

Abstract
RNA interference-mediated depletion of phospholipase D2 (PLD2), but not PLD1, inhibited recycling of transferrin receptors in HeLa cells, whereas the internalization rate was unaffected by depletion of either PLD. Although reduction of both PLD isoforms inhibits PLD activity stimulated by phorbol 12-myristic 13-acetate, only depletion of PLD2 decreased nonstimulated activity. Cells with reduced PLD2 accumulated a greater fraction of transferrin receptors in a perinuclear compartment that was positive for Rab11, a marker of recycling endosomes. EFA6, an exchange factor for Arf6, has been proposed to stimulate the recycling of transferrin receptors. Thus, one consequence of EFA6 overexpression would be a reduction of the internal pool of receptors. We confirmed this observation in control HeLa cells; however, overexpression of EFA6 failed to decrease the internal pool of transferrin receptors that accumulate in cells previously depleted of PLD2. These observations suggest that either PLD2 is required for a constitutive Arf6-mediated recycling pathway or in the absence of PLD2 transferrin receptors accumulate in recycling endosomes that are not responsive to overexpression of EFA6.
INTRODUCTION
Intracellular membrane traffic events require complex molecular machinery. We know a great deal about proteins such as small GTPases, coat components, and a variety of adaptors that play key roles during the formation of vesicles. Recently, it has been recognized that membrane lipids are also important for this process (De Camilli et al., 1996; Roth, 1999). Several mutations that block secretion or endocytosis were identified in enzymes that modify lipids, and many proteins involved in vesicle formation contain lipid-binding domains. A very important group of such lipid-modifying enzymes are those that modify phosphatidylinositides. Type I phosphatidylinositol phosphate kinases (PIP5KIs), which produce phosphatidylinositol bisphosphate (PIP2), have the ability to regulate rates of receptor-mediated endocytosis (Barbieri et al., 2001; Padron et al., 2003; Di Paolo et al., 2004). Another lipid-modifying enzyme known to affect membrane traffic both in secretion and in endocytosis is phospholipase D (PLD) (Bi et al., 1997; Roth et al., 1999; Hughes and Parker, 2001; Shen et al., 2001; Du et al., 2003, 2004; Pathre et al., 2003). PLD cleaves the head group from phosphatidylcholine, the major membrane phospholipid in animal cells, and produces phosphatidic acid (PA). Before PLD genes were identified, Arf1, a key regulator of the formation of COPI vesicles at the Golgi complex, was purified as a cytosolic factor that stimulates PLD activity (Brown et al., 1993; Cockcroft et al., 1994; Frohman et al., 1999). Later, it was shown that PLD activity in Golgi membranes could stimulate the formation of COPI vesicles in vitro (Ktistakis et al., 1996). Once the genes encoding PLD1 and PLD2 in mammals were identified and inactive mutants of them were overexpressed, they were found to inhibit vesicle transport originating at the Golgi complex in live cells (Hammond et al., 1995; Colley et al., 1997; Denmat-Ouisse et al., 2001).
Ligand binding to growth factor or hormone receptors that lead to their internalization are frequently accompanied by stimulated PLD activity. Although such ligand-induced activity could be part of a signaling cascade and only concurrent with endocytosis, it is also possible that PLD functions directly in endocytosis. Both PLD1 and 2 are found, at least in part, on Golgi membranes, endosomes, and at the plasma membrane, localization consistent with a possible role in endocytosis (Colley et al., 1997; Toda et al., 1999; Freyberg et al., 2001; Hughes and Parker, 2001; Lucocq et al., 2001; Du et al., 2003, 2004; Hiroyama and Exton, 2005). Overexpression of catalytically inactive mutants of PLD1 and PLD2 inhibited the down-regulation of epidermal growth factor (EGF) receptors in response to EGF (Shen et al., 2001), and expression of catalytically inactive or truncated PLD2 perturbed agonist-induced internalization of the angiotensin (Du et al., 2004) and the μ opioid receptors (Koch et al., 2004). Phagocytosis in macrophages was also inhibited by expression of truncated PLD2 (Iyer et al., 2004). These observations are consistent with PLD being required for at least some step in the endocytic pathway.
Agonist-induced endocytosis and phagocytosis differ in important ways and/or use additional components compared with the constitutive endocytosis of nutrient receptors such as the transferrin receptor. The rate of endocytosis of growth factors and hormones is acutely regulated and phagocytosis is a very specialized form of endocytosis that is primarily actin driven. Thus, we were interested in determining whether PLD enzymes are required for constitutive endocytosis, and, if so, in what step. Depleting each isoform of PLD by RNA interference (RNAi), we found that in HeLa cells PLD2, but not PLD1, inhibited the rate of recycling of transferrin receptors but not the rate of internalization. Our results suggest that PLD2 is required for an Arf6-mediated transport of membrane receptors from endosomes back to the plasma membrane.
MATERIALS AND METHODS
Reagents and Antibodies
Alexa-labeled transferrin was purchased from Invitrogen (Carlsbad, CA). [3H]Palmitic acid and [32P]orthophosphate were from PerkinElmer Life and Analytical Sciences (Boston, MA), and leupeptin was from Roche Diagnostics (Indianapolis, IN). Thin layer chromatography (TLC) plates were from Whatman (Maidstone, United Kingdom) for the PLD activity assay and from J. T. Baker (Phillipsburg, NJ) for the assay of production of PIP2. Lipid standards were purchased from Avanti Polar Lipids (Alabaster, AL). All other chemicals were from Sigma-Aldrich (St. Louis, MO) unless otherwise specified. Monoclonal anti-transferrin receptor and polyclonal anti-Rab11 (Zymed Laboratories, South San Francisco, CA), polyclonal anti-myc (Santa Cruz Biotechnology, Santa Cruz, CA), monoclonal anti-α tubulin (Sigma-Aldrich), monoclonal antibody recognizing both PLDs (Upstate Biotechnology, Charlottesville, VA), and monoclonal anti-early endosome antigen 1 (EEA1) (BD Transduction Laboratories, Lexington, LY) were purchased from commercial sources. Polyclonal antibodies against PLD1, Arf6, and green fluorescent protein (GFP) were kindly provided by M. Frohman (SUNY Stony Brook, Brooklyn, NY), J. Donaldson (National Institutes of Health, Bethesda, MD), and J. Seemann (University of Texas Southwestern, Dallas, TX), respectively. The plasmid pEGFPC3 was from Clonetech (Mountain View, CA), and pEG-FPC3-EFA6 plasmid was provided by J. Donaldson. pCMV5 plasmids encoding HA-hPIP5KIα, myc-hPIP5KIβ were from Dr. H. Yin (University of Texas Southwestern), and a plasmid encoding HA-hPIP5KIγ was provided by Dr. P. De Camilli (Yale University, New Haven, CT). Plasmid encoding PLD2 was from M. Frohman (SUNY Stony Brook).
RNA Interference
Small interference RNA (siRNA) oligonucleotides were designed according to the protocols provided by the laboratory of T. Tuschl (Rockefeller University, New York, NY) (http://www.rockefeller.edu/labheads/tuschl/sirna.html). RNA oligonucleotides encoding both the sense and antisense of the target were from the RNA oligonucleotide synthesis core at UT Southwestern. siRNA oligonucleotides were annealed by heating equal amounts of complementing oligonucleotides at 90°C for 1 min at a final concentration of 20 μM in 2 mM Mg-acetate, 100 mM K-acetate, 30 mM HEPES-KOH, pH 7.4, buffer, followed by 1-h incubation at 37°C. The siRNA sequences targeting human PLD1 (GenBank accession no. NM_002662) were 1) aaa auc ugg aca cgc ggg aac and 2) aag gaa acc uag uaa cug agc. For human PLD2 (GenBank accession no. NM_002663) were 1) aag agg tgg cug gug gug uug and 2) aau ggg gca ggu uac uuu gcu. An oligonucleotide corresponding to nucleotides 695–715 of the firefly luciferase (U31240) was used as a negative control. On day 1, HeLa or SV589 cells were plated in six-well plates (or glass coverslips in 4-well plates) at 40–50% confluence in antibiotic-free DMEM supplemented with 10% (vol/vol) fetal bovine serum (FBS), 1 mM Na-pyruvate, and 10 mM HEPES. Two hours later, siRNA were introduced into cells using Oligofectamine reagent (Invitrogen) according to the manufacturer's instructions, with 10 μl of 20 μM siRNA and 4 μl of transfection reagent/well (amounts for coverslips in 4-well plates were 0.25 these volumes) and left for 14–16 h. Medium containing siRNA complexes was removed and replaced with normal growth medium. On day 3 or 4, cells were lysed and analyzed by Western blotting or were used for experiments.
Quantitative Real-Time PCR
Total RNA was extracted from HeLa cells transfected with siRNAs using RNA STAT-60 (Tel-Test, Friendswood, TX) isolation reagent. RNA samples were treated with DNAse I (RNAse-free; Roche Diagnostics), and reverse-transcribed with random hexamers using SuperScript II RNase H-reverse transcriptase (Invitrogen) to generate cDNA. Primer Express software (PerkinElmer Life and Analytical Sciences) was used to design primers for cyclophilin (GenBank accession no. XM_057194), which was used as the internal control, and for PLD1 and 2. The primers for cyclophilin were forward TGC CAT CGC CAA GGA GTAG and reverse TGC ACA GAC GGT CAC TCA AA; for PLD1, forward GCA GCC CCT TTG CTT TTA CT and reverse TAC CCG TGG CTC GTT TTTC; and for PLD2, forward CAA CCG TCT CTT GAC CAT GTC and reverse ACT GAC TTC CAG GAA CTC TGT CAT. Primers were validated through analysis of template titration and dissociation curves to establish both linearity of the reaction and production of a single product. PCR assays were conducted on an Applied Biosystems Prism 7000 sequence detection system. The 20-μl final reaction volume contained 50 ng of reverse-transcribed RNA, 150 nM of each primer, and 10 μl of SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA). PCR reactions were performed in triplicate and relative RNA levels were determined by the comparative Ct method (User Bulletin No. 2, PerkinElmer Life and Analytical Sciences).
Plasmids Transfections and Microinjection
Plasmids encoding either green fluorescent protein alone (GFP) pEGFPC3 (Clontech) or GFP fused to the exchange factor for Arf6 (pEGFPC3-EFA6) were transfected into HeLa cells using LipofectAMINE 2000 according to the manufacturer's instructions (Invitrogen). The only modification was that the amount of transfection reagent was 0.9 μl, instead of the 1.5 μl suggested, per well of a 24-well plate. Microinjection into HeLa cells was performed using a semiautomatic system consisting of the Transjector 5246 and Micromanipulator 5171 (Eppendorf, Hamburg, Germany) connected to an upright ECLIPSE TE300 microscope (Nikon, Tokyo, Japan). Needles were pulled from 1.2-mm-diameter glass capillaries (Warner Instruments, Hamden, CT) with the use of a P-97 needle puller (Sutter Instruments, Novato, CA). Plasmids were injected into the nucleus at 0.1 μg/μl and incubated for 3 h at 37°C for expression.
Phospholipase D Activity
HeLa cells transfected with siRNAs were labeled for 16 h with 2.5 μCi of [3H]palmitic acid in normal medium. Cells were then washed, and fresh medium without radioactive label but containing 1% (vol/vol) ethanol and phorbol 12-myristate 13-acetate (PMA) in the concentrations indicated was added for 30 min. At the end of 30 min, cellular lipids were extracted (Bligh and Dyer, 1959). Cells were washed with ice-cold phosphate-buffered saline (PBS) and extracted with 1.2 ml of methanol/phosphate-buffered saline solution at a 2:1 ratio. The cell extracts were scraped and transferred into a 2-ml Microfuge tube, and 0.4 ml of chloroform was added for 30 min at room temperature. After centrifugation for 5 min at 14,000 rpm to pellet cell debris, the clear supernatant was transferred to a glass tube, and 0.5 ml of chloroform and 0.4 ml of 23 mM HCl was added to produce an aqueous and an organic phase. The phases were separated by a spin at 3000 rpm in a Sorvall RT6000B centrifuge for 5 min, and then the aqueous phase was discarded and the organic phase containing the lipids was evaporated under a stream of nitrogen. Lipid extracts were redissolved in 50 μl of chloroform, and 5-μl aliquots were measured in a scintillation counter. Samples containing equal radioactivity were loaded onto TLC plates (LK6DF; Whatman) and resolved in a solvent system of chloroform:methanol:acetic acid:acetone:water at 270:54:54: 108:27 (vol/vol) ratios. The lipid bands were visualized by autoradiography and analyzed using a scanning densitometer (GE Healthcare, Little Chalfont, Buckinghamshire, United Kingdom).
PIP2 Analysis
Cells were labeled with 40 μCi/ml [32P]ortophosphoric acid for 4 h in phosphate-free DMEM containing 0.5% dialyzed FBS. Cellular lipids were extracted with a 4:10:5 mixture of CHCl3:CH3OH:1 N HCl and were resolved by TLC [Si250F (19C); J. T. Baker] in a solvent system composed of n-propanol: H2O:NH4OH(30%) in 65:15:20 (vol/vol) ratios. Lipids were visualized by autoradiography and quantified by densitometry. Lipid standards were detected with iodine vapors.
Endocytosis and Recycling of Transferrin
Cells transfected with siRNA oligonucleotides were rinsed and incubated in serum-free medium for 30 min to remove any residual transferrin and then were exposed to 50 μg/ml transferrin conjugated with Alexa Fluor 488 or 633 (Invitrogen) at 37°C for the times indicated. Internalization was stopped by chilling the cells on ice. External transferrin was removed by washing with ice-cold serum-free DMEM and PBS, whereas bound transferrin was removed by an acid wash in PBS at pH 5.0 followed by a wash with PBS at pH 7.0. The fluorescence intensity of internalized transferrin was measured for 10,000 cells by flow cytometry using an FACSCalibur (BD Biosciences, San Jose, CA) instrument, and the average intensity of the cell population was recorded for each time point. Data are normalized to the maximum increase in mean fluorescence of control cells. We confirmed that the uptake of fluorescent transferrin was receptor-mediated by measuring the uptake of fluorescent transferrin in the presence of a 100-fold excess of nonfluorescent holo-transferrin. No increase in cell-associated fluorescence was obtained in the presence of the excess competing nonfluorescent transferrin. Cell viability was measured by staining with propidium iodide. To measure recycling, cells were incubated first in medium lacking transferrin to free the receptors from ligand and subsequently in medium containing fluorescent transferrin at 37°C for 1 h to completely label the receptor population. Bound transferrin was removed from the cell surface by an acid wash in PBS at pH 5.0 followed by a wash with PBS at pH 7.0. Recycling was then measured as the loss of cell-associated fluorescence by cells incubated in PBS containing 0.2% bovine serum albumin (BSA), 200 μg/ml deferoxamine, and 600 μg/ml unlabeled transferrin for the indicated times at 32 or 37°C. The release of transferrin was stopped by chilling cells on ice and samples were washed and fluorescence was measured as indicated above.
Fluorescence Microscopy
Cells on coverslips were washed with PBS and fixed in 3.7% formaldehyde for 15 min. The fixative was removed, and cells were washed with DMEM and permeabilized with 50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.25% gelatin, 5 mM EDTA, 0.02% NaN3, 0.05% NP-40, and 0.05% Triton X-100, or for the experiments labeling with Alexa-transferrin in addition to antibody, with 0.1% saponin in PBS containing 5% FBS. Nonspecific binding sites were blocked with PBS containing 1% BSA before incubating samples with primary and secondary antibodies for 1 h at room temperature. Coverslips were mounted on glass slides with Aqua Poly/Mount (Polysciences, Warrington, PA) and visualized with a Zeiss Axiovert 200M microscope or with a Zeiss 510 confocal microscope. For quantification of transferrin, parameters were set on the Axiovert 200M microscope and ORCA-285 (Hamamatsu, Bridgewater, NJ) camera such that no area of any image was saturated (out of the linear range), and a series of images were taken with these settings. The outlines of cells to be quantified were delineated and the average pixel intensity within each cell obtained using Openlab software version 4.0.2 (Improvision, Lexington, MA)
RESULTS
Phospholipase D1 and -2 Are Effectively Knocked Down by RNA Interference
To specifically knock down the expression of PLD genes, we transfected double-stranded siRNA oligonucleotides into HeLa cells. To determine the extent of knock down, we measured PLD proteins by immunoblotting samples collected ∼48 h after transfection. PLD1 was reduced 83 and 90% using two different sets of siRNAs, whereas PLD2 was knocked down 66 and 95% for two sets of siRNA oligonucleotides (Figure 1A). In a previous study, in which we reduced the three isoforms of the type I phosphatidylinositol phosphate kinases with siRNA, we found that their expression was coordinately regulated (Padron et al., 2003). Suppression of one isoform led to changes in mRNA and protein expression of the others. Thus, we tested whether this was the case for PLD isoforms. We transfected HeLa cells with the pair of oligonucleotides that was the most effective in suppressing each isoform, and 48 h later we measured mRNA for both PLD1 and PLD2 by real-time quantitative PCR. We did not observe coordinated regulation of PLD isoforms; siRNA oligonucleotides only decreased the targeted isoform (Figure 1B). In this study, we primarily used the set of oligonucleotides that was most effective in suppressing the target protein. However, in certain experiments, as indicated, we performed experiments with the other, slightly less effective set of oligonucleotides as well, as a control for off-target effects.
Figure 1.
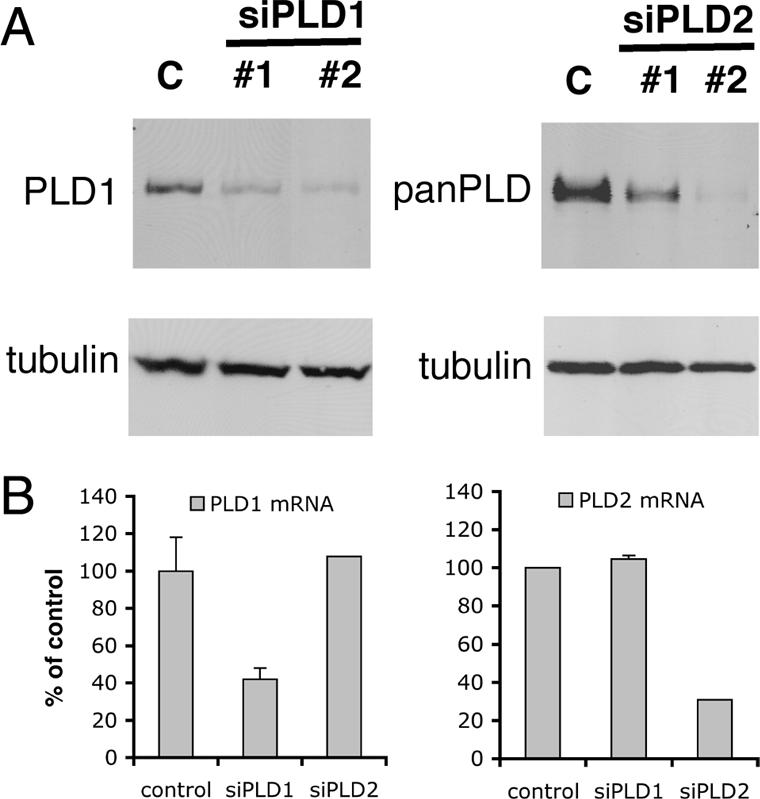
Expression of phospholipases D1 and D2 is effectively decreased by RNA interference. Two distinct siRNAs complementary to mRNA sequences of each of PLD1 or PLD2, and one siRNA targeting luciferase (control, C), were transfected into HeLa cells. Forty-eight hours later immunoblots with PLD1, panPLD, and tubulin antibodies were performed, and a representative is shown (A). mRNA for each PLD isoform was measured in cells treated with siRNA specific for one isoform by real-time quantitative PCR. The mean with SD (SD) from three experiments is shown normalized to the control (B).
Depletion of PLD2 protein was assessed with a panPLD antibody that recognized both PLD isoforms when they were expressed in Sf9 cells. Thus, because cells treated with siRNA against PLD2 contained very little protein recognized by the pan-PLD antibody, our results suggest that HeLa cells might express little PLD1 compared with PLD2. However, we cannot be certain of this because of possible differences in affinity of the antibody for each PLD isoform.
PLD2 Is Required for Efficient Recycling of Transferrin Receptors
We examined the distribution of transferrin receptors in control cells and in cells depleted of PLD by immunofluorescence. Cells with reduced PLD2, but not those with reduced PLD1 (our unpublished data), showed a distinct pattern compared with control cells, most cells having accumulated receptors in a perinuclear, internal compartment (Figure 2A). Accumulation of receptors inside the cell can occur, in principle, by either increasing the rate of internalization or by decreasing the rate of recycling back to the cell surface. Thus, using fluorescently labeled transferrin, we measured both internalization and recycling. Internalization of Alexa-labeled transferrin (50 μg/ml) was followed for 60 min at 37°C, whereas recycling was followed for 30 min at 32°C or for 60 min at 37°C after loading cells for 1 h with Alexa-labeled transferrin at 37°C. Experiments conducted at either temperature showed a reduction in the recycling of transferrin receptors in cells treated with siRNA against PLD2. However, because transferrin recycling at 37°C is very fast, we used a lower temperature, 32°C, to measure initial recycling rates, an approach used by other investigators (Paleotti et al., 2005) (Figure 2B, recycling). After fitting the data to one-exponential decay (Prism 3.0cx), recycling rate constants were calculated to be 0.087 (R2 = 0.93) and 0.005 (R2 = 0.76) for control and siRNA for PLD2 (siPLD2), respectively. Transferrin recycling assays at 37°C for up to 60 min showed that siPLD2 cells had the same extent of transferrin recycling as control cells (Supplemental Figure 1A). A second pair of PLD2 siRNA oligonucleotides reduced recycling significantly as well, whereas siPLD1 oligonucleotides did not (Supplemental Figure 1B).
Figure 2.

PLD2 is required for efficient recycling of transferrin receptors. Transferrin receptors were labeled by indirect immunofluorescence 2 d after transfection with siPLD2 or with a control siRNA (A). Recycling and internalization of transferrin (see Materials and Methods) were also measured in cells treated with siPLD. Recycling and Internalization graphs present the mean and SD of three experiments.
Transferrin binding assays on cells at low temperature showed that depleting PLD2 decreased transferrin receptors at the cell surface, suggesting that reduced recycling resulted in a reduction of transferrin receptors on the cell surface (our unpublished data). Consistent with having fewer receptors on the cell surface, transferrin uptake experiments showed that, initially, cells treated with PLD2 siRNA internalize transferrin slower than control cells. However, at later times the difference is reduced and by 1 h the siPLD2 cells have accumulated as much transferrin as control cells. (Figure 2B, internalization). We observed that after the 1-h period to allow fluorescent transferrin to fill the internal pool, the accumulated fluorescence of internalized transferrin at the beginning of the chase (time 0) was similar in both control and treated cells. This is explained by the relatively small fraction of HeLa cell transferrin receptors that is on the cell surface at steady state. To determine the proportion of transferrin receptors at the cell surface, we labeled cells with transferrin for 1 h at 37°C, and then cells were chilled on ice before washing with and without PBS at pH 5.0, a treatment that releases bound transferrin. Cells were detached with a nonenzymatic reagent to prevent removal of surface receptors, and fluorescence was measured by flow cytometry. The pH 5.0 wash released 10.2 ± 0.4% (n = 3) of the fluorescence. If 90% of transferrin receptors in HeLa cells are internal, then from the data shown in Figure 2B, in control cells 54% of all transferrin receptors (0.6 × 0.9) are internalized in 5 min, which means that the 10% of the transferrin receptor population that resides on the cell surface is internalized in approximately a minute. Our combined results suggest that depletion of PLD2, but not PLD1, reduces the recycling rate of transferrin receptors while the internalization rate is essentially unchanged. Although the cell surface proportion of receptors decreases in PLD2-depleted cells, this is not reflected in a substantial increase in the internal population, because such a small fraction of receptors in control HeLa cells is on the cell surface at steady state.
We also depleted PLD2 and measured transferrin receptor recycling in a second human cell line to exclude the possibility that the observed effects in HeLa cells were cell type specific. PLD2 siRNA oligonucleotides were transfected into the transformed fibroblast cell line SV589 (Yamamoto et al., 1984), using the same protocol as in HeLa and PLD2 protein was measured 2 and 3 d after transfection. In SV589 cells, PLD2 protein was considerably decreased 3 d after transfection, and the kinetics of transferrin recycling was significantly reduced (Figure 3). Recycling rate constant values of 0.085 (R2 = 0.99) and 0.007 (R2 = 0.89) for control and PLD2-depleted cells, respectively, were obtained using the curve-fitting software Prism 3.0cx (GraphPad Software, San Diego, CA).
Figure 3.
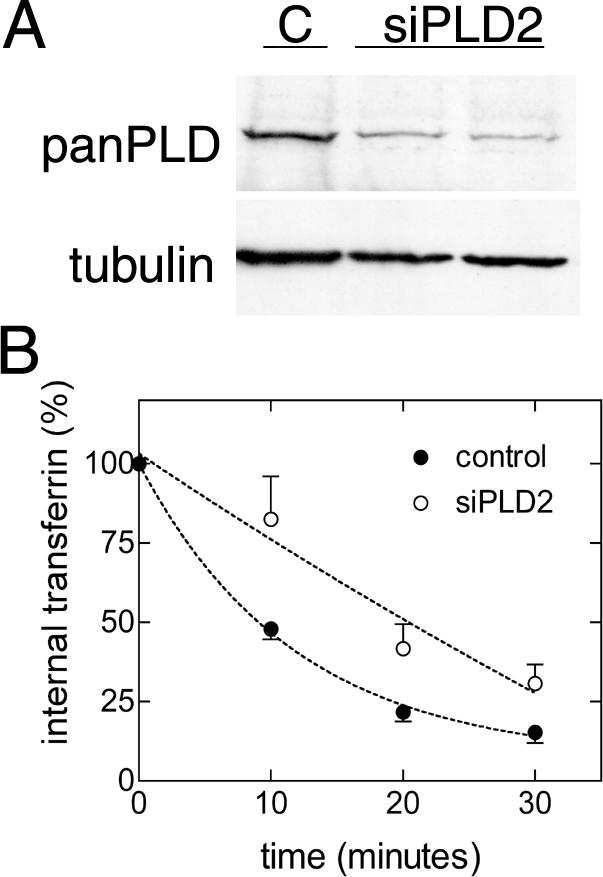
Depletion of PLD2 inhibits transferrin receptor recycling in fibroblasts. SV589 cells were transfected with control and siPLD2 oligonucleotides and 72 h later immunoblots with panPLD and tubulin antibodies were performed. A representative blot is shown (A). Recycling of transferrin was measured as in Figure 2; the mean and SD of three experiments are plotted (B).
Because a reduction in PLD activity by depleting PLD2 inhibited transferrin receptor recycling, we tested whether increasing PLD2 expression would accelerate recycling by transiently transfecting a plasmid encoding PLD2. However, overexpression of PLD2 in HeLa cells, which in fact resulted in elevated PLD activity, did not increase transferrin receptor recycling in our experiments (Supplemental Figure 2). This negative result could mean that even though basal PLD activity is necessary to maintain constitutive transferrin receptor recycling (siRNA data), PLD activity is not rate limiting for this process. Nevertheless, it is possible that the lack of an effect of PLD2 overexpression in recycling is because of other reasons, for example, that the additional PA is not produced at the right location.
Although Reduction of Each of the PLD Isoforms Reduced PLD Activity Stimulated by Phorbol 12-Myristic 13-Acetate, Only PLD2 Depletion Decreases the Basal Activity
PLD activity was measured with and without addition of PMA. PMA stimulates PLD activity by activating protein kinase C (PKC), which is a direct activator of PLD (Singer et al., 1996). Controls and cells treated with PLD siRNAs were labeled with [3H]myristic acid, and lipids were extracted and resolved by TLC. PLD1 siRNA reduced PLD activity stimulated by PMA by 45%, but it had no effect on the basal activity (Figure 4). Interestingly, a reduction of PLD2 not only produced a substantial decrease in PMA-stimulated PLD activity (57%) but also produced a reduction of 68.1% in basal PLD activity (Figure 4). These results suggest that, in HeLa cells, PLD2 is the main contributor of nonstimulated PLD activity, and this is consistent with a role for PLD2 in a constitutive membrane traffic pathway, such as endocytic recycling of transferrin receptors.
Figure 4.

Decreased expression of PLD1 reduces PMA-induced PLD activity but not basal PLD activity, whereas decreased expression of PLD2 decreases both activities. 3H-Labeled lipids were extracted from duplicate samples of cells treated with the reagents indicated and were analyzed by TLC. An autoradiograph of a representative experiment is shown (A). Autoradiographs of three experiments were quantified by densitometry (arrow), and the average values and SD are plotted on the graph (B).
Transferrin Receptors Accumulate Abnormally in Rab11-positive Endosomes in Cells with Reduced PLD2
After internalized transferrin receptors fuse with early/sorting endosomes, they return to the cell surface either directly in a fast cycle or through a second compartment, the recycling endosome, in a slower cycle (Maxfield and McGraw, 2004). To determine in which of these compartments transferrin receptors accumulated, we performed experiments labeling the transferrin receptor and EEA1 or Rab11 antibodies, which are markers for the early/sorting, and recycling endosomes, respectively. We observed that the internal compartment where the transferrin receptor accumulated stained strongly with Rab11 (Figure 5A). Interestingly, we found that in 20–30% of cells depleted of PLD2, the Rab11-positive compartment contained tubules of various sizes that seemed largely devoid of transferrin receptors. However, at this point we do not understand the nature or significance of this phenomenon. To label transferrin receptors and early/sorting endosomes, we incubated cells with Alexa 488 transferrin (50 μg/ml for1 h at 37°C) before staining with a monoclonal anti-EEA1 antibody. Transferrin colocalized with EEA1 to some extent as expected. However, in contrast to the previous result, there was no apparent enrichment of EEA1 in the precise location where transferrin accumulated in cells in which PLD2 was reduced (Figure 5B). It is worth noting that, because the panPLD antibody was not suitable for immunofluorescence labeling in conditions compatible with Rab11 labeling, we cannot demonstrate that PLD2 is knocked down in a given cell in the PLD2 siRNA-treated population sample. However, because the phenotype presented in Figure 1 was prevalent and reduced recycling was confirmed in the population, we conclude that the most likely interpretation is that the cells with the typical phenotype after PLD2 depletion in fact had reduced PLD2.
Figure 5.

Transferrin receptors accumulate abnormally in Rab11-positive endosomes in cells with reduced PLD2 expression. Control and siPLD2-treated cells were labeled with transferrin receptor and Rab11 antibodies (A) or with fluorescent transferrin and EEA1 antibody (B), and confocal micrographs were taken. Most cells in the siPLD2 samples had a prominent perinuclear accumulation of transferrin receptors; typical cells with this phenotype are shown.
In addition to Rab11 and EEA1, we also performed indirect immunofluorescence against GM130 and CD63, markers for Golgi and lysosomes, in siPLD2 cells as a control to test whether the gross morphology of other organelles changes (Supplemental Figure 3). We did not observe morphological changes in organelles other than in the Rab11-positive compartment.
PLD2 Is Required for EFA6-mediated Membrane Recycling
The small GTPase Arf6 has been reported to be involved during internalization and recycling of membrane and to activate PLD2 in vivo (Radhakrishna and Donaldson, 1997; D'Souza-Schorey et al., 1998; Franco et al., 1999; Hiroyama and Exton, 2005; Paleotti et al., 2005; Tanabe et al., 2005). Interestingly, SMAP1 and EFA6, which are regulators for Arf6, have been proposed to control, respectively, the internalization and recycling steps of the transferrin receptor (Franco et al., 1999; Tanabe et al., 2005). Thus, we tested the possibility that PLD2 might be acting downstream of an EFA6/Arf6 recycling pathway. We expressed GFP-tagged EFA6, or GFP alone as control, by transient transfection and measured recycling of Alexa 633 transferrin by flow cytometry. In this way, we were able to measure fluorescent transferrin (red) only in those cells that expressed EFA6 (green). We observed that EFA6 increased the recycling of transferrin receptors (Figure 6). The rate constant in GFP-expressing cells was 0.064 ± 0.01, R2 = 0.94 (n = 4), which was ∼20% lower than in untransfected HeLa cells (Figure 2), possibly because of the stress produced by the transfection procedure. However, in GFP-EFA6-expressing cells, the recycling rate increased to 0.11, R2 = 0.81 (n = 4). We noticed that cells previously transfected with PLD2 siRNA did not tolerate subsequent transfection with the LipofectAMINE 2000 reagent. Thus, to determine whether EFA6 required PLD2, we turned to microinjection to express EFA6 and used microscopy to follow the effects on transferrin endocytosis. PLD2-depleted and control cells that had been microinjected with plasmid DNA encoding GFP-tagged EFA6 were incubated for 1 h in Alexa 568 transferrin. After surface transferrin was removed by a pH 5.0 wash, cells were fixed and prepared for microscopy. As reported previously (Franco et al., 1999) and consistent with having an increased recycling rate (Figure 6), in cells not treated with PLD2 siRNA and injected with the plasmid expressing EFA6 (n = 21), internal transferrin was reduced by 56% compared with uninjected cells (n = 34) (Figure 7A, top, and B). Thus, the data shown in Figures 6 and 7 indicate that overexpressing EFA6 shifts the steady-state distribution of transferrin receptors by increasing the recycling rate. In contrast, cells with reduced PLD2 (n = 15), which accumulated transferrin in the recycling endosome, failed to export transferrin when EFA6 was subsequently overexpressed (Figure 7A, bottom, and B). The combined results suggest that either PLD2 is downstream of EFA6/Arf6 in the regulation of a recycling pathway for transferrin receptors, or in the absence of PLD2, transferrin receptors enter a compartment that is not affected by EFA6.
Figure 6.
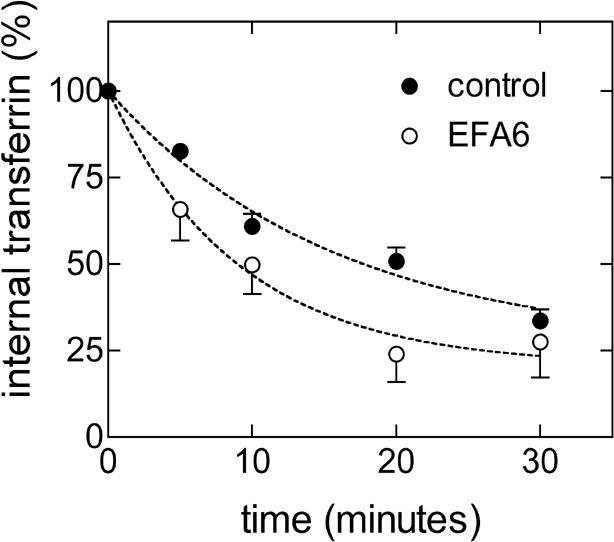
EFA6 increases the recycling of transferrin receptors. Cells transfected with GFP only (control) or GFP-EFA6 were labeled with Alexa 633 transferrin at 37°C to fill the endocytic recycling pathway, and then the surface population of transferrin was removed. Cells were then incubated for various intervals at 32°C to allow the internalized transferrin to recycle to the cell surface and be released. The mean fluorescence (Alexa 633) of the cell population expressing GFP at each time point was measured by FACS and plotted as percentage of the value measured with no chase. Curves were fit to the data using the curve-fitting software Prism 3.0cx. Values are the means and SEs of four experiments.
Figure 7.

PLD2 acts downstream of the EFA6 mediated recycling of transferrin receptors. Cells were transfected with siRNA for PLD2 or a control siRNA, and 2 d later they were microinjected with a plasmid encoding GFP-EFA6. Cells were allowed to internalize fluorescent transferrin and were incubated in the absence of transferrin to allow internalized transferrin to recycle. Cells were fixed and stained with antibody to GFP to identify those overexpressing the exchange factor (A). Cells overexpressing EFA6 and treated with siPLD2 did not show the accelerated loss of fluorescent transferrin seen in cells treated with control siRNA. For quantification of transferrin, micrographs were taken using identical settings. Graph (B) shows average pixel intensity and SD of EFA6-expressing cells in control (n = 21) and siPLD2 (n = 15)-treated cells as well as that of neighboring uninjected cells (n = 34 from control samples and n = 14 from siPLD2 samples).
Depletion of PLD2 Reduces PIP2 Production, but Expression of PIP5KI Enzymes Does Not Relieve Inhibition of Transferrin Recycling
An effector of Arf6 for stimulating PLD activity is hPIP5KIβ (Honda et al., 1999). PIP2, the product of PIP5KI enzymes, activates PLD2 activity and the product of PLD, PA, in turn activates PIP5KI (Moritz et al., 1992; Jenkins et al., 1994; Colley et al., 1997). We measured PIP2 production and found that cells depleted of PLD2 produced less PIP2 (49 ± 13.1% compared with control cells. Surprisingly, we observed that PIP, the precursor for PIP2, was also reduced (55 ±13.1%) (Figure 8, A–C). Thus, we tested whether the effect of PLD2 on transferrin recycling could be rescued by overexpressing hPIP5KIβ, an enzyme that, when overexpressed, has been shown to increase PIP2 levels in cells (Yamamoto et al., 2001; Padron et al., 2003). Cells previously treated with siRNA for PLD2 were microinjected with a plasmid encoding myc-tagged hPIP5KIβ and 3 h later labeled with fluorescent transferrin and anti-myc antibody. However, hPIP5KIβ failed to restore transferrin recycling in PLD2-depleted cells; cells treated with PLD2 siRNA that overexpressed hPIP5KIβ maintained the increased accumulation of internal transferrin typical of cells with reduced PLD2 (Figure 8D). The other PIP5KI isoforms, α and γ, also failed to restore recycling of transferrin (our unpublished data).
Figure 8.

Depletion of PLD2 reduces PIP2 production, but overexpression of PIP5KI does not relieve inhibition of transferrin recycling. Cells were treated with siRNAs to PLD1, PLD2, or a control. Cells were labeled with [32P]phosphoric acid and then lipids were extracted and analyzed by TLC. An autoradiogram with duplicate samples of a representative experiment is shown. (A) PIP and PIP2 are indicated by the arrows. The average (±SD) of three experiments quantified by densitometry are plotted as percentage of control and shown in B and C. Cells depleted of PLD2 were microinjected with myc-tagged hPIP5KIβ, labeled with fluorescent transferrin as in Figure 7, and stained with anti-myc antibody (D).
DISCUSSION
Previous work on the role of PLD enzymes for endocytosis of growth factor or hormone receptors is complicated by the fact that PLD may play a role in signaling pathways that recruit specialized endocytic machinery to the receptor as well as having effects on constitutive endocytic machinery. In addition, the endocytic pathways taken by such receptors can be complicated and depend upon the degree to which the receptor is activated (Sigismund et al., 2005). Expression of inactive mutants of both PLD1 and PLD2 inhibited EGF receptor (EGFR) down-regulation in fibroblasts expressing the EGFR (Shen et al., 2001). Overexpression of a truncated PLD2 or its depletion by RNAi, but not PLD1, inhibited the agonist-induced endocytosis of μ opioid and angiotensin receptors as well as phagocytosis in macrophages (Du et al., 2004; Iyer et al., 2004; Koch et al., 2004). RNAi of PLD2 also inhibited constitutive internalization of glutamate receptors transiently expressed in human embryonic kidney 293 cells (Bhattacharya et al., 2004). Now, we show that depletion of PLD2, but not PLD1, inhibits constitutive endocytic recycling of the transferrin receptor. In contrast to the results obtained by Bhattacharya et al. (2004) for the glutamate receptor, we observed that depleting PLD2 inhibited the recycling step, and not internalization. The effect on internalization at steady-state was secondary to the effect seen in recycling. In contrast to endocytosis of the transferrin receptor, constitutive internalization of glutamate receptors has been reported to be insensitive to dynamin mutations, require binding to proteins of the arrestin family, and, in the case of glutamate receptor 5, even be independent of clathrin (Dale et al., 2001; Fourgeaud et al., 2003; Bhattacharya et al., 2004). Thus, important differences exist in the endocytic machinery required for these two types of receptors, which may well include differences in their dependence on PLD enzymes.
PLD activity is stimulated by receptors for a variety of ligands such as hormones or growth factors. In our assay conditions, no activators are used, so the PLD activity is constitutive. Indeed, we did see a reduction in this constitutive activity after PLD2 depletion (Figure 4). In contrast, PLD1 depletion did not decrease constitutive activity, even though it did reduce the PMA-stimulated activity. Thus, in HeLa cells PLD2 is the primary, if not the sole, contributor of steady-state PLD activity. PA produced by this activity could be used for a “housekeeping” function of PLD, such as recycling in the endocytic pathway, in addition to the previously known activities of PLDs after stimulation.
Our colabeling experiments suggest that cells in which PLD2 is depleted accumulate transferrin receptors in a Rab11-positive compartment, probably the recycling endosome. Thus, PLD2 might be acting in a recycling pathway between recycling endosomes and the plasma membrane. Alternatively, PLD2 could be acting in the fast recycling pathway in the early endosomes, and cells depleted of PLD2 would miss-target transferrin receptors to the slow (Rab11) recycling pathway. The latter possibility combined with the involvement of EFA6 in recycling described here is consistent with a proposed functional role of Arf6 in the endosomal sorting processes where clathrin-dependent and independent internalized cargo seem to converge (Naslavsky et al., 2003, 2004). Little is known about the molecular machinery involved in constitutive return of material from either early or recycling endosomes to the cell surface, but the small GTPase Arf6 seems to mediate one recycling pathway. Whether Arf6 regulates clathrin-dependent or independent endocytosis or both is still controversial. Arf6 mutants have been reported to affect endocytosis of membrane proteins internalized independent of clathrin, such as interleukin-2 receptor α subunit Tac and major histocompatability complex I, as well as the transferrin receptor (D'Souza-Schorey et al., 1995; Radhakrishna and Donaldson, 1997; Caplan et al., 2002; Paleotti et al., 2005). One possibility is that Arf6 could receive inputs from different regulators and in turn activate different sets of effectors both in time and space to control distinct pathways as well as different steps of a given pathway. For example, SMAP1, a recently discovered GTPase-activating protein preferential for Arf6, seems to control internalization but not recycling of transferrin receptors (Tanabe et al., 2005), whereas expression of EFA6 decreased the internal accumulation of transferrin receptors, apparently because of increased membrane recycling (Franco et al., 1999). Recombinant Arf6 could activate partially pure rat brain PLD activity but whether Arf6 can activate PLD2 directly has not been demonstrated (Massenburg et al., 1994). However, PA was required for Arf6 to activate hPIP5KIβ in vitro and tagged-PLD2 colocalized with Arf6 and hPIP5KIβ in membrane ruffles produced after EGF treatment in HeLa cells (Honda et al., 1999). Thus, one possibility is that at least part of the constitutive recycling of membrane is mediated by an EFA6/Arf6 pathway and requires basal PLD2 activity. Our observation that EFA6-mediated recycling is blocked by PLD2 RNAi supports this view. Recently, it was found that sec10, a subunit of the exocyst complex, binds to Arf6-GTP and its depletion by RNAi also inhibited recycling of transferrin receptors (Prigent et al., 2003). The exocyst is an eight-subunit complex involved in the docking of vesicles to the plasma membrane (Finger et al., 1998; Guo et al., 1999). The exocyst seems to integrate inputs from different small GTPase-dependent processes such as Arf6, Rabs, Rho, and Ral (Lipschutz and Mostov, 2002; Prigent et al., 2003). Interestingly, both Rho and Ral can bind to PLD and activate or participate in its activation (Hammond et al., 1995; Jiang et al., 1995; Luo et al., 1997). It will be interesting to learn whether Arf6 bound to sec10 is capable of activating PLD2, suggesting that PA may be produced on vesicles during tethering, or whether binding to sec10 inhibits the stimulation of PLD2 by Arf6.
We observed basal production of PIP2 to be reduced by depletion of PLD2. Because Arf6 is a direct activator and PA is a coactivator of PIP5KI enzymes, PIP2 may well be a critical lipid in an Arf6-mediated recycling pathway. However, overexpression of PIP5KI enzymes did not rescue recycling. Therefore, if PIP2 is involved in recycling it does so in combination with other factors, or exogenous PIP5K enzyme activity requires normal PLD2 activity. Unexpectedly, PLD2 depletion also decreased phosphatidylinositol phosphate (PIP) (Figure 8), the precursor of PIP2 that is synthesized by phosphatidylinositol 4-kinases. It is currently not known whether PA has direct or indirect effects on this class of enzymes.
Supplementary Material
Acknowledgments
We thank those that provided reagents used in this study. We thank J. Repa for the use of sequence detection system, M. Porteus for use of FACSCalibur, and J. Seemann for advice in the microinjection experiments and for the use of microscope. This work was supported by fellowships from the Pew Foundation Latin American Fellows Program and the American Heart Association (to D. P.) and by Diane and Hal Brierley Chair in Biomedical Research (to M.G.R.). This investigation was conducted in a facility constructed with support from the Research Facilities Improvement Program Grant C06 RR-15437.
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E05–05–0389) on November 16, 2005.
Abbreviations used: PA, phosphatidic acid; PIP, phosphatidylinositol phosphate; PIP2, phosphatidylinositol bisphosphate; PIP5KI, phosphatidylinositol phosphate kinase I; PLD, phospholipase D.
The online version of this article contains supplemental material at MBC Online (http://www.molbiolcell.org).
References
- Barbieri, M. A., Heath, C. M., Peters, E. M., Wells, A., Davis, J. N., and Stahl, P. D. (2001). Phosphatidylinositol-4-phosphate 5-kinase-1beta is essential for epidermal growth factor receptor-mediated endocytosis. J. Biol. Chem. 276, 47212-47216. [DOI] [PubMed] [Google Scholar]
- Bhattacharya, M., Babwah, A. V., Godin, C., Anborgh, P. H., Dale, L. B., Poulter, M. O., and Ferguson, S. S. (2004). Ral and phospholipase D2-dependent pathway for constitutive metabotropic glutamate receptor endocytosis. J. Neurosci. 24, 8752-8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi, K., Roth, M. G., and Ktistakis, N. T. (1997). Phosphatidic acid formation by phospholipase D is required for transport from the endoplasmic reticulum to the Golgi complex. Curr. Biol. 7, 301-307. [DOI] [PubMed] [Google Scholar]
- Bligh, E. G., and Dyer, W. J. (1959). A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37, 911-917. [DOI] [PubMed] [Google Scholar]
- Brown, H. A., Gutowski, S., Moomaw, C. R., Slaughter, C., and Sternweis, P. C. (1993). ADP-ribosylation factor, a small GTP-dependent regulatory protein, stimulates phospholipase D activity. Cell 75, 1137-1144. [DOI] [PubMed] [Google Scholar]
- Caplan, S., Naslavsky, N., Hartnell, L. M., Lodge, R., Polishchuk, R. S., Donaldson, J. G., and Bonifacino, J. S. (2002). A tubular EHD1-containing compartment involved in the recycling of major histocompatability complex class I molecules to the plasma membrane. EMBO J. 21, 2557-2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cockcroft, S., Thomas, G. M., Fensome, A., Geny, B., Cunningham, E., Gout, I., Hiles, I., Totty, N. F., Truong, O., and Hsuan, J. J. (1994). Phospholipase D: a downstream effector of ARF in granulocytes. Science 263, 523-526. [DOI] [PubMed] [Google Scholar]
- Colley, W. C., Sung, T. C., Roll, R., Jenco, J., Hammond, S. M., Altshuller, Y., Bar-Sagi, D., Morris, A. J., and Frohman, M. A. (1997). Phospholipase D2, a distinct phospholipase D isoform with novel regulatory properties that provokes cytoskeletal reorganization. Curr. Biol. 7, 191-201. [DOI] [PubMed] [Google Scholar]
- D'Souza-Schorey, C., Li, G., Colombo, M. I., and Stahl, P. D. (1995). A regulatory role for ARF6 in receptor-mediated endocytosis. Science 267, 1175-1178. [DOI] [PubMed] [Google Scholar]
- D'Souza-Schorey, C., van Donselaar, E., Hsu, V. W., Yang, C., Stahl, P. D., and Peters, P. J. (1998). ARF6 targets recycling vesicles to the plasma membrane: insights from an ultrastructural investigation. J. Cell Biol. 140, 603-616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale, L. B., Bhattacharya, M., Seachrist, J. L., Anborgh, P. H., and Ferguson, S. S. (2001). Agonist-stimulated and tonic internalization of metabotropic glutamate receptor 1a in human embryonic kidney 293 cells: agonist-stimulated endocytosis is beta-arrestin1 isoform-specific. Mol. Pharmacol. 60, 1243-1253. [DOI] [PubMed] [Google Scholar]
- De Camilli, P., Emr, S. D., McPherson, P. S., and Novick, P. (1996). Phosphoinositides as regulators in membrane traffic. Science 271, 1533-1539. [DOI] [PubMed] [Google Scholar]
- Denmat-Ouisse, L. A., Phebidias, C., Honkavaara, P., Robin, P., Geny, B., Min do, S., Bourgoin, S., Frohman, M. A., and Raymond, M. N. (2001). Regulation of constitutive protein transit by phospholipase D in HT29-cl19A cells. J. Biol. Chem. 276, 48840-48846. [DOI] [PubMed] [Google Scholar]
- Di Paolo, G., Moskowitz, H. S., Gipson, K., Wenk, M. R., Voronov, S., Obayashi, M., Flavell, R., Fitzsimonds, R. M., Ryan, T. A., and De Camilli, P. (2004). Impaired PtdIns(4,5)P2 synthesis in nerve terminals produces defects in synaptic vesicle trafficking. Nature 431, 415-422. [DOI] [PubMed] [Google Scholar]
- Du, G., Altshuller, Y. M., Vitale, N., Huang, P., Chasserot-Golaz, S., Morris, A. J., Bader, M. F., and Frohman, M. A. (2003). Regulation of phospholipase D1 subcellular cycling through coordination of multiple membrane association motifs. J. Cell Biol. 162, 305-315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du, G., Huang, P., Liang, B. T., and Frohman, M. A. (2004). Phospholipase D2 localizes to the plasma membrane and regulates angiotensin II receptor endocytosis. Mol. Biol. Cell 15, 1024-1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finger, F. P., Hughes, T. E., and Novick, P. (1998). Sec3p is a spatial landmark for polarized secretion in budding yeast. Cell 92, 559-571. [DOI] [PubMed] [Google Scholar]
- Fourgeaud, L., Bessis, A. S., Rossignol, F., Pin, J. P., Olivo-Marin, J. C., and Hemar, A. (2003). The metabotropic glutamate receptor mGluR5 is endocytosed by a clathrin-independent pathway. J. Biol. Chem. 278, 12222-12230. [DOI] [PubMed] [Google Scholar]
- Franco, M., Peters, P. J., Boretto, J., van Donselaar, E., Neri, A., D'Souza-Schorey, C., and Chavrier, P. (1999). EFA6, a sec7 domain-containing exchange factor for ARF6, coordinates membrane recycling and actin cytoskeleton organization. EMBO J 18, 1480-1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freyberg, Z., Sweeney, D., Siddhanta, A., Bourgoin, S., Frohman, M., and Shields, D. (2001). Intracellular localization of phospholipase D1 in mammalian cells. Mol. Biol. Cell 12, 943-955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frohman, M. A., Sung, T. C., and Morris, A. J. (1999). Mammalian phospholipase D structure and regulation. Biochim. Biophys. Acta 1439, 175-186. [DOI] [PubMed] [Google Scholar]
- Guo, W., Roth, D., Walch-Solimena, C., and Novick, P. (1999). The exocyst is an effector for Sec4p, targeting secretory vesicles to sites of exocytosis. EMBO J. 18, 1071-1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond, S. M., Altshuller, Y. M., Sung, T. C., Rudge, S. A., Rose, K., Engebrecht, J., Morris, A. J., and Frohman, M. A. (1995). Human ADP-ribosylation factor-activated phosphatidylcholine-specific phospholipase D defines a new and highly conserved gene family. J. Biol. Chem. 270, 29640-29643. [DOI] [PubMed] [Google Scholar]
- Hiroyama, M., and Exton, J. H. (2005). Localization and regulation of phospholipase D2 by ARF6. J. Cell. Biochem. [DOI] [PubMed]
- Honda, A., et al. (1999). Phosphatidylinositol 4-phosphate 5-kinase alpha is a downstream effector of the small G protein ARF6 in membrane ruffle formation. Cell 99, 521-532. [DOI] [PubMed] [Google Scholar]
- Hughes, W. E., and Parker, P. J. (2001). Endosomal localization of phospholipase D 1a and 1b is defined by the C-termini of the proteins, and is independent of activity. Biochem. J. 356, 727-736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer, S. S., Barton, J. A., Bourgoin, S., and Kusner, D. J. (2004). Phospholipases D1 and D2 coordinately regulate macrophage phagocytosis. J. Immunol. 173, 2615-2623. [DOI] [PubMed] [Google Scholar]
- Jenkins, G. H., Fisette, P. L., and Anderson, R. A. (1994). Type I phosphatidylinositol 4-phosphate 5-kinase isoforms are specifically stimulated by phosphatidic acid. J. Biol. Chem. 269, 11547-11554. [PubMed] [Google Scholar]
- Jiang, H., Luo, J. Q., Urano, T., Frankel, P., Lu, Z., Foster, D. A., and Feig, L. A. (1995). Involvement of Ral GTPase in v-Src-induced phospholipase D activation. Nature 378, 409-412. [DOI] [PubMed] [Google Scholar]
- Koch, T., Brandenburg, L. O., Liang, Y., Schulz, S., Beyer, A., Schroder, H., and Hollt, V. (2004). Phospholipase D2 modulates agonist-induced mu-opioid receptor desensitization and resensitization. J. Neurochem. 88, 680-688. [DOI] [PubMed] [Google Scholar]
- Ktistakis, N. T., Brown, H. A., Waters, M. G., Sternweis, P. C., and Roth, M. G. (1996). Evidence that phospholipase D mediates ADP ribosylation factor-dependent formation of Golgi coated vesicles. J. Cell Biol. 134, 295-306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipschutz, J. H., and Mostov, K. E. (2002). Exocytosis: the many masters of the exocyst. Curr. Biol. 12, R212-R214. [DOI] [PubMed] [Google Scholar]
- Lucocq, J., Manifava, M., Bi, K., Roth, M. G., and Ktistakis, N. T. (2001). Immunolocalisation of phospholipase D1 on tubular vesicular membranes of endocytic and secretory origin. Eur. J. Cell Biol. 80, 508-520. [DOI] [PubMed] [Google Scholar]
- Luo, J. Q., Liu, X., Hammond, S. M., Colley, W. C., Feig, L. A., Frohman, M. A., Morris, A. J., and Foster, D. A. (1997). RalA interacts directly with the Arf-responsive, PIP2-dependent phospholipase D1. Biochem. Biophys. Res. Commun. 235, 854-859. [DOI] [PubMed] [Google Scholar]
- Massenburg, D., Han, J. S., Liyanage, M., Patton, W. A., Rhee, S. G., Moss, J., and Vaughan, M. (1994). Activation of rat brain phospholipase D by ADP-ribosylation factors 1, 5, and 6, separation of ADP-ribosylation factor-dependent and oleate-dependent enzymes. Proc. Natl. Acad. Sci. USA 91, 11718-11722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxfield, F. R., and McGraw, T. E. (2004). Endocytic recycling. Nat. Rev. Mol. Cell. Biol. 5, 121-132. [DOI] [PubMed] [Google Scholar]
- Moritz, A., De Graan, P. N., Gispen, W. H., and Wirtz, K. W. (1992). Phosphatidic acid is a specific activator of phosphatidylinositol-4-phosphate kinase. J. Biol. Chem. 267, 7207-7210. [PubMed] [Google Scholar]
- Naslavsky, N., Weigert, R., and Donaldson, J. G. (2003). Convergence of non-clathrin- and clathrin-derived endosomes involves Arf6 inactivation and changes in phosphoinositides. Mol. Biol. Cell 14, 417-431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naslavsky, N., Weigert, R., and Donaldson, J. G. (2004). Characterization of a nonclathrin endocytic pathway: membrane cargo and lipid requirements. Mol. Biol. Cell 15, 3542-3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padron, D., Wang, Y. J., Yamamoto, M., Yin, H., and Roth, M. G. (2003). Phosphatidylinositol phosphate 5-kinase Ibeta recruits AP-2 to the plasma membrane and regulates rates of constitutive endocytosis. J. Cell Biol. 162, 693-701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paleotti, O., Macia, E., Luton, F., Klein, S., Partisani, M., Chardin, P., Kirchhausen, T., and Franco, M. (2005). The small G-protein ARF6GTP recruits the AP-2 adaptor complex to membranes. J. Biol. Chem. 280, 21661-21666. [DOI] [PubMed] [Google Scholar]
- Pathre, P., Shome, K., Blumental-Perry, A., Bielli, A., Haney, C. J., Alber, S., Watkins, S. C., Romero, G., and Aridor, M. (2003). Activation of phospholipase D by the small GTPase Sar1p is required to support COPII assembly and ER export. EMBO J. 22, 4059-4069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prigent, M., Dubois, T., Raposo, G., Derrien, V., Tenza, D., Rosse, C., Camonis, J., and Chavrier, P. (2003). ARF6 controls post-endocytic recycling through its downstream exocyst complex effector. J. Cell Biol. 163, 1111-1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radhakrishna, H., and Donaldson, J. G. (1997). ADP-ribosylation factor 6 regulates a novel plasma membrane recycling pathway. J. Cell Biol. 139, 49-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth, M. G. (1999). Lipid regulators of membrane traffic through the Golgi complex. Trends Cell Biol. 9, 174-179. [DOI] [PubMed] [Google Scholar]
- Roth, M. G., Bi, K., Ktistakis, N. T., and Yu, S. (1999). Phospholipase D as an effector for ADP-ribosylation factor in the regulation of vesicular traffic. Chem. Phys. Lipids 98, 141-152. [DOI] [PubMed] [Google Scholar]
- Shen, Y., Xu, L., and Foster, D. A. (2001). Role for phospholipase D in receptor-mediated endocytosis. Mol. Cell. Biol. 21, 595-602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigismund, S., Woelk, T., Puri, C., Maspero, E., Tacchetti, C., Transidico, P., Di Fiore, P. P., and Polo, S. (2005). Clathrin-independent endocytosis of ubiquitinated cargos. Proc. Natl. Acad. Sci. USA 102, 2760-2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer, W. D., Brown, H. A., Jiang, X., and Sternweis, P. C. (1996). Regulation of phospholipase D by PKC is synergistic with ADP-ribosylation factor and independent of protein kinase activity. J. Biol. Chem. 271, 4504-4510. [DOI] [PubMed] [Google Scholar]
- Tanabe, K., Torii, T., Natsume, W., Braesch-Andersen, S., Watanabe, T., and Satake, M. (2005). A Novel GTPase-activating protein for ARF6 directly interacts with clathrin and regulates clathrin-dependent endocytosis. Mol. Biol. Cell 16, 1617-1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toda, K., Nogami, M., Murakami, K., Kanaho, Y., and Nakayama, K. (1999). Colocalization of phospholipase D1 and GTP-binding-defective mutant of ADP-ribosylation factor 6 to endosomes and lysosomes. FEBS Lett. 442, 221-225. [DOI] [PubMed] [Google Scholar]
- Yamamoto, M., Hilgemann, D. H., Feng, S., Bito, H., Ishihara, H., Shibasaki, Y., and Yin, H. L. (2001). Phosphatidylinositol 4,5-bisphosphate induces actin stress-fiber formation and inhibits membrane ruffling in CV1 cells. J. Cell Biol. 152, 867-876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto, T., Davis, C. G., Brown, M. S., Schneider, W. J., Casey, M. L., Goldstein, J. L., and Russell, D. W. (1984). The human LDL receptor: a cysteine-rich protein with multiple Alu sequences in its mRNA. Cell 39, 27-38. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.