sci 28: 625–628 [DOI] [PubMed] [Google Scholar]
- Kim CA, Gingery M, Pilpa RM, Bowie JU (2002) The SAM domain of polyhomeotic forms a helical polymer. Nat Struct Biol 9: 453–457 [DOI] [PubMed] [Google Scholar]
- Kim CA, Phillips ML, Kim W, Gingery M, Tran HH, Robinson MA, Faham S, Bowie JU (2001) Polymerization of the SAM domain of TEL in leukemogenesis and transcriptional repression. EMBO J 20: 4173–4182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CA, Sawaya MR, Cascio D, Kim W, Bowie JU (2005) Structural organization of a sex-comb-on-midleg/polyhomeotic copolymer. J Biol Chem 280: 27769–27775 [DOI] [PubMed] [Google Scholar]
- Klambt C (1993) The Drosophila gene pointed encodes two ETS-like proteins which are involved in the development of the midline glial cells. Development 117: 163–176 [DOI] [PubMed] [Google Scholar]
- Lai ZC, Rubin GM (1992) Negative control of photoreceptor development in Drosophila by the product of the yan gene, an ETS domain protein. Cell 70: 609–620 [DOI] [PubMed] [Google Scholar]
- Lee T, Hoofnagle AN, Kabuyama Y, Stroud J, Min X, Goldsmith EJ, Chen L, Resing KA, Ahn NG (2004) Docking motif interactions in MAP kinases revealed by hydrogen exchange mass spectrometry. Mol Cell 14: 43–55 [DOI] [PubMed] [Google Scholar]
- Mackereth CD, Scharpf M, Gentile LN, MacIntosh SE, Slupsky CM, McIntosh LP (2004) Diversity in structure and function of the Ets family PNT domains. J Mol Biol 342: 1249–1264 [DOI] [PubMed] [Google Scholar]
- O'Neill EM, Rebay I, Tjian R, Rubin GM (1994) The activities of two Ets-related transcription factors required for Drosophila eye development are modulated by the Ras/MAPK pathway. Cell 78: 137–147 [DOI] [PubMed] [Google Scholar]
- Qiao F, Bowie JU (2005) The many faces of SAM. Sci STKE 2005: re7. [DOI] [PubMed] [Google Scholar]
- Qiao F, Song H, Kim CA, Sawaya MR, Hunter JB, Gingery M, Rebay I, Courey AJ, Bowie JU (2004) Derepression by depolymerization: structural insights into the regulation of Yan by Mae. Cell 118: 163–173 [DOI] [PubMed] [Google Scholar]
- Rainey MA, Callaway K, Barnes R, Wilson B, Dalby KN (2005) Proximity-induced catalysis by the protein kinase ERK2. J Am Chem Soc 127: 10494–10495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachander R, Bowie JU (2004) SAM domains can utilize similar surfaces for the formation of polymers and closed oligomers. J Mol Biol 342: 1353–1358 [DOI] [PubMed] [Google Scholar]
- Rebay I (2002) Keeping the receptor tyrosine kinase signaling pathway in check: lessons from Drosophila. Dev Biol 251: 1–17 [DOI] [PubMed] [Google Scholar]
- Rebay I, Rubin GM (1995) Yan functions as a general inhibitor of differentiation and is negatively regulated by activation of the Ras1/MAPK pathway. Cell 81: 857–866 [DOI] [PubMed] [Google Scholar]
- Robinson MJ, Harkins PC, Zhang J, Baer R, Haycock JW, Cobb MH, Goldsmith EJ (1996) Mutation of position 52 in ERK2 creates a nonproductive binding mode for adenosine 5′-triphosphate. Biochemistry 35: 5641–5646 [DOI] [PubMed] [Google Scholar]
- Schwede T, Kopp J, Guex N, Peitsch MC (2003) SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res 31: 3381–3385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidel JJ, Graves BJ (2002) An ERK2 docking site in the pointed domain distinguishes a subset of ETS transcription factors. Genes Dev 16: 127–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharrocks AD (2001) The ETS-domain transcription factor family. Nat Rev Mol Cell Biol 2: 827–837 [DOI] [PubMed] [Google Scholar]
- Sharrocks AD, Yang SH, Galanis A (2000) Docking domains and substrate-specificity determination for MAP kinases. Trends Biochem Sci 25: 448–453 [DOI] [PubMed] [Google Scholar]
- Shindyalov IN, Bourne PE (1998) Protein structure alignment by incremental combinatorial extension (CE) of the optimal path. Protein Eng 11: 739–747 [DOI] [PubMed] [Google Scholar]
- Slupsky CM, Gentile LN, Donaldson LW, Mackereth CD, Seidel JJ, Graves BJ, McIntosh LP (1998) Structure of the Ets-1 pointed domain and mitogen-activated protein kinase phosphorylation site. Proc Natl Acad Sci USA 95: 12129–12134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song H, Nie M, Qiao F, Bowie JU, Courey AJ (2005) Antagonistic regulation of Yan nuclear export by Mae and Crm1 may increase the stringency of the Ras response. Genes Dev 19: 1767–1772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan PB, Kim SK (1999) Signaling specificity: the RTK/RAS/MAP kinase pathway in metazoans. Trends Genet 15: 145–149 [DOI] [PubMed] [Google Scholar]
- Tootle TL, Lee PS, Rebay I (2003) CRM1-mediated nuclear export and regulated activity of the Receptor Tyrosine Kinase antagonist YAN require specific interactions with MAE. Development 130: 845–857 [DOI] [PubMed] [Google Scholar]
- Tootle TL, Rebay I (2005) Post-translational modifications influence transcription factor activity: a view from the ETS superfamily. Bioessays 27: 285–298 [DOI] [PubMed] [Google Scholar]
- Vivekanand P, Tootle TL, Rebay I (2004) MAE, a dual regulator of the EGFR signaling pathway, is a target of the Ets transcription factors PNT and YAN. Mech Dev 121: 1469–1479 [DOI] [PubMed] [Google Scholar]
- Voas MG, Rebay I (2004) Signal integration during development: insights from the Drosophila eye. Dev Dyn 229: 162–175 [DOI] [PubMed] [Google Scholar]
- Yamada T, Okabe M, Hiromi Y (2003) EDL/MAE regulates EGF-mediated induction by antagonizing Ets transcription factor Pointed. Development 130: 4085–4096 [DOI] [PubMed] [Google Scholar]
- Yang SH, Sharrocks AD, Whitmarsh AJ (2003) Transcriptional regulation by the MAP kinase signaling cascades. Gene 320: 3–21 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures