

Abstract
Objective:
To determine the role of tumor necrosis factor α (TNF-α) and Fas ligand (FasL, CD95L) in superantigen-induced and endotoxin-induced liver injury.
Summary Background Data:
Gram-positive bacteria are increasingly common causes of sepsis and multiorgan failure, but the pathophysiologic mechanisms of superantigen-provoked hepatotoxicity remain elusive.
Methods:
Intravital fluorescence microscopy was used to study the liver microcirculation in mice challenged with superantigen (staphylococcal enterotoxin A, SEA) or endotoxin (lipopolysaccharide, LPS) combined with d-galactosamine.
Results:
Administration of 10 μg LPS and 50 μg SEA caused similar hepatocellular damage as determined by liver enzymes and apoptosis. Notably, TNF-α–deficient mice were completely protected against hepatic injury provoked by LPS, whereas no protection was observed in response to SEA. On the other hand, FasL-deficient mice were protected against liver injury induced by SEA, but no protection was found when challenged with LPS. LPS increased clear-cut leukocyte recruitment, whereas SEA had no significant effect on leukocyte responses in the liver microcirculation. Leukocyte responses to LPS were decreased by >56% in TNF-α gene-targeted animals. Moreover, antiadhesive therapy, ie, immunoneutralization of P-selectin, which is an effective inhibitor of leukocyte recruitment, protected against LPS-induced but not against SEA-induced hepatic damage.
Conclusions:
These novel findings demonstrate that the mechanisms of hepatic injury in endotoxin-induced and superantigen-induced sepsis are principally different. On one hand, SEA-provoked hepatotoxicity is mediated by FasL and is not associated with leukocyte recruitment. On the other hand, liver damage provoked by LPS is mediated by TNF-α and characterized by prominent leukocyte responses. These data may facilitate development of more specific therapies against sepsis of different origins.
The molecular mechanisms of hepatotoxicity induced by enterotoxin (superantigen) from Gram-positive bacteria and endotoxin (lipopolysaccharide) from Gram-negative bacteria were examined. It was found that staphylococcal enterotoxin A-induced liver injury is primarily mediated by Fas ligand, whereas endotoxin-provoked hepatic damage is dependent on tumor necrosis factor α.
Most experimental studies of sepsis have focused on the mechanisms of endotoxin-induced organ injury. This complex macromolecule is common to the outer membrane of most clinically relevant Gram-negative bacteria found in human infections.1 Indeed, Gram-negative bacteria dominated the microbial etiology of sepsis in the late 1970s and throughout the 1980s.2 However, the resurgence of Gram-positive bacterial infections has marked a dramatic change in the prevalence pattern of sepsis. In fact, at present, close to 50% of all cases of sepsis are caused by Gram-positive bacteria.2 Nonetheless, the mechanisms behind organ injury and systemic inflammation induced by Gram-positive bacteria remain largely unknown. Liver injury is a common feature of bacterial toxemia and constitutes a key component in the development of severe shock and multiple organ failure. Hepatic damage is the result of the actions exerted by multiple proinflammatory mediators, including cytokines and chemokines, acting directly or indirectly (through leukocyte recruitment) to cause hepatocyte necrosis and apoptosis. Numerous studies have shown that hepatocytes are particularly sensitive to tumor necrosis factor α (TNF-α) and Fas ligand (FasL)-mediated apoptosis in different disease models.3–5 Indeed, it is well documented that TNF-α plays a dominant role in endotoxin-induced hepatotoxicity,6 but the relative role of TNF-α and FasL in Gram-positive sepsis remains elusive.
Gram-positive bacteria may activate the host immune system by several different mechanisms. On one hand, major cell wall components of Gram-positive bacteria, including peptidoglycan and lipoteichoic acid, have been shown to be capable of stimulating inflammatory responses.7 On the other hand, some Gram-positive bacteria, in particular Staphylococcus aureus and Streptococcus pyogenes, secrete powerful toxins, commonly referred to as superantigens because of their capacity to induce intensive T-cell activation and proliferation independently of classic antigen presentation.8 Superantigens bind to major histocompatibility complex-encoded class II proteins outside the normal antigen-presenting groove and to variable regions of T-cell receptor β-chains.1 In fact, S. aureus is one of the most commonly isolated bacteria from patients with sepsis,9,10 and produces a wide array of toxins, such as staphylococcal enterotoxin A (SEA), belonging to a family of related enterotoxins.8,11 It has been shown that SEA has the capacity to potentiate endotoxin-induced toxicity by more than 50,000 times.12
The purpose of this study was to examine the microvascular effector mechanisms of SEA and the relative contributions of TNF-α and FasL to SEA-induced liver injury. For this purpose, intravital fluorescence microscopy was used in the liver of TNF-α gene-deficient and FasL gene-deficient mice.
MATERIALS AND METHODS
Animals
Adult male wild-type C57BL/6, TNF-α–deficient (B6;129S6-Tnf∥tm1Gkl), and FasL-deficient (B6Smn.C3H-Fasl∥gld) mice (Jackson Laboratory, Bar Harbor, ME) weighing 23 g to 28 g were kept on a 12–12 hour light-dark cycle with free access to food and tap water. Animals were anesthetized by intraperitoneal administration of 7.5 mg ketamine hydrochloride (Hoffman-La Roche, Basel, Switzerland) and 2.5 mg xylazine (Janssen Pharmaceutica, Beerse, Belgium) per 100 mg body weight. The right jugular vein was cannulated with a polyethylene catheter for intravenous administration of fluorescent dyes and additional anesthesia. The local ethics committee at Lund University approved all the experiments of this study.
Experimental Protocol
Seven hours before surgery and intravital observation, mice were challenged with SEA (50 μg/mouse; Sigma Chemical Co., St. Louis, MO) or lipopolysaccharide (LPS; 10 μg/mouse, LPS was from Escherichia coli serotype 0111:B4, Sigma) combined with d-galactosamine (Gal; 18 mg/mouse, Sigma). In separate experiments, a monoclonal antibody against P-selectin (RB40.34, 40 μg/mouse; Pharmingen, San Diego, CA) was administered intravenously immediately before intraperitoneal challenge with LPS/Gal or SEA/Gal. In another set of experiments, wild-type animals received either an agonistic anti-Fas receptor antibody (Jo-2, 10 μg/mouse intraperitoneal, Pharmingen) or recombinant TNF-α (50 μg/mouse intraperitoneal, R&D Systems Europe Ltd., Abingdon, Oxon, United Kingdom) combined with Gal (14 mg/mouse) 4 hours before surgery. In anesthetized animals, a transverse subcostal incision was performed, and the ligamentous attachments from the liver to the diaphragm and the abdominal wall were gently released. The animals were positioned on their left side, and the left liver lobe was carefully exteriorized onto an adjustable stage for analysis of hepatic microcirculation by use of intravital fluorescence microscopy. The liver surface was covered with a circular glass to avoid tissue drying and exposure to ambient oxygen. An equilibration period of 5 minutes was allowed before starting microscopical observation. After intravital observations, animals were killed by exsanguination, and blood was drawn from the heart for analysis of liver function test, ie, alanine aminotransferase (ALT) and aspartate aminotransferase (AST) using standard spectrophotometric procedures, and systemic leukocyte counts, including polymorphonuclear leukocytes and mononuclear leukocytes using a hematocytometer.
Intravital Microscopy
The liver microcirculation was studied by use of a modified Olympus microscope (BX50WI, Olympus Optical Co. GmbH, Hamburg, Germany) equipped with different water immersion lenses (×40 NA 0.75/×63 NA 0.9). The image was televised using a charge-coupled device video camera (FK 6990 Cohu, Pieper GmbH, Schwerte, Germany) and recorded on videotape for subsequent off-line evaluation. Vascular perfusion within individual microvessels was studied after contrast enhancement by injection of fluroescein isothiocyanate (FITC)-dextran (0.1 mL, 2 μmol/kg, Sigma). In vivo labeling of leukocytes with rhodamine-6G (0.1 mL, 0.05 mg/mL, Sigma) allowed quantitative analysis of leukocyte flow behavior in the liver microcirculation. Quantification of microcirculatory parameters was performed off-line by frame-to-frame analysis of the videotaped images. Five postsinusoidal venules with connecting sinusoids were evaluated in each animal. Microcirculatory analysis included determination of the number of perfused sinusoids given as a percentage of the total number of sinusoids observed (ie, sinusoidal perfusion). Within postsinusoidal venules, leukocyte rolling was measured by counting the number of cells rolling during 30 seconds and is expressed as cells per minute. Leukocyte adhesion was measured by counting the number of cells that adhered along the venular endothelium and remained stationary during the observation period of 30 seconds, and is expressed as cells per millimeter venule length. Leukocyte sequestration in the sinusoids was evaluated by counting the number of trapped leukocytes per 20-30 high-power fields per animal and is given as leukocytes per 10 high-power fields. The diameter of the venules was not different between the experimental groups. Hepatocyte apoptosis was measured in the same microscopic setup as described. For this purpose, the fluorochrome Hoechst 33342 (0.02 mL, 0.2 μg/mL, Molecular Probes, Leiden, The Netherlands) was topically applied onto the liver surface for staining of hepatocyte DNA. Hoechst is a fluorescent dye that has been widely used for analysis of nuclear morphology, eg, nuclear condensation and fragmentation in cultured hepatocytes and endothelial cells.13 After exsanguination and 10 minutes of incubation, 6 microscopic fields (using the ×63 objective) were recorded for off-line quantification of hepatocyte nuclei showing signs of apoptosis (chromatin condensation and fragmentation). Hepatocyte apoptosis is given as the percentage of hepatocytes showing apoptotic features of the total number of apoptotic hepatocytes. We have previously shown a strong correlation between caspase-3 levels and apoptosis determined by Hoechst staining.14
Reverse-Transcription Polymerase Chain Reaction
Total RNA was extracted from whole liver tissue using an acid guanidinium-phenol-chloroform method (TRIzol Reagent) and treated with RNase-free DNase (DNase I) according to the manufacturer's protocol to remove potential contamination from genomic DNA. RNA concentrations were determined by measuring the absorbance spectrophotometrically at 260 nm. Reverse-transcription polymerase chain reaction (RT-PCR) was performed with the SuperScript One-Step RT-PCR system. Each reaction contained 500 ng total liver RNA as template and 0.2 μM of each primer in a final volume of 50 μL. Mouse β-actin served as an internal control gene. The RT-PCR profile was 1 cycle of DNA synthesis at 50°C for 30 minutes followed by 40 cycles of denaturation at 94°C for 30 seconds, annealing at 55°C, and extension at 72°C for 1 minute, followed by 1 cycle of final extension at 72°C for 10 minutes. After RT-PCR, aliquots of the RT-PCR products were separated on 2% agarose gel containing ethidium bromide and photographed. The primer sequences of TNF-α, FasL, and β-actin were as follows: TNF-α (f) 5′-GGC AGG TCT ACT TTG GAG TCA TTG-3′, TNF-α (r) 5′-ACA TTC GAG GCT CCA GTG AAT TCG-3′; FasL (f) 5′ CAG CTC TTC CAC CTG CAG AAG G-3′, FasL (r) 5′-AGA TTC CTC AAA ATT GAT CAG AGA GAG-3′; β-actin (f) 5′-ATG TTT GAG ACC TTC AAC ACC-3′, β-actin (r) 5′-TCT CCA GGG AGG AAG AGG AT-3′.
Enzyme-Linked Immunosorbent Assay (ELISA)
The right liver lobe was weighed and washed in PBS. The liver sample was homogenized in an antiprotease buffer and centrifuged, and the amount of cytokine-induced neutrophil chemoattractant (KC) in the supernatant was analyzed by use of a double antibody Quantikine ELISA kit (R & D Systems) using recombinant murine KC as standard. Data are given as pg KC per milligram of liver tissue. The minimal detectable protein concentrations were less than 0.5 pg/mL.
Statistical Analyses
Data are presented as mean values ± SEMs. Statistical evaluations were performed using Kruskal-Wallis one-way analysis of variance on ranks followed by multiple comparisons versus control group (Dunn method). P < 0.05 was considered significant, and n represents the number of animals.
RESULTS
Hepatocellular Damage and Apoptosis
Initially, doses of LPS and SEA were titrated out to find specific regimens inducing similar liver damage. It was found that administration of 10 μg LPS or 50 μg SEA in combination with 18 mg d-galactosamine caused an extensive and similar damage to the liver, demonstrated by the increase in liver enzymes and apoptosis (Fig. 1 and 2). For example, it was found that LPS and SEA increased ALT by 29-fold and 24-fold, respectively (Fig. 1A; P < 0.05 vs. control; n = 5–7). Moreover, LPS and SEA enhanced the level of apoptosis in the liver by more than 17-fold and 12-fold, respectively (Fig. 2; P < 0.05 vs. control; n = 5–7). Interestingly, FasL-deficient mice were protected against the increase in liver enzymes and apoptosis induced by SEA, ie, liver enzymes were reduced by more than 63% (Fig. 1; P < 0.05 vs. wild-types; n = 5–6), and apoptosis was decreased by 57% (from 29.8% ± 4.4% to 13.1% ± 2.7%; Fig. 2; P < 0.05 vs. wild-types; n = 5–6). However, mice lacking TNF-α were not protected from SEA-induced hepatocellular injury and apoptosis (Fig. 1 and 2). Conversely, we observed that TNF-α–deficient but not FasL gene-targeted animals were protected against hepatic injury provoked by LPS challenge (Fig. 1 and 2). Thus, liver enzymes and apoptosis were reduced by 88% and 69%, respectively, in LPS-treated TNF-α–deficient mice (P < 0.05 vs. wild-types; n = 5–6). Next, we analyzed the hepatic gene expression of TNF-α and FasL in response to LPS and SEA treatment. Total RNA was isolated from the liver, reverse transcribed into cDNA, and PCR-amplified with specific primers for TNF-α and FasL. The results showed that the expressions of TNF-α and FasL mRNA were low in negative controls, and that LPS challenge caused a clear-cut expression of TNF-α mRNA, but only a moderate expression of FasL. In contrast, administration of SEA had only a minor effect on the gene-expression of TNF-α in the liver, whereas SEA induced a strong expression of FasL mRNA (Fig. 3A and B).

FIGURE 1. Liver enzymes 7 hours after treatment with LPS (10 μg) or SEA (50 μg) in combination with Gal (18 mg). Negative control animals received only PBS (control). Mice were wild-types (WT), TNF-α–deficient and FasL-deficient (–/–). The levels of (A) ALT and (B) AST were determined spectrophotometrically. *P < 0.05 versus LPS + WT and #P < 0.05 versus SEA + WT. Data represent means ± SEMs, and n = 5–6.

FIGURE 2. Apoptosis of hepatocytes 7 hours after treatment with LPS (10 μg) or SEA (50 μg) in combination with Gal (18 mg). Negative control animals received only PBS (control). Mice were wild-types (WT), TNF-α–deficient and FasL-deficient (–/–). Hepatocyte apoptosis is given as the percentage of observed hepatocyte nuclei with morphologic signs of apoptosis, ie, chromatin condensation and fragmentation, after administration of the fluorochrome Hoechst 33342. *P < 0.05 versus LPS + WT and #P < 0.05 versus SEA + WT. Data represent means ± SEMs, and n = 5–6.

FIGURE 3. Expression of (A) TNF-α mRNA and (B) FasL in the liver. β-Actin serves as an housekeeping gene. RNA was isolated from the liver 7 hours after treatment with LPS (10 μg) or SEA (50 μg) in combination with Gal (18 mg). Negative control animals received PBS. The results presented are from 1 experiment, which is representative of 3 others performed.
Microvascular Perfusion
Deterioration of microvascular perfusion is a key feature of liver failure. Herein, we found that the reduction of sinusoidal perfusion in LPS-treated mice was abolished in TNF-α–deficient mice, but not in mice lacking FasL (Fig. 4; n = 6–7). Conversely, SEA-induced perfusion failure was significantly reversed in FasL gene-targeted mice but not in TNF-α–deficient mice (Fig. 4; n = 5–6).
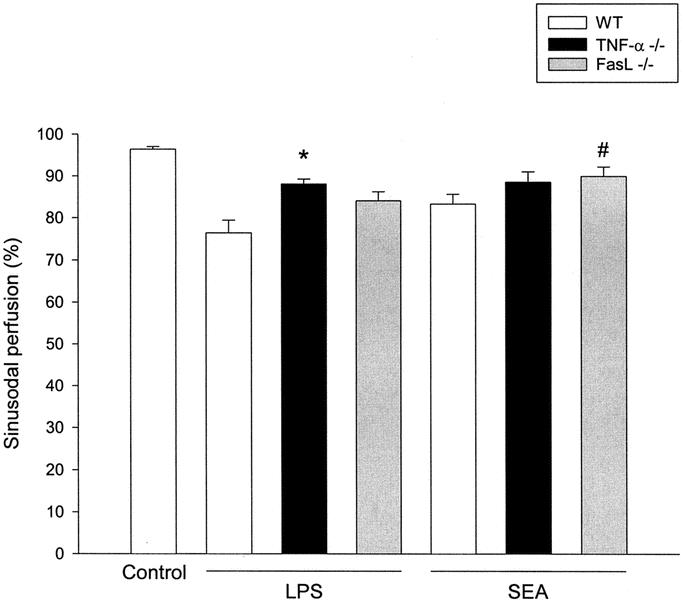
FIGURE 4. Sinusoidal perfusion in the murine liver 7 hours after treatment with LPS (10 μg) or SEA (50 μg) in combination with Gal (18 mg). Negative control animals received only PBS (control). Mice were wild-types (WT), TNF-α–deficient and FasL-deficient (–/–). Sinusoidal perfusion is given as the percentage of observed sinusoids with functional perfusion. *P < 0.05 versus LPS + WT and #P < 0.05 versus SEA + WT. Data represent means ± SEMs, and n = 5–6.
Leukocyte Recruitment
The number of rolling and adherent leukocytes was 2.0 ± 0.4 cells/min and 1.6 ± 0.5 cells/mm, respectively, in control animals (Fig. 5; n = 5). Challenge with LPS markedly increased the leukocyte responses in the postsinusoidal venules. In fact, administration of LPS increased leukocyte rolling by 9-fold, ie, to 17.8 ± 3.2 cells/min (Fig. 5A; P < 0.05 vs. control; n = 5–6). Moreover, LPS treatment enhanced firm leukocyte adhesion by 18-fold, ie, to 29.4 ± 3.5 cells/mm (Fig. 5B; P < 0.05 vs. control; n = 5–6). In TNF-α–deficient mice, LPS-induced leukocyte rolling and adhesion were reduced down to 7.8 ± 2.2 cells/min and 11.1 ± 0.7 cells/mm, corresponding to a more than 56% and 62% decrease, respectively (Fig. 5; P < 0.05 vs. wild-types; n = 5–6). Leukocytes trapped in sinusoids can also exert damaging impact on the liver.15,16 Indeed, LPS markedly augmented the number of sequestered leukocytes in the hepatic sinusoids (Fig. 5C; P < 0.05 vs. control; n = 5–6). The LPS-induced trapping of leukocytes in the sinusoids was reduced by 66% in TNF-α–deficient mice (Fig. 5C; P < 0.05 vs. wild-types; n = 5–6), but no change was observed in FasL gene-targeted animals compared with wild-types (Fig. 5C; n = 5–7). In contrast with LPS, it was found that challenge with SEA did not increase accumulation of leukocytes in postsinusoidal venules nor in sinusoids (Fig. 5C; P > 0.05 vs. control; n = 5–6). In separate experiments, we found that challenge with TNF-α and Jo-2 (a Fas receptor agonist) caused clear-cut liver injury, but only TNF-α provoked significant leukocyte recruitment. In fact, in TNF-α–challenged mice, ALT and AST were increased by more than 8-fold and 14-fold, respectively (to 7.0 ± 1.9 and 14.6 ± 2.5 μKat/L; P < 0.05 vs. PBS control; n = 5). Leukocyte rolling and firm adhesion increased by more than 11-fold and 14-fold, respectively, to 25.2 ± 1.5 cells/min and 23.7 ± 1.8 cells/mm in response to TNF-α challenge (P < 0.05 vs. PBS control; n = 5). Interestingly, after challenge with Jo-2, ALT and AST were increased by more than 13-fold and 10-fold, respectively (to 10.6 ± 2.9 and 11.7 ± 3.9 μKat/L; P < 0.05 vs. PBS control; n = 5), whereas leukocyte rolling and firm adhesion were not significantly increased from 2.1 ± 1.0 to 5.1 ± 1.8 cells/min and from 1.7 ± 1.1 to 4.7 ± 0.7 cells/mm, respectively (P > 0.05 vs. PBS control; n = 5).

FIGURE 5. Leukocyte (A) rolling and (B) firm adhesion in hepatic postsinusoidal venules, and (C) sinusoidal trapping of leukocytes 7 hours after treatment with LPS (10 μg) or SEA (50 μg) in combination with Gal (18 mg). Negative control animals received only PBS (control). Mice were wild-types (WT), TNF-α–deficient and FasL-deficient (–/–). *P < 0.05 versus LPS + WT and #P < 0.05 versus control. Data represent means ± SEMs, and n = 5–6.
Next, we examined the effect of inhibiting leukocyte recruitment on septic liver injury, by immunoneutralization of P-selectin. As shown previously,14 we found that pretreatment with the anti–P-selectin antibody significantly reduced leukocyte rolling and adhesion in livers of LPS-treated mice (Table 1; P < 0.05; n = 5). In contrast, it was observed that inhibition of P-selectin had no effect on the increase in liver enzymes and apoptosis provoked by SEA (Table 1; P > 0.05; n = 4). Moreover, considering that CXC chemokines such as KC play an important role in endotoxin-induced leukocyte extravasation and hepatic injury,17 it was of interest to examine the liver production of KC. It was found that baseline expression of KC was 3.7 ± 2.4 pg/mg of liver tissue in control mice (n = 5). Challenge with LPS significantly increased the KC content of the livers to 12.8 ± 1.4 pg/mg (P < 0.05 vs. control; n = 5). However, the KC levels in SEA-treated mice were not different from baseline levels, ie, 4.0 ± 0.8 pg/mg liver tissue (P > 0.05 vs. control; n = 5). The number of circulating leukocytes was not different between the groups (not shown).
TABLE 1. Role of P-Selectin in Endotoxin-Induced and Superantigen-Induced Leukocyte Recruitment and Liver Injury

DISCUSSION
This study points to important mechanistic differences underlying the pathogenesis of Gram-positive and Gram-negative sepsis. Our data demonstrate that FasL is a key mediator in superantigen-induced liver injury, whereas endotoxin-induced liver damage is independent of FasL. Instead, it was found that TNF-α plays a critical role in the hepatic damage provoked by endotoxin. In contrast with endotoxin, superantigen did not stimulate leukocyte accumulation or expression of the chemokine KC in the liver. Knowing that Gram-positive bacteria constitute an increasingly frequent etiology in sepsis, these findings may be important when considering specific molecular therapy in septic patients.
A major shift in the bacterial etiology of sepsis has occurred during the last 2 decades,2 ie, an increasing prevalence of sepsis caused by Gram-positive bacteria has been noted. It has been widely held that the initiation of the systemic inflammatory response with respect to downstream release of proinflammatory cytokines results in a similar pathophysiologic process, regardless of the causative microbial pathogen. However, such a paradigm is directly challenged by the present results. In addition, an accumulating body of scientific data indicates fundamental differences in the host response to Gram-negative and Gram-positive organisms.1 Considering that traditional studies of sepsis have mainly focused on endotoxin-mediated effects on the immune system, it is of critical importance to define potential differences in the molecular mechanisms behind sepsis caused by Gram-negative and Gram-positive organisms.1 Therefore, the purpose of the present study was to determine the relative contribution of TNF-α and FasL to the liver damage induced by endotoxin and superantigen. We found that administration of our titrated doses of LPS and SEA provoked a similar hepatic injury as measured by the increase in serum levels of ALT and AST in addition to hepatocyte apoptosis, which is pertinent when comparing disease-causing mechanisms of different microbial stimuli. Notably, it was observed that FasL-deficient mice were markedly protected from SEA-provoked liver injury, whereas no protection was observed when these mice were challenged with LPS, suggesting that FasL is an important mediator in superantigen-induced but not LPS-induced hepatic injury. In this context, it is worth noting that TNF-α gene-targeted animals were completely protected from LPS-induced liver injury, which is in line with previous data in the literature.6 However, we found no significant difference in liver enzymes between TNF-α–deficient and wild-type mice challenged with SEA. Hepatocyte apoptosis is a prominent and important pathophysiologic feature in septic liver injury. We used the fluorochrome Hoechst 33342 to determine nuclear condensation and fragmentation, which we have validated for quantitative analyses of apoptosis by demonstrating an intimate correlation with changes in caspase-3 levels in the liver.14 It was found that both LPS and SEA significantly increased the level of apoptosis in the liver. LPS-induced apoptosis was reduced by 69% in TNF-α–deficient but not in FasL-deficient mice, adding further evidence for the important role of TNF-α in the liver damage associated with endotoxemia. Our observation that FasL-deficient mice are sensitive to LPS challenge is in line with a previous report showing that administration of a Fas fusion protein had no effect on LPS-induced hepatocyte apoptosis and liver injury.6 Conversely, we observed that FasL gene-targeted but not TNF-α–deficient animals were significantly protected from SEA-provoked apoptosis in the liver. Considered together, these findings suggest that FasL is a dominant mediator of superantigen-induced liver injury, whereas LPS-provoked hepatic damage is predominately mediated by TNF-α. This notion is indirectly supported by in vitro data showing that the spectrum of cytokine secretion from leukocytes stimulated with LPS and SEA are different—eg, it was demonstrated that LPS is much more potent than SEA in inducing TNF-α secretion.18,19 Indeed, we found that the gene expression of TNF-α in the liver was substantially higher in response to LPS than SEA in the present study. Moreover, mRNA expression of FasL in the liver was markedly increased in SEA-challenged mice, but only moderate in mice challenged with LPS. In this context, it is interesting to note that patients with Gram-negative sepsis have higher levels of circulating TNF-α than those with Gram-positive sepsis.20,21
Numerous studies have shown that infiltration and activation of leukocytes constitute a rate-limiting step in endotoxin-induced liver injury.22–24 It was recently shown that endotoxin-induced leukocyte recruitment is a multistep process, initiated by selectin-mediated rolling,24 mainly P-selectin,14 in postsinusoidal venules. Moreover, it has been documented that CD18 mediates stationary adhesion of leukocytes to the endothelium in hepatic venules.24 In this context, it should be mentioned that some studies have suggested that the associated sinusoidal sequestration of leukocytes also may contribute to hepatocellular liver injury, although a definitive contribution of sinusoidal leukocytes to the tissue damage remains controversial.15,24 As expected, LPS provoked a clear-cut leukocyte response in the liver, including a marked increase in leukocyte rolling and adhesion in postsinusoidal venules and substantial trapping of leukocytes in the sinusoids. Indeed, LPS-induced leukocyte responses were significantly attenuated in TNF-α–deficient mice but intact in mice lacking FasL, suggesting that TNF-α–mediated leukocyte recruitment is a key feature in the pathophysiology of endotoxin-induced liver injury. This notion is also in line with the fact that TNF-α per se can provoke hepatic leukocyte recruitment.24,25 Moreover, immunoneutralization of P-selectin, which markedly protects against LPS-induced leukocyte recruitment and tissue injury in the liver,14 had no effect on SEA-induced liver damage. This lack of effect of inhibiting P-selectin may be related to the observation herein that SEA is weak inducer of TNF-α gene expression. In addition, we observed in separate experiments that activation of the Fas receptor with a monoclonal antibody (Jo-2) caused a marked hepatocellular damage but no significant leukocyte response in the liver. In contrast, TNF-α challenge induced both liver injury and leukocyte recruitment. These findings suggest that superantigen-induced and FasL-dependent liver injury is independent of leukocyte recruitment. This conclusion is also supported by the observation that inhibition of P-selectin (which protects against LPS-induced liver injury) had no effect on the hepatic damage provoked by SEA challenge. Furthermore, we found that, in contrast with LPS, SEA did not increase the local production of KC, which has been shown to be a key chemokine regulating extravascular recruitment of leukocytes in the liver.17 Considered collectively, our data indicate that the hepatic damage caused by SEA is effectuated in the absence of significant leukocyte recruitment. However, it should be noted that leukocyte responses were significantly higher in FasL-deficient mice after treatment with SEA compared with controls. The reason for this small difference is not known at present but may be related to increased leukocyte survival (ie, decreased apoptosis) in the FasL-deficient mice.
Taken together, these novel results demonstrate a key role of FasL, but not TNF-α, in superantigen-induced hepatotoxicity in the mouse. Moreover, our data indicate that, in contrast with LPS, SEA-induced liver injury is independent of TNF-α and KC function and leukocyte recruitment. Thus, this study supports the emerging paradigm that sepsis caused by different microbial organisms may display distinct pathophysiologic mechanisms, which may be not only important for an increased understanding of the disease mechanisms but also critical as a basis for the development of more specific therapies in septic patients.
ACKNOWLEDGMENTS
This work was supported by grants from the Swedish Research Council (2001–6576, 2002–955, 2002–8012, 2003–4661), Crafoordska stiftelsen, Blanceflors stiftelse, Einar och Inga Nilssons stiftelse, Harald och Greta Jaenssons stiftelse, Greta och Johan Kocks stiftelser, Fröken Agnes Nilssons stiftelse, Magnus Bergvalls stiftelse, Mossfelts stiftelse, Nanna Svartz stiftelse, Ruth och Richard Julins stiftelse, Svenska Läkaresällskapet and Teggers stiftelse, Allmäna sjukhusets i Malmö stiftelse för bekämpande av cancer, MAS fonder, and Lund University.
Discussions
Dr. Rogiers: I would like to thank you for sending me the paper, and I would like to congratulate you on the clean experiment, very nicely and adequately presented. I am not an expert in this field, but I will still ask you some questions.
You demonstrate very elegantly 2 different pathways for the hepatotoxicity. But there is a difference in the inflammatory reaction and the leukocyte addition. Can you speculate what the reason behind this is? The second question I have is this. Both in your paper and in your talk, you speak about the potential of developing specific therapies from this. Can you also speculate in which direction this goes?
Dr. Thorlacius: The mechanism is that to get leucocytes out into the tissue, appropriate inflammatory mediators must cooperate. One critical mediator is TNF-α, and as you have seen in our data, superantigen is not an effective inducer of TNF-α. The reason on the mechanistic level is that superantigens do not provoke the correct mediators to provoke leukocyte recruitment. It has also been shown in Gram-negative and Gram-positive sepsis in the clinic that TNF-α levels are much higher in Gram-negative sepsis than in Gram-positive sepsis. Our data are in line with the clinical observations in patients. I think that explains the reason that superantigens do not provoke leukocyte recruitment in the liver.
On your question of how this may affect the development of specific therapies, I think that starting back from the studies where we have seen the failures of inhibiting TNF-α, most people have come to the conclusion that it was a time-dependent effect, ie, one comes too late with anti–TNF-α treatment. That might be true, but also, anti–TNF-α compounds were not very effective in inhibiting the function of TNF-α. Moreover, in light of our present findings, it may be suggested that an additional mechanism could be that the patients were not very well characterized. Most septic patients actually have combined Gram-positive and Gram-negative sepsis. Considering that Gram-positive and Gram-negative bacteria potentiate each other by a factor higher than 50,000, one can easily imagine that small amounts of superantigen are needed to cause very severe disease. If this holds true, it is apparent that we have to define patients better for clinical studies. It is also apparent that different people respond differently to specific antigens. This is a very complex problem. You have to define not only the exact bacteriologic etiology of the patients but also patients' molecular response to specific microbes.
Dr. Clavien: Dr. Thorlacius, congratulations for this beautiful study. The data are novel and convincingly show that staphylococcal A antigen causes liver injury by a much different mechanism than LPS. I have 3 questions. First, the data set would be bolstered significantly if FasL was also measured by PCR, optimally real-time PCR, as presented for TNF-α. Did you measure and quantify FasL? Second, you mentioned only the Hoechst stain to assess apoptosis. Did you look at other markers such as Tunel staining, caspase activity, or cytochrome C release into the cytoplasm, which might better document apoptotic cell death? My last question deals with the source of FasL. There are emerging data suggesting that NK/NKT cells are the source of FasL in various models of liver injury. Therefore, it is tempting to speculate that staphylococcal A antigen activates these cells to produce Fas ligands. Did you consider experiments deleting NK/NKT cells, for example by anti-NK 1.1 antisera? This would provide further insight into the mechanism of injury caused by Gram-positive organisms. Like any fine studies, this work has generated further important questions.
Dr. Thorlacius: Thank you very much for these insightful questions. These are of course all critical points. The first point on the FasL expression: our hypothesis was that when we give superantigen, it does not activate liver but intestinal T cells. We collected blood but could not detect FasL in the blood. The next step, which answers your third question, is that we now think that this is a local effect in the liver, either through the NKT cells or the T cells, cells that express T-cell receptors, perhaps not secreting it but rather expressing FasL on the surface of the cells. We are now isolating different subpopulations of the T-cell receptor-positive cells in the liver for measuring FasL expression. Apoptosis: I agree with you. More methods are always better, but we have just recently validated this morphologic method, and it correlates very well with levels of caspase-3 in the liver. Of course, it would have added more information to determine the levels of caspase-3 also. Actually, I like this morphologic method, considering the original description of apoptosis, which was based on morphologic features.
Footnotes
Reprints: Henrik Thorlacuis, MD, PhD, Department of Surgery, Malmö University Hospital, S-205 02, Malmö, Sweden. E-mail: Henrik.Thorlacius@kir.mas.lu.se.
*These authors contributed equally to this work.
REFERENCES
- 1.Opal SM, Cohen J. Clinical gram-positive sepsis: does it fundamentally differ from gram-negative bacterial sepsis? Crit Care Med. 1999;27:1608–1616. [DOI] [PubMed] [Google Scholar]
- 2.Martin GS, Mannino DM, Eaton S, et al. The epidemiology of sepsis in the United States from 1979 to 2000. N Engl J Med. 2003;348:1546–1554. [DOI] [PubMed] [Google Scholar]
- 3.Bradham C, Plumpe J, Manns M, et al. Mechanisms of hepatic toxicity. Am J Physiol. 1998;275:G387–G392. [DOI] [PubMed] [Google Scholar]
- 4.Wallach D, Kovalenko A, Varfolomeev E, et al. Death-inducing functions of ligands of the tumor necrosis factor family: a Sanhedrin verdict. Curr Opin Immunol. 1998;10:279–288. [DOI] [PubMed] [Google Scholar]
- 5.Rudiger HA, Clavien PA. Tumor necrosis factor α, but not Fas, mediates hepatocellular apoptosis in the murine ischemic liver. Gastroenterology. 2002;122:202–210. [DOI] [PubMed] [Google Scholar]
- 6.Josephs MD, Bahjat R, Fukuzuka K, et al. Lipopolysaccharide and D-galactosamine-induced hepatic injury is mediated by TNF-α and not by Fas ligand. Am J Physiol. 2000;278:R1196–R1201. [DOI] [PubMed] [Google Scholar]
- 7.Wang JE, Jorgensen PF, Almlof M, et al. Peptidoglycan and lipoteichoic acid from Staphylococcus Aureus induce tumor necrosis factor alpha, interleukin 6 (IL-6), and IL-10 production in both T cells and monocytes in a human whole blood model. Infect Immun. 2000;68:3965–3970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marrack P, Kappler J. The staphylococcal enterotoxins and their relatives. Science. 1990;248:705–711. [DOI] [PubMed] [Google Scholar]
- 9.Vincent JL, Bihari DJ, Suter PM, et al. The prevalence of nosocomial infections in intensive care units in Europe: results of the European Prevalence of Infection in Intensive Care (EPIC) Study. EPIC International Advisory Committee. JAMA. 1995;274:639–644. [PubMed] [Google Scholar]
- 10.Richards MJ, Edwards JR, Culver DH, et al. Nosocomial infections in medical intensive care units in the United States. National Nosocomial Infections Surveillance System. Crit Care Med. 1999;27:887–892. [DOI] [PubMed] [Google Scholar]
- 11.Tarkowski A, Collins LV, Jonsson I-M, et al. Microbial superantigens as virulence factors and ways to counteract their actions. Scand J Infect Dis. 2003;35:642–646. [DOI] [PubMed] [Google Scholar]
- 12.Schlievert PM. Enhancement of host susceptibility to lethal endotoxin shock by staphylococcal pyrogenic exotoxin type C. Infect Immun 1982;36:123–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rauen U, Polzar B, Stephan H, et al. Cold-induced apoptosis in cultured hepatocytes and liver endothelial cells: mediation by reactive oxygen species. FASEB J. 1999;13:155–168. [DOI] [PubMed] [Google Scholar]
- 14.Klintman D, Li X, Thorlacius H. Important role of P-selectin in leukocyte recruitment, hepatocellular injury and apoptosis in endotoxemic mice. Clin Diagn Lab Immunol. 2004;11:56–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chosay JG, Essani NA, Dunn CJ, et al. Neutrophil margination and extravasation in sinusoids and venules of liver during endotoxin-induced injury. Am J Physiol. 1997;272:G1195–G1200. [DOI] [PubMed] [Google Scholar]
- 16.Yadav S, Howell DN, Gao W, et al. L-selectin and ICAM-1 mediate reperfusion injury and neutrophil adhesion in the warm ischemic mouse liver. Am J Physiol. 1998;275:G1341–1352. [DOI] [PubMed] [Google Scholar]
- 17.Li X, Klintman D, Wang Y, et al. Critical role of CXC chemokines in the extravasation process of leukocytes in endotoxemia. J Leukoc Biol. 2004;75:443–452. [DOI] [PubMed] [Google Scholar]
- 18.Bjork L, Andersson J, Ceska M, et al. Endotoxin and Staphylococcus aureus enterotoxin A induce different patterns of cytokines. Cytokine 1992;4:513–519. [DOI] [PubMed] [Google Scholar]
- 19.Muller-Alouf H, Alouf JE, Gerlach D, et al. Comparative study of cytokine release by human peripheral blood mononuclear cells stimulated with Streptococcus pyogenes superantigenic erythrogenic toxins, heat-killed streptococci, and lipopolysaccharide. Infect Immun. 1994;62:4915–4921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cohen J, Carlet J. INTERSEPT: an international, multicenter, placebo-controlled trial of monoclonal antibody to human tumor necrosis factor-alpha in patients with sepsis. International Sepsis Trial Study Group. Crit Care Med. 1996;24:1431–1440. [DOI] [PubMed] [Google Scholar]
- 21.Fisher CJ Jr., Opal SM, Dhainaut JF, et al. Influence of an anti-tumor necrosis factor monoclonal antibody on cytokine levels in patients with sepsis. The CB0006 Sepsis Syndrome Study Group. Crit Care Med. 1993;21:318–327. [DOI] [PubMed] [Google Scholar]
- 22.Holman JM Jr, Saba TM. Hepatocyte injury during post-operative sepsis: activated neutrophils as potential mediators. J Leukoc Biol. 1988;43:193–203. [DOI] [PubMed] [Google Scholar]
- 23.Jaeschke H, Farhood A, Smith CW. Neutrophil-induced liver cell injury in endotoxin shock is a CD11b/CD18-dependent mechanism. Am J Physiol. 1991;261:G1051–G1056. [DOI] [PubMed] [Google Scholar]
- 24.Klintman D, Schramm R, Menger MD, et al. Leukocyte recruitment in hepatic injury: selectin-mediated leukocyte rolling is a prerequisite for CD18-dependent firm adhesion. J Hepatol. 2002;36:53–59. [DOI] [PubMed] [Google Scholar]
- 25.Fox-Robichaud A, Kubes P. Molecular mechanisms of tumor necrosis factor alpha-stimulated leukocyte recruitment into the murine hepatic circulation. Hepatology. 2000;31:1123–1127. [DOI] [PubMed] [Google Scholar]