

Abstract
Objective:
To assess the effects of inhibiting both tumor necrosis factor (TNF)-α production and xanthine oxidase activity on the inflammatory response, mitogen-activated protein kinase (MAPK) activation and mortality in necrotizing acute pancreatitis in rats.
Summary Background Data:
Pancreatic injury triggers 2 major pathways involved in the systemic effects of severe acute pancreatitis: pro-inflammatory cytokines and oxidative stress.
Methods:
Pancreatitis was induced by intraductal infusion of 3.5% sodium taurocholate. We examined whether treatment with oxypurinol, a specific inhibitor of xanthine oxidase, and/or pentoxifylline, an inhibitor of TNF-α production, affects pancreatic damage, ascites, lung inflammation, and MAPK phosphorylation.
Results:
Oxypurinol prevented p38 phosphorylation in the pancreas and partially avoided the rise in lung myeloperoxidase activity. Pentoxifylline prevented erk 1/2 and JNK phosphorylation in the pancreas, and it partially reduced ascites and the rise in lung myeloperoxidase activity. Combined treatment with oxypurinol and pentoxifylline almost completely abolished ascites, MAPK phosphorylation in the pancreas, and the increase in lung myeloperoxidase activity. Histology revealed a reduction in pancreatic and lung damage. These changes were associated with a significant improvement of survival.
Conclusions:
Simultaneous inhibition of TNF-α production and xanthine oxidase activity greatly reduced local and systemic inflammatory response in acute pancreatitis and decreased mortality rate. These effects were associated with blockade of the 3 major MAPKs.
Pancreatic injury triggers two major pathways involved in systemic effects of acute pancreatitis: pro-inflammatory cytokines and oxidative stress. Simultaneous inhibition of TNF-α production and xanthine oxidase greatly reduced local and systemic inflammatory response in acute pancreatitis, and decreased mortality rate. These effects were associated with blockade of the three MAP kinases.
Systemic complications are the most important contributors to multiple organ failure and death during the first stages of severe acute pancreatitis (AP).1 The relationship between pancreatic injury and this uncontrolled systemic response is not completely understood. However, experimental and clinical evidence has shown the involvement of different mechanisms triggering the systemic response in AP: cytokines,2 platelet activating factor,3 oxygen free radicals,4,5 proteolytic enzymes,6 phospholipase A2,7 and the complement system.8 Among them, pro-inflammatory cytokines and oxidative stress seem to be critically involved in the development of local and systemic complications associated with severe AP.9,10
Serum levels of pro-inflammatory cytokines, such as TNF-α, IL-1β, and IL-6, increase during the course of AP, and these levels appear to be correlated with the severity of pancreatic inflammation.11–15 TNF-α may be an initiator of the cytokine cascade, because it induces the synthesis and release of other cytokines and the activation of alveolar macrophages.16 Pretreatment with an antibody against TNF-α or blockade of TNF-α production with pentoxifylline ameliorates experimental AP.17–19 Moreover, in knockout mice deficient in TNF-α receptors, the rate of mortality due to necrotizing AP decreased because the systemic response was restrained.10
The role of oxidative stress in AP has been evidenced indirectly by the beneficial effects of antioxidants20–24 as well as directly by pancreatic glutathione depletion and increased lipid peroxidation.22,23 Furthermore, circulating xanthine oxidase (XO) released by the damaged pancreas acts as a source of systemic oxidative stress contributing to lung inflammation through a mechanism mediated by up-regulation of P-selectin.25
Therefore, pancreatic injury seems to trigger at least 2 different pathways: pro-inflammatory cytokines and oxidative stress; both are involved in the systemic effects of AP. Those treatments blocking exclusively 1 of these pathways have shown only slight beneficial effects in experimental AP. Nevertheless, combined treatment by simultaneous blocking of both pathways has not been tested so far.
Mitogen-activated protein kinase (MAPK) signaling cascades are induced by pro-inflammatory cytokines and stress stimuli, mediating most of their effects on the immune response and cell death.26–28 Three major families of MAPKs have been found so far in mammalian cells: p38, extracellular signal-regulated kinase (erk 1/2), and c-Jun NH2-terminal protein kinase (JNK). Cholecystokinin and other pancreatic secretagogues, such as cerulein, activate all these MAPKs in the pancreas.29–32 Among them, p38 has been involved in the severity of pancreatitis and in the respiratory distress syndrome associated with AP. Indeed, inhibition of p38 decreased pancreatic and pulmonary injury in severe AP in rats.33–34
The aim of the present study was to assess the effects of simultaneous inhibition of TNF-α production and XO activity on the pancreatic and systemic inflammatory response and MAPK activation in necrotizing AP in rats.
MATERIALS AND METHODS
Experimental Model
Male Wistar rats (250–300 g body weight) were used. All animals received human care according to the criteria outlined in the “Guide for the Care and Use of Laboratory Animals” prepared by the National Academy of Sciences and published by the National Institutes of Health (NIH publication 86-23, revised 1985). The Research Committee of the School of Medicine (University of Valencia, Spain) approved the study protocol. They were fed on a standard laboratory diet and tap water ad libitum and were subjected to a 12 hours light-dark cycle.
Animals were anesthetized with an intraperitoneal administration of ketamine (80 mg/kg body weight) and acepromazine (2.5 mg/kg body weight). The biliopancreatic duct was cannulated through the duodenum, and the hepatic duct was closed by a small bulldog clamp. Pancreatitis was induced by retrograde injection into the biliopancreatic duct of sodium taurocholate (3.5%; Sigma, St Louis, MO) in a volume of 0.1 mL/100 g body weight using an infusion pump (Harvard Instruments).
Study Design
In a first series of experiments, the development of local and systemic effects in this particular model of experimental AP was investigated to determine a single time point for assessing the effects of treatments. Therefore, the time course of the following inflammatory and oxidative stress parameters was performed: serum TNF-α, lung myeloperoxidase (MPO) activity, and plasma XO activity. Samples were obtained at 0, 1, 3, 6, and 9 hours after intraductal infusion of taurocholate, immediately frozen and maintained at −80° until assayed. Blood samples were obtained from the inferior cava vein by direct puncture. For histologic studies, pieces of the lung and of the central body of pancreas were also obtained. Serum lipase activity was measured to confirm the appropriate induction of pancreatitis.
In the second series of experiments, we assessed the effects of treatment with oxypurinol, as an inhibitor of XO, and pentoxifylline, as an inhibitor of TNF-α production, at a time point when the inflammatory process was firmly established in both pancreas and lung. This time point was selected according to results of the first series of experiments. Treatments were administered immediately after taurocholate infusion. Animals were distributed in the following groups:
Control (C): Infusion of 0.9% NaCl into the biliopancreatic duct (0.1 mL/100 g) and into the femoral vein (0.066 mL/min for 30 minutes).
AP: Infusion of 3.5% sodium taurocholate into the biliopancreatic duct and saline solution into the femoral vein (0.066 mL/min for 30 minutes).
AP + Oxypurinol (O): Infusion of 3.5% sodium taurocholate into the biliopancreatic duct and 5 mM oxypurinol into the femoral vein (0.066 mL/min for 30 minutes).
AP + Pentoxifylline (P): Infusion of 3.5% sodium taurocholate into the biliopancreatic duct and pentoxifylline (12 mg/kg body weight) into the femoral vein (0.066 mL/min for 30 minutes).
AP + Oxypurinol + Pentoxifylline (OP): Infusion of 3.5% sodium taurocholate into the biliopancreatic duct and 5 mM oxypurinol and pentoxifylline (12 mg/kg body weight) into the femoral vein (0.066 mL/min for 30 minutes).
Volume of ascites as well as all the same parameters studied in the first series of experiments were measured.
In the third series of experiments, we assessed the effects of treatment with oxypurinol and pentoxifylline on phosphorylation of the 3 major families of MAPKs, ie, p38, erk 1/2, and JNK, as an index of their activation. Animals were distributed in the same groups as the previous series.
Eventually, a fourth series of experiments was performed to evaluate if the treatment showing maximal response was able to reduce mortality in our model of AP. Hence, the mortality rate was determined in 2 groups of animals (30 rats each one), ie, AP and AP plus treatment.
Assays
Lipase Activity
Serum lipase activity was determined by the LIPASE-PS kit (Sigma Diagnostics, Lyon) according to the supplier's specifications.
Measurement of Ascites
Ascites was evaluated by measuring the liquid removed from the peritoneal cavity. This procedure was performed immediately after re-laparotomy, just before obtaining blood and tissue samples.
Total TNF-α
Total tumor necrosis factor (TNF)-α levels were measured in serum using the ChemiKine TNF-α EIA Kit (Chemicon International, Temecula, CA) that measures both TNF-α free and bound to proteins.
Myeloperoxidase
MPO was measured photometrically using 3,3′,5,5′-tetramethylbenzidine as a substrate.35 Samples were homogenized with 0.5% hexadecyl-trimethylammonium bromide in 50 mM phosphate buffer, pH 6.0. Homogenates where then processed as previously described by Closa et al.16 Enzyme activity was assessed photometrically at 630 nm. The assay mixture consisted of 20 mL of supernatant, 10 mL of tetramethylbenzidine (final concentration, 1.6 mM) dissolved in DMSO, and 70 mL H2O2 (final concentration, 3.0 mM) dissolved in 80 mM phosphate buffer, pH 5.4.
Xanthine Oxidase
Plasma XO activity was measured in plasma by fluorometry as described by Beckman et al.36 This enzyme activity was determined at 37°C using 1 mM pterin as substrate in 50 mM phosphate buffer, pH 7.4, containing 0.1 mM EDTA. Excitation and emission wavelength were 345 nm and 390 nm, respectively.
Phosphorylation of MAPKs
Phosphorylation of p38, erk 1/2 (p42/p44), and JNK were determined by Western blotting and chemiluminescence detection using the Phototope-HRP Detection kit (Cell Signaling Technology, Inc., Beverly, MA). The following antibodies purchased from Cell Signaling Technology were used: Phospho-p38 MAPK (Thr 180/Tyr 182), phospho-p44/42 MAPK (Thr 202/Tyr 204), and phospho-SAPK/JNK (Thr183/Tyr 185) antibodies.
Histologic Studies
Pancreas pieces were rapidly removed and fixed in 10% buffered formalin. The left lung was first perfused by its main bronchus with 10% buffered formalin up to a standard volume, according to the thoracic cavity, to avoid pulmonary collapse; lung pieces were then rapidly removed and placed in 10% buffered formalin. Pancreas and lung pieces were subsequently embedded in paraffin, cut, and stained with hematoxylin and eosin. Assessment of tissue alterations with light microscopy was conducted by an experienced pathologist who was unaware of the treatments used. A combined histologic scoring of severity associated with AP was obtained for pancreas and lung as previously described by Norman et al.37
Statistical Analyses
Results are expressed as mean ± SD with the number of experiments given in parentheses. Statistical analyses was performed in 2 steps. One-way analysis of variance (ANOVA) was performed first. When the overall comparison of groups was significant, differences between individual groups were investigated by the Tukey method. Differences were considered to be significant at P < 0.05.
Statistical analyses of the mortality study were performed with the Kaplan-Meier curve and log-rank test. Differences were considered to be significant at P < 0.05.
RESULTS
Time Course
The time course of inflammatory and oxidative stress parameters was evaluated in taurocholate-induced AP to determine a single time point for assessing the effects of treatments. The appropriate induction of AP was demonstrated by histology and elevation of serum lipase activity (results not shown). Figure 1A shows that serum total TNF-α levels increased at 1 hour after the induction of AP (P = 0.002) and remained high at 3, 6, and 9 hours (P = 0.013, 0.001, and 0.015, respectively). Lung MPO activity was measured as a marker of pulmonary inflammatory response. Figure 1B shows that MPO activity was significantly elevated at 3, 6, and 9 hours after induction (P = 0.049, 0.02, 0.031, respectively). Accordingly, histology showed a marked neutrophil infiltration in the lung.
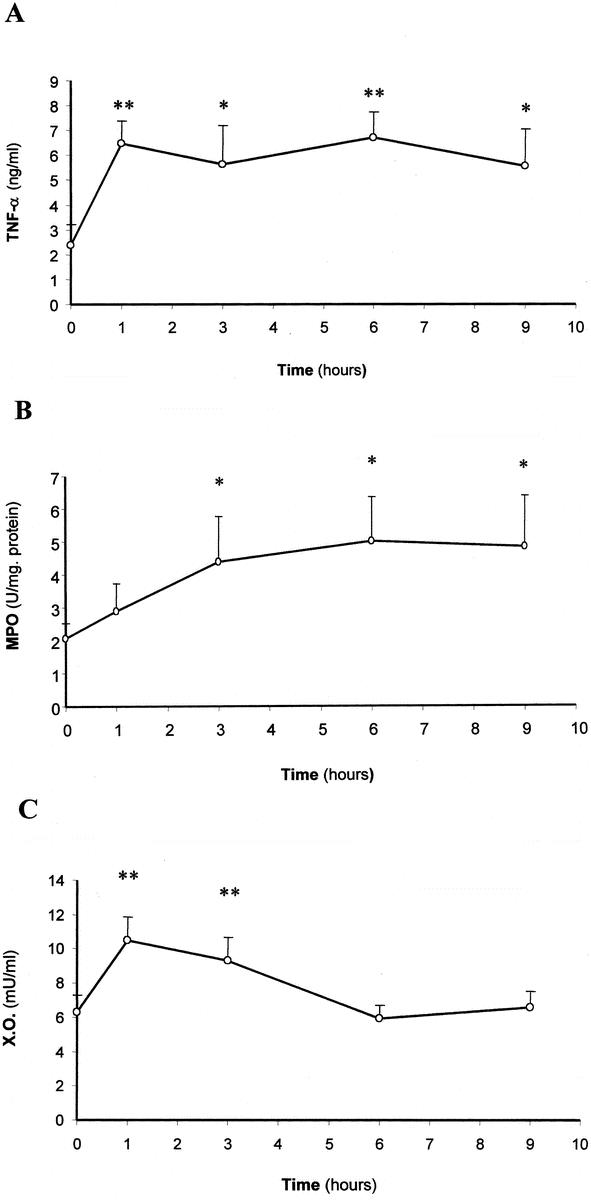
FIGURE 1. Serum TNF-α levels (A), lung MPO activity (B), and plasma XO activity (C) in the course of taurocholate-induced AP in rats. The number of experiments was 3–7. Results are expressed as mean ± SD. Statistical difference is indicated as follows: * P < 0.05, ** P < 0.01 versus 0 hours.
Regarding oxidative stress parameters, XO activity was determined in plasma in the course of AP as an important pro-oxidant mechanism involved in this disease. Figure 1C shows that plasma XO activity was significantly higher at 1 and 3 hours after the induction of AP (P = 0.0001 and 0.008, respectively), whereas it returned to control values at 6 hours and thereafter.
Serum TNF-α levels, lung MPO activity and plasma XO activity were also measured in sham-operated rats 1, 3, 6, and 9 hours after surgery, and they were not significantly different from those obtained in control rats at 0 hours (results not shown).
AP Treatment by Inhibition of XO and TNF-α Production
The effects of AP treatment with oxypurinol, a specific inhibitor of XO, and pentoxifylline, an inhibitor of TNF-α production, were evaluated 6 hours after pancreatitis induction, when the systemic inflammatory process was firmly established in both pancreas and lung.
Oxypurinol administration, either alone or in combination with pentoxifylline, completely inhibited plasma XO activity in our experimental conditions (ie, it was non-detectable). On the other hand, pentoxifylline completely prevented the rise in total TNF-α levels in plasma at 1 hour after AP induction (6.5 ± 0.9 in the AP group versus 2.5 ± 1.3 ng/ml with pentoxifylline; n = 4–5) and at 6 hours (6.7 ± 1.0 versus 2.7 ± 1.6 ng/ml; n = 4). As expected, this level of prevention was also observed with the combined treatment [TNF levels were 1.5 ± 1.0 ng/ml at 1 hour and 1.4 ± 1.0 ng/ml (n = 3) at 6 hours; P = 0.0001 versus the AP group in both cases].
Serum lipase activity increased markedly (22-fold; P < 0.01) at 6 hours after AP induction [47 ± 19 U/L (n = 5) in controls versus 1050 ± 203 U/L (n = 8) in the AP group; P = 0.0001]. This rise was not prevented by either oxypurinol treatment [1062 ± 391 U/L (n = 4)] or pentoxifylline [945 ± 356 U/L (n = 7)] when given alone. However, when rats were treated simultaneously with oxypurinol and pentoxifylline, serum lipase activity was significantly lower, 64% less (P = 0.0001), than in the AP group without therapy. Indeed, serum lipase activity was only 375 ± 82 (n = 5) in rats with AP receiving the combined treatment.
The volume of ascites was 2.9 ± 0.9 mL at 6 hours after AP induction (P = 0.0001 versus the control group), and this was not modified by oxypurinol treatment (Fig. 2 A). However, pentoxifylline administration significantly reduced the volume of ascites by 53% (P = 0.008), and the simultaneous treatment with oxypurinol and pentoxifylline reduced ascites almost completely (90%; P = 0.0001).
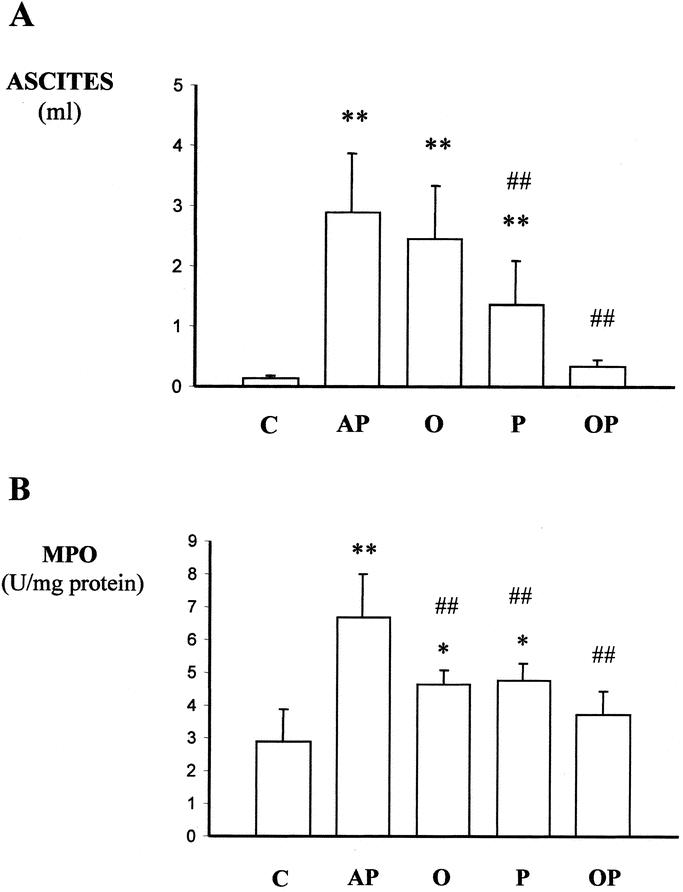
FIGURE 2. Effects of treatment with oxypurinol and pentoxifylline on the volume of ascites (A) and on lung MPO activity (B) 6 hours after induction of AP in rats. Abbreviations: C, control rats; AP, rats with taurocholate-induced AP; O, rats with AP treated with oxypurinol; P, rats with acute pacreatitis treated with pentoxifylline; OP, rats with AP receiving the combined treatment with oxypurinol and pentoxifylline. The number of experiments was 5–7. Results are expressed as mean ± SD. Statistical difference is indicated as follows: * P < 0.05, ** P < 0.01 versus control group; ## P < 0.01 versus taurocholate group.
The effects of treatments on MPO activity in lung are depicted in Figure 2B. While the sole administration of pentoxifylline or oxypurinol did not abolish the increase in this enzyme activity, the combined therapy was able to prevent the increase in lung MPO activity associated with AP (P = 0.0001).
Histology (Table 1; Figs. 3 and 4) showed that administration of oxypurinol or pentoxifylline reduced partially interstitial edema in pancreas as well as polymorphonuclear (PMN) infiltrate in both pancreas and lung. The combined treatment with oxypurinol and pentoxifylline almost completely prevented all the inflammatory features studied, ie, interstitial edema, hyperemia, and PMN infiltrate. It is noteworthy that this combined treatment abolished the enlargement of alveolar walls in the lung as well as the inflammatory infiltrate in the surrounding areas of acinar necrosis (Fig. 3). Consequently, the combined treatment markedly reduced the combined histologic scoring of severity associated with AP both in pancreas and lung (Fig. 4).
TABLE 1. Histology of Pancreas and Lung from Rats Treated With Taurocholate and Oxypurinol Plus Pentoxifylline

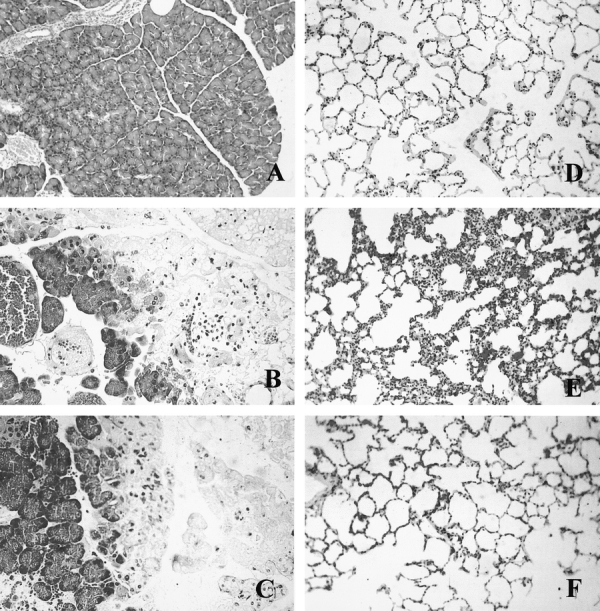
FIGURE 3. Representative photographs of pancreatic and lung histology in control rats (A, D), 6 hours after taurocholate-induced AP (B, E), and in rats with AP receiving combined treatment with oxypurinol and pentoxifylline (C, F).
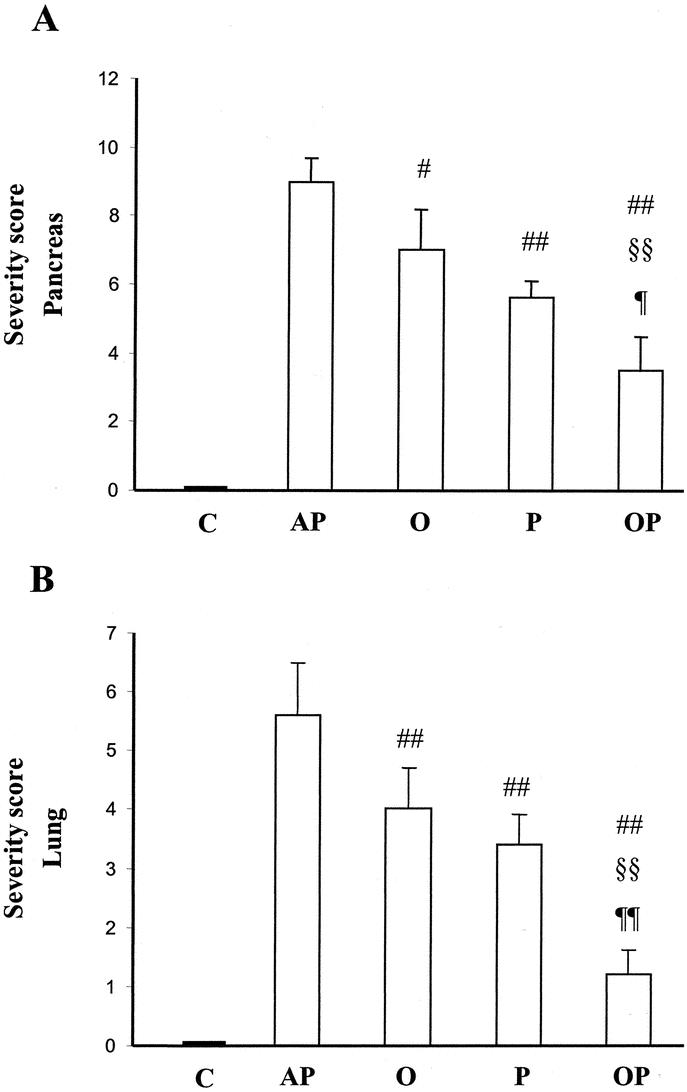
FIGURE 4. Effects of treatment with oxypurinol and pentoxifylline on the combined histologic scoring of severity in AP. The scoring from assessment of acinar necrosis, interstitial edema, and infiltrate of inflammatory cells are combined, as indicated in Methods, to show histologic severity in pancreas (A). The scoring from assessment of PMN infiltrate and hyperemia are combined to show histologic severity in lung (B). C, control rats; AP, rats with taurocholate-induced AP; O, rats with AP treated with oxypurinol; P, rats with acute pancreatitis treated with pentoxifylline; OP, rats with AP receiving the combined treatment with oxypurinol and pentoxifylline. The number of experiments was 4–5. Results are expressed as mean ± SD. Statistical difference is indicated as follows: # P = 0.04, ## P < 0.01 versus taurocholate group; §§ P = 0.001 versus oxypurinol group; ¶ P = 0.04 and ¶¶ P = 0.0001 versus pentoxifylline group.
AP Treatment and MAPK Phosphorylation
The effects of treatments on MAPK phosphorylation in the pancreas were assessed 30 minutes after AP induction, when the maximal phosphorylation of the 3 major MAPKs was found.
Our results show that oxypurinol prevented p38 phosphorylation (P = 0.001) (Fig. 5A), whereas pentoxifylline avoided phosphorylation of JNK (P = 0.001) (Fig. 5B) and erk 1/2 (P = 0.019) (Fig. 5C). The combined treatment with pentoxifylline and oxypurinol led to simultaneous blockade of these 3 MAPKs (P = 0.0001 for p38 and JNK; P = 0.049 for erk) (Figs. 5, A–C).
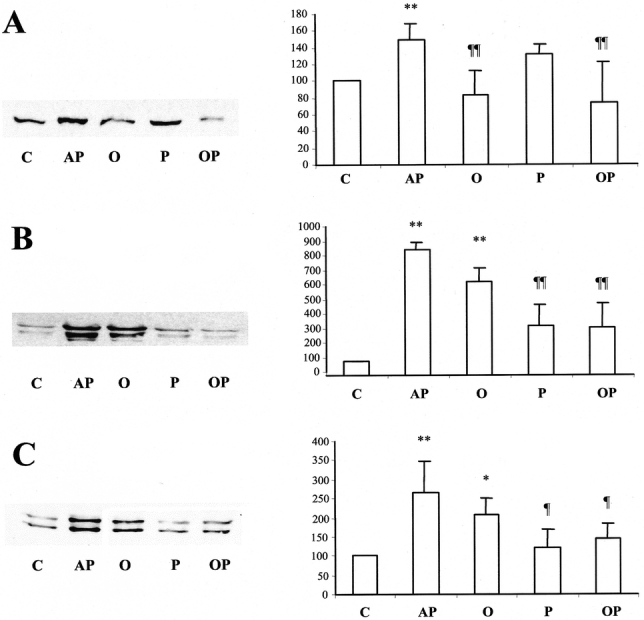
FIGURE 5. Effects of treatment with oxypurinol and pentoxifylline on phosphorylation of p38 (A), JNK (p46/p54) (B), and erk (p42/44) (C) 30 minutes after induction of AP in rats. On the left, representative Western blots of phosphorylated MAPKs are shown. On the right, densitometry histograms of Western blots of phospho-p38 (A), phospho-p46 (B), and phospho-p44 (C) are shown. Abbreviations: JNK, c-Jun NH2-terminal kinase; erk, extracellular signal-regulated kinase; C, control rats; AP, rats with taurocholate-induced AP; O, rats with AP treated with oxypurinol; P, rats with acute pancreatitis treated with pentoxifylline; OP, rats with AP receiving the combined treatment with oxypurinol and pentoxifylline. The number of experiments was 3–4. Results are expressed as mean ± SD. Statistical difference is indicated as follows: * P < 0.05, ** P < 0.01 versus control group; ¶ P < 0.05 and ¶¶ P < 0.01 versus taurocholate group.
Mortality Study
The survival curve shows that the combined treatment increased the survivorship significantly (P = 0.047), at 4 days after taurocholate-induced pancreatitis (Fig. 6). Indeed, the percentage of survival was 80% (n = 30) after taurocholate-induced pancreatitis, whereas it was 96.7% (ie, only 1 rat died out of 30) when the treatment with oxypurinol and pentoxifylline was administered after taurocholate infusion. Hence, this combined treatment significantly reduced the mortality associated with AP in our experimental model.
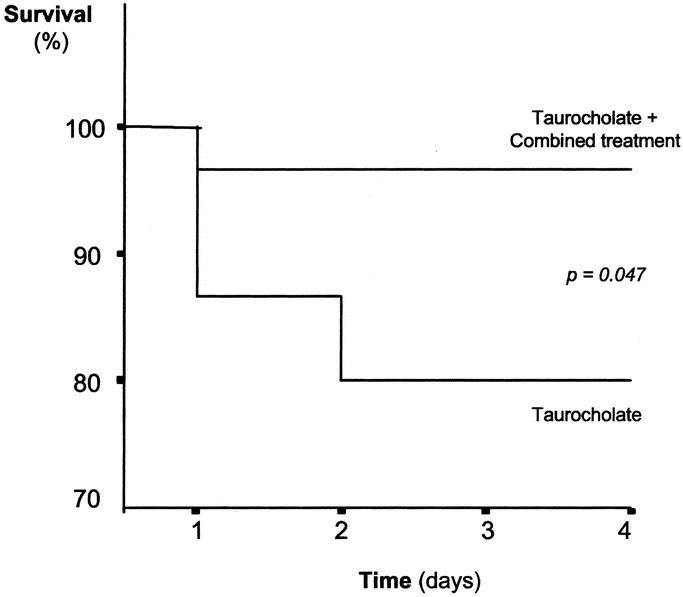
FIGURE 6. Kaplan-Meier survival curve of rats with taurocholate-induced pancreatitis and rats with AP receiving the combined treatment with oxypurinol and pentoxifylline. Thirty rats were used in each group.
DISCUSSION
The role of oxidative stress in the pathogenesis of AP and the potential benefits of antioxidants have been the subjects of numerous studies.22,23 However, recent evidence indicates that oxygen free radicals act as mediators of tissue damage rather than being the initiation event in AP.5
On the other hand, oxygen free radicals have been found to be involved in the systemic response associated with AP, especially in lung inflammation. It has been reported that oxygen free radicals generated by XO mediate the up-regulation of P-selectin in lung during AP,25,38 which triggers leukocyte recruitment.
Regarding the profile of plasma XO activity in the course of AP, we have found that this activity increases initially but decreases toward control values at 6 hours and thereafter. This subsequent reduction in plasma enzyme activity may be caused by XO binding to endothelial cell glycosaminoglycans.39
In the present study, inhibition of XO with oxypurinol has no effect on serum lipase activity, which indicates that free radicals generated by XO have a little effect on this parameter. In contrast, oxypurinol treatment partially reduced the rise in lung MPO activity and PMN infiltration. These results agree with previous reports indicating a role for XO triggering neutrophil infiltration into the lung.4
Much experimental evidence obtained so far demonstrates that pro-inflammatory cytokines, such as TNF-α, are involved in the pathogenesis of AP.9,10 TNF-α is detected in plasma early in the course of AP, and pretreatment of rats with an antibody against TNF-α reduced high serum amylase in AP.17,18 Pentoxifylline exhibits marked anti-inflammatory properties mediated by inhibition of TNF-α production.40–42 Blockade of TNF-α production with pentoxifylline has already been shown to partially prevent glutathione depletion and pancreatic inflammation in cerulein-induced AP.19
In the present study, pentoxifylline completely prevented the increase in serum total TNF-α levels found in taurocholate-induced pancreatitis. This effect was associated with a partial reduction of ascites, lung MPO activity, and inflammatory infiltrate. Nevertheless, inhibition of TNF-α production did not prevent the increase in serum lipase activity. Simultaneous inhibition of XO and TNF-α production with oxypurinol and pentoxifylline abolished the inflammatory changes associated with taurocholate-induced pancreatitis. This combined treatment almost completely prevented ascites as well as the increase in lung MPO activity. In addition, histology revealed a large reduction in the inflammation and damage both in the pancreas and lung when rats received this treatment. It is noteworthy that the combined treatment eventually resulted in a significant decrease in the mortality rate.
Oxidative stress and TNF-α seem to potentiate each other, generating a vicious circle in the course of AP. TNF-α intensifies oxidative stress through different mechanisms: 1) conversion of xanthine dehydrogenase to XO in endothelial cells;43 2) increasing the mitochondrial production of reactive oxygen species;44 and 3) promoting chemotaxis and activation of neutrophils.13 On the other hand, oxidative stress causes activation of MAPK,45–49 which are subsequently responsible for induction of TNF-α production.34,50,51 Thus, the cross-talk between oxidative stress and pro-inflammatory cytokines leads to an amplification of the local and systemic inflammatory response. In accordance, our results establish that blockade of only 1 of these pathways is not enough to achieve an effective treatment.
Oxidative stress and pro-inflammatory cytokines trigger common signal transduction pathways involved in the inflammatory cascade, particularly through activation of MAPK, such as c-Jun N-terminal protein kinase, p38 and extracellular signal-regulated kinases.26,47 We have consequently studied the effects of oxypurinol and/or pentoxifylline on phosphorylation in the 3 major families of MAPKs as an index of their activation. Our results show that oxypurinol reduces taurocholate-induced p38 phosphorylation in the pancreatic tissue, whereas pentoxifylline diminishes taurocholate-induced erk 1/2 and JNK phosphorylation. Therefore, the combined treatment decreased activation of the 3 major MAPK families in pancreas. Accordingly, abrogation of the pancreatic inflammatory process may prevent triggering of the systemic response. Nevertheless, since our treatment is systemic, the blockade of MAPKs might also occur in other tissues, such as lung and liver, thus contributing to block the systemic inflammatory process.
Our results suggest that MAPK signaling cascades might constitute major mechanisms leading to pancreatic and systemic inflammation in AP. The p38 pathway would be related to oxidative stress, while erk 1/2 and JNK pathways would apparently be related to pro-inflammatory cytokines. Thus, an effective prevention of acute inflammation is attained only by a simultaneous blockade of these 3 signaling cascades.
In conclusion, simultaneous inhibition of TNF-α production and XO abolished the link between oxidative stress and pro-inflammatory cytokines that leads to an amplification of the inflammatory response. This combined treatment largely reduced local and systemic effects in AP and decreased the mortality rate. These effects were associated with reduced activation of the 3 major MAPKs. Any potential therapy should be directed toward simultaneous blockade of both pro-inflammatory cytokines and oxidative stress.
ACKNOWLEDGMENTS
The authors thank Mrs. Juana Belloch for her skillful technical assistance and Mrs. Marilyn R. Noyes for revising the manuscript.
Footnotes
Reprints: Dr. Juan Sastre, Department of Physiology, School of Medicine, Avda. Blasco Ibañez 17, 46010 Valencia, Spain. E-mail: juan.sastre@uv.es.
This work was supported by the following Grants: Grant GV01-137 from Generalitat Valenciana, 1FD1997-1616 from Ministerio de Ciencia y Tecnología, and the 1rst Grant for research on exocrine pancreas from Solvay Pharma-Spain funded by Solvay Pharma and sponsored by ANEP to J. S.; and Grant BFI 2001-2849 from CICYT to J.V.
REFERENCES
- 1.Halonen KI, Leppäniemi AK, Puolakkainen PA, et al. Severe acute pancreatitis: Prognosis factors in 270 consecutive patients. Pancreas. 2000;21:266–271. [DOI] [PubMed] [Google Scholar]
- 2.Norman JG, Fink GW, Denham W, et al. Tissue-specific cytokine production during experimental acute pancreatitis: A probable mechanism for distant organ dysfunction. Dig Dis Sci. 1997;42:1783–1788. [DOI] [PubMed] [Google Scholar]
- 3.Hofbauer B, Saluja AK, Bhatia M, et al. Effect of recombinant platelet-activating factor acetylhydrolase on two models of experimental acute pancreatitis. Gastroenterology. 1998;115:1238–1247. [DOI] [PubMed] [Google Scholar]
- 4.Folch E, Gelpí E, Roselló-Catafau J, et al. Free radicals generated by xanthine oxidase mediate pancreatitis-associated organ failure. Dig Dis Sci. 1998;43:2405–2410. [DOI] [PubMed] [Google Scholar]
- 5.Rau B, Poch B, Gansauge F, et al. Pathophysiologic role of oxygen free radicals in acute pancreatitis. Initiating event or mediator of tissue damage? Ann Surg. 2000;231:352–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nakae Y, Naruse S, Kitagawa M, et al. Activation of trypsinogen in experimental models of acute pancreatitis in rats. Pancreas. 1995;10:306–313. [DOI] [PubMed] [Google Scholar]
- 7.Edelson JD, Vadas P, Villar J, et al. Acute lung failure induced by phospholipase A2. Am Rev Resp Dis. 1991;143:1102–1109. [DOI] [PubMed] [Google Scholar]
- 8.Whicher JT, Barnes MP, Brown A, et al. Complement activation and complement control proteins in acute pancreatitis. Gut. 1982;23:944–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Beaux AC, Fearon KCH. Circulating endotoxin, tumor necrosis factor-alpha, and their natural antagonists in the pathophysiology of acute pancreatitis. Scand J Gastroenterol. 1996;31(Suppl 219):43–46. [DOI] [PubMed] [Google Scholar]
- 10.Denham W, Yang J, Fink G, et al. Gene targeting demonstrates additive detrimental effects of interleukin 1 and tumor necrosis factor during pancreatitis. Gastroenterology. 1997;113:1741–1746. [DOI] [PubMed] [Google Scholar]
- 11.Grewal HP, Kotb M, el Din AM, et al. Induction of tumor necrosis factor in severe acute pancreatitis and its subsequent reduction after hepatic passage. Surgery. 1994;115:213–21. [PubMed] [Google Scholar]
- 12.Norman JG, Fink GW, Franz MG. Acute pancreatitis induces intrapancreatic tumor necrosis factor gene expression. Arch Surg. 1995;130:966–970. [DOI] [PubMed] [Google Scholar]
- 13.Kusske AM, Rongione AJ, Reber HA. Cytokines and acute pancreatitis. Gastroenterology. 1996;110:639–642. [DOI] [PubMed] [Google Scholar]
- 14.McKay CJ, Gallagher G, Brooks B, et al. Increased monocyte cytokine production in association with systemic complications in acute pancreatitis. Br J Surg. 1996;83:919–923. [DOI] [PubMed] [Google Scholar]
- 15.Mayer J, Rau B, Gansauge F, et al. Inflammatory mediators in human acute pancreatitis: clinical and pathophysiological implications. Gut. 2000;47:546–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Closa D, Sabater L, Fernandez-Cruz L, et al. Activation of alveolar macrophages in lung injury associated with experimental acute pancreatitis is mediated by the liver. Ann Surg. 1999;229:230–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grewal HP, Mohey el Din A, et al. Amelioration of the physiologic and biochemical changes of acute pancreatitis using an anti-TNF-alpha polyclonal antibody. Am J Surg. 1994;167:214–219. [DOI] [PubMed] [Google Scholar]
- 18.Hughes CB, Grewal HP, Gaber LW, et al. Anti-TNFα therapy improves survival and ameliorates the pathophysiologic sequelae in acute pancreatitis in the rat. Am J Surg. 1996;171:274–280. [DOI] [PubMed] [Google Scholar]
- 19.Gómez-Cambronero LG, Camps B, García de la Asunción J, et al. Pentoxifylline ameliorates caerulein-induced pancreatitis in rats. Role of glutathione and nitric oxide. J Pharmacol Exp Ther. 2000;293:670–676. [PubMed] [Google Scholar]
- 20.Guice KS, Miller DE, Oldham KT, et al. Superoxide dismutase and catalase. A possible role in established pancreatitis. Am J Surg. 1986;151:163–169. [DOI] [PubMed] [Google Scholar]
- 21.Sandilands D, Jeffrey IJM, Haboubi NY, et al. Abnormal drug metabolism in chronic pancreatitis. Treatment with antioxidants. Gastroenterology. 1990;98:766–772. [DOI] [PubMed] [Google Scholar]
- 22.Schoenberg MH, Büchler M, Beger HG. The role of oxygen radicals in experimental acute pancreatitis. Free Radic Biol Med. 1992;12:515–522. [DOI] [PubMed] [Google Scholar]
- 23.Sweiry JH, Mann GE. Role of oxidative stress in the pathogenesis of acute pancreatitis. Scand J Gastroenterol. 1996;31(Suppl 219):10–15. [DOI] [PubMed] [Google Scholar]
- 24.Sweiry JH, Shibuya I, Asada N, et al. Acute oxidative stress modulates secretion and repetitive Ca 2+ spiking in rat exocrine pancreas. Biochim Biophys Acta. 1999;1454:19–30. [DOI] [PubMed] [Google Scholar]
- 25.Folch E, Salas A, Prats N, et al. H(2)O(2) and PARS mediate lung P-selectin upregulation in acute pancreatitis. Free Radic Biol Med. 2000;28:1286–1294. [DOI] [PubMed] [Google Scholar]
- 26.Raingeaud J, Gupta S, Rogers JS, et al. Pro-inflammatory cytokines and environmental stress cause p38 mitogen-activated protein kinase activation by dual phosphorylation on tyrosine and threonine. J Biol Chem. 1995;270:7420–7426. [DOI] [PubMed] [Google Scholar]
- 27.Conze D, Krahl T, Kennedy N, et al. C-Jun NH(2)-terminal kinase (JNK)1 and JNK2 have distinct roles in CD8(+) T cell activation. J Exp Med. 2002;195:811–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dong C, Davis RJ, Flavell RA. MAP kinases in the immune response. Annu Rev Immunol. 2002;20:55–72. [DOI] [PubMed] [Google Scholar]
- 29.Duan RD, Williams JA. Cholecystokinin rapidly activates mitogen-activated protein kinase in rat pancreatic acini. Am J Physiol. 1994;267:G401–G408. [DOI] [PubMed] [Google Scholar]
- 30.Dabrowski A, Grady T, Logsdon CD, et al. Jun kinases are rapidly activated by cholecystokinin in rat pancreas both in vitro and in vivo. J Biol Chem. 1996;271:5686–5690. [DOI] [PubMed] [Google Scholar]
- 31.Schafer C, Ross SE, Bragado MJ, et al. A role for the p38 mitogen-activated protein kinase/Hsp 27 pathway in cholecystokinin-induced in the actin cytoskeleton in rat pancreatic acini. J Biol Chem. 1998;273:24173–24180. [DOI] [PubMed] [Google Scholar]
- 32.Wagner AC, Metzler W, Hofken T, et al. p38 map kinase is expressed in the pancreas and is immediately activated following cerulein hyperstimulation. Digestion. 1999;60:41–47. [DOI] [PubMed] [Google Scholar]
- 33.Yang J, Denham W, Tracey KJ, et al. The physiologic consequences of macrophage pacification during severe acute pancreatitis. Shock. 1998;10:169–175. [DOI] [PubMed] [Google Scholar]
- 34.Yang J, Murphy C, Denham W, et al. Evidence of a central role for p38 map kinase induction of tumor necrosis factor alpha in pancreatitis-associated pulmonary injury. Surgery. 1999;126:216–222. [PubMed] [Google Scholar]
- 35.Trush MA, Egner PA, Kensler TW. Myeloperoxidase as a biomarker of skin irritation and inflammation. Food Chem Toxicol. 1994;32:143–147. [DOI] [PubMed] [Google Scholar]
- 36.Beckman JS, Parks DA, Pearson JD, et al. A sensitive fluorometric assay for measuring xanthine dehydrogenase and oxidase in tissues. Free Radic Biol Med. 1989;6:607–615. [DOI] [PubMed] [Google Scholar]
- 37.Norman JG, Franz MG, Fink GS, et al. Decreased mortality of severe acute pancreatitis after proximal cytokine blockade. Ann Surg. 1995;221:625–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Folch E, Salas A, Panes J, et al. Role of P-selectin and ICAM-1 in pancreatitis-induced lung inflammation in rats: significance of oxidative stress. Ann Surg. 1999;230:792–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tan S, Yokoyama Y, Dickens E, et al. Xanthine oxidase activity in the circulation of rats following hemorrhagic shock. Free Radic Biol Med. 1993;15:407–414. [DOI] [PubMed] [Google Scholar]
- 40.Sullivan GW, Carper HT, Novick WJ, et al. Inhibition of the inflammatory action of interleukin-1 and tumor necrosis factor (α) on neutrophil function by pentoxifylline. Infect Immunol. 1988;56:1722–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Edwards MJ, Abney DL, Miller FN. Pentoxifylline inhibits interleukin-2-induced leukocyte-endothelial adherence and reduce systemic toxicity. Surgery. 1991;110:199–204. [PubMed] [Google Scholar]
- 42.Schandené L, Vandenbussche P, Crusiaux A, et al. Differential effects of pentoxifylline on the production of tumor necrosis factor-α (TNF-α) and interleukin-6 (IL-6) by monocyte and T cells. Immunology. 1992;76:30–34. [PMC free article] [PubMed] [Google Scholar]
- 43.Friedl HP, Till GO, Ryan US, et al. Mediator-induced activation of xanthine oxidase in endothelial cells. FASEB J. 1989;3:2512–2518. [DOI] [PubMed] [Google Scholar]
- 44.Adamson GM, Billings RE. Tumor necrosis factor induced oxidative stress in isolated mouse hepatocytes. Arch Biochem Biophys. 1992;294:223–229. [DOI] [PubMed] [Google Scholar]
- 45.Mendelson KG, Contois LR, Tevosian SG, et al. Independent regulation of JNK/p38 mitogen-activated protein kinases by metabolic oxidative stress in the liver. Proc Natl Acad Sci U S A. 1996;93:12908–12913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Aikawa R, Komuro I, Yamazaki T, et al. Oxidative stress activates extracellular signal-regulated kinases through Src and Ras in cultured cardiac myocytes of neonatal rats. J Clin Invest. 1997;100:1813–1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang X, Martindale JL, Liu Y, et al. The cellular response to oxidative stress: influences of mitogen-activated protein kinase signalling pathways on cell survival. Biochem J. 1998;333:291–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Uchida K, Shiraishi M, Naito Y, et al. Activation of stress signaling pathways by the end product of lipid peroxidation. 4-hydroxy-2-nonenal is a potential inducer of intracellular peroxide production. J Biol Chem. 1999;274:2234–2242. [DOI] [PubMed] [Google Scholar]
- 49.Kurata S. Selective activation of p38 MAPK cascade and mitotic arrest caused by low level oxidative stress. J Biol Chem. 2000;275:23413–23416. [DOI] [PubMed] [Google Scholar]
- 50.Blinman TA, Gukovsky I, Mouria M, et al. Activation of pancreatic acinar cells on isolation from tissue: cytokine upregulation via p38 MAP kinase. Am J Physiol. 2000;279:C1993–C2003. [DOI] [PubMed] [Google Scholar]
- 51.Denham W, Yang J, Wang H, et al. Inhibition of p38 mitogen activate kinase attenuates the severity of pancreatitis-induced adult respiratory distress syndrome. Crit Care Med. 2000;28:2567–2572. [DOI] [PubMed] [Google Scholar]