

Abstract
As an alternative to the scanning mechanism of initiation, the direct-internal-initiation mechanism postulates that the translational machinery assembles at the AUG start codon without traversing the entire 5′ untranslated region (5′-UTR) of the mRNA. Although the existence of internal ribosome entry sites (IRESs) in viral mRNAs is considered to be well established, the existence of IRESs in cellular mRNAs has recently been challenged, in part because when testing is carried out using a conventional dicistronic vector, Northern blot analyses might not be sensitive enough to detect low levels of monocistronic transcripts derived via a cryptic promoter or splice site. To address this concern, we created a new promoterless dicistronic vector to test the putative IRES derived from the 5′-UTR of an mRNA that encodes the translation initiation factor eIF4G. Our analysis of this 5′-UTR sequence unexpectedly revealed a strong promoter. The activity of the internal promoter relies on the integrity of a polypyrimidine tract (PPT) sequence that had been identified as an essential component of the IRES. The PPT sequence overlaps with a binding site for transcription factor C/EBPβ. Two other transcription factors, Sp1 and Ets, were also found to bind to and mediate expression from the promoter in the 5′-UTR of eIF4G mRNA. The biological significance of the internal promoter in the eIF4G mRNA might lie in the production of an N-terminally truncated form of the protein. Consistent with the idea that the cryptic promoter we identified underlies the previously reported IRES activity, we found no evidence of IRES function when a dicistronic mRNA containing the eIF4G sequence was translated in vitro or in vivo. Using the promoterless dicistronic vector, we also found promoter activities in the long 5′-UTRs of human Sno and mouse Bad mRNAs although monocistronic transcripts were not detectable on Northern blot analyses. The promoterless dicistronic vector might therefore prove useful in future studies to examine more rigorously the claim that there is IRES activity in cellular mRNAs.
Translational control plays an important role in regulating gene expression in eukaryotic cells (9, 24, 30, 52, 54, 69, 73). Most of the translational regulation occurs at the level of initiation, which is usually the rate-limiting step in protein synthesis (15, 29, 44, 50, 68). Initiation of translation in eukaryotes normally depends on the m7GpppN 5′-cap structure of mRNA, which recruits the 43S ribosome preinitiation complex via interaction with the cap binding protein eIF4E (69). The translational machinery then migrates downstream until it meets the first AUG codon in the optimal context (41, 43). This scanning model implies that the migration of the 43S preinitiation complex is inhibited by any long and highly structured 5′-untranslated regions (5′-UTRs). About 90% of 5′-UTRs of vertebrate mRNAs have a length in the range between 100 and 300 bases, which is compatible with the ribosome-scanning model. However, the remaining mRNAs have longer atypical 5′-UTRs which may also contain one or more AUG codons. These mRNAs may not be efficiently translated using the cap-dependent initiation and ribosome-scanning mechanism (73). Furthermore, cap-dependent translation initiation is compromised under many cellular conditions including mitosis (5), apoptosis (10, 49), cellular stresses such as hypoxia and heat shock (55, 60, 64), and viral infection (45, 53, 56).
Under the aforementioned cellular conditions, many proteins still need to be synthesized possibly via the internal ribosome entry site (IRES)-mediated initiation mechanism (36). In this mechanism, the ribosome preinitiation complex is recruited directly to the IRES element independent of the 5′-cap structure and begins to scan within the vicinity of the initiator AUG codon by effectively avoiding a large part of the 5′-UTR sequence. The IRES-mediated initiation was first demonstrated a decade ago in picornaviruses (3, 19, 27, 71). Picornavirus mRNAs are naturally uncapped at their 5′ ends. Their 5′-UTRs usually have significant secondary structure spanning approximately over 500 bases and are punctuated by multiple AUG codons. Such a structure is predicted to inhibit the ribosome recruitment and linear ribosome scanning. However, these 5′-UTRs can effectively confer a cap-independent translation by directly recruiting ribosomes.
Recently, it has been suggested that some cellular mRNAs may also use the IRES-mediated initiation mechanism (27, 61, 71). Indeed, over 40 mRNAs have been reported to use IRESs to direct translation initiation, and the list is growing rapidly. An IRES database (http://www.rangueil.inserm.fr/IRESdatabase) has been created to reflect their potential importance and to categorize the cellular IRES elements. However, the existence of such cellular IRESs has been questioned recently due to the inevitable drawbacks of the conventional dicistronic test and the fact that dicistronic mRNA assays do not work for the known cellular IRESs (42). This has generated debate over the existence of cellular IRESs and how studies should be conducted on future cellular IRES candidates (63). In most of the previous cellular IRES studies, an artificial dicistronic construct was used in which a candidate IRES element is inserted between the two cistrons. The upstream cistron acts as an indicator of cap-dependent translation, and the downstream cistron indicates the IRES activity. This approach is considered a “gold standard” for characterizing cellular IRESs (61). However, it is acknowledged by both sides of the debate that one of the major questions is whether cryptic promoter or potential splice sites exist in the “cellular IRES” sequence of the dicistronic construct that may generate monocistronic mRNA of the second cistron. Although RNA analyses such as Northern blotting were conducted to rule out the existence of monocistronic mRNA in most previous studies. Kozak (42, 63) argued that the RNA analysis approaches used in these studies were not sensitive enough to rule out the existence of monocistronic mRNA present at 5% or less of the dicistronic mRNA level. Thus, some of the previously reported cellular IRESs might be falsely identified, and it is clear that relevant rigorous experimental methods are needed to clarify these disputes.
In this study, we created a new promoterless dicistronic vector to rule out the existence of cryptic promoters in any potential cellular IRES elements. This analysis also relies on enzyme assays in the same way as the conventional dicistronic test for IRES activity and therefore is more compatible with IRES study than any existing RNA analysis approaches. Using this vector, we first tested the 5′-UTR of eukaryotic translation initiation factor 4GI (eIF4G), the largest subunit of eIF4F. eIF4G is important for translational control by acting as a molecular scaffold and coordinating the activities of eIF3, eIF4A, eIF4E, and poly(a)-binding protein (PABP) (22, 28, 39). The translation of eIF4G is cap independent under conditions of viral infection (37) and involves an IRES-dependent mechanism (20, 21, 37). The 5′-UTR of eIF4G has been functionally characterized and shown to have a polypyrimidine tract (PPT) region that is essential for IRES activity (20, 21).
Using the novel promoterless dicistronic vector, we demonstrated that a strong promoter activity exists in the 5′-UTR sequence of eIF4G that can explain the enhanced expression of the second cistron observed in the conventional dicistronic tests. Further mapping of the promoter sequence in the 5′-UTR of eIF4G showed that the PPT sequence was absolutely required for the promoter activity. Using gel shift and supershift analysis, we showed that transcription factor C/EBPβ binds to this PPT sequence. In addition, the 5′-UTR of eIF4G could not direct IRES-dependent translation in rabbit reticulocyte lysate (RRL) or in HeLa cells when dicistronic RNA transcripts were analyzed. Thus, the cryptic promoter in the 5′-UTR of eIF4G underlies the enhanced expression of the second cistron observed in the conventional dicistronic test. Using the same promoterless dicistronic vector, we also observed promoter activities in the 5′-UTR sequences of human Sno and mouse Bad, although no monocistronic transcripts were detected by northern blot. Hence, cryptic promoter activities in the 5′-UTR sequences of cellular mRNAs are more prevalent than anticipated, and they can be misinterpreted to be IRES sequences in the conventional dicistronic analysis due to the low intrinsic sensitivity of Northern blot analysis for ruling out the existence of monocistronic transcripts. We propose that the promoterless dicistronic vector may be used to establish claims of cellular IRES in future studies.
MATERIALS AND METHODS
Materials.
Restriction enzymes, m7GpppG cap analogue, Pfu polymerase, and SL2 cell line were purchased from New England Biolabs, Amersham/Pharmacia Biotech, Stratagene, and the American Type Culture Collection, respectively. T7 and Sp6 RNA polymerases, RNasin, RNase-free DNase, rabbit reticulocyte lysate, Luciferase assay ‘Stop & Glo' kit, pGL3-Promoter plasmid, and pSP64 poly(A) plasmid were from Promega. RNeasy Mini Kit and Oligotex mRNA Mini Kit were from Qiagen. Rediprime II Random Prime Labeling System and [32P]-dCTP were from Amersham Biosciences. The Sephadex G-25 Quick Spin Columns (TE) for radiolabeled DNA purification were from Roche. MAGNA nylon transfer membrane was from Osmonics Inc. Wild-type and mutant C/EBPβ oligonucleotides used in the electrophoretic mobility shift assay (EMSA) and antibodies to nuclear proteins Sp1, Sp3, and C/EBPβ were obtained from Santa Cruz Biotechnology. Schneider's Drosophila culture medium, Lipofectamine Plus, and Lipofectin transfection reagent were purchased from Invitrogen. Oligonucleotides were synthesized by Sigma-Genosys. I.M.A.G.E. expressed sequence tag EST clones were obtained from either the American Type Culture Collection or Research Genetics.
Construction of plasmids.
To engineer dicistronic constructs containing the IRES element of human rhinovirus (HRV) or the 5′-UTR of eIF4G, the dicistronic vector pRF (70) was used. Plasmid pGL2/CAT/4G/LUC containing the 5′-UTR of eIF4G (20) was used as template for PCR to amplify the 5′-UTR of eIF4G by using the following two primers containing SpeI and NcoI sites: 5′-CAAACTAGTTCTAGATGGGGGTCCT-3′ and 5′-CAACCATGGTGATATCCTTTCCTCC-3′. The PCR product was cloned into the SpeI and NcoI sites of pRF vector to obtain plasmid pR-eIF4G-F (see Fig. 1A). The IRES sequence of HRV in pGL3-R-HRV plasmid (70) was released by digestion with SpeI and NcoI and cloned into pRF, resulting in pR-HRV-F.
FIG. 1.

Stimulation of the second-cistron expression by the 5′-UTR sequence of eIF4G. (A) Schematic diagram of dicistronic constructs without insert (pRF), or with the IRES of HRV (pR-HRV-F) and the 5′-UTR of eIF4G (pR-eIF4G-F) in the intergenic region. The locations of several relevant restriction enzyme sites are shown by arrows. (B) Relative luciferase activity generated by the dicistronic constructs. HeLa cells were transfected with pRF, pR-HRV-F, and pR-eIF4G-F constructs. At 24 h following transfection, the cells were harvested, the Renilla and firefly luciferase (R. Luc. and F. Luc.) activities were measured, and the relative ratios were calculated and normalized to that of the vector-transfected cells (RF). The data are from six independent experiments.
To engineer promoterless dicistronic constructs that allow analysis of promoter activity of the DNA insert in the intergenic region, the simian virus 40 (SV40) promoter sequence including the chimeric intron between SmaI and Eco RV was removed from pRF, pR-eIF4G-F, and pR-HRV-F (see Fig. 1A), resulting in pRF (−P), pR-eIF4G-F(−P), and pR-HRV-F(−P), respectively. For all the promoter analysis studies, the promoterless dicistronic constructs were used. In some studies, the SV40 enhancer was deleted by cloning the NheI-HpaI fragment from the promoterless dicistronic plasmid pR-eIF4G-F(−P) into the pGL3-promoter vector (Promega), which contains an SV40 poly (A) signal but does not contain the SV40 enhancer. The resulting construct is named pR-eIF4G-F(−PE). Systematic deletions of the 5′-UTR sequence of eIF4G were generated by PCR. The eIF4G linker-scanning constructs were made by the PCR-based overlap extension technique, which is similar to the PCR-based site-directed mutagenesis method described previously (25). A 10-base linker with the sequence ACTCTAGACT was used to replace wild-type sequences.
To generate poly(A)-tailed in vitro transcripts for RNA transfection study, constructs containing poly(A) were engineered using the vector pSP64 PolyA (Promega), which has a 30-bp (dA-dT) sequence. The EcoRV-XbaI fragment of pRF vector that contains the Renilla luciferase gene was first cloned into the pSP64 PolyA vector at the XbaI and blunted HindIII sites to generate plasmid pSP-RA30. The XbaI fragment of pR-HRV-F that contains the IRES of HRV and the firefly luciferase gene were then isolated and cloned into pSP-RA30 at the XbaI site to generate pSP-R-HRV-FA30. The pSP-RFA30 plasmid was obtained by removing the IRES sequence of HRV from pSP-R-HRV-FA30 by digestion with SpeI and NcoI. To engineer pSP-R-eIF4G-FA30, the 5′-UTR sequence (from −368 to −44) of eIF4G that has been reported to contain full IRES activity (20) was amplified by PCR using the following primers with SpeI and NcoI sites: 5′-CAAACTAGTCTAGATGGGGGTCCT-3′ and 5′-ACACCATGGATTCGGATCTGGGGA-3′. The PCR products were used to replace the IRES sequence of HRV in pSP-R-HRV-FA30.
To clone the 5′-UTRs of mouse Bad, human Sno, and human inhibitor apoptosis 1 (hIAP-1, referred to hereafter as HIAP) in both pRF and pRF(−P) vectors, I.M.A.G.E. EST clones were used as templates. The 405-bp 5′-UTR sequence of mouse Bad (accession no. NM_007522) was amplified using mouse I.M.A.G.E. clone 948554 (accession no. AA544696) with the primers 5′-GACTAGTCGCACACCTATCCTGGCA-3′ and 5′-GCCATGGTTGGATCCTGGAGGCCTG-3′. The 709-bp 5′-UTR sequence of human Sno (accession no: U70730) was assembled using I.M.A.G.E. clones 1713010 (accession no. AI129968) and 211285 (accession no. H69011), and finally amplified by the following two primers: 5′-AACTAGTGGTTTCAAATTGGCCCT-3′ and 5′-GGTTTTCCATGGTACACTCT-3′. The 1.3-kb 5′-UTR sequence of human HIAP (accession no. AF070674) was assembled using human I.M.A.G.E. clones 2349482 (accession no. AI827488) and 428231 (accession no.AA002125) by a combination of PCR and restriction digestion. All of the 5′-UTR cDNA fragments were finally cloned between the SpeI and NcoI sites of the pRF and pRF(−P) vectors, resulting in pR-Bad-F, pR-Sno-F, pR-HIAP-F, pR-Bad-F(−P) and pR-Sno-F(−P). All the above constructs were confirmed by DNA sequencing.
Cell culture and DNA and RNA transfection.
HeLa and H1299 cells were maintained at 37°C under 5% CO2 in Dulbecco modified Eagle medium and RPMI 1640 medium, respectively, supplemented with 10% fetal bovine serum. Schneider's Drosophila cell line 2 (SL2) was maintained at room temperature under atmospheric CO 2 in Schneider's Drosophila medium supplemented with 10% fetal bovine serum.
DNA transfection in both HeLa and H1299 cells was performed with Lipofectamine Plus reagents as specified by the manufacturer. In a 24-well plate, approximately 105 cells/well were plated and were transfected with 0.4 μg of DNA the next day. Cells were harvested for the luciferase assay 24 h following transfection. SL2 cells were transfected using the calcium phosphate method as previously described (25). Briefly, 4 μg of pR-eIF4G-F(−P) or pR-eIF4G-F(−PE) was cotransfected with 2 μg of Drosophila expression plasmids for Sp1, Ets-1 (38), or combination of Sp1 plus Ets-1. Equal amounts of DNA were obtained in each transfection by adding empty vector PacO. Cells were incubated with calcium phosphate-precipitated DNA for 48 h and then harvested for the luciferase assay.
RNA transfection was performed using the cationic liposome-mediated method as described by Dwarki et al (16). Briefly, approximately 2 × 105 cells/well were seeded onto six-well plates on the day before transfection. Opti-MEM I medium (1 ml) in a 12-by 75-mm polystyrene snap-cap tube was mixed with 12.5 μg of Lipofectin reagent and 5 μg of capped mRNA. The liposome-RNA-medium mixture was immediately added to cells. At 8 h following transfection, the cells were harvested and processed for luciferase analysis.
Preparation of cytoplasmic extract (S100).
Cytoplasmic extract (S100) was prepared as previously described (31). Briefly, HeLa cells were collected using rubber policeman, washed with phosphate-buffered saline and then resuspended at a density of 5 × 107 cells/ml in homogenization buffer H100 (10 mM Tris-HCl [pH 7.4], 1.5 mM MgCl2, 10 mM KCl, 0.5 mM dithiothreitol, 0.1 mM phenylmethylsulfonyl fluoride, 10 μg of leupeptin per ml). The cells were homogenized with a Dounce homogenizer for 20 strokes on ice. Cell nuclei were pelleted by centrifugation at 2,000 × g for 10 min. The supernatant was then adjusted to 150 mM KCl and centrifuged at 100,000 × g at 4°C for 90 min. The supernatant was recovered as S100 extract. The extract was flash frozen with 5% glycerol and stored at −70°C.
In vitro transcription and translation.
In vitro transcription and translation were performed as previously described (75). DNA templates with a poly(A) tail were linearized with EcoRI, while DNA templates without a poly(A) tail were linearized with BamHI. Capped transcripts were synthesized in the presence of 1 mM m7GpppG and purified using the Qiagen RNeasy mini kit. A 50-ng portion of capped RNA transcripts was used to program cell-free translation in rabbit reticulocyte lysate (RRL) in a final volume of 10 μl containing 6.5 μl of RRL. The translation mixture contained 2 μl of either H100 buffer or cytoplasmic extract S100 (8.0 μg/μl).
Nuclear extract preparation, EMSA, and UV cross-linking.
HeLa nuclear extract preparation, EMSA, and supershift analysis were done as previously described (25). The two strands of oligonucleotides used as EMSA probes were annealed prior to labeling. The sequences of the sense strand of the oligonucleotide probes are 5′-TAGCTTTCTTTCCCCAGATCC-3′ (eIF4G C/EBPβ [−68 to −48]), 5′-GAGGTGGGCTCTTCCTGCTTCC-3′ (eIF4G Ets-1 [−101 to −80]), and 5′-GCTGGGGGGTGGGGAGTTGG-3′ (eIF4G Sp1 [−151 to −132]). The oligonucleotides used for competition analysis are wild-type MRG1 Sp1 (5′-TTAAGCTTCGCTCCGCCCTTCC-3′), mutant MRG1 Sp1 (5′-TTAAGCTTCGCTTTGCCCTTCC-3′) (25), wild-type C/EBPβ (5′-TGCAGATTGCGCAATGTGCA-3′), and mutant C/EBPβ (5′-TGCAGAGACTAGTCTCTGCA-3′). For UV cross-linking, the reaction mixture containing probe and nuclear extract was incubated at room temperature for 20 min and then irradiated with UV at 254 nm at a distance of 5 cm for 30 min by using a UV Stratalinker 1800 (Stratagene). The mixture was then separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (10% polyacrylaide).
Northern blot analysis.
Subconfluent H1299 cells in 10-cm plates were transfected with constructs (4 μg/plate) using Lipofectamine Plus. The total RNAs were extracted using an RNeasy mini kit 48 h following transfection. Residual plasmid DNA in the total RNA was digested with RNase-free DNase. The poly(A) RNAs were then isolated from 250 μg of total RNAs using the Oligotex mRNA mini kit. One-fifth of the mRNAs were separated in 1% agarose gels in the presence of formaldehyde and morpholinepropanesulfonic acid (MOPS) buffer and blotted onto MAGNA nylon membranes. The blots were hybridized with a 32P-labeled firefly luciferase DNA probe (1,656 bp), which was isolated from pRF by cleaving with NcoI and XbaI and labeled using Rediprime II random-prime labeling system.
RESULTS
The 5′-UTR of eIF4G directs expression of the second cistron in a dicistronic construct.
To further analyze the IRES of eIF4G, we reengineered the dicistronic plasmids by cloning the cDNA encoding the 5′-UTR of eIF4G into a widely used dicistronic vector, pRF (70) (see also Fig. 1A). This vector contains the SV40 promoter to direct the transcription of dicistronic RNA encoding Renilla luciferase as the first cistron and firefly luciferase as the second cistron. Following the SV40 poly(A) signal, there is an SV40 enhancer that enhances the promoter activity. The IRES sequence of HRV was engineered in the same way as eIF4G and was used as a control (Fig. 1A). These plasmids were transfected into HeLa cells, and both Renilla and firefly luciferase activities were measured. As shown in Fig. 1B, the 5′-UTR of eIF4G displays an unusually high activity in directing the expression of firefly luciferase. The activity is ∼850-fold higher than that of the control vector pRF and ∼40-fold higher than that of the HRV IRES. Similar results were observed after transfecting other cell lines such as H1299 cells (data not shown). This observation is consistent with previous studies which suggested that the stimulation of the second cistron expression was due to IRES activities in the 5′-UTR of eIF4G (20, 21).
Translation of dicistronic RNA transcripts in HeLa cells and RRL.
The above results in Fig. 1 suggest that (i) the 5′-UTR of eIF4G contains IRES activity which enhances the translation of firefly luciferase from the dicistronic mRNA by internal initiation, as suggested previously (20, 21); (ii) the 5′-UTR of eIF4G may contain a cryptic promoter which directs transcription of the firefly luciferase gene; and/or (iii) the 5′-UTR of eIF4G contains a cryptic splicing acceptor site which creates a splicing variant with only the second cistron of the firefly luciferase gene. To distinguish between these possibilities, we first generated dicistronic RNAs in vitro from the dicistronic constructs and used them to program translation both in HeLa cells and in RRL. RNA transfection is one of the major methods for characterizing translation efficiency and identifying eukaryotic regulatory factors influencing IRES activity. This method bypasses the complex issue of transcriptional regulation and requires only the cytoplasmic delivery of the transcripts. For purposes of RNA transfection, pSP-RF A30, pSP-R-eIF4G-FA30, and pSP-R-HRV-FA30 were engineered and used for producing dicistronic transcripts containing the m 7GpppG cap and polyadenylated tail in vitro (Fig. 2A). Transcripts were introduced into HeLa cells by Lipofectin encapsulation (16). At 8 h following transfection, cell lysates were prepared for luciferase activity measurement. As expected, the firefly luciferase of the vector RNA was very poorly translated and its activity (arbitrary units) represented only about 0.16% of that of Renilla luciferase (data not shown). It increased to about 6.7% with the dicistronic RNAs containing HRV IRES. Therefore, HRV IRES significantly stimulated the translation of firefly luciferase, about 40-fold higher than that of the vector control (Fig. 2B). However, no stimulation of firefly luciferase expression was observed with the 5′-UTR of eIF4G. We next used the dicistronic RNA to program translation in RRL. As shown in Fig. 2C, the IRES of HRV did not show any enhancement of firefly luciferase expression in RRL (solid bar), indicating that RRL may lack factors necessary for IRES-dependent translation initiation. The addition of HeLa S100 cytoplasmic extract to RRL stimulated ∼40-fold expression of firefly luciferase under the control of IRES of HRV (Fig. 2C). However, no stimulation of firefly luciferase expression was observed by the 5′-UTR of eIF4G in either the absence or presence of HeLa S100 extract (Fig. 2C), confirming the results of RNA transfection studies. Thus, the 5′-UTR of eIF4G may not contain an IRES element to direct cap-independent translation from a dicistronic RNA transcript.
FIG. 2.

Translation of dicistronic mRNA in HeLa cells and in RRL. (A) Schematic diagram of the dicistronic mRNA used for translation in HeLa cells. In vitro transcripts with 5′-cap (m 7G) and 3′-poly(A) tail (A30) were synthesized using T7 RNA polymerase from linearized vector alone (RFA30), constructs containing the IRES of HRV (R-HRV-FA30), and the 5′-UTR of eIF4G (R-eIF4G-FA30). R., Renilla; F., firefly. (B) Relative luciferase activity from dicistronic mRNAs in HeLa cells. HeLa cells were transfected with the dicistronic mRNAs, and 8 h following transfection, Renilla and firefly luciferase (R. Luc. and F. Luc.) activities were measured and the relative ratios were calculated and normalized to that of the vector-transfected cells (RF). (C) Relative luciferase activity from dicistronic RNA in RRL. Capped dicistronic transcripts were synthesized using T7 RNA polymerase from linearized pRF, pR-HRV-F, and pR-eIF4G-F (Fig. 1). The in vitro transcripts were then used to program translation in RRL in the presence of buffer alone (H100) or the HeLa cytoplasmic extract (S100). Following the cessation of translation, Renilla and firefly luciferase activities were measured, the ratio of firefly to Renilla activity was determined, and the relative ratios were calculated and normalized to the vector control in the presence of buffer (H100). The data are representative of three independent experiments. The gels shown at the bottom are in vitro transcripts (500 ng each) separated on a 1% agarose gel that were used for in vitro and in vivo studies, respectively.
Cryptic promoter activity in the 5′-UTR of eIF4G.
To analyze whether the 5′-UTR of eIF4G contains any promoter activity, we simply removed the unique SV40 promoter together with the intron sequence from the dicistronic constructs. These promoterless dicistronic constructs (Fig. 3A) were then transfected into HeLa cells for determination of both Renilla and firefly luciferase activities (Fig. 3B). As expected, both the Renilla and firefly luciferase activities were minimal but could be detected for the pRF(−P) vector control. Only a twofold increase in firefly luciferase activity was observed with the pR-HRV-F(−P) construct. This small increase is in dramatic contrast to the 20-fold increase when the pR-HRV-F construct was used, as shown in Fig. 1B. Thus, the enhanced expression of firefly luciferase from pR-HRV-F constructs (Fig. 1) was not due to production of monocistronic transcript by a cryptic promoter in the HRV sequence. Surprisingly, the pR-eIF4G-F(−P) construct generated more than 900-fold-higher firefly luciferase activity than that of the vector control. This increase was similar to that generated by the pR-eIF4G-F construct shown in Fig. 1B, suggesting that the significant enhancement in the firefly luciferase expression by the 5′-UTR sequence of eIF4G is probably due to the presence of a strong promoter in this region.
FIG. 3.

Cryptic promoter activity of the 5′-UTR of eIF4G. (A) Schematic diagram of promoterless dicistronic construct of pRF(−P), pR-HRV-F(−P) and pR-eIF4G-F(−P). The sequences of the SV40 promoter and chimeric intron were removed from the parental dicistronic constructs shown in Fig. 1A. (B) Relative luciferase activity generated from the promoterless constructs. The promoterless constructs were transfected into HeLa cells, and 24 h following the transfection, cells were harvested for determination of Renilla and firefly luciferase (R. Luc. and F. Luc.) activity. The relative ratios between firefly and Renilla luciferase activities were calculated and normalized to that of the vector-transfected cells [pRF(−P)]. The data were from six independent experiments performed in duplicate.
Mapping of the eIF4G promoter in the 5′-UTR.
The above studies suggest that the 5′-UTR sequence of eIF4G contain a cryptic promoter which may have been thought to be an IRES element in previous studies. To further validate our study, we mapped the cryptic promoter by deleting DNA sequences systematically from both the 5′ and 3′ ends of the 5′-UTR of eIF4G in the promoterless dicistronic vector (Fig. 4A). As shown in Fig 4B, the promoter activity increased significantly when 50 bases was deleted from the 5′ end of the 5′-UTR (D368-318), indicating that there is a repressor element within this region. The promoter activity did not significantly change after further deletion up to 200 bases from the 5′ end (D368-168). However, deletion of an additional 50 bases from the 5′ end resulted in a dramatic decrease in promoter activity (D368-118), suggesting that the 168 bases upstream of the translation start site contains the full promoter activity of the 5′-UTR of eIF4G. Furthermore, deletion of the region from −49 to −1 with respect to the 3′ end did not significantly affect promoter activity, whereas deletion of the region from −69 to −1 resulted in complete loss of the promoter activity. Thus, the region from −69 to −49 upstream of the translation start site probably contains the critical elements for the promoter in the 5′-UTR sequence of eIF4G. Interestingly, this region corresponds to the PPT region that was shown previously to be critical for the IRES activity of eIF4G (20, 21) (see Fig. 5A for the sequence).
FIG. 4.

Effects of deletions on the promoter activity of the 5′-UTR of eIF4G. (A) Schematic diagram of the deletions in the 5′-UTR of eIF4G. The positions of the 5′ and 3′ ends of each deletion are indicated on the left and right, respectively. These mutant 5′-UTRs were engineered into the promoterless dicistronic vector at the intergenic region. (B) Relative luciferase activity generated from the wild-type and mutant 5′-UTR sequences of eIF4G. HeLa cells were transfected with the constructs shown in panel A, and 24 h following transfection, Renilla and firefly luciferase (R. Luc. and F. Luc.) activities were measured and the ratio of firefly to Renilla luciferase was calculated and normalized to that of the wild type (WT) control.
FIG. 5.

Mapping of sequences important for the promoter activity in the 5′-UTR of eIF4G. (A) Schematic demonstration of the linker-scanning mutations. Linker-scanning mutations were made onto the full-length or truncated D168 constructs. The putative transcription factor binding sites predicted by MatInspector software are shown above the sequence. The dashed line indicates the polypyrimidine tract region important for IRES activity. WT, wild type. (B and C) Relative luciferase activity of wild-type and mutant full-length (B) or D368-168 (C) constructs in HeLa cells. At 24 h following transfection of the constructs into HeLa cells, the cells were harvested for determination of Renilla and firefly luciferase activity. The ratio of firefly to Renilla luciferase activity was calculated and normalized to that of the wild-type sequences. Data are from three independent experiments.
Linker-scanning analysis of the cryptic promoter in the 5′-UTR of eIF4G.
To identify potential transcription factor binding sites required for the cryptic promoter activity in the 5′-UTR of eIF4G, we engineered a series of linker-scanning constructs near the translation start site. Linker-scanning constructs were engineered into the full-length 5′-UTR sequence by site-directed mutagenesis in which 10 successive nucleotides were replaced with the sequence ACTCTAGACT (Fig. 5A). This sequence was chosen because it contains no sequence homologous to any known transcription factor binding sites. These constructs were then transfected into HeLa cells, and the promoter activity was measured using cell lysate following transfection. As shown in Fig. 5B, replacement with L4, L5, or L6 (corresponding to −79 to −49 upstream of the translation start site) resulted in significant decreases in promoter activity. This observation was consistent with the results of the deletion-mapping study shown in Fig. 4B (construct D69-1). However, the results in Fig. 4B (compare the results for constructs D368-168, D368-118, and D368-68) also suggested that this region (−79 to −49) itself did not contain the full promoter activity. To identify other cis elements, we next engineered the same linker-scanning constructs into the D368-168 truncated constructs since it contains minimal sequence for the full promoter activity. As shown in Fig. 5C, two additional regions, −149 to −139 (L13) and −89 to −69 (L7 and L6), were shown to be important for the proximal promoter activity.
Identification of transcription factors for the cryptic promoter in the 5′-UTR of eIF4G.
The above results suggest that there are several critical regions in the cryptic promoter of the 5′-UTR of eIF4G. To determine the biochemical composition of protein complexes binding to these regions, nuclear extracts from HeLa cells were analyzed by EMSA using labeled probes corresponding to regions from −151 to −132, −101 to −80, and −68 to −48. These sequences were chosen because they are overlapped with the critical regions identified by promoter-mapping analysis shown in Fig. 4 and 5. In addition, sequence analysis (57) showed that they contain Sp1/Sp3, Ets-1, and C/EBPβ binding sites, respectively (Fig. 5A). As shown in Fig. 6A, the probe from −151 to −132 formed three protein complexes in EMSA (lanes 2 and 5). Addition of cold Sp1 oligonucleotides prevented the formation of the three complexes (lane 3). In contrast, addition of cold mutant Sp1 oligonucleotides had no effect on the complex formation (lane 4). Furthermore, addition of Sp1 antibody to the reaction mixture inhibited the formation of the uppermost complex (lane 6) while addition of Sp3 antibody inhibited the formation of the two lower complexes (lane 7). Addition of both antibodies completely inhibited the formation of all three complexes (lane 8), while addition of C/EBPβ antibody had no effect (lane 9). These results clearly indicate that the region from −151 to −132 contains a typical Sp1/Sp3 binding site.
FIG. 6.

Identification of nuclear proteins binding to the regions from −151 to −132, −101 to −80, and −68 to −48 in the 5′-UTR of eIF4G. (A) EMSA and supershift analysis of a probe with the sequence from −151 to −132. HeLa nuclear extract (N. E.) (10 μg) was incubated with the probe alone (lane 2) or in the presence of unlabeled Sp1 (lane 3), mutant Sp1 (lane 4) oligonucleotide as competitors or in the absence (lane 5) or presence of anti-Sp1 (lane 6), anti-Sp3 (lane 7), both anti-Sp1 and anti-Sp3 (lane 8), or anti-C/EBPβ (lane 9) antibodies (Ab) for the supershift assay. The DNA-protein complexes were separated by PAGE. The DNA-protein complexes generated are indicated by arrows. The gel of lanes 5 to 9 was run longer than the gel of lanes 1 to 4 to better separate the protein complexes. (B) EMSA and UV cross-linking of a probe with the sequence from −101 to −80. HeLa nuclear extract (10 μg) was incubated with the probe alone (lane 2) or in the presence of the unlabeled probe (lane 3) or Sp1 oligonucleotide (lane 4) as competitors. Cross-linking of DNA-protein complexes was performed by incubating 10 μg of nuclear extract with the labeled probe followed by UV irradiation and separation by SDS-PAGE (lane 5). (C) EMSA, supershift, and UV cross-linking of a probe with the sequence from −68 to −48. HeLa nuclear extract (10 μg) was incubated with the probe alone (lane 2), or in the presence of the unlabeled probe (lane 3), wild-type C/EBPβ (lane 4) or mutant C/EBPβ (lane 5) oligonucleotides as competitors, or in the absence (lane 6) or presence (lane 7) of anti-C/EBPβ antibody for the supershift assay. The DNA-protein complexes were separated by PAGE. UV cross-linking was performed in the way same as described for panel B, with the probe from −68 to −48. (D) Sequence comparison of the consensus C/EBPβ binding site with the probe from −68 to −48. Also shown are the wild-type (w) and mutant (m) C/EBPβ oligonucleotides used for the EMSA in panel C. Conserved nucleotides are aligned by three dots. N = A, C, G, or T. The PPT of eIF4G is underlined. Lane 1 in all experiments (panels A to C) is a control consisting of the probe alone incubated without nuclear extract.
The region from −101 to −80 was predicted to contain an Ets-1 binding sequence (Fig. 5A). EMSA analysis showed that two specific protein complexes were formed (Fig. 6B, lane 2). Addition of cold probe inhibited the formation of these complexes (lane 3), while addition of nonrelevant cold Sp1 probe had no effect on complex formation (lane 4). As shown by UV cross-linking (lane 5), at least six major proteins with different molecular weights bound to this sequence, indicating that this sequence is recognized by a large number of different proteins. The identities of these proteins remain to be determined.
The region from −68 to −48 was predicted to contain a C/EBPβ binding sequence. As shown in Fig. 6D, C/EBPβ binding sequence involves 12 bases with a core sequence of (A/G)N(A/G)T(T/G)NNG(A/C)AA(T/G). The homologous sequence of C/EBP-β in the 5′-UTR of eIF4G is located in the region from −61 to −50 that overlaps the PPT. EMSA analysis showed that a single specific protein complex was formed with this probe (Fig. 6C, lane 2). This complex is specific, since addition of cold probe prevented the formation of this complex (lane 3). The addition of cold probe corresponding to the consensus C/EBPβ binding sequence also competed for the formation of the complex (lane 4). In contrast, a cold mutant C/EBPβ probe had no effect on complex formation (lane 5). Hence, the protein involved in binding to the sequences of −68 to −48 is probably C/EBPβ. This conclusion was further confirmed using C/EBPβ antibody, which inhibited the formation of the complex (lane 7). UV cross-linking analysis also showed a single protein with the same molecular weight as C/EBPβ (lane 8). Taken together, these results demonstrated that the region from −68 to −48 contains a C/EBPβ binding sequence.
Transcription factors Sp1 and Ets-1 synergistically trans-activate the activity of eIF4G promoter in SL2 cells.
To determine directly whether the above transcription factors could functionally modulate the promoter activity in the 5′-UTR sequence of eIF4G, Drosophila SL2 cells, which are deficient in Sp1-, Sp3-, and Ets-related proteins (12, 13), were used. The reason for using insect instead of mammalian cells is that Sp1-, Sp3-, and Ets-related factors are expressed in virtually all mammalian cells, which could affect the interpretation of this experiment. We introduced the pR-eIF4G-F(−P) construct along with Drosophila expression plasmid pPacSp1 or pPacUEts-1 into Drosophila SL2 cells. As shown in Fig. 7A, both pPacSp1 and pPacUEts-1 stimulated the promoter activity in the 5′-UTR sequence of eIF4G. To test the possible functional interplay between the Sp1- and Ets-related proteins, we performed cotransfection experiments using combinations of plasmids expressing Ets-1 and Sp1. As shown in Fig. 7A, pPacSp1 and pPacUEts-1 synergistically stimulated the promoter activity in the 5′-UTR sequence of eIF4G. Since our promoterless construct pR-eIF4G-F(−P) contains an SV40 enhancer downstream of the poly(A) signal sequence that may complicate the interpretation of the above results (Fig. 3A), we removed the SV40 enhancer region from pR-eIF4G-F(−P) by cloning its NheI-HpaI fragment into the pGL3-promoter vector, which contains intact SV40 poly(A) signal but without SV40 enhancer. The same pattern of results was observed (Fig. 7B). Thus, the trans activation of promoter by Sp1 and Ets-1 occurs through the cis elements present in the 5′-UTR sequence of eIF4G.
FIG. 7.
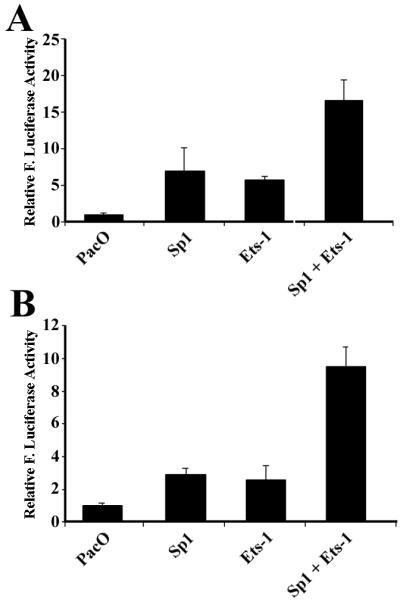
Sp1 and Ets-1 proteins activate the promoter in the 5′-UTR sequence of eIF4G in Drosophila SL2 cells. pR-eIF4G-F(−P) (A) and pR-eIF4G-F(−PE) (B) were transfected into SL2 cells together with Drosophila expression vector (PacO), or plasmids expressing transcription factors Sp1 and Ets-1. At 48 h following transfection, the cells were harvested for determination of firefly (F.) luciferase activity, which was normalized to that of vector-transfected cells. The data are from three independent experiments.
Analysis of 5′-UTRs of Sno, Bad, and HIAP by conventional and promoterless dicistronic test.
To determine how robust the promoterless dicistronic test is in differentiating cryptic promoters from IRES activities, we analyzed three additional cellular 5′-UTRs by using both conventional and promoterless dicistronic tests (Fig. 8A). The sno gene encodes a component of the histone deacetylase complex and can act as a tumor suppressor in mice (67), although paradoxically it also promotes oncogenic transformation of chicken embryo fibroblasts and differentiation of quail embryo fibroblasts (6). The human sno gene has a 5′-UTR of 700 bases that is rich in AT. Bad is a member of the Bcl-2 family of proapoptotic proteins that is thought to exert a death-promoting effect by blocking the prosurvival function of Bcl-XL through its heterodimerization with Bcl-XL (7). The mouse Bad mRNA has a 5′-UTR of 470 bases which contains a small upstream open reading frame encoding a peptide of 60 amino acids. Human HIAP is a member of the IAP protein family that is evolutionally conserved and can block apoptosis when expressed in cells derived from multiple tissues (14). HIAP mRNA has a 5′-UTR of ∼3 kb, which would severely inhibit cap-dependent translation initiation (33).
FIG. 8.

Conventional and promoterless dicistronic test of the 5′-UTRs of Sno, Bad, and HIAP. (A) Schematic diagram of conventional and promoterless dicistronic constructs of the 5′-UTRs of Sno, Bad and HIAP. (B) Conventional dicistronic and promoterless dicistronic constructs were transfected into HeLa cells. At 24 h following transfection, cells were harvested for enzyme assay and the ratio of Renilla (R.) to firefly (F.) luciferase activity was determined and normalized to that of the respective vector controls. The data are from three independent experiments.
As shown in Fig. 8B, the 5′-UTRs of human Sno and mouse Bad both significantly increased the expression of the second cistron in the conventional dicistronic test (open bars), suggesting that these 5′-UTRs have either IRES and/or cryptic promoter activity. In contrast, no increase in the second-cistron expression was observed with the 5′-UTR of human HIAP, indicating that it does not contain either IRES or cryptic promoter activity. Interestingly, the 5′-UTRs of Sno and Bad stimulated even higher expression of the second cistron in the promoterless dicistronic vector (solid bars). This observation strongly argues that the enhanced expression of the second cistron observed in the conventional dicistronic test is due to the promoter rather than IRES activity in the 5′-UTRs of Sno and Bad.
Northern blot analysis.
To determine whether the transcript derived from the cryptic promoter of eIF4G, Sno, and Bad can be detected by Northern blotting using the firefly luciferase gene as a probe, poly(A) RNAs were isolated for Northern blot analysis 48 h following transfection of dicistronic constructs pRF, pR-HRV-F, pR-eIF4G-F, pR-Sno-F, pR-Bad-F, and pR-eIF4G-F(−P). As a control for monocistronic transcript, the pRF(−R) construct was engineered by removing DNA sequences encoding Renilla luciferase (the 0.95-kb EcoRV-PvuII fragment [Fig 1A]). This plasmid uses the SV40 promoter to direct the synthesis of the firefly luciferase transcript without the Renilla luciferase sequence. As shown in Fig 9A, the dicistronic transcript from control pRF (lane 1) and the monocistronic transcript from control pRF(−R) (lane 4) were detected as expected. The transcript produced from pR-eIF4G-F(−P) has a size similar to that from pRF(−R) (compare lanes 4 and 7) as expected, suggesting that it is a monocistronic mRNA. The same monocistronic transcript was also observed with pR-eIF4G-F (lane 3), These observations confirm that the 5′-UTR of eIF4G has a strong promoter activity for expressing the second cistron of firefly luciferase. Surprisingly, no dicistronic transcript was found with pR-eIF4G-F (lane 3).
FIG. 9.

Northern blot analysis of eIF4G, Sno, and Bad. (A) Poly(A) RNAs were isolated following transfection with pRF (lane 1), pR-HRV-F (lane 2), pR-eIF4G-F (lane 3), pRF(−F) (lane 4), pR-Sno-F (lane 5), pR-Bad-F (lane 6), and pR-eIF4G-F(−P) (lane 7) and used for Northern blot analysis as described in Materials and Methods. The asterisks indicate the dicistronic transcript, the arrowhead indicate the monosistronic transcript and the pound sign indicates the minor transcript. (B) Small fraction of the cells transfected with various constructs for Northern analysis in panel A were used to prepare cell lysates for determination of both firefly and luciferase activities.
To determine whether the lack of intact dicistronic mRNA from pR-eIF4G-F was due to the low sensitivity of Northern blot analysis, lysates were prepared for the Renilla luciferase activity assay from the same cells that were used for the Northern blot analysis in Fig. 9A. As shown in Fig. 9B, pR-eIF4G-F displayed about 12% of the Renilla luciferase activity of pRF or pR-HRV-F (compare column 3 with columns 1 and 2), suggesting that the level of the dicistronic transcript containing Renilla luciferase gene is relatively low. This low level of Renilla luciferase activity was consistently observed for pR-eIF4G-F, and the low level of the dicistronic transcript is probably due to alternative splicing events as proposed previously (see Discussion). This event would splice out Renilla luciferase sequences from the dicistronic transcripts and generate a monocistronic transcript that encodes only firefly luciferase. The minor transcript, with a size slightly larger than the monocistronic transcript generated from pR-eIF4G-F (Fig. 9A, lane 3), may represent the alternatively spliced products.
Intact dicistronic mRNAs were produced from pR-HRV-F (lane 2), pR-Sno-F (lane 5), and pR-Bad-F (lane 6) with the expected sizes. No monocistronic transcripts were detected with these constructs. These observations are consistent with the conclusion that the HRV sequence is an IRES element that does not contain a cryptic promoter. However, the lack of detectable monocistronic transcript from Sno and Bad would have suggested that they, too, do not have cryptic promoters, had we not performed the promoterless dicistronic test (Fig. 8). To determine whether the firefly luciferase was expressed in the cells used for Northern blot analysis, we measured the firefly luciferase activity. As shown in Fig. 9B, the firefly luciferase activity of Sno and Bad is, respectively, ∼16- and ∼9-fold higher than that generated from pRF and similar to that of pR-HRV-F. Based on these observations, we conclude that Northern blot analysis is not sensitive enough to detect monocistronic transcripts generated from moderate cryptic promoters and thus the promoterless dicistronic assay is a better approach to confirm the existence of IRES in 5′-UTR sequences. It is noteworthy that two unknown bands of high mobility were also detected in all samples on Northern blotting. The similar products have also been observed previously (11). However, because they are smaller than the monocistronic mRNA, they are not expected to be translated into full-length firefly luciferase proteins and do not affect the interpretation of the data.
DISCUSSION
The recent rapid increase in the number of cellular IRES elements discovered has raised great interest in the field of gene regulation research (27, 61, 71). However, because the conventional dicistronic test inherits inevitable drawbacks and the dicistronic RNA assays do not work for cellular IRES elements, a debate still exists about the validity of cellular IRESs, although viral IRESs are well established (36, 42, 63). In this study, we created and tested a promoterless dicistronic vector which can be used easily to safeguard the claims of cellular IRES in future studies.
One of the more than 40 cellular mRNAs that have been shown to have IRES activities to date is eIF4G (see http://www.rangueil.inserm.fr/IRESdatabase). As a major subunit of eukaryotic translation initiation machinery, eIF4G plays a major role in cell growth regulation and cell transformation (2, 18, 26). However, regulation of eIF4G expression is poorly understood. Two different isoforms of cDNA for eIF4G have been isolated (35, 37, 74). The first sequence has a 5′-UTR of 368 bases which contains an active IRES (20, 21). The IRES activity of this 5′-UTR has been well characterized and mapped to a polypyrimidine tract upstream of the AUG start codon (20). The second cDNA sequence (extended eIF4G) encodes a protein that has 156 more amino acids at the amino terminus than the first one and contains a new 5′-UTR, which also contains IRES activity, although the activity is much lower than that of the first 5′-UTR (35, 37). Interestingly, both the truncated and the extended versions of eIF4G function similarly in promoting protein synthesis and cell transformation (18, 26). Currently, it is not known which protein isoforms are expressed in cells. The 5′-UTR of the truncated eIF4G (74) was thought to be an intron of the extended eIF4G gene (23, 37). Analysis of the human genome sequence shows that the 5′-UTR is located in intron 4 of the extended eIF4G gene (unpublished observation). In this study, we showed that the 5′-UTR of the truncated eIF4G clearly contains promoter sequence which may be used to generate transcripts for expression of truncated eIF4G. It is therefore tempting to speculate that at least two alternative promoters may exist before exon 5 to generate transcripts with various length of the 5′-UTR responsible for production of the truncated eIF4G protein. Indeed, sequence analysis of the eIF4G genome showed that there may be another promoter in the GC-rich region about 1.6 kb upstream of the 5′-UTR of truncated eIF4G (unpublished observation).
The promoter activity associated with the 5′-UTR of eIF4G in this study was probably thought previously to be an IRES activity. First, expression of firefly luciferase of the second cistron driven by the 5′-UTR of eIF4G in a promoterless dicistronic test is at the same level as that observed in a conventional dicistronic test. Second, the cryptic promoter sequence has been mapped and the transcription factor binding sites in the DNA encoding the 5′-UTR have been delineated. Third, some of the transcription factors which are responsible for promoter activation of the 5′-UTR of eIF4G have also been identified using EMSA and complementation assays in insect cells. Fourth, the Northern blot analysis demonstrated that monocistronic transcripts encoding firefly luciferase were produced from both the conventional and the promoterless dicistronic plasmid. Finally, as another alternative strategy to bypass the complex issue of transcription or splicing possibly presented by 5′-UTRs in the conventional dicistronic DNA test, we also performed RNA transfection and in vitro translation using dicistronic RNA transcripts. However, the 5′-UTR of eIF4G failed to show any IRES activity. In contrast, the IRES of HRV in dicistronic RNAs can direct the translation of the second cistron both in HeLa cells and in RRL supplemented with HeLa extract. Although similar observations have been made with some other cellular IRESs in previous studies (8, 70), this observation significantly undermines the claim of cellular IRES activity despite the possibility that nuclear experience of the transcripts other than transcription or splicing may contribute to the prerequisites for internal initiation mediated by the cellular IRESs (70).
Interestingly, we showed that the promoter activity in the 5′-UTR sequence of eIF4G relies on the existence of the region from −68 to −48, which corresponds to the PPT that was previously described to be essential for IRES activity of eIF4G. Either deletion or point mutation of this region resulted in dramatic loss of the promoter activity. This region binds to transcription factor C/EBPβ, a member of the CCAAT/enhancer binding protein (C/EBP) family that is expressed in proliferating cells (46, 51). Although essential, the C/EBPβ binding element itself displays minimal promoter activity. We also showed by deletion and linker-scanning analysis that the regions from −168 to −68, −151 to −132, and −101 to −80 are also important elements for this promoter activity. While we demonstrated that the region from −151 to −132 binds the Sp1/Sp3 transcription factor, more work is needed to identify the proteins that bind to the region from −101 to −80, which was shown to be highly homologous to the Ets-1 consensus binding sequence. This region binds to at least six proteins in HeLa nuclear extracts, which remain to be identified. Since different Ets proteins exhibit low selectivity in binding-site preference (66, 72) and since Ets-1 proteins do not exist in Drosophila SL2 cells, we analyzed the possible effect of Ets-1 on the promoter activity in the 5′-UTR sequence of eIF4G in SL2 cells. Our results clearly demonstrated that Ets-1 could trans-activate the promoter activity in the 5′-UTR sequence of eIF4G. We also demonstrated that Sp1 and Ets-1 work synergistically on this promoter and stimulate the promoter activity by about 10 to 20-fold. The apparent disparity between this number and the nearly 1,000-fold expression noted in Fig. 2 suggests that other transcription factors, C/EBPβ in particular, may also play a major role in the activation of the 5′-UTR promoter. Based on the results of our promoter analysis, we conclude that the transcription factors C/EBPβ, Sp1/Sp3, and Ets-1 functionally interact to trans-activate the cryptic promoter in the 5′-UTR sequence of eIF4G and therefore may be responsible for the transcriptional regulation of the truncated version of eIF4G.
Interestingly, pR-eIF4G-F generated an additional minor monocistronic transcript (Fig. 9A) that is slightly larger than the normal monocistronic transcript (Fig. 9A), generated by the cryptic promoter in pR-eIF4G-F(−P). This transcript is unlikely to be derived from transcription by the promoter in the 5′-UTR sequence of eIF4G because it is not produced by pR-eIF4G-F(−P). It is probably generated by other mechanisms related to the 5′-UTR sequence of eIF4G because such a transcript was not observed with the vector control and other constructs containing the 5′-UTR of HRV, Bad, and Sno. One such possible mechanism is alternative splicing. The PPT region in the 5′-UTR of eIF4G is followed by a perfect splicing acceptor sequence (TTTCTTTCCCCAGA) which has been suggested to be used for splicing to generate the extended form of eIF4G (23). Use of this acceptor sequence and the upstream donor sequence in the chimeric intron (see Fig. 1) would result in the production of a transcript without the Renilla luciferase gene sequence. Furthermore, although the dicistronic transcript was not detected by Northern blot analysis, pR-eIF4G-F produced about 10% Renilla luciferase activity compared to other constructs that produced the dicistronic transcripts (Fig. 9), suggesting that about 10% of the dicistronic transcripts exist in the cells transfected with pR-eIF4G-F. The other 90% of the dicistronic transcripts may have been processed to smaller transcripts by alternative splicing. Based on the intensity of the bands on Northern blots (Fig. 9A), the minor monocistronic transcript produced by alternative splicing (lane 3) is about one-third as common as that produced by transcription (lane 3) using the promoter in the 5′-UTR sequence of eIF4G. Therefore, alternative splicing may represent another mechanism responsible for the apparently high firefly luciferase activity observed with the 5′-UTR sequence of eIF4G in the conventional dicistronic test. In light of these findings, it is imperative to rule out the alternative splicing as well as the cryptic promoter when examining a candidate IRES of cellular mRNAs, possibly by testing with an intronless dicistronic construct.
Heterogeneity of transcription initiation has been documented previously for some proto-oncogenes such as c-myc (47, 48). Transcription of c-myc involves P0, P1, P2, and P3 promoters. Differential promoter usage has been observed in a variety of cell types, and atypical usage of the promoter has generally been associated with abnormal or deregulated control of cell growth. The transcription starting point of P0, P1, and P2 c-myc mRNAs are located 1,172, 524, and 363 bases upstream from the translation start codon CUG, respectively, while that of P3 resides in an intron. P1 and P2 promoter sequences are therefore located in the 5′-UTR of P0 transcripts. Similarly, the 1,038 bases of the 5′-UTR sequence of vascular endothelial growth factor were also demonstrated to contain an alternative transcription initiation site (1).
While it is recognized that the long 5′-UTRs present at the majority of the proto-oncogenes inhibit cap-dependent translation (73) and that IRES-mediated translation initiation may be used for these mRNAs, it has not been well appreciated that these long 5′-UTR DNA sequences may contain promoters for transcription of a less abundant mRNA species with significantly shorter 5′-UTRs. These alternative transcripts with shorter 5′-UTRs are compatible with cap-dependent translation initiation and thus do not require an IRES-mediated translation mechanism. For example, the major transcript of platelet-derived growth factor B chain (PDGF B/c-sis) has a 5′-UTR of 1,023 bases that is transcribed by an upstream TATA-containing promoter (17, 40, 58). However, this transcript cannot be translated due to the large size of the 5′-UTR (34, 58, 59). Recently, it has been suggested that IRES-mediated translation initiation may play an important role in the translational regulation of PDGF B/c-sis mRNA during cell differentiation (4), and the 630-base sequence within the central portion of the 5′-UTR is important for the IRES activity (65). Interestingly, earlier studies of the PDGF B/c-sis gene showed that an additional transcript with a 5′-UTR of only 15 bases was produced in cultured cells on phorbol myristate acetate or transforming growth factor β1 stimulation (17) and in developing rat brain (62). The production of this shorter transcript was postulated to derive from internal transcription initiation, as suggested by DNase I hypersensitivity analysis of the first exon sequences (17). Sequence analysis indeed revealed multiple Sp1 binding sites in the 5′-UTR sequence of PDGF B/c-sis (reference 63 and unpublished observations). It is therefore possible that the alternative transcription for a shorter and less abundant 5′-UTR may be important in regulating PDGF B/c-sis expression during cell differentiation, which would undermine the reported IRES activity of the long 5′-UTR of PDGF B/c-sis. This possibility certainly merits attention in future studies of the translational regulation of any long cellular 5′-UTRs.
The promoterless dicistronic vector created in this study is clearly a very sensitive approach to detect any potential cryptic promoters in long cellular 5′-UTRs. Promoterless vector has been used in one earlier study for investigation of possible IRES activity in the 5′-UTR from XIAP (X-linked inhibitor of apoptosis protein) (32). The advantage of our vector is that it was derived from parental dicistronic vectors, and therefore data from two sets of vectors are readily comparable. As we have also shown with the 5′-UTRs of Bad, Sno, and HIAP, this vector should be applicable to any cellular 5′-UTRs and can be used as a good control for analysis of cellular IRES elements. Despite the differences in nucleotide composition, the 5′-UTRs of Bad, Sno, and HIAP are all long and punctuated with multiple AUGs that are incompatible with the cap-dependent ribosome-scanning mechanism of translation initiation; therefore, they are potential candidates as IRES elements. However, using the promoterless dicistronic vector, we found that the 5′-UTRs of Bad and Sno contain promoter activity at a moderate level with no IRES activity whereas the 5′-UTR of HIAP does not contain either promoter or IRES activities. This observation, together with the findings of eIF4G in this study, argues that cryptic promoter activities prevail in the 5′-UTR of cellular mRNAs, which may generate alternative transcripts with shorter 5′-UTRs compatible with cap-dependent translation initiation. Furthermore, our failure to detect the less abundant monocistronic transcripts generated from the promoter in the 5′-UTR sequence of Bad and Sno by Northern blot analysis undermines the usefulness of this method in IRES studies. If not carefully ruled out by the promoterless dicistronic test, these 5′-UTRs would otherwise be considered to have IRES activities. Therefore, we propose that the promoterless dicistronic vector should be used as a control to safeguard the claim of cellular IRES in future studies.
Acknowledgments
This work was supported in part by National Institutes of Health grants CA64539 and GM59475 and by Department of Defense grant DAMD17-02-1-0073. J.-T.Z. is a recipient of a Career Investigator Award from the American Lung Association.
We are indebted to Robert E. Rhoads for plasmid pGL2/CAT/4G/LUC, A. E. Willis for plasmids pRF and pGL3RHRV, Guntram Suske and Etty Benveniste for plasmids pPacO and pPacSp1, and Philip Marsden for plasmid pPac-UEts-1. We also thank David Ohannesian and Chow Hwee Lee for their critical comments on the manuscript.
REFERENCES
- 1.Akiri, G., D. Nahari, Y. Finkelstein, S. Y. Le, O. Elroy-Stein, and B. Z. Levi. 1998. Regulation of vascular endothelial growth factor (VEGF) expression is mediated by internal initiation of translation and alternative initiation of transcription. Oncogene 17:227-236. [DOI] [PubMed] [Google Scholar]
- 2.Bauer, C., I. Diesinger, N. Brass, H. Steinhart, H. Iro, and E. U. Meese. 2001. Translation initiation factor eIF-4G is immunogenic, overexpressed, and amplified in patients with squamous cell lung carcinoma. Cancer 92:822-829. [DOI] [PubMed] [Google Scholar]
- 3.Belsham, G. J., and N. Sonenberg. 1996. RNA-protein interactions in regulation of picornavirus RNA translation. Microbiol. Rev. 60:499-511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bernstein, J., O. Sella, S. Y. Le, and O. Elroy-Stein. 1997. PDGF2/c-sis mRNA leader contains a differentiation-linked internal ribosomal entry site (D-IRES). J. Biol. Chem. 272:9356-9362. [DOI] [PubMed] [Google Scholar]
- 5.Bonneau, A. M., and N. Sonenberg. 1987. Involvement of the 24-kDa cap-binding protein in regulation of protein synthesis in mitosis. J. Biol. Chem. 262:11134-11139. [PubMed] [Google Scholar]
- 6.Boyer, P. L., C. Colmenares, E. Stavnezer, and S. H. Hughes. 1993. Sequence and biological activity of chicken snoN cDNA clones. Oncogene 8:457-466. [PubMed] [Google Scholar]
- 7.Chao, D. T., and S. J. Korsmeyer. 1998. BCL-2 family: regulators of cell death. Annu. Rev. Immunol. 16:395-419. [DOI] [PubMed] [Google Scholar]
- 8.Chiang, P. W., L. E. Carpenter, and P. J. Hagerman. 2001. The 5′-untranslated region of the FMRI message facilitates translation by internal ribosome entry. J. Biol. Chem. 276:37916-37921. [DOI] [PubMed] [Google Scholar]
- 9.Clemens, M. J., and U. A. Bommer. 1999. Translational control: the cancer connection. Int. J. Biochem. Cell Biol. 31:1-23. [DOI] [PubMed] [Google Scholar]
- 10.Clemens, M. J., M. Bushell, and S. J. Morley. 1998. Degradation of eukaryotic polypeptide chain initiation factor (eIF) 4G in response to induction of apoptosis in human lymphoma cell lines. Oncogene 17:2921-2931. [DOI] [PubMed] [Google Scholar]
- 11.Coldwell, M. J., S. A. Mitchell, M. Stoneley, M. MacFariane, and A. E. Willis. 2000. Initiation of Apaf-1 translation by internal ribosome entry. Oncogene 19:899-905. [DOI] [PubMed] [Google Scholar]
- 12.Courey, A. J., and R. Tjian. 1988. Analysis of Sp1 in vivo reveals multiple transcriptional domains, including a novel glutamine-rich activation motif. Cell 55:887-898. [DOI] [PubMed] [Google Scholar]
- 13.Dennig, J., M. Beato, and G. Suske. 1996. An inhibitor domain in Sp3 regulates its glutamine-rich activation domains. EMBO J. 15:5659-5667. [PMC free article] [PubMed] [Google Scholar]
- 14.Deveraux, Q. L., and J. C. Reed. 1999. IAP family proteins—suppressors of apoptosis. Genes Dev. 13:239-252. [DOI] [PubMed] [Google Scholar]
- 15.Donahue, T. F. 2000. Genetic approaches to translation initiation in Saccharomyces cerevisiae, p. 487-502. In N. Sonenberg, J. W. B. Hershey, and M. B. Mathews (ed.), Translational control of gene expression. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 16.Dwarki, V. J., R. W. Malone, and I. M. Verma. 1993. Cationic liposome-mediated RNA transfection. Methods Enzymol. 217:644-654. [DOI] [PubMed] [Google Scholar]
- 17.Fen, Z., and T. O. Daniel. 1991. 5′ untranslated sequences determine degradative pathway for alternate PDGF B/c-sis mRNA's. Oncogene 6:953-959. [PubMed] [Google Scholar]
- 18.Fukuchi-Shimogori, T., I. Ishii, K. Kashiwagi, H. Mashiba, H. Ekimoto, and K. Igarashi. 1997. Malignant transformation by overproduction of translation initiation factor eIF4G. Cancer Res. 57:5041-5044. [PubMed] [Google Scholar]
- 19.Gale, M., Jr., S. L. Tan, and M. G. Katze. 2000. Translational control of viral gene expression in eukaryotes. Microbiol. Mol. Biol. Rev. 64:239-280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gan, W., M. LaCelle, and R. E. Rhoads. 1998. Functional characterization of the internal ribosome entry site of eIF4G mRNA. J. Biol. Chem. 273:5006-5012. [DOI] [PubMed] [Google Scholar]
- 21.Gan, W., and R. E. Rhoads. 1996. Internal initiation of translation directed by the 5′-untranslated region of the mRNA for eIF4G, a factor involved in the picornavirus-induced switch from cap-dependent to internal initiation. J. Biol. Chem. 271:623-626. [DOI] [PubMed] [Google Scholar]
- 22.Gingras, A. C., B. Raught, and N. Sonenberg. 1999. eIF4 initiation factors: effectors of mRNA recruitment to ribosomes and regulators of translation. Annu. Rev. Biochem. 68:913-963. [DOI] [PubMed] [Google Scholar]
- 23.Gradi, A., H. Imataka, Y. V. Svitkin, E. Rom, B. Raught, S. Morino, and N. Sonenberg. 1998. A novel functional human eukaryotic translation initiation factor 4G. Mol. Cell. Biol. 18:334-342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hake, L. E., and J. D. Richter. 1997. Translational regulation of maternal mRNA. Biochim. Biophys. Acta 1332:M31-M38. [DOI] [PubMed] [Google Scholar]
- 25.Han, B., N. Liu, X. Yang, H. B. Sun, and Y. C. Yang. 2001. MRG1 expression in fibroblasts is regulated by Sp1/Sp3 and an Ets transcription factor. J. Biol. Chem. 276:7937-7942. [DOI] [PubMed] [Google Scholar]
- 26.Hayashi, S., K. Nishimura, T. Fukuchi-Shimogori, K. Kashiwagi, and K. Igarashi. 2000. Increase in cap- and IRES-dependent protein synthesis by overproduction of translation initiation factor eIF4G. Biochem. Biophys. Res. Commun. 277:117-123. [DOI] [PubMed] [Google Scholar]
- 27.Hellen, C. U., and P. Sarnow. 2001. Internal ribosome entry sites in eukaryotic mRNA molecules. Genes Dev. 15:1593-1612. [DOI] [PubMed] [Google Scholar]
- 28.Hentze, M. W. 1997. eIF4G: a multipurpose ribosome adapter? Science 275:500-501. [DOI] [PubMed] [Google Scholar]
- 29.Hershey, J. W. 1991. Translational control in mammalian cells. Annu. Rev. Biochem. 60:717-755. [DOI] [PubMed] [Google Scholar]
- 30.Hershey, J. W. B., and S. Miyamoto. 2000. Translational control and cancer, p. 637-654. In N. Sonenberg, J. W. B. Hershey, and M. B. Mathews (ed.), Translational control of gene expression. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 31.Holcik, M., and R. G. Korneluk. 2000. Functional characterization of the X-linked inhibitor of apoptosis (XIAP) internal ribosome entry site element: role of La autoantigen in XIAP translation. Mol. Cell. Biol. 20:4648-4657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Holcik, M., C. Lefebvre, C. Yeh, T. Chow, and R. G. Korneluk. 1999. A new internal-ribosome-entry-site motif potentiates XIAP-mediated cytoprotection. Nat. Cell. Biol. 1:190-192. [DOI] [PubMed] [Google Scholar]
- 33.Horrevoets, A. J., R. D. Fontijn, A. J. van Zonneveld, C. J. de Vries, J. W. ten Cate, and H. Pannekoek. 1999. Vascular endothelial genes that are responsive to tumor necrosis factor-alpha in vitro are expressed in atherosclerotic lesions, including inhibitor of apoptosis protein-1, stannin, and two novel genes. Blood 93:3418-3431. [PubMed] [Google Scholar]
- 34.Horvath, P., A. Suganuma, M. Inaba, Y. B. Pan, and K. C. Gupta. 1995. Multiple elements in the 5′ untranslated region down-regulate c-sis messenger RNA translation. Cell Growth Differ. 6:1103-1110. [PubMed] [Google Scholar]
- 35.Imataka, H., A. Gradi, and N. Sonenberg. 1998. A newly identified N-terminal amino acid sequence of human eIF4G binds poly(A)-binding protein and functions in poly(A)-dependent translation. EMBO J. 17:7480-7489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jackson, R. J. 2000. A comparative view of initiation site selection mechanisms, p. 637-654. In N. Sonenberg, J. W. B. Hershey, and M. B. Mathews (ed.), Translational control of gene expression. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 37.Johannes, G., and P. Sarnow. 1998. Cap-independent polysomal association of natural mRNAs encoding c-myc, BiP, and eIF4G conferred by internal ribosome entry sites. RNA 4:1500-1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Karantzoulis-Fegaras, F., H. Antoniou, S. L. Lai, G. Kulkarni, C. D'Abreo, G. K. Wong, T. L. Miller, Y. Chan, J. Atkins, Y. Wang, and P. A. Marsden. 1999. Characterization of the human endothelial nitric-oxide synthase promoter. J. Biol. Chem. 274:3076-3093. [DOI] [PubMed] [Google Scholar]
- 39.Keiper, B. D., W. Gan, and R. E. Rhoads. 1999. Protein synthesis initiation factor 4G. Int. J. Biochem. Cell Biol. 31:37-41. [DOI] [PubMed] [Google Scholar]
- 40.Khachigian, L. M., J. W. Fries, M. W. Benz, D. T. Bonthron, and T. Collins. 1994. Novel cis-acting elements in the human platelet-derived growth factor B-chain core promoter that mediate gene expression in cultured vascular endothelial cells. J. Biol. Chem. 269:22647-22656. [PubMed] [Google Scholar]
- 41.Kozak, M. 1999. Initiation of translation in prokaryotes and eukaryotes. Gene 234:187-208. [DOI] [PubMed] [Google Scholar]
- 42.Kozak, M. 2001. New ways of initiating translation in eukaryotes? Mol. Cell. Biol. 21:1899-1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kozak, M. 1989. The scanning model for translation: an update. J. Cell Biol. 108:229-241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kozak, M. 1991. Structural features in eukaryotic mRNAs that modulate the initiation of translation. J. Biol. Chem. 266:19867-19870. [PubMed] [Google Scholar]
- 45.Lamphear, B. J., R. Kirchweger, T. Skern, and R. E. Rhoads. 1995. Mapping of functional domains in eukaryotic protein synthesis initiation factor 4G (eIF4G) with picornaviral proteases. Implications for cap-dependent and cap-independent translational initiation. J. Biol. Chem. 270:21975-21983. [DOI] [PubMed] [Google Scholar]
- 46.Lekstrom-Himes, J., and K. G. Xanthopoulos. 1998. Biological role of the CCAAT/enhancer-binding protein family of transcription factors. J. Biol. Chem. 273:28545-28548. [DOI] [PubMed] [Google Scholar]
- 47.Marcu, K. B., S. A. Bossone, and A. J. Patel. 1992. myc function and regulation. Annu. Rev. Biochem. 61:809-860. [DOI] [PubMed] [Google Scholar]
- 48.Marcu, K. B., A. J. Patel, and Y. Yang. 1997. Differential regulation of the c-MYC P1 and P2 promoters in the absence of functional tumor suppressors: implications for mechanisms of deregulated MYC transcription. Curr. Top. Microbiol. Immunol. 224:47-56. [DOI] [PubMed] [Google Scholar]
- 49.Marissen, W. E., and R. E. Lloyd. 1998. Eukaryotic translation initiation factor 4G is targeted for proteolytic cleavage by caspase 3 during inhibition of translation in apoptotic cells. Mol. Cell. Biol. 18:7565-7574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mathews, M. B., N. Sonenberg, and J. W. B. Hershey. 2000. Origins and principles of translational control, p. 1-32. In N. Sonenberg, J. W. B. Hershey, and M. B. Mathews (ed.), Translational control of gene expression. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 51.McKnight, S. L. 2001. McBindall—a better name for CCAAT/enhancer binding proteins? Cell 107:259-261. [DOI] [PubMed] [Google Scholar]
- 52.Morris, D. R. 1995. Growth control of translation in mammalian cells. Prog. Nucleic Acid Res. Mol. Biol. 51:339-363. [DOI] [PubMed] [Google Scholar]
- 53.Ohlmann, T., M. Ran, V. M. Pain, and S. J. Morley. 1996. The C-terminal domain of eukaryotic protein synthesis initiation factor (eIF) 4G is sufficient to support cap-independent translation in the absence of eIF4E. EMBO J. 15:1371-1382. [PMC free article] [PubMed] [Google Scholar]
- 54.Pain, V. M. 1996. Initiation of protein synthesis in eukaryotic cells. Eur. J. Biochem. 236:747-771. [DOI] [PubMed] [Google Scholar]
- 55.Panniers, R. 1994. Translational control during heat shock. Biochimie 76:737-747. [DOI] [PubMed] [Google Scholar]
- 56.Pestova, T. V., I. N. Shatsky, and C. U. Hellen. 1996. Functional dissection of eukaryotic initiation factor 4F: the 4A subunit and the central domain of the 4G subunit are sufficient to mediate internal entry of 43S preinitiation complexes. Mol. Cell. Biol. 16:6870-6878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Quandt, K., K. Frech, H. Karas, E. Wingender, and T. Werner. 1995. MatInd and MatInspector: new fast and versatile tools for detection of consensus matches in nucleotide sequence data. Nucleic Acids Res. 23:4878-4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rao, C. D., M. Pech, K. C. Robbins, and S. A. Aaronson. 1988. The 5′ untranslated sequence of the c-sis/platelet-derived growth factor 2 transcript is a potent translational inhibitor. Mol. Cell. Biol. 8:284-292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ratner, L., B. Thielan, and T. Collins. 1987. Sequences of the 5′ portion of the human c-sis gene: characterization of the transcriptional promoter and regulation of expression of the protein product by 5′ untranslated mRNA sequences. Nucleic Acids Res. 15:6017-6036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rhoads, R. E., and B. J. Lamphear. 1995. Cap-independent translation of heat shock messenger RNAs. Curr. Top. Microbiol. Immunol. 203:131-153. [DOI] [PubMed] [Google Scholar]
- 61.Sachs, A. B. 2000. Cell cycle-dependent translation initiation: IRES elements prevail. Cell 101:243-245. [DOI] [PubMed] [Google Scholar]
- 62.Sasahara, M., S. Amano, H. Sato, J. G. Yang, Y. Hayase, M. Kaneko, I. Sato, M. Suzaki, and F. Hazama. 1998. Normal developing rat brain expresses a platelet-derived growth factor B chain (c-sis) mRNA truncated at the 5′ end. Oncogene 16:1571-1578. [DOI] [PubMed] [Google Scholar]
- 63.Schneider, R., and M. Kozak. 2001. New ways of initiating translation in eukaryotes? Mol. Cell. Biol. 21:8238-8246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schneider, R. J. 2000. Translational control during heat shock, p. 581-593. In N. Sonenberg, J. W. B. Hershey, and M. B. Mathews (ed.), Translational control of gene expression. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 65.Sella, O., G. Gerlitz, S. Y. Le, and O. Elroy-Stein. 1999. Differentiation-induced internal translation of c-sis mRNA: analysis of the cis elements and their differentiation-linked binding to the hnRNP C protein. Mol. Cell. Biol. 19:5429-5440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sharrocks, A. D., A. L. Brown, Y. Ling, and P. R. Yates. 1997. The ETS-domain transcription factor family. Int. J. Biochem. Cell Biol. 29:1371-1387. [DOI] [PubMed] [Google Scholar]
- 67.Shinagawa, T., H. D. Dong, M. Xu, T. Maekawa, and S. Ishii. 2000. The sno gene, which encodes a component of the histone deacetylase complex, acts as a tumor suppressor in mice. EMBO J. 19:2280-2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sonenberg, N. 1994. mRNA translation: influence of the 5′ and 3′ untranslated regions. Curr. Opin. Genet. Dev. 4:310-315. [DOI] [PubMed] [Google Scholar]
- 69.Sonenberg, N. 1994. Regulation of translation and cell growth by eIF-4E. Biochimie 76:839-846. [DOI] [PubMed] [Google Scholar]
- 70.Stoneley, M., T. Subkhankulova, J. P. Le Quesne, M. J. Coldwell, C. L. Jopling, G. J. Belsham, and A. E. Willis. 2000. Analysis of the c-myc IRES; a potential role for cell-type specific trans-acting factors and the nuclear compartment. Nucleic Acids Res. 28:687-694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vagner, S., B. Galy, and S. Pyronnet. 2001. Irresistible IRES. Attracting the translation machinery to internal ribosome entry sites. EMBO Rep. 2:893-898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wasylyk, B., S. L. Hahn, and A. Giovane. 1993. The Ets family of transcription factors. Eur. J. Biochem. 211:7-18. [DOI] [PubMed] [Google Scholar]
- 73.Willis, A. E. 1999. Translational control of growth factor and proto-oncogene expression. Int. J. Biochem. Cell Biol. 31:73-86. [DOI] [PubMed] [Google Scholar]
- 74.Yan, R., W. Rychlik, D. Etchison, and R. E. Rhoads. 1992. Amino acid sequence of the human protein synthesis initiation factor eIF-4 gamma. J. Biol. Chem. 267:23226-23231. [PubMed] [Google Scholar]
- 75.Zhang, J. T., and V. Ling. 1991. Study of membrane orientation and glycosylated extracellular loops of mouse P-glycoprotein by in vitro translation. J. Biol. Chem. 266:18224-18232. [PubMed] [Google Scholar]