

Abstract
Objective:
To determine the mechanism by which gut-derived factors present in mesenteric lymph from rats subjected to trauma-hemorrhagic shock (T/HS) induce endothelial cell death.
Summary Background Data:
Intestinal ischemia after hemorrhagic shock results in gut barrier dysfunction and the subsequent production of biologically active and tissue injurious factors by the ischemic gut. These factors are carried in the mesenteric lymph and reach the systemic circulation via the mesenteric lymph, thereby ultimately resulting in distant organ injury. Although studies have established that trauma-hemorrhagic (T/HS) shock but not trauma-sham-shock (T/SS) mesenteric lymph is cytotoxic to endothelial cells, whether T/HS lymph-induced endothelial cell death occurs via an apoptotic or a necrotic pathway is unknown. The mechanisms underlying T/HS lymph-induced cytotoxicity are likewise unknown.
Methods:
Human umbilical vein endothelial cell (HUVEC) monolayers were incubated with medium, sham-shock, or post shock mesenteric lymph (5%) for 4 hours, after which the mode of cell death (ie, apoptosis versus necrosis) was determined using morphologic (confocal microscopy), biochemical (nucleosomal release), and DNA-based (gel electrophoresis) assays. To clarify the cellular pathways involved in T/HS lymph-induced HUVEC cell death, caspase-3, caspase-9, caspase-8, and BID activity was measured as was the ability of the pan-caspase inhibitor z-VAD-fmk to prevent T/HS lymph-induced cell death.
Results:
T/HS, but not T/SS, mesenteric lymph or medium was cytotoxic and caused the appearance of the classic morphologic signs of apoptosis, including membrane blebbing, cell shrinkage, and apoptotic body formation. Nucleosomal release and a DNA laddering pattern was also observed in the HUVECs incubated with T/HS lymph. These signs of apoptosis were associated with increased caspase activity as reflected in activation of the pro-apoptotic caspases, caspase-8, -9, and -3, as well as the pro-apoptotic bcl-2-related protein BID. However, since the broad-spectrum caspase inhibitor z-VAD-fmk delayed T/HS lymph-induced HUVEC cell death, but did not prevent it fully, it appears that other factors besides caspases are involved in the endothelial cell toxicity of T/HS lymph.
Conclusions:
Gut-derived factors in T/HS, but not T/SS, mesenteric lymph cause endothelial cell death via an apoptotic mechanism that involves both caspase-dependent and caspase-independent pathways.
Gut-derived factors produced during an episode of trauma hemorrhagic shock and carried in the mesenteric lymph are toxic for endothelial cells. These experiments investigated the mode and mechanisms of endothelial cell death and found that lymph-induced endothelial cell death occurs via an apoptotic mechanism that involves both caspase-dependent and caspase-independent mechanisms.
It has been recognized for over 25 years that the development of a dysregulated inflammatory response as well as acute lung injury and the development of the multiple organ dysfunction syndrome complicates the recovery of patients with severe trauma.1 Although this relationship between trauma-hemorrhagic shock (T/HS) and pulmonary dysfunction has been recognized for many years and studied extensively in both patients and animal models, the pathogenesis of T/HS-induced pulmonary microvascular damage is not well understood. Nevertheless, it is clear that hemorrhagic shock and major trauma are associated with intestinal ischemia, loss of gut barrier function and the intestine becoming a pro-inflammatory organ. Most recently, we have shown experimentally that T/HS-induced lung injury can be prevented by ligating the main mesenteric lymph duct exiting the intestine.2–4 On the basis of these studies and results showing that mesenteric lymph from T/HS, but not trauma-sham shock lymph (T/SS), rats injures endothelial cells and activates neutrophils,5–10 we have proposed that early post T/HS-induced lung injury is related to gut-derived factors carried in the mesenteric lymph. Having shown that gut-derived factors contained in T/HS lymph appear to be necessary for T/HS-induced lung injury and that the endothelium plays an important role in this process, we believed that studies directed at understanding the mechanisms responsible for T/HS intestinal lymph-induced endothelial cell injury are crucial to elucidating the pathophysiology of T/HS-induced lung injury. We therefore investigated the cellular and molecular mechanisms associated with T/HS lymph-induced endothelial cell cytotoxicity. The results of these studies indicate that T/HS lymph kills endothelial cells via an apoptotic process that involves both caspase-dependent and caspase-independent processes.
MATERIALS AND METHODS
Animals
Adult male specific pathogen-free Sprague-Dawley rats (Charles River Laboratories, Portage, MI) weighing 350 to 450 g were used after a minimum 7-day acclimation period. The animals were housed under barrier conditions and kept at 25°C with a 12-hour light/dark cycle. Rats were allowed free access to water and chow (Teklad 22/5 Rodent Diet W-8640, Harlan Teklad, Madison, WI). All animals were maintained in accordance with the recommendations of the “Guide for the Care and Use of Laboratory Animals,” and the New Jersey Medical School Animal Care and Use Committee approved all experiments.
Experimental Protocol for Induction of T/HS and the Collection of Mesenteric Lymph
The induction of T/HS and collection of mesenteric lymph was performed as previously described.2,3,5,6 Briefly, the femoral artery was isolated using aseptic techniques and cannulated with polyethylene (PE-50) tubing containing 0.1 mL of heparinized saline (10 units/mL). The catheter was attached in line to a blood-pressure recorder (Columbus Instruments BP2, Columbus, OH), which was used to measure the animal's blood pressure continuously during the shock period. Next, aseptic cannulation of the internal jugular vein was performed using a 50-guage-silicone catheter. Blood was withdrawn from the internal jugular vein into a syringe containing 10 units of heparin suspended in 0.3 mL of 0.9% normal saline solution to prevent clotting. A laparotomy was performed, and a catheter was placed into the efferent lymph duct draining the mesenteric lymph node complex. The mean arterial pressure was reduced to 30 mmHg and maintained at this level for 90 minutes by withdrawing or re-infusing shed blood (kept at 37°C) as needed. Mesenteric lymph collected during the postshock period was centrifuged to remove the cellular elements, then frozen and stored at −80°C until assayed. T/SS rats were anesthetized, underwent a laparotomy and had their mesenteric lymph ducts cannulated for lymph collection. However, no blood or fluid was withdrawn or infused.
Experimental Design
Our Trauma/Hemorrhagic shock model first involves laparotomy. The addition of laparotomy is important, because tissue injury is a component of clinical traumatic hemorrhage and the combined insult of hemorrhage plus tissue injury results in an inflammatory result that more closely reflects trauma patients, all of whom have soft tissue injury and require instrumentation with vascular cannulation.11–13
The first goal of these experiments was to determine the relative contribution of apoptosis to T/HS lymph-induced endothelial cell death, while the second goal was to investigate the potential molecular pathways involved in T/HS lymph-induced endothelial cell death. To accomplish these goals, human umbilical endothelial cells (HUVECs) were incubated with lymph collected from rats subjected to T/HS or T/SS. In the first group of experiments, several methods of assessing cell death were used. Specifically, morphology (confocal microscopy), biochemical (nucleosomal release) and DNA-based (gel electrophoresis) assays were used to differentiate between apoptosis and necrosis. Having established that T/HS lymph induces HUVEC apoptosis, activation of several members of the pro-apoptotic caspase family of proteins (caspase-3, caspase-8, and caspase-9) was measured in the second group of experiments as was the activation of the pro-apoptotic protein BID. Lastly, we tested the ability of the pan-caspase inhibitor z-VAD-fmk to limit the cytotoxic effects of T/HS lymph.
In these studies, T/HS and T/SS lymph, collected between 1 and 3 hours after T/HS or T/SS, was tested at a 5% (vol/vol) concentration for the following reason. The blood volume of a rat is about 6% of its body weight (ie, 18 mL in a 300-g rat). During the 90 minutes shock period, approximately 0.3 mL of lymph is produced; during the postshock period, lymph production is approximately 0.5 mL/h (ie, 1.5 mL over 3 hours). Since lung injury is present at 3 hours after the end of the shock period (ie, 4.5 hours after the induction of T/HS), approximately 1.8 mL of lymph would have been produced during this time period. Viewed simply, the 1.8-mL of lymph produced would represent about 10% of the 18-mL blood volume of the rat. Thus, testing T/HS at a 5% vol/vol concentration in vitro seemed clinically and biologically reasonable.
Cell Culture
HUVECs were obtained from BioWhittaker and cultured in endothelial growth medium. The HUVECs used for all experiments were from a white female umbilical cord. The cells were grown in a humidified atmosphere of 95% air and 5% CO2 at 37°C. Second to fourth passage HUVECs were used in all experiments.
Assessing Morphologic Changes
The fluorescent-labeled phallotoxin, phalloidin (Molecular Probes, Eugene, OR), was used to assess endothelial cell cytoskeletal changes after incubation with T/SS or T/HS lymph. Phallotoxins are F-actin–specific binding peptides that can be used to visualize cytoskeletal structures. Briefly, HUVECs were grown in 6-well plates on Poly-D-Lysine–coated 1-mm glass coverslips. After 1-, 2-, and 3-hour incubation periods with T/HS lymph, cells were washed twice with prewarmed phosphate-buffered saline (PBS; pH 7.4). Cells were then fixed in 3.7% formaldehyde solution in PBS. The cells were then permeabilized with 0.1% Triton X-100 in PBS for 5 minutes, followed by staining with fluorescent phallotoxin for 20 minutes. The coverslips were then mounted on slides and examined using Nikon PCM 2000 laser scanning confocal microscopy.
DNA Fragmentation
Detection by Gel Electrophoresis
To demonstrate the presence or absence of DNA laddering, approximately 2 × 106 cells were incubated with T/HS or T/SS lymph for 4 hours. Adherent cells were removed with a cell scraper and combined with any “free-floating” supernatant cells. Using a DNA Isolation Kit (Oncogene Research Products, Boston MA) low molecular weight DNA extracts were prepared. Approximately 2 μg of DNA extract were fractionated on 1.5% agarose gel in 1× Tris Borate-EDTA (TBE) buffer and stained with ethidium bromide.
Detection by Nucleosome ELISA
Approximately 2 × 105 cells were treated and harvested as described above. The number of free nucleosomes generated during DNA fragmentation was estimated using ELISA (Oncogene Research Products). Briefly, DNA extracts are prepared, and free nucleosomes are detected by an antihistone primary antibody followed by incubation with a streptavidin-linked horseradish peroxidase secondary antibody. Addition of a stop solution results in a color change. A plate reader (Dynatech MR500) was used to measure absorbance at 450/595 nm. Color change intensity (optical density) correlates with the number of free nucleosomes.
Caspase Activity
Caspase-3 Fluorometric Assay
Caspase-3 activity was measured using the ApoAlert Caspase-3 fluorescent assay kit (Clontech Laboratories, Palo Alto, CA). After a 4-hour incubation period with T/HS or T/SS lymph, adherent and supernatant HUVECs were collected, combined, and resuspended in 50 μL of cold lysis buffer (supplied by manufacturer) and then incubated on ice for 30 minutes.
Next, 10 μg/sample of the resulting whole cell extracts were incubated with 50 μL of 2× reaction buffer (supplied by manufacturer) containing 10 mM DTT and 5 μL of the fluorogenic caspase-3 substrate DEVD-AFC at 37°C for 1 hour. The samples were transferred to 96-well plates (CoStar; Corning Inc., Corning, NY). The fluorescence of each well was read using a plate reader (FL500 Bio-Tex Instruments) with excitation and emission filters set at 410 nm and 508 nm, respectively. Fluorescence correlates with the amount of caspase activity.
Caspase Levels by Western Blot Analysis
After a 4-hour incubation period with mesenteric lymph, HUVEC whole cell extracts were prepared by homogenizing cells in lysis buffer (1% [vol/vol] Nonidet P-40, 240 mM NaCl, 50 mM Tris-HCL [pH 8.0], 0.5 mM EDTA, 1 mM DTT, 0.5 mM phenylmethylsufonyl fluoride, 1 mM NaVO3, and 1 μg/ml of aprotinin, leupeptin, and pepstatin). Protein extracts (10 μg/sample) were resolved on 12% SDS-polyacrylamide gels, and blotted on nitrocellulose membranes. Membranes were subsequently incubated in a solution containing 1× Tris–buffered saline (TBS), 0.1% Tween, supplemented with 5% nonfat dry milk or 5% bovine serum albumin. For immunodetection the following antibodies were used: anticaspase-9 polyclonal antibody, anticleaved caspase-3 polyclonal antibody, anti-BID polyclonal antibody (Cell Signaling Technology, Beverly, MA); and anticaspase-8 monoclonal antibody (PharMingen, San Diego, CA). After overnight incubation at 4°C with primary antibody at dilutions of 1:1000, the membranes were washed in TBS (0.1% Tween) followed by 1 hour of incubation at room temperature with horseradish peroxidase–conjugated goat-anti mouse or goat anti-rabbit antibody. Enhanced chemiluminescence (ECL-Plus; Amersham Pharmacia Biotech, UK) was used for protein detection.
Caspase Inhibition
HUVECs were pretreated for 1 hour with N-benzyloxycarbonyl Val-Ala-DL-ASP-fluoromethylketone (z-VAD-fmk; Alexis Biochemicals, San Diego, CA), which is a known irreversible pan-caspase inhibitor. Z-VAD-fmk was dissolved in fresh DMSO (Sigma, St. Louis MO) at 100 mM concentration and then added to the endothelial cultures 1 hour before treatment with shock lymph. The final concentration of z-VAD-fmk was 100 μm.
Assessment of the Effect of z-VAD-fmk on the Percentage of Apoptotic Appearing Cells Over Time
HUVECs seeded at 20 × 104 cells per well of a 12-well plate were grown to confluence. In the first group, HUVECs were preincubated with 100 μm of z-VAD-fmk followed by 2, 4, and 6-hour incubation periods with medium containing 5% (vol/vol) mesenteric lymph. The second group was incubated only with T/HS lymph. An inverted light microscope (Olympus CK2) was used to count the number of apoptotic appearing cells per high-powered field (hpf). Morphologic criteria used included cell shrinkage and membrane blebbing. For each well, cells were counted in 5 random high-powered views. Cell counts were performed in a blinded fashion.
Statistical Analysis
All data were analyzed by analysis of variance followed by Tukey-Kramer multiple comparisons test and are expressed as the mean ± SD. Statistical significance was considered to be reached when P ≤ 0.05.
RESULTS
The morphologic appearance of HUVECS incubated in T/SS lymph was normal and similar to that of HUVECs incubated in medium (Fig. 1A). In contrast, after a 3-hour incubation with T/HS lymph, the HUVECs showed morphologic evidence of apoptosis with shrinking of the cytoplasm, nuclear condensation, and membrane blebbing (Fig. 1B).

FIGURE 1. Laser scanning confocal images of HUVECs stained with phalloidin (60× magnification). (A) HUVECs incubated with T/SS lymph appear normal with well-organized actin fibers (arrow). (B) HUVECs incubated with T/HS lymph display cytoplasmic shrinkage with accompanying disorganization of actin cytoskeleton, nuclear condensation, and cell surface protrusions. (Inset-B) Image of HUVEC incubated with T/HS lymph demonstrates apoptotic body formation (arrow).
Apoptosis is associated with a characteristic ladder-type pattern of DNA degradation as well as internucleosomal cleavage resulting in the generation of free nucleosomes; therefore, we next examined HUVECs incubated in T/HS or T/SS lymph for these changes. Genomic DNA from HUVECs incubated in T/HS lymph, but not T/SS lymph, demonstrated the “hallmark” DNA step-laddering pattern of apoptosis (Fig. 2A). Likewise, free nucleosome release was increased in the DNA extracts from the T/HS lymph-treated HUVECs compared with HUVECs incubated in T/SS lymph or medium (Fig. 2B; P < 0.001).
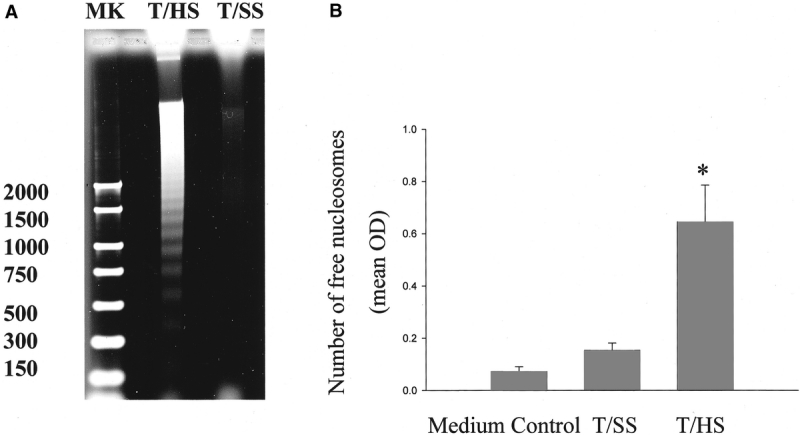
FIGURE 2. (A) T/HS lymph but not T/SS lymph or medium induced DNA fragmentation. HUVECs were incubated with lymph for 4 hours, after which genomic DNA was isolated, fractionated on a 1.5% agarose gel and stained with ethidium bromide. The hallmark DNA step-laddering pattern characteristic of apoptosis is present only in the HUVECS incubated with T/HS lymph. These results are representative of 3 independent studies (M, marker). (B) The number of free nucleosomes generated in HUVECs after 4-hour incubation with T/HS lymph was increased compared with HUVECs incubated in medium or T/SS lymph. Nuclear extracts were prepared using a Nucleosome ELISA kit (Oncogene Research). Optical density (OD) correlates with the number of free nucleosomes. Data expressed as mean ± SD. *P < 0.001 versus all other groups; n = 5 per group.
Consistent with an apoptotic pattern of cell death, HUVECs incubated in T/HS lymph, but not medium or T/SS lymph, increased caspase-3 activity (Fig. 3A; P < 0.05). Further support for the activation of caspase-3 in the T/HS lymph-treated HUVECs was the observation that the activated cleaved capase-3 p17 subunit was increased in the HUVECs incubated in T/HS lymph but not T/SS lymph (Fig. 3B).

FIGURE 3. (A) Caspase-3 activity is increased in HUVECs after 4-hour incubation with T/HS lymph. Whole cell extracts of HUVECS incubated in medium, T/HS, or T/SS lymph were prepared and standardized to 10 μg of protein. An ApoAlert kit (Clontech) was used to detect Caspase-3 activity. The amount of fluorescence correlates with caspase-3 activity. Data expressed as mean ± SD. *P < 0.05 versus all groups; n = 5 for T/HS and T/SS, and n = 3 for medium control. (B) Incubation with T/HS lymph but not T/SS lymph results in cleavage of caspase 3. Equal amounts of whole cell extracts (10 μg) prepared from cells treated with T/SS and T/HS lymph were resolved by SDS-PAGE and probed with anticleaved caspase-3 antibody. These results are representative of 3 independent studies.
T/HS lymph-induced HUVEC apoptosis was associated with increased caspase-3 activity; therefore, we tested whether the broad-spectrum pan-caspase inhibitor, z-VAD-fmk would reverse T/HS lymph-induced apoptosis of HUVECs exposed to T/HS lymph. Pretreatment of HUVECs for 1 hour with z-VAD-fmk (100 μm) before the addition of T/HS lymph significantly decreased T/HS lymph-induced caspase-3 activity to levels even below those observed in HUVECs incubated with medium or T/SS lymph only (Fig. 4). Pretreatment of HUVECs with Z-VAD-fmk decreased the percentage of apoptotic appearing cells present 2 and 4 hours after the HUVECs were incubated with T/HS lymph. However, z-VAD-fmk's protective effect was lost when the HUVECs were incubated with T/HS lymph for 6 hours (Fig. 5).

FIGURE 4. Preincubation of HUVECs with z-VAD-fmk for 1 hour before a 4-hour exposure to T/HS lymph reduces caspase-3 activity to levels comparable to those observed when HUVECs are incubated in T/SS lymph or medium. n = 4 per group. Data expressed as mean ± SD. *P < 0.001 versus T/SS and medium controls; **P < 0.001 versus all groups.

FIGURE 5. Preincubation of HUVECs with Z-VADfmk for 1 hour before the addition of T/HS lymph reduced the percentage of apoptotic appearing HUVECs after 2-hour and 4-hour, but not 6-hour, incubation periods. These results illustrate that Z-VADfmk delays the appearance of T/HS lymph-induced apoptosis. Data expressed as mean ± SD. *P < 0.05; n = 3 for each group.
Since caspase-3 activity is increased in both the mitochondrial/caspase-9 and the death receptor/caspase-8 apoptotic pathways, we next tested whether T/HS lymph would activate either of these pathways. Western blot analysis demonstrated cleavage of both procaspase-8 and procaspase-9 to their respective 43- and 37-kDa active subunits in HUVECs incubated in T/HS lymph (Fig. 6). The cleavage of neither procaspase-8 nor procaspase-9 was observed when the HUVECs were incubated in T/SS lymph. Pretreatment of the HUVECs with Z-VAD-fmk abrogated the T/HS lymph-induced cleavage of procaspase-8, procaspase-9, and caspase-3 to their active subunits (Fig. 6).

FIGURE 6. T/HS lymph increases the conversion of procaspase-8, procaspase-9, and caspase-3 to their active subunits and this conversion is abrogated by z-VAD-fmk. Equal amounts of whole cell extracts (10 μg) prepared from cells treated with T/SS and T/HS lymph with or without z-VAD-fmk pretreatment were resolved by SDS-PAGE and probed with anticaspase-8 and -9 antibodies. Activation of caspase-8 and -9 was characterized by the appearance of cleaved subunits at 43 kDa and 38 kDa, respectively. The cleaved caspase-3 p17/19 subunits were only present in the T/HS lymph treated group. These results are representative of 3 independent studies.
To further investigate mitochondrially related apoptotic signaling pathways in T/HS lymph-induced HUVEC apoptosis, we tested whether the pro-apoptotic bcl-2 family protein BID is activated by T/HS lymph. Western blot analysis demonstrated that HUVECs incubated in T/HS lymph, but not T/SS lymph, activated Bid as reflected in the cleavage of the 22-kDa native form into its activated 15-kDa truncated form (t-Bid). Pretreatment of cells with z-VAD-fmk abrogated this cleavage (Fig. 7).
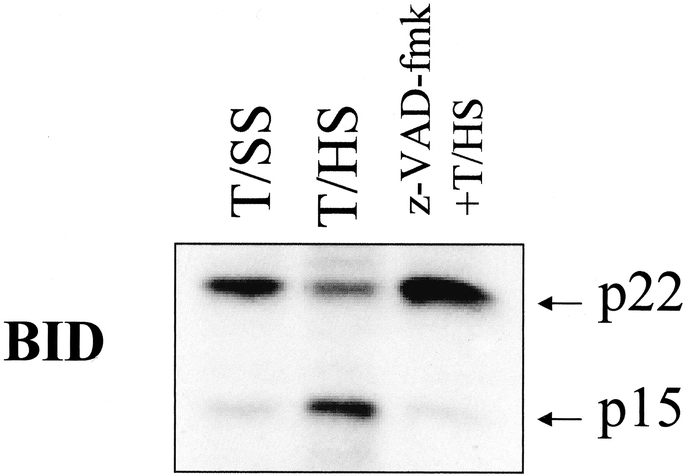
FIGURE 7. T/HS lymph activates Bid and this activation is abrogated by z-VAD-fmk. Equal amounts of whole cell extracts (10 μg) prepared from cells treated with T/SS and T/HS lymph with or without z-VAD-fmk pretreatment were resolved by SDS-PAGE and probed with an anti-BID antibody. Increased expression of the 15-kDa (t-BID) subunit is demonstrated in the T/HS but not the T/SS lymph treated cells. Pretreatment of cells with z-VAD-fmk significantly decreases t-BID expression in T/HS lymph treated cells.
DISCUSSION
It is well accepted that host-derived endogenous factors play a critical role in the induction and perpetuation of the systemic inflammatory response as well as in the development of ARDS and MODS.1 However, neither the source nor the exact identities of these critical pro-inflammatory organ and cell injury-inducing factors are completely known. This lack of knowledge has contributed to the failure of human clinical trials testing antiinflammatory therapies to prevent or treat ARDS and MODS.14 Our previous work showing that T/HS-induced lung injury, neutrophil activation as well as endothelial cell activation, and injury are due to gut-derived factors carried in the mesenteric lymph and not the portal blood2,3,7–9 has begun to address one of these issues; it has identified the ischemic gut as a source of factors leading to early post-T/HS lung injury as well as the induction of a systemic inflammatory state. That is, T/HS mesenteric lymph appears to be a biologically relevant fluid that is directly involved in transducing the hypotensive effects of trauma-hemorrhage into a proinflammatory and tissue injurious event. Accordingly, the ability to collect, analyze, and test the effects of T/HS lymph in controlled in vitro studies provides a unique opportunity to investigate the potential mechanisms and cellular pathways involved in the development of ARDS and MODS. The exact factors responsible for T/HS mesenteric lymph's cytotoxic effects are yet to be identified. However, previous work demonstrates that neither endotoxin or bacteria are present in T/HS mesenteric lymph,15 thus bacteria and their products are not responsible for the cytotoxic effects of T/HS lymph. In addition, T/HS lymph prepared for all in vitro studies is acellular; it is therefore unlikely that activated cells are responsible for T/HS lymph's cytotoxic effects. Various protein and lipid isolation techniques are currently being used to identify the toxic factors present in lymph. Although the exact factors responsible for T/HS toxicity remain to be determined, our previous15 and ongoing (unpublished) results indicate that these toxic effects are not due to TNF or other cytokines.
In the current study, we investigated the potential cellular pathways and mechanisms involved in T/HS lymph-induced HUVEC cytotoxicity, since T/HS lymph has profound effects on endothelial cells and the endothelium is a central effector of the immuno-inflammatory response and endothelial injury/activation contributes to organ dysfunction.16–18 Furthermore, as regulators of coagulation and inflammation, endothelial cells in conjunction with circulating neutrophils are able to promote tissue ischemia, injury, and capillary leak. In fact, endothelial-leukocyte interactions resulting in tissue injury appear to be a common pathway by which a diverse number of initiating factors, such as bacteria, endotoxin, cytokines, and ischemia can lead to organ failure and MODS.1 This notion appears to be especially true in the pathogenesis of pulmonary microvascular injury after intestinal ischemia-reperfusion injury.1
Our results showing that T/HS lymph-induced HUVEC cytotoxicity occurs through an apoptotic process is consistent with recent in vivo studies implicating apoptosis as one of the events involved in trauma/hemorrhagic shock as well as sepsis-induced organ failure.19–23 Recent advances in the field of endothelial cell biology have led to new insights in the mechanisms associated with endothelial cell apoptosis. Conceptually, apoptosis can be viewed as occurring through one of 3 major pathways. These are the caspase-dependent receptor-mediated pathway, the caspase-dependent mitochondrial-mediated pathway, and the noncaspase-dependent mitochondrial-mediated pathway24–26 (Fig. 8). Studies of TNF-induced apoptosis have resulted in a large body of information on the molecular mechanisms and pathways associated with receptor-mediated, caspase-dependent apoptosis.26 In this process, TNF binds to the death receptor, which activates the inducer caspase, caspase-8, which in turn leads to activation of the pro-apoptotic Bcl-2 family of proteins (ie, Bid) and the release of cytochrome c from the mitochondria. The effector caspase, caspase-3, is subsequently activated, ultimately resulting in apoptosis. In nonreceptor-mediated, caspase-dependent apoptosis (ie, mitochondrial-mediated), the apoptotic signal leads to the release of cytochrome c from the mitochondria, which activates the inducer caspase, caspase 9, which in turn activates caspase 3, thereby leading to an apoptotic response. The critical role of caspase activation in the induction of apoptosis, in both of these pathways, has been documented by transfection studies as well as by studies documenting that pharmacologic inhibition of caspase induction abrogates the apoptotic response.24,25 Consequently, since T/HS lymph caused HUVEC apoptosis, we measured whether caspase 3, caspase 8, and/or caspase 9 as well as Bid would be activated in HUVECs exposed to T/HS lymph. Our observations that T/HS, but not T/SS, lymph activated all of these pro-apoptotic proteins suggested that T/HS lymph-induced HUVEC apoptosis was mediated, at least in part, by both caspase-dependent pathways. However, the observation that the pan-caspase inhibitor Z-VAD-fmk, while preventing activation of these caspases, did not ultimately prevent T/HS lymph-induced apoptosis suggests that caspase-independent apoptotic pathways are also involved. Less is known about the caspase-independent, mitochondrial-mediated pathway of apoptosis than the caspase-dependent apoptotic pathways.25 As illustrated in Figure 8, caspase-independent apoptosis is believed to be induced by apoptogenic factors released from the mitochondria, such as apoptosis inducing factor (AIF), Omi/Htra2, or endonuclease G, which translocate to the nucleus.25,27,28 In most systems, Z-VAD-fmk does block the release of these mitochondrial apoptogenic factors;25 this may explain why Z-VAD-fmk delayed the appearance of T/HS lymph-induced HUVEC apoptosis but did not prevent it.

FIGURE 8. Schematic diagram illustrating the major pro-apoptotic components of the caspase-dependent and caspase-independent apoptotic pathways as well as the sites where they are inhibited by Z-VAD-fmk. Caspase-dependent apoptosis occurs either through the death receptor or the mitochondrial-mediated pathway. In the death receptor pathway, the initiator caspase, caspase-8 is activated while, in the mitochondrial pathway, the initiator caspase, caspase-9 is activated. Both caspase-8 and caspase-9 ultimately turn on the effector caspase arm of the pathway as reflected in activated caspase-3. In caspase-independent apoptosis, mitochondrial membrane permeabilization triggers the release of death-promoting factors such as AIF or the serine protease Omni/htra2, which cause caspase-independent death signaling downstream from the mitochondria. As illustrated, z-VAD-fmk, the pan-caspase inhibitor, blocks both caspase-dependent apoptotic pathways at multiple sites. However, it has no effect on the caspase-independent apoptotic pathway. The results of the current study indicate that T/HS lymph-induced HUVEC apoptosis is occurring through all 3 apoptotic pathways and thus involves caspase-dependent and caspase-independent processes.
The fact that Z-VAD-fmk delayed the onset of T/HS lymph-induced HUVEC apoptosis by about 2 hours suggests that both caspase-dependent and caspase-independent apoptotic pathways are involved. Moreover, since caspase inhibition delayed HUVEC apoptosis, it appears that caspase-dependent apoptotic signaling may precede caspase-independent signaling, although ultimately T/HS lymph-induced HUVEC apoptosis involves caspase-independent pathways. This observation that T/HS lymph-induced HUVEC apoptosis activates caspases but is not fully caspase-dependent is consistent with other recent studies29–32 and highlights the ever-emerging complexity of the apoptotic process. These studies have shown that the inhibition of caspase activation will delay but not prevent apoptosis and other variants of programmed cell death induced by certain insults in some cell populations. In fact, caspase inhibition has been documented to uncover, or even enhance, underlying caspase-independent apoptotic pathways.27,32
One potential limitation of this study is that rat lymph was tested on human endothelial cells, raising the question of whether these observations are confounded by the cross-species nature of the studies. However, although HUVECs are of human origin, we have previously shown that their response to rat T/HS lymph is similar to that of rat pulmonary microvascular endothelial cells (RPMVEC).6 Furthermore, in pilot studies, we have found that RPMVECs exposed to T/HS lymph have an apoptotic morphology (unpublished data).
In summary, on the basis of biochemical, morphologic, and DNA-based assays, it appears that T/HS lymph kills HUVECs by an apoptotic process. Caspase-8, caspase-9, and caspase-3 were activated by T/HS lymph, but caspase inhibition did not completely rescue the HUVECs from T/HS lymph-induced apoptosis; therefore, it appears that T/HS lymph-induced endothelial cell apoptosis involves both caspase-dependent and caspase-independent pathways. These in vitro studies are consistent with our recent in vivo studies (unpublished), which show that T/HS lymph causes endothelial cell apoptosis in vivo and thus provides important basic information on the pathogenesis of trauma/hemorrhagic shock-induced organ injury.
Footnotes
Reprints: Edwin A. Deitch, MD, Professor and Chairman, Department of Surgery, MSB G506, UMDNJ-New Jersey Medical School, 185 South Orange Avenue Newark, NJ 07103; E-mail: edeitch@umdnj.edu.
Sponsored by NIH grant R01 GM 59841 (Dr. Deitch).
REFERENCES
- 1.Deitch EA. Multiple organ failure: Pathophysiology and potential future therapy. Ann Surg. 1992;216:117–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Magnotti LJ, Upperman JS, Xu DZ, et al. Gut-derived mesenteric lymph but not portal blood increases endothelial cell permeability and potentiates lung injury following hemorrhagic shock. Ann Surg. 1998;228:518–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Deitch EA, Adams CA, Lu Q, et al. A time course study of the protective effect of mesenteric lymph duct ligation on hemorrhagic shock-induced pulmonary injury and the toxic effects of lymph from shocked rats on endothelial cell monolayer permeability. Surgery. 2001;129:39–47. [DOI] [PubMed] [Google Scholar]
- 4.Sambol JT, Xu DZ, Adams CA, et al. Mesenteric lymph duct ligation provides long-term protection against hemorrhagic shock-induced lung injury. Shock. 2000;14:416–420. [PubMed] [Google Scholar]
- 5.Upperman JS, Deitch EA, Guo W, et al. Post-hemorrhagic shock mesenteric lymph is cytotoxic to endothelial cells and activates neutrophils. Shock. 1998;10:407–414. [DOI] [PubMed] [Google Scholar]
- 6.Deitch EA, Adams CA, Lu Q, et al. Mesenteric lymph from rats subjected to trauma-hemorrhagic shock are injurious to rat pulmonary microvascular endothelial cells as well as human umbilical vein endothelial cells. Shock. 2001;16:290–293. [DOI] [PubMed] [Google Scholar]
- 7.Dayal SD, Hasko G, Lu Q, et al. Trauma-hemorrhagic shock mesenteric lymph upregulates adhesion molecule expression and IL-6 production in human umbilical vein endothelial cells. Shock. 2002;17:491–495. [DOI] [PubMed] [Google Scholar]
- 8.Adams JM, Hauser CJ, Adams CA, et al. Entry of gut lymph into the circulation primes rat neutrophil respiratory burst in hemorrhagic shock. Crit Care Med. 2001;29:2194–2198. [DOI] [PubMed] [Google Scholar]
- 9.Caruso JM, Feketeova E, Dayal SD, et al. Factors in intestinal lymph after shock increase neutrophil adhesion molecule expression and pulmonary leukosequestration. J Trauma. 2003;55:727–723. [DOI] [PubMed] [Google Scholar]
- 10.Deitch EA. Role of the gut lymphatic system in multiple organ failure. Curr Opin Crit Care. 2001;7:92–98. [DOI] [PubMed] [Google Scholar]
- 11.Wang P, Zheng F, Burkhardt J, et al. Trauma-hemorrhage and resuscitation in the mouse: effects on cardiac output and organ blood flow. Am J Physiol. 1993;264:H1166–H1173. [DOI] [PubMed] [Google Scholar]
- 12.Deitch EA. Animal models of sepsis and shock. A review and lessons learned. Shock. 1998;9:1–11. [DOI] [PubMed] [Google Scholar]
- 13.Stephan RN, Saizawa M, Conrad PJ, et al. Depressed antigen presentation function and membrane interleukin-1 activity of peritoneal macrophages after laparotomy. Surgery. 1987;102:147–154. [PubMed] [Google Scholar]
- 14.Zeni F, Freeman B, Natanson C. Anti-inflammatory therapies to treat sepsis and septic shock: a reassessment. Crit Care Med. 1997;25:1095–1100. [DOI] [PubMed] [Google Scholar]
- 15.Adams CA, Xu D, Lu Q, et al. Factors larger than 100 kDa in post-hemorrhagic shock mesenteric lymph are toxic for endothelial cells. Surgery. 2001;129:351–362. [DOI] [PubMed] [Google Scholar]
- 16.Osborn L. Leukocyte adhesion to endothelium in inflammation. Cell. 1990;62:3–6. [DOI] [PubMed] [Google Scholar]
- 17.Prober JS, Cotran RS. The role of endothelial cells in inflammation. Transplantation. 1990;50:537–544. [DOI] [PubMed] [Google Scholar]
- 18.Vane JR, Anggard EE, Botting RM. Regulatory functions of the vascular endothelium. New Engl J Med. 1990;323:27–36. [DOI] [PubMed] [Google Scholar]
- 19.Hotchkiss RS, Swanson PE, Freeman BD, et al. Apoptotic cell death inpatients with sepsis, shock, and multiple organ dysfunction. Crit Care Med. 1999;27:1230–51. [DOI] [PubMed] [Google Scholar]
- 20.Guan J, Jin DD, Jin LJ, et al. Apoptosis in organs of rats in early stage after polytrauma combined with shock. J Trauma. 2002;52:104–111. [DOI] [PubMed] [Google Scholar]
- 21.Assaly R, Olson D, Hammersly J, et al. Initial evidence of endothelial cell apoptosis as a mechanism of systemic capillary leak syndrome. Chest. 2001;120:1301–1308. [DOI] [PubMed] [Google Scholar]
- 22.Oberholzer A, Oberholzer C, Moldawer LL. Sepsis syndromes: understanding the role of innate and acquired immunity. Shock. 2001;16:83–96. [DOI] [PubMed] [Google Scholar]
- 23.Stefanec T. Endothelial apoptosis; could it have a role in the pathogenesis and treatment of disease? Chest. 2000;117:841–854. [DOI] [PubMed] [Google Scholar]
- 24.Mallat Z, Tedgui A. Apoptosis in the vasculature: mechanisms and functional importance. Brit J Pharm. 2000;130:947–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Green DR, Reed JC. Mitochondrion and Apoptosis. Science. 1998;281:1309–1312. [DOI] [PubMed] [Google Scholar]
- 26.Denecker G, Vercammen D, Declercq W, et al. Apoptotic and necrotic cell death induced by death domain receptors. Cell Mol Life Sci. 2001;58:356–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mathiasen IS, Jaattela M. Triggering caspase-independent cell death to combat cancer. Trens Mol Med. 2002;8:212–220. [DOI] [PubMed] [Google Scholar]
- 28.Daugas E, Nochy D, Ravagnan L, et al. Apoptosis-inducing factor (AIF): a ubiquitous mitochondrial oxidoreductase involved in apoptosis. FEBS Letters. 2000;476:118–123. [DOI] [PubMed] [Google Scholar]
- 29.Belmokhtar CA, Hillion J, Segal-Bendirdjian E. Staurosporine induces apoptosis through both caspase-dependent and caspase-independent mechanisms. Oncogene. 2001;20:3354–3362. [DOI] [PubMed] [Google Scholar]
- 30.Joseph B, Marchetti P, Formstecher P, et al. Mitochondrial dysfunction is an essential step for killing of non-small cell lung carcinomas resistant to conventional treatment. Oncogene. 2002;21:65–77. [DOI] [PubMed] [Google Scholar]
- 31.Skulachev PV. Mitochondrial physiology and pathology: concepts of programmed death of organelles, cells and organisms. Mol Asp Med. 1999;20:139–184. [DOI] [PubMed] [Google Scholar]
- 32.Leist M, Jaattela M. Four deaths and a funeral: from caspases to alternatives. Nat Rev Mol Cell Biol. 2001;2:589–598. [DOI] [PubMed] [Google Scholar]