

Abstract
During granule-mediated killing by cytotoxic T lymphocytes or natural killer cells, the serine protease granzyme B enters the target cell by endocytosis and induces apoptosis. Previous studies suggested a role for the mannose 6-phosphate receptor, but further experiments with purified granzyme B indicated this was not essential. Additionally, it is now clear that grB is exocytosed from killer cells in a high-molecular-weight complex with the proteoglycan serglycin. Here granzyme B was delivered as a purified monomer, or in complex with either glycosaminoglycans or serglycin, and killing was evaluated. When granzyme B was a monomer, soluble mannose 6-phosphate had a limited impact, whereas apoptosis induced by the complexed grB was effectively inhibited by mannose 6-phosphate. Most importantly, when granzyme B and perforin were delivered together from granules, inhibition by mannose 6-phosphate was also observed. In pulldown assays mediated by the cation-independent mannose 6-phosphate receptor, granzyme B bound to the receptor more intensely in the presence of immobilized heparan sulfate. We therefore propose the model that under physiological conditions serglycin-bound granzyme B is critically endocytosed by a mannose 6-phosphate receptor, and receptor binding is enhanced by cell surface heparan sulfate.
INTRODUCTION
Granzyme B (grB) is an important effector molecule in the granule-mediated killing pathway by cytotoxic T lymphocytes (CTL) and natural killer (NK) cells (Lord et al., 2003; Roberts et al., 2003; Trapani and Sutton, 2003). There is intense interest in grB-related mechanisms since killer cell cytotoxicity and thus grB are implicated in both organism homeostasis and pathology, through elimination of virally or oncogenically transformed cells and through autoimmunity or graft versus host disease, respectively (Barry and Bleackley, 2002; Russell and Ley, 2002; Trapani and Smyth, 2002). In the target cell, grB activates an apoptotic pathway by cleaving key substrates in the cytoplasm, though these events are critically codependent on the pore-forming protein perforin (pfn). Because grB must access the substrates within the cell, a key step in grB function must be entry into the target cell.
The current hypothesis for grB uptake into the target cell proposes an endocytic mechanism (Froelich et al., 1996), but there has been considerable discussion about this pathway. The first model proposed that the cation-independent mannose 6-phosphate receptor (CI-MPR) was a critical grB receptor (Motyka et al., 2000). However concerns were raised when it was found that grB released from cytotoxic granules was complexed with the large proteoglycan serglycin and that this would be the predominant physiological form of grB (Galvin et al., 1999; Metkar et al., 2002). Notably, uptake studies that identified the CI-MPR as the grB receptor had been performed with a purified free form of grB (Motyka et al., 2000). Because free grB is small (32 kDa; Poe et al., 1991), whereas a grB-serglycin complex is at least eight times larger (serglycin alone from the NK-like cell line YT-Indy is ∼250 kDa; Raja et al., 2002), the concern was that complexing grB to serglycin might sterically hinder or altogether block binding to the CI-MPR.
As an alternative to the CI-MPR, cell surface heparan sulfate has been implicated in grB uptake through four independent studies (Bird et al., 2005; Kurschus et al., 2005; Raja et al., 2005; Shi et al., 2005). In addition, evidence has been presented to suggest that grB may electrostatically exchange from serglycin to cell surface heparan sulfate (Raja et al., 2005). Notably, three of the reports suggested that alternate receptors or mechanisms for grB likely also exist (Bird et al., 2005; Kurschus et al., 2005; Raja et al., 2005). Even so, these models contribute to further skepticism over the CI-MPR-mediated grB uptake model.
Finally, several lines of evidence have argued directly against a role for the CI-MPR in grB uptake and killing. For instance, some data indicated that a CI-MPR-independent pathway was sufficient to mediate grB uptake leading to apoptosis, so though a CI-MPR-mediated grB uptake pathway existed, the pathway was not critical (Trapani et al., 2003). Further, using target cell lines (immortalized fibroblasts and concanavalin A (ConA)-stimulated lymphoblasts) derived from CI-MPR knockout mice, CTL effectively induced chromium release and DNA fragmentation irrespective of CI-MPR expression (Dressel et al., 2004a, 2004b). The data have been interpreted to indicate that the CI-MPR is not critical in grB-mediated killing and have thus driven focus away from this model.
To date, studies of grB uptake, including most CI-MPR-directed analyses, have largely ignored the contribution of serglycin. For this reason we decided to evaluate the impact of serglycin on the CI-MPR-mediated uptake pathway. The effects of heparan sulfate and other glycosaminoglycans (GAGs) on grB-CI-MPR binding were also tested through a series of pulldown assays. Finally, the killing mechanism for ConA-stimulated lymphoblast targets has been characterized to understand the meaning and implications of the CI-MPR knockout studies.
MATERIALS AND METHODS
Cells and Reagents
Unless otherwise specified, chemical reagents were available from Sigma-Aldrich Canada (Oakville, Ontario, Canada). Maintenance of Jurkat cells and L1210 (H-2d; a subclone of the original mouse lymphocyte leukemia line, selected for resistance to Fas stimulation), as well as maintenance and isolation of human CTL and alloreactive CTL from wild type, grB-/-, granzyme A-/-/grB-/-, or pfn-/- mice (all with a mixed genetic background of C57BL/6 and 129 (H-2b)) were as described (Atkinson et al., 1998), except that before killing assays, alloreactive CTL were not stimulated with anti-CD3 antibody. ConA-stimulated lymphoblasts were splenocytes derived from BALB/c mice (H-2d) and were stimulated 48 h with 5 μg/ml ConA in RMPI 1640 media (Invitrogen Canada, Burlington, Ontario, Canada) supplemented with 10% fetal bovine serum (Hyclone, Logan, UT), 90 U/ml interleukin-2 (Chiron, Emeryville, CA) and 100 U/ml each of penicillin and streptomycin (Invitrogen). CI-MPR-deficient (MS) and -overexpressing (MS9II; both H-2k) cells were maintained as described (Gabel et al., 1983; Watanabe et al., 1990). Human grB was purified from the cytotoxic granules of YT-Indy cells (Caputo et al., 1999). Pfn and isolated granules, prepared from the rat NK-like line RNK-16 (Winkler et al., 1996), were kindly given by Dr. D. Hudig (University of Nevada, Reno, NV). Notably, both pfn and isolated granule stocks contain 1 M sodium chloride. Human replication deficient AD type 5 d170-3 (AdV) was purified in our laboratory as described (Bett et al., 1994). Streptolysin O (SLO) was kindly obtained from Dr. S. Bhakdi (Institute of Medical Microbiology and Hygiene, Mainz, Germany; Bhakdi et al., 1985, 1993).
To form GAG-grB complexes, heparan sulfate (250 μg/ml), heparin (100 μg/ml; Hepalean, Organon Teknika, St. Laurent, Quebec, Canada), or chondroitin sulfate A (50 μg/ml) were incubated with grB in DMEM (Invitrogen) and 0.1% (wt/vol) bovine serum albumin (BSA) for 60–90 min at 37°C. When complexes were filter fractionated, materials were loaded on a YM-100 Microcon centrifugal filter (Fisher Scientific International, Nepean, Ontario, Canada) and centrifuged at 12 000 × g until the filter was dry. Retained materials were then recovered in fresh media, in a volume equivalent to the starting material. To estimate grB in each fraction, enzymatic activity as measured by the colorimetric substrate BAADT (butoxylcarbonyl-Ala-Ala-Asp-thiobenzyl ester; Enzyme Systems, Livermore, CA) was compared with a standard curve (Odake et al., 1991). Alternatively, fractions were analyzed by nondenaturing Agarose gel, as previously described (Veugelers et al., 2004).
To obtain the CTL degranulate material, 107 cells/ml human CTL, in either DMEM with 0.1% BSA, or in phenol Red-free RPMI (Invitrogen) with 20% (vol/vol) phosphate-buffered saline (PBS) were layered into wells coated with anti-human CD3, either clone OKT3 (received from Dr. K. Kane, University of Alberta, Edmonton, Alberta, Canada) or clone HIT3a (BD Biosciences, Mississauga, Ontario, Canada) and then incubated 4 h at 37°C. The culture medium was recovered to collect the degranulate material. As needed, grB levels were estimated by BAADT assay. Filter fractionation of CTL material to obtain high-molecular-weight degranulate material has been described previously (Veugelers et al., 2004).
Killing Assays
Jurkat suspensions (2 × 105) were incubated in DMEM and 0.1% BSA with grB from various sources (purified, isolated granules, CTL degranulate), with or without additional factors, for 3 h at 37°C. Where indicated, AdV was used at 100 plaque-forming units per cell; pfn was routinely added at a 10-fold dilution (assessed as sublytic by 7-amino-actinomycin D exclusion; BD Biosciences), unless otherwise stated; and SLO was added at 300 ng/ml. Where indicated, 20 mM mannose 6-phosphate (M6P; disodium salt) was added to cells at room temperature for 15 min before addition of apoptotic stimuli. Because pfn and granule stocks contain 1 M sodium chloride and there are extra sodium ions associated with M6P disodium salt, the Na+ concentration was adjusted to within 140–160 mM with HEPES buffer. The Ca2+ concentration was maintained at 2 mM. DNA fragmentation was monitored by TUNEL labeling (terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling; Gavrieli et al., 1992) using fluorescein-conjugated dUTP (Roche Diagnostics, Laval, Quebec, Canada) as the labeling reagent, according to the supplier's protocol. The percentage of TUNEL-positive cells was determined by flow cytometry (Heibein et al., 1999). Percent specific TUNEL-positive cells were calculated by the following equation: (% specific TUNEL positive cells) = 100% × ([sample % TUNEL positive cells] - [untreated sample % TUNEL positive cells])/(100% - [untreated sample % TUNEL positive cells]).
Two-day ConA-stimulated mouse lymphoblasts and L1210 were treated with alloreactive mouse CTL (stimulated with irradiated BALB/c (H-2d) splenocytes; 5:1 effector/target); MPR-deficient and -overexpressing cells were treated with alloreactive mouse CTL (stimulated with irradiated C3H (H-2k) splenocytes). DNA fragmentation was assessed by [3H]thymidine release or cell permeabilization by chromium release (Garner et al., 1994).
bCI-MPR and GAG Pulldown Assays
For CI-MPR-mediated pulldown, biotinylated soluble CI-MPR (bCI-MPR; 7.5 μg/ml; received from Dr. P. Lobel, Center for Advanced Biotechnology and Medicine, Piscataway, NJ) was allowed to prebind to Neutravidin-Agarose beads (12.5% [vol/vol]; Pierce Biotechnology, Rockford, IL) in PBS with 0.75% BSA. Mock samples lacked bCI-MPR and prebinding was carried out for 30 min at room temperature with constant agitation. For pulldowns with GAGs, to cyanogen bromide-activated Sepharose 4B, chondroitin sulfate A, heparan sulfate, and heparin (Sigma) were coupled (at 1 μmol/ml medium), according to the supplier's directions. To estimate conjugated GAG, Azure A binding by GAG-Sepharose less mock-coupled Sepharose was compared with a GAG-specific standard curve (Thuy and Nyhan, 1992). For pulldown assays, GAG-Sepharose (total GAG in 10-fold molar excess to grB) was resuspended under conditions similar to above. Stocks of purified grB, or of fresh high-molecular-weight degranulate (∼1 μg/ml grB final concentration) were added to the bead mixtures, and the additional volume created a bead slurry of 10%; notably, for bCI-MPR pulldowns, grB was in excess. In some samples, M6P or soluble GAGs (in 500-fold molar excess of grB) were present. Samples were allowed to incubate 30 min at room temperature with constant agitation, including for sequential bead incubations (Figure 4C). Beads were washed five times with PBS and then eluted directly into SDS-sample buffer. For analysis by immunoblot, grB was detected with mouse anti-human grB (clone 2C5; Santa Cruz Biotechnology, Santa Cruz, CA; 400 ng/ml), followed by goat anti-mouse IgG (Bio-Rad Laboratories, Hercules, CA; 1:3000 dilution). To detect serglycin by sensitivity-enhanced silver stain, samples were first resolved on a Tris 4–15% SDS-PAGE gradient gel (Bio-Rad). To stain, the protocol was essentially as described (Min and Cowman, 1986; Ikegami-Kawai and Takahashi, 2002). Briefly, gels were sequentially stained with 0.05% (wt/vol) Alcian Blue 8GX (electrophoresis grade) in 2% acetic acid and then with a silver stain kit (Bio-Rad) based on the procedure of Merril et al. (1981), beginning from the oxidation step. To prepare standard controls to identify serglycin, high-molecular-weight degranulate (diluted to a final concentration of 50%, vol/vol), was treated with or without chondroitinase ABC (0.5 U/ml) in reaction buffer (50 mM Tris-HCl, pH 8.0, 1 M sodium acetate, 0.02% BSA) overnight at 37°C.
Figure 4.

Granzyme B binding to CI-MPR is inhibited by mannose 6-phosphate and soluble heparin, but is enhanced by immobilized GAGs. (A) To obtain a concentrated source of predominantly serglycin-complexed grB, CTL degranulate material was obtained as the supernatant of a CTL culture and incubated for 4 h at 37°C in the presence of immobilized anti-CD3 antibody. Subsequently the degranulate was fractionated over a 100-kDa cutoff filter, and the high molecular retentate was utilized. Degranulate retentate was incubated with biotinylated, soluble CI-MPR (bCI-MPR) prebound to Neutravidin-Agarose beads. In some samples M6P (0.2–20 mM) was present during incubation. (i) The bead fraction was probed by immunoblot to detect grB. (ii) The bead fraction was resolved on a 4–15% gradient SDS-PAGE, and sensitivity-enhanced silver staining was performed to detect proteoglycans. As a control to identify serglycin, degranulate retentate was treated with or without chondroitinase ABC, and was analyzed in titrated amounts. (iii) The soluble fraction of samples was recovered and fractionated over a 100-kDa cutoff filter. Equal volumes of samples were analyzed by immunoblot to detect grB. (B) Purified grB was incubated with GAG-Sepharose beads (CS, chondroitin sulfate A; HS, heparan sulfate; Hep, heparin), in the presence (+) or absence (-) of 20 mM M6P. The bead fraction was probed by immunoblot to detect grB. (C) Purified grB (i) or degranulate retentate (ii) was incubated with free bCI-MPR in the presence or absence of GAG-Sepharose beads (total GAG estimated at 10-fold molar excess of grB). The soluble fraction was then applied to Neutravidin-Agarose beads in order to immobilize bCI-MPR. Subsequently, the bead fraction was probed by immunoblot to detect grB. Band intensity was quantified by densitometry and is expressed as the normalized mean ± SD (i; n = 3) or ± range (ii; n = 2) (* 0.01 < p < 0.05). (D) Purified grB (i) or degranulate retentate (ii) was incubated with bCI-MPR prebound to Neutravidin-Agarose beads. In the indicated samples, GAG was present at 500-fold molar excess of grB. The bead fraction was probed by immunoblot to detect grB. Notably, chondroitin sulfate and heparan sulfate had inconsistent effects on grB binding to the bCI-MPR, therefore cannot be interpreted reliably. On the other hand, consistently heparin effectively blocked grB binding to bCI-MPR; when band intensity was quantified by densitometry, compared with the std in the absence of GAG (normalized to 1.0), heparin diminished grB recovery to (i) 0.23 ± 0.15 (0.001 < p < 0.01; n = 3) and (ii) 0.26 ± 0.21 (0.01 < p < 0.05; n = 3).
RESULTS
MPR-dependence of grB-induced Cell Death Is Affected by Conditions of Delivery
To understand the basis of the disparate results found with grB-mediated killing, we tested grB delivered by both adenovirus (AdV) and pfn. In Jurkat cells treated with grB and AdV, DNA fragmentation was blocked by soluble M6P (Figure 1A). However, when Jurkats were incubated with grB and pfn, M6P had a minimal impact (Figure 1B). Thus, grB-mediated killing was dependent on a M6P receptor (MPR) when delivered by AdV, but not when delivered by pfn. This suggested that though AdV could functionally mimic pfn to deliver grB, it did not do so by an identical mechanism. Further, this highlighted the importance of choosing appropriate lytic agents, particularly in uptake studies of granzymes.
Figure 1.
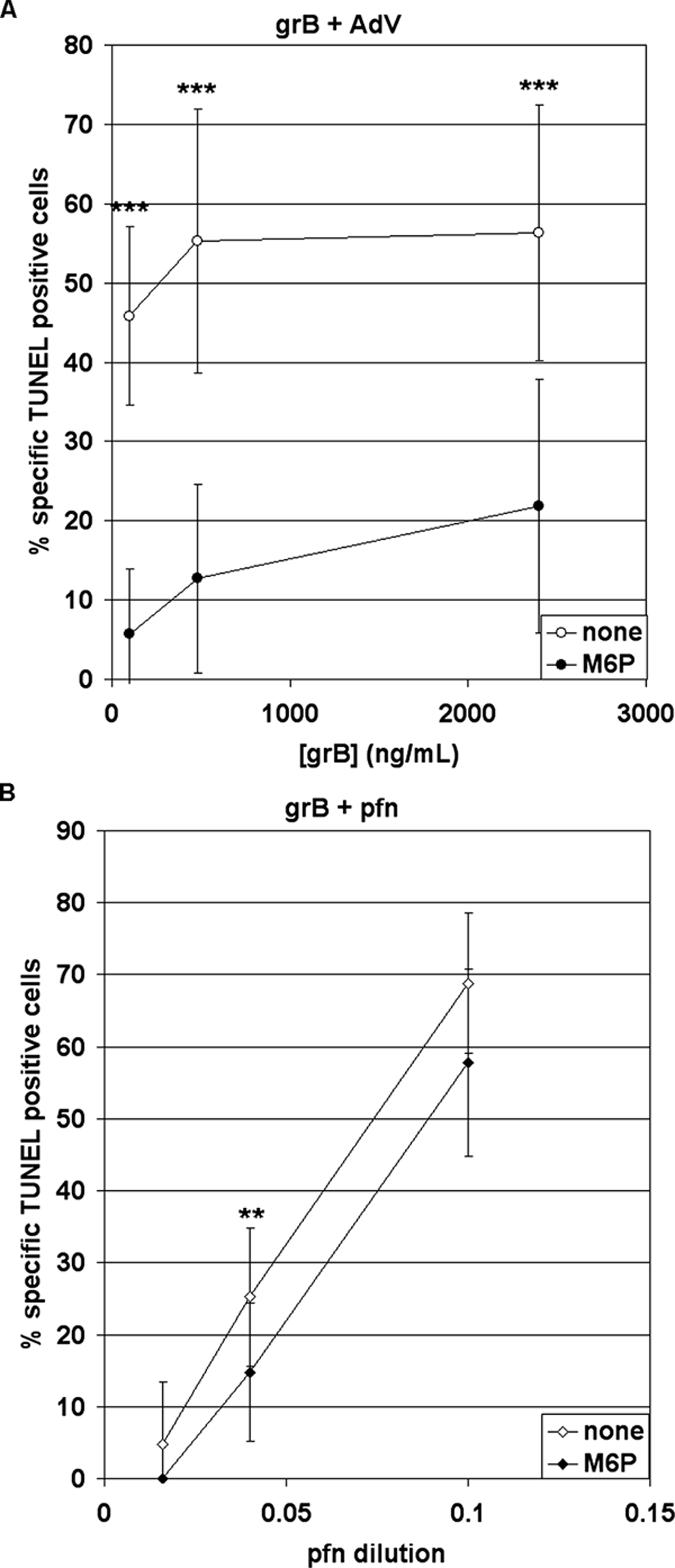
GrB and pfn-mediated DNA fragmentation is MPR-independent, but grB- and AdV-mediated DNA fragmentation is MPR-dependent. Jurkat cells were treated with apoptotic stimuli, in the presence or absence of 20 mM M6P. DNA fragmentation was assessed after 3 h, by TUNEL labeling and flow cytometry analysis. (A) Cells were treated with the indicated concentrations of grB, along with AdV. (B) Cells were treated with 500 ng/ml purified grB and the indicated dilutions of purified pfn. Data represent the mean ± SD of six (A) or five (B) separate experiments (** 0.001 < p < 0.01; *** p < 0.001).
Glycosaminoglycan-complexed grB Directs pfn-assisted Killing to an MPR
The in vitro results above contradict evidence that grB-induced DNA fragmentation during CTL killing was dependent on the CI-MPR (Motyka et al., 2000; Trapani et al., 2003). This implied that another cytotoxic granule factor might direct the grB and pfn killing pathway through the CI-MPR. The most likely candidate was serglycin, because grB secreted from killer cells is in a stable complex with this proteoglycan (Galvin et al., 1999). To test the hypothesis that serglycin-complexed grB preferentially relied on the CI-MPR for pfn-assisted apoptosis, an in vitro system was developed. Purified grB was incubated with one of two GAGs, either heparan sulfate or heparin (Figure 2A) in order to form a complex. The grB-heparan sulfate/heparin material was then used in killing assays along with pfn to assess the CI-MPR dependence of grB killing.
Figure 2.

GrB-heparan sulfate complexes rely on MPRs to induce apoptosis in the presence of pfn. (A) The basic chemical units of chondroitin sulfate A (serglycin-modifying GAG) and heparin/heparan sulfate. Heparin is the most densely sulfated glycosaminoglycan, whereas heparan sulfate includes variations with fewer sulfate groups (adapted from Kolset et al., 2004). (B) Purified grB (25 μg/ml) was incubated with PBS (none), heparan sulfate (HS; 125 μg/ml), heparin (Hep; 100 μg/ml), or chondroitin sulfate A (CS, 50 μg/ml) in media for 90 min at 37°C. Samples were resolved on a nondenaturing Agarose gel and immunoblotted for grB (f, free grB; *, complexed grB). Data are representative of at least three separate experiments. (C and D) Purified grB (20 μg/ml) was mock-treated with PBS or treated with heparin (Hep; 100 μg/ml) for a 60-min incubation in media at 37°C. Samples were then fractionated over a 100-kDa cutoff centrifugal filter. Final fractions were readjusted back to the original volume with media. (C) GrB enzymatic activity was assessed in precomplexed samples (S) and in fractions (F, filtrate; R, retentate) by cleavage of the colorimetric substrate BAADT. The shown relative absorbance readings were within the linear range of the assay, as determined by a standard curve. (D) GrB proapoptotic activity was assessed in a killing assay. Jurkat cells were treated with a 200-fold dilution of precomplexed sample or fraction in the presence of AdV. DNA fragmentation was assessed after 3 h, by TUNEL labeling and flow cytometry. (E and F) Jurkat cells were treated with apoptotic stimuli and inhibitors at 37°C. DNA fragmentation was assessed after 3 h, by TUNEL labeling and flow cytometry analysis. (E) grB (2.4 μg/ml) was preincubated for 1 h at 37°C in the presence or absence of heparin (Hep, 100 μg/ml) and then was added to cells (diluted about fourfold) along with pfn in the presence or absence of 20 mM M6P. Data are representative of two separate experiments. (F) grB (500 ng/ml) was preincubated for 1 h at 37°C in the presence or absence of heparin (Hep, 100 μg/ml) and then was added to cells (diluted about fourfold) along with AdV in the presence or absence of 20 mM M6P. Data represent the mean ± SD of three separate experiments (* 0.01 < p < 0.05).
The formation of grB-heparan sulfate/heparin complexes was monitored after grB was incubated either alone or in the presence of the GAG. First, complex formation was analyzed by a gel shift assay, and grB was detected by immunoblot analysis (Figure 2B). The mock-treated grB had a band near the origin, which is consistent with migration of the positively charged molecule toward the cathode at the top of the gel. It was therefore unexpected to also observe a band that migrated strongly toward the anode. Though this band has not been characterized, it has been speculated that it might represent aggregated grB material. Regardless, when grB was incubated with chondroitin sulfate A as a control (Galvin et al., 1999), a shift in band mobility toward the anode was observed, suggesting the negative charge of the chondroitin sulfate A had induced this shift in grB migration. Similarly, coincubation of grB with heparan sulfate or heparin resulted in a band shift toward the anode, though the effect of heparin was greater. This enhanced grB association is likely due to the stronger negative charge of heparin, compared with both heparan sulfate and chondroitin sulfate A (Figure 2A). Overall these data suggest that at least a portion of the grB had formed a stable complex with either GAG.
The effect of complex formation on grB activity was monitored; for heparin-treated grB, enzymatic and apoptotic activities were detected by cleavage of a colorimetric substrate (Figure 2C) or when delivered by AdV to induce DNA fragmentation in Jurkat targets (Figure 2D), respectively. By either assay, grB activity was not affected by heparin. When samples were fractionated over a 100-kDa cutoff centrifugal filter to separate complexed grB from free, specifically heparin-treated grB was retained on the filter. Similar results were observed when grB was coincubated with heparan sulfate (unpublished data). This further indicated formation of a grB-heparan sulfate/heparin complex.
Next, the grB-heparan sulfate/heparin complexes were evaluated for their dependence on the CI-MPR in pfn-assisted killing assays (Figure 2E). Although pfn-mediated delivery of free grB was insensitive to M6P, delivery of heparin-(Figure 2E) or heparan sulfate-complexed (unpublished data) grB was effectively blocked by M6P. Thus, in contrast to pfn and free grB, apoptosis induced by pfn and grB-heparan sulfate/heparin complexes was critically MPR-dependent. In a control experiment, grB and AdV induced DNA-fragmentation in Jurkat targets whether or not grB was complexed to heparin; notably, this killing was always competed by M6P (Figure 2F). Similar results were observed when grB was heparan sulfate-bound (unpublished data). These data indicated that binding of heparan sulfate/heparin to grB did not have a deleterious effect with respect to the proapoptotic activity of grB. In contrast, grB and pfn-induced apoptosis was reduced in the presence of heparin (Figure 2E) and heparan sulfate (unpublished data), but this was consistent with the fact that GAGs have been found to inhibit the membranolytic activity of pfn (Tschopp and Masson, 1987).
Ultimately, the data support the hypothesis that when grB is electrostatically complexed to a GAG, killing assisted by pfn is preferentially directed through an MPR. If, on the other hand, heparan sulfate/heparin or serglycin are absent, then grB may induce killing following cell entry from numerous pathways.
Apoptosis Induced by grB-Serglycin Is MPR-dependent
Because apoptosis induced by pfn and GAG-complexed grB was critically dependent on an MPR, the hypothesis was tested in a more physiological manner with grB-serglycin complexes obtained in the form of CTL degranulate (Galvin et al., 1999). We have shown previously that degranulate collection by this method yields grB in a predominantly serglycin-complexed state (Veugelers et al., 2004). The degranulate material was further characterized by filter fractionation over a 100-kDa cutoff filter to partition free and serglycin-complexed grB. The grB-serglycin complex was stable for at least 24 h (Figure 3A) and showed a slight tendency to dissociate from serglycin as the pH was raised to pH 7.0 or greater (Figure 3B). Also, the complex was stable over two freeze-thaw cycles (Figure 3C), allowing limited storage of materials at -80°C.
Figure 3.

Killing by serglycin-bound grB is MPR-dependent. CTL degranulate material was obtained as the supernatant of a CTL culture that was incubated for 4 h at 37°C in the presence (or absence, in the case of spontaneous degranulate) of immobilized anti-CD3 antibody. (A–C) After the indicated treatments, degranulate samples were fractionated over a 100-kDa cutoff filter. Equal volumes of samples were analyzed by immunoblot to detect grB. Data are representative of at least three separate experiments. (A) Time-course. Degranulate was incubated at 37°C in a 5% CO2 atmosphere for the indicated hours and fractionated at time points. (B) pH effect. To degranulate samples, the following buffers were added to a final concentration of 50 mM: sodium acetate, pH 4.5; MES, pH 5.5; HEPES, pH 7.0; Tris, pH 8.0; boric acid, pH 9.0. Samples were incubated 1 h at 37°C. (C) freeze-thaw effect. Samples were subjected to the indicated number of freeze-thaw cycles. One cycle allowed freezing for at least 30 min at -80°C, followed by rapid thaw in a 37°C water bath. (D–F) Jurkat cells were treated with apoptotic stimuli and inhibitor at 37°C. DNA fragmentation was assessed after 3 h, by TUNEL labeling and flow cytometry analysis. Data are representative of at least three separate experiments. (D) grB, either from purified sources (100 ng/ml) or from supernatants of anti-CD3-stimulated CTL (∼100 ng/ml), was added to cells along with AdV or SLO, in the presence or absence of 20 mM M6P. (E) grB, either from purified sources (500 ng/ml) or from CTL supernatants (untreated, ∼25 ng/ml; anti-CD3-treated, ∼400 ng/ml), was added to cells with or without AdV. (F) Cells were treated with isolated granules at the indicated dilutions. Data represent the mean ± SD of five separate experiments (* 0.01 < p < 0.05; *** 0.001 < p < 0.01).
To compare the killing pathways of free, purified grB versus serglycin-bound grB in CTL degranulate, these were codelivered with SLO, in the presence or absence of M6P. Though we have shown previously that pfn was present in degranulate material (Veugelers et al., 2004), it was inactivated because of calcium in the medium. Thus exogenous SLO, or a functional analogue, was required to induce killing in the presence of degranulated grB. Both the purified grB and serglycin-bound grB were able to induce DNA fragmentation in the presence of SLO. Strikingly, although M6P had no effect on SLO-assisted delivery of the purified grB, it efficiently blocked DNA fragmentation in response to SLO and degranulate material (Figure 3D). Notably, SLO was likely behaving in a manner similar to pfn, because grB-mediated killing displayed similar pathway profiles whether delivered by pfn or SLO. Thus these data further suggested that apoptosis induced by serglycin-bound grB and pfn is directed preferentially through an MPR. Importantly, AdV-assisted delivery confirmed the proapoptotic potential of either grB material (Figure 3, D and E). When supernatants were taken from CTL that had not been stimulated with immobilized anti-CD3, very little proapoptotic activity was detected, suggesting induction of apoptosis was dependent on release of cytotoxic granule materials and that degranulation was specifically dependent on CD3 stimulation (Figure 3E). Also, in the absence of AdV, all sources of grB induced negligible levels of apoptosis (Figure 3E).
A physiologically relevant model of granule-mediated killing was achieved through the use of isolated granules. In granules, serglycin formed a mixed complex carrying grB and pfn, at least (Metkar et al., 2002). To induce death in targets, isolated granules deliver a complex of serglycin with grB and pfn. When Jurkat cells were challenged with grB and pfn delivered in the form of intact granules, the DNA fragmentation response was M6P-sensitive (Figure 3F). This suggested that serglycin-complexed grB and pfn were critically dependent on an MPR for entry into the cell. Taken together, these data imply that the CI-MPR would in fact be critically involved in grB-induced killing under physiological conditions.
Immobilized GAGs Enhance Granzyme Binding to the CI-MPR
Recently four independent studies have pointed to a role for cell surface heparan sulfate in grB binding and uptake (Bird et al., 2005; Kurschus et al., 2005; Raja et al., 2005; Shi et al., 2005). Such findings for purified grB are consistent with the binding and uptake of monomeric grB, as suggested by the model in the current report. Intriguingly, however, evidence has been presented that grB exchanged from serglycin to Sepharose-heparan sulfate (Raja et al., 2005). To reconcile the heparan sulfate-mediated uptake model with the MPR-mediated uptake model, a series of pulldown assays were performed to consider grB interactions with the CI-MPR and with GAGs.
First, it was important to establish the specificity with which grB bound to GAGs versus MPRs, especially with respect to M6P-sensitivity because this has been the primary indicator of a pathway involving MPRs. This was tested by incubating grB with immobilized CI-MPR or GAGs in the presence of M6P. Concentrated grB-serglycin complexes were obtained from CTL degranulate material by fractionating over a 100-kDa cutoff filter and using the high-molecular-weight retentate. The degranulate retentate was incubated with a biotinylated, soluble form of the CI-MPR (bCI-MPR) that was prebound to Neutravidin-Agarose beads. The bead fraction was analyzed for grB by immunoblot. GrB associated with the beads in a bCI-MPR-dependent manner, and these interactions were M6P-dependent, because increasing concentrations of M6P sugar in the sample prevented grB from binding to the bCI-MPR beads (Figure 4Ai). The bCI-MPR-associated materials were further characterized, and revealed a proteoglycan of >250 kDa (Figure 4Aii). Consistent with evidence that serglycin is the predominant proteoglycan in cytotoxic granules (Grujic et al., 2005), this high-molecular-weight band was likely serglycin, both because of its size and sensitivity to chondroitinase ABC in control samples. The interaction was specific because serglycin was retrieved in the precipitated material only in the presence of bCI-MPR. In addition, 20 mM M6P in the sample prevented serglycin from partitioning with the beads, suggesting serglycin associated with the CI-MPR in a M6P-dependent manner. Notably, the sample soluble pools were fractionated over the 100-kDa cutoff filter, and because grB partitioned preferentially to the retentate fraction, this revealed that even after sample incubation, grB was predominantly bound to serglycin (Figure 4Aiii). The most likely interpretation of these data are that serglycin is coprecipitated with granzymes, implying that the trimer serglycin-grB-CI-MPR may form. Any other CI-MPR-binding factor present in degranulate could also behave as the molecular bridge between serglycin and the receptor.
To characterize the interactions of GAGs and grB, chondroitin sulfate, heparan sulfate, or heparin was conjugated to Sepharose beads. GrB, whether from purified (Figure 4B) or degranulate (unpublished data) sources, bound stably to the GAG-Sepharose, which was consistent with previous findings (Bird et al., 2005; Kurschus et al., 2005; Raja et al., 2005; Shi et al., 2005). However, M6P did not block the interaction (Figure 4B). Importantly, this indicated that grB bound to the CI-MPR and to GAGs by distinct mechanisms, and this provided a practical means for distinguishing these binding events. In particular, with respect to the killing assays, this strengthened the interpretation that M6P inhibited grB-serglycin-mediated killing was inhibited by blocking MPR association. Further, this challenged an interpretation that suggested M6P blocked the charge-charge interaction of grB and cell surface heparan sulfate (Shi et al., 2005).
The findings in this and other (Bird et al., 2005; Kurschus et al., 2005; Raja et al., 2005) reports suggest that when grB binds to cell surface heparan sulfate, this is not the physiologically critical step in grB entry into the target cell. Intriguingly, it was found that immobilized heparan sulfate, as well as chondroitin sulfate and heparin, potentiated grB-CI-MPR interactions. Either purified grB (Figure 4Ci) or degranulate retentate (Figure Cii) was incubated in the presence of both GAG-Sepharose beads and soluble bCI-MPR. Subsequently, the bCI-MPR and associated molecules were recovered in the soluble fraction and then immobilized by incubating with Neutravidin-Agarose beads. GrB bound to bCI-MPR was monitored by immunoblotting the bead fraction. As important controls, parallel-treated samples were prepared in the absence of any Sepharose beads, or with Sepharose beads lacking any bound GAG. In these samples, the grB bound to bCI-MPR was comparable. However, the presence of GAG-coated Sepharose beads in the first incubation step enhanced bCI-MPR-mediated recovery of grB. These findings contrasted pulldown experiments in the presence of soluble GAGs. Purified grB (Figure 4Di) or degranulate retentate (Figure 4Dii) was incubated with bCI-MPR prebound to Neutravidin-Agarose beads in the presence or absence of excess GAGs (chondroitin sulfate A, heparan sulfate, or heparin). To detect grB binding to the bCI-MPR, the bead fraction was analyzed by immunoblot. Remarkably, heparin was highly effective at preventing a bCI-MPR-grB interaction, whether grB was derived from a purified source or degranulate. Chondroitin sulfate A or heparan sulfate did not have reproducible effects on grB binding to the receptor and had at best a partially inhibitory effect. Overall, this was consistent with and implied a new interpretation of previous findings that heparin effectively inhibited both grB cell surface binding (Raja et al., 2005), as well as killing using isolated cytotoxic granules (Shi et al., 2005). But more importantly the effect of GAGs on grB-bCI-MPR interactions was dependent on whether the GAG was soluble or immobilized. Because in vivo, heparan sulfate is immobilized on the surface of cells, this suggested that cell surface heparan sulfate can promote grB binding to the CI-MPR.
Alloreactive CTL Induce Granzyme A- and B-independent Death in Lymphoblast Targets
The data presented thus far argue compellingly that under physiological conditions CI-MPR-mediated uptake of grB critically precedes apoptosis induction. Even so, these data are in strong disagreement with a study of CTL killing in CI-MPR knockout cell lines. In ConA-stimulated lymphoblasts (Dressel et al., 2004a) and in immortalized fibroblasts (Dressel et al., 2004b), CTL-induced DNA fragmentation was comparable between CI-MPR-deficient and wild-type cells. Because the induction of DNA fragmentation was sensitive to treatment with EGTA (Dressel et al., 2004a, 2004b) or caspase inhibitors (Dressel et al., 2004a), it was concluded that this killing was grB-mediated and did not critically rely on the CI-MPR. Notably, these approaches did not conclusively rule out a Fas-mediated killing mechanism. Fas-mediated killing is indeed dependent on caspases (Enari et al., 1995; Los et al., 1995; Tewari and Dixit, 1995). Also, though activation of the Fas pathway is calcium-independent (Rouvier et al., 1993), Fas-mediated killing by CTL is EGTA-sensitive, because Fas-ligand localizes to cytotoxic granules, and its surface expression is regulated by the calcium-dependent granule-release event (Bossi and Griffiths, 1999).
Therefore killing of ConA-stimulated lymphoblasts was assessed with respect to grB dependence. Two-day ConA-stimulated lymphoblasts were challenged with alloreactive CTL derived from grB+/+, grB-/-, granzyme A-/-/grB-/-, or pfn-/- mice, in the presence or absence of EGTA, and tritium- and chromium-release responses were monitored (Figure 5, A and B). As controls, L1210, either untreated (Figure 5, C and D) or treated with ConA (Figure 5, E and F), were similarly challenged and monitored. Strikingly, neither DNA fragmentation (Figure 5A) nor cell permeabilization (Figure 5B) responses in ConA-stimulated lymphoblasts required granzyme A and/or grB. Further, the death responses were only partially dependent on pfn. Even so, CTL-induced killing was effectively blocked by EGTA (Figure 5, A and B). Most notably, EGTA inhibited the pfn-independent component of killing, which may have been mediated by either the Fas pathway or other pfn-independent cytotoxic granule factors. Thus EGTA is not an effective means of distinguishing granzyme and pfn-activated pathways from alternate killing mechanisms in these targets.
Figure 5.

Allogeneic CTL attack induces grB-independent DNA fragmentation in ConA-stimulated lymphoblast targets. Two-day ConA-stimulated lymphoblasts (A and B), L1210 (C and D), or 24-h ConA-treated L1210 (E and F) were challenged with alloreactive CTL (5:1 effector/target ratio). ConA-treated targets were washed extensively before CTL attack. DNA fragmentation in targets was monitored by tritium-release at 2 h (A, C, and E), or cell permeabilization by chromium-release at 4 h (B, D, and F). CI-MPR-deficient (G) and CI-MPR-overexpressing (H and I) fibroblast cell lines were challenged with alloreactive CTL, and DNA fragmentation in targets was monitored by tritium release at 3 h (G and H) or 6 h (I). E/T, effector to target ratio; grA, granzyme A; KO, knockout; WT, wild type. Data represent the mean ± SD of three separate experiments (A–F and I); or represent the mean ± SD of triplicate samples, and are representative of at least three separate experiments (G and H; * 0.01 < p < 0.05; ** 0.001 < p < 0.01; *** p < 0.001).
This finding could not simply be attributed to abnormal behavior of the effector cells. In L1210 a normal response was observed (Simon et al., 1997), because DNA fragmentation was specifically grB- and pfn-dependent (Figure 5C), whereas cell permeabilization was dependent on either granzyme A or grB in cooperation with pfn (Figure 5D). The findings also could not be attributed to ConA treatment, because the L1210 response was similar when cells were preincubated in both the presence (Figure 5, E and F) and absence (Figure 5, C and D) of ConA.
This issue was also addressed in CI-MPR-deficient (Figure 5G) and CI-MPR-overexpressing (Figure 5, H and I) fibroblast cell lines. As previously reported (Motyka et al., 2000), fragmentation was exclusively CI-MPR-dependent (Figure 5, G and H). Though the death was primarily attributable to the activity of grB, a grB-independent component was also detectable (Figure 5H). Even so, both grB-dependent and -independent killing was abrogated by EGTA treatment. More strikingly, at a later time point, cell death was no longer grB-dependent, yet remained EGTA-sensitive (Figure 5I).
Together these results argue that the previously observed CI-MPR-independent killing mechanism was likely not mediated by grB (Dressel et al., 2004a). As such, there is no basis to conclude that grB-mediated uptake and killing must be CI-MPR-independent.
DISCUSSION
Recently, there have been concerns regarding the model that predicts the CI-MPR is a specific grB receptor. After the initial evidence (Motyka et al., 2000), recent data have suggested that this model might be irrelevant. First, a CI-MPR-independent pathway was sufficient for grB-mediated killing (Trapani et al., 2003). Second, CTL-mediated killing was effectively induced irrespective of CI-MPR expression (Dressel et al., 2004a, 2004b). Thirdly, cell surface heparan sulfate has been identified as a functional receptor for grB (Bird et al., 2005; Kurschus et al., 2005; Raja et al., 2005; Shi et al., 2005). Despite these dissenting claims, new data support the model that, during granule-mediated killing, a grB-serglycin complex is released and, with the cooperation of pfn, induces apoptosis in a CI-MPR-dependent manner.
Renewed support for the CI-MPR-mediated grB uptake model was inspired by the observation that the pathway leading to grB-mediated killing was affected by specific cofactors during delivery to target cells. When grB was complexed to heparan sulfate/heparin and delivered by pfn (Figure 2E) or was complexed to serglycin and delivered by SLO or pfn (Figures 3, D and F, respectively), then grB-mediated killing was critically sensitive to M6P inhibition. In contrast, when grB was delivered in its free form by pfn or SLO (Figures 1B and 3D, respectively), then grB-induced apoptosis was unaffected by M6P. However, the latter likely represents nonphysiological conditions, given that granule-released grB was predominantly serglycin-bound (Figure 3A and Veugelers et al., 2004; Keefe et al., 2005). Rather, these data suggest that under physiological conditions, grB-induced killing will require CI-MPR-mediated uptake. Because CI-MPR-mediated endocytosis is dynamin-dependent (Willingham et al., 1981), this is consistent with previous indications that the grB-serglycin uptake pathway specifically involves dynamin (Veugelers et al., 2004). Also, against expectations (Metkar et al., 2002), serglycin does not mask the M6P binding determinant on grB, nor does it create a steric barrier. Evidence even suggests that serglycin may remain bound to grB when the latter associates with the CI-MPR (Figure 4Aii), though it is unknown whether this binding would occur in vivo.
Initial experiments used heparan sulfate/heparin as a substitute for serglycin as a number of reports provide evidence that it is a good replacement. First, free GAG chains can function in a manner similar to an intact proteoglycan. Complexing grB to chondroitin sulfate A chains (Galvin et al., 1999), like complexing to serglycin (Metkar et al., 2002), decreased the rate at which grB cleaved in vitro-translated caspases 3 and 7. Second, different systems have suggested that grB will not only bind to chondroitin sulfate A chains, but also to heparin or heparan sulfate. Purification of grB has been assisted by its preferential binding to heparin-derived columns (Hanna et al., 1993), and grB has been found to bind to cell surfaces via heparan sulfate chains (Bird et al., 2005; Kurschus et al., 2005; Raja et al., 2005; Shi et al., 2005). The validity of this initial model system was confirmed by similar findings with grB-serglycin from degranulate material and granules. The latter result with isolated granules is very important because grB, pfn, and serglycin are delivered together in a complex, similar to release from live killer cells.
Though the present data appear to contradict some previous studies, in fact, the sum of the data supply interesting details to the model. This study was the first to address the impact of serglycin on grB-mediated killing and its dependence on the CI-MPR. These new data also confirm prior detection of CI-MPR-independent uptake in the absence of serglycin (Trapani et al., 2003), which underscores the contribution of serglycin in focusing grB and pfn killing via the CI-MPR.
Further insights can be gained from studies that have implied cell surface heparan sulfate is a receptor assisting in grB-mediated killing (Bird et al., 2005; Raja et al., 2005; Shi et al., 2005). In fact, killing by monomeric grB and pfn, which was not critically dependent on MPRs, would be consistent with such a model. However our data suggest that serglycin critically directs grB and pfn killing through the CI-MPR. This is consistent with the fact that killing was not abolished by removing cell surface heparan sulfate (Kurschus et al., 2005; Raja et al., 2005). Strikingly, the results presented here provide evidence that cell surface heparan sulfate may actually help coordinate grB-CI-MPR interactions (Figure 4C). A potentiating role for heparan sulfate in CI-MPR-directed grB killing is consistent with findings that decreased cell surface heparan sulfate reduced killing (Raja et al., 2005). The cooperative mechanisms of serglycin and cell surface heparan sulfate are unclear. The simplest model to describe serglycin function is that the electrostatically based interaction prevents positively charged grB from associating with negatively charged cell surface heparan sulfate. GrB-serglycin could then preferentially bind to the CI-MPR via the M6P moiety on grB. Some grB may exchange from serglycin to cell surface heparan sulfate (Raja et al., 2005), but our results suggest this serves to focus the granzyme to the MPR in a manner analogous to a large number of growth factors and cytokines. For example, transforming growth factor-β (Lyon et al., 1994), vascular endothelial growth factor (Cohen et al., 1995), hepatocyte growth factor (Zioncheck et al., 1995), and basic fibroblast growth factor (Yayon et al., 1991) utilize heparan sulfate as a cofactor for binding and signaling from high-affinity receptors.
The CI-MPR-mediated grB-uptake model was also challenged by the finding that CTL-mediated killing was equivalent in both wild-type and CI-MPR knockout cell lines (Dressel et al., 2004a, 2004b). Implicit in these experiments was the assumption that killing by granzymes was calcium dependent, whereas death induced by Fas/Fas-ligand occurred in the presence of EGTA. To address this concern, the CTL-mediated killing of similar target cells, ConA-stimulated lymphoblasts, as well as of CI-MPR-deficient and -overexpressing cells was characterized to clearly define the effector molecules. Remarkably, CTL-induced DNA fragmentation, though EGTA-sensitive, was not always dependent on grB (Figure 5, A, H, and I). The most likely pathway used was Fas/Fas-ligand, which in vitro is calcium independent, but when mediated by CTL is sensitive to EGTA (Bossi and Griffiths, 1999). This finding highlights the redundancy of CTL-mediated killing to ensure successful induction of apoptosis in target cells. But most importantly, this argues that the previously detected CI-MPR-independent killing cannot be attributed to grB activity (Dressel et al., 2004a, 2004b).
The physiological relevance of further grB uptake studies must be judged based on the inclusion of serglycin or a suitable GAG analogue. The source of grB is also an important matter. It is becoming common practice to utilize granzymes from recombinant sources because of more efficient recovery of materials, but this grB usually lacks the M6P moiety critical for binding to the CI-MPR. Thus in studies of granzyme uptake, recombinant materials should not be used as a measure of physiological events. This having been said, great insight could be gained through comparative analysis of grB from natural and recombinant sources. Further, alternative uptake routes may prove useful for therapeutic approaches.
In summary, this report presents data that provide new support for a much criticized grB uptake model, namely the CI-MPR-dependent pathway. A novel finding is that this is the critical mechanism of uptake when grB is bound to serglycin. Further, there is a compromise with recent seemingly contradictory data through evidence that cell surface heparan sulfate promotes grB binding to the CI-MPR. Though GAGs have typically been regarded as inert molecules, our results suggest that they play a key accessory role to influence the pathway of grB and pfn-mediated killing.
Acknowledgments
We thank Dr. Darren Roberts, Dr. Simonetta Sipione, and Catherine Ewen for helpful discussion, and Dr. Peter Lobel for the biotinylated soluble cation-independent mannose 6-phosphate receptor. This work was supported by grants from the Canadian Institute of Health Research (CIHR), the Canadian Cancer Society, and the Howard Hughes Medical Institute. KV. received Studentships from the Alberta Heritage Foundation for Medical Research (AHFMR) and the Natural Sciences and Engineering Research Council of Canada. R.C.B. is a CIHR Distinguished Scientist, a Medial Scientist of the AHFMR, a Canada Research Chair, and a Howard Hughes International Scholar.
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E05–07–0631) on November 9, 2005.
Abbreviations used: AdV, adenovirus; bCI-MPR, biotinylated CI-MPR; CI-MPR, cation-independent mannose 6-phosphate receptor; ConA, concanavalin A; CTL, cytotoxic T lymphocyte; GAG, glycosaminoglycan; grB, granzyme B; M6P, mannose 6-phosphate; MPR, mannose 6-phosphate receptor; NK, natural killer; SLO, streptolysin O; TUNEL, terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling.
References
- Atkinson, E. A., Barry, M., Darmon, A. J., Shostak, I., Turner, P. C., Moyer, R. W., and Bleackley, R. C. (1998). Cytotoxic T lymphocyte-assisted suicide: caspase 3 activation is primarily the result of the direct action of granzyme B. J. Biol. Chem. 273, 21261-21266. [DOI] [PubMed] [Google Scholar]
- Barry, M., and Bleackley, R. C. (2002). Cytotoxic T lymphocytes: all roads lead to death. Nat. Rev. Immunol. 2, 401-409. [DOI] [PubMed] [Google Scholar]
- Bett, A. J., Haddara, W., Prevec, L., and Graham, F. L. (1994). An efficient and flexible system for construction of adenovirus vectors with insertions or deletions in early regions 1 and 3. Proc. Natl. Acad. Sci. USA 91, 8802-8806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhakdi, S., Tranum-Jensen, J., and Sziegoleit, A. (1985). Mechanism of membrane damage by streptolysin-O. Infect. Immun. 47, 52-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhakdi, S., Weller, U., Walev, I., Martin, E., Jonas, E., and Palmer, M. (1993). A guide to the use of pore-forming toxins for controlled permeabilization of cell membranes. Med. Microbiol. Immunol. 182, 167-175. [DOI] [PubMed] [Google Scholar]
- Bird, C. H., Sun, J., Ung, K., Karambalis, D., Whisstock, J. C., Trapani, J. A., and Bird, P. I. (2005). Cationic sites on granzyme B contribute to cytotoxicity by promoting its uptake into target cells. Mol. Cell. Biol. 25, 7854-7867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossi, G., and Griffiths, G. M. (1999). Degranulation plays an essential part in regulating cell surface expression of Fas ligand in T cells and natural killer cells. Nature Med. 5, 90-96. [DOI] [PubMed] [Google Scholar]
- Caputo, A., Parrish, J. C., James, M.N.G., Powers, J. C., and Bleackley, R. C. (1999). Electrostatic reversal of serine proteinase substrate specificity. Proteins 35, 415-424. [PubMed] [Google Scholar]
- Cohen, T., Gitay-Goren, H., Sharon, R., Shibuya, M., Halaban, R., Levi, B.-Z., and Neufeld, G. (1995). VEGF121, a vascular endothelial growth factor (VEGF) isoform lacking heparin binding ability, requires cell-surface heparan sulfates for efficient binding to the VEGF receptors of human melanoma cells. J. Biol. Chem. 270, 11322-11326. [DOI] [PubMed] [Google Scholar]
- Dressel, R., Raja, S. M., Höning, S., Seidler, T., Froelich, C. J., von Figura, K., and Günther, E. (2004a). Granzyme-mediated cytotoxicity does not involve the mannose 6-phosphate receptors on target cells. J. Biol. Chem. 279, 20200-20210. [DOI] [PubMed] [Google Scholar]
- Dressel, R., von Figura, K., and Günther, E. (2004b). Unimpaired allorejection of cells deficient for the mannose 6-phosphate receptors Mpr300 and Mpr46. Transplantation 78, 758-761. [DOI] [PubMed] [Google Scholar]
- Enari, M., Hug, H., and Nagata, S. (1995). Involvement of an ICE-like protease in Fas-mediated apoptosis. Nature 375, 78-81. [DOI] [PubMed] [Google Scholar]
- Froelich, C. J. et al. (1996). New paradigm for lymphocyte granule-mediated cytotoxicity: target cells bind and internalize granzyme B, but an endosomolytic agent is necessary for cytosolic delivery and subsequent apoptosis. J. Biol. Chem. 271, 29073-29079. [DOI] [PubMed] [Google Scholar]
- Gabel, C. A., Goldberg, D. E., and Kornfeld, S. (1983). Identification and characterization of cells deficient in the mannose 6-phosphate receptor: evidence for an alternate pathway for lysosomal enzyme targeting. Proc. Natl. Acad. Sci. USA 80, 775-779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvin, J. P., Spaeny-Dekking, L.H.A., Wang, B., Seth, P., Hack, C. E., and Froelich, C. J. (1999). Apoptosis induced by granzyme B-glycosaminoglycan complexes: implications for granule-mediated apoptosis in vivo. J. Immunol. 162, 5345-5350. [PubMed] [Google Scholar]
- Garner, R. et al. (1994). Characterization of a granule-independent lytic mechanism used by CTL hybridomas. J. Immunol. 153, 5413-5421. [PubMed] [Google Scholar]
- Gavrieli, Y., Sherman, Y., and Ben-Sasson, S. (1992). Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J. Cell Biol. 119, 493-501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grujic, M., Braga, T., Lukinius, A., Eloranta, M., Knight, S., Pejler, G., and Åbrink, M. (2005). Serglycin-deficient cytotoxic T lymphocytes display defective secretory granule maturation and granzyme B storage. J. Biol. Chem. 280, 33411-33418. [DOI] [PubMed] [Google Scholar]
- Hanna, W., Zhang, X., Turbov, J., Winkler, U., Hudig, D., and Froelich, C. J. (1993). Rapid purification of cationic granule proteases: application to human granzymes. Protein Exp. Purif. 4, 398-404. [DOI] [PubMed] [Google Scholar]
- Heibein, J. A., Barry, M., Motyka, B., and Bleackley, R. C. (1999). Granzyme B-induced loss of mitochondrial inner membrane potential (ΔΨm) and cytochrome c release are caspase independent. J. Immunol. 163, 4683-4693. [PubMed] [Google Scholar]
- Ikegami-Kawai, M., and Takahashi, T. (2002). Microanalysis of hyaluronan oligosaccharides by polyacrylamide gel electrophoresis and its application to assay of hyaluronidase activity. Analyt. Biochem. 311, 157-165. [DOI] [PubMed] [Google Scholar]
- Keefe, D., Shi, L., Feske, S., Massol, R., Navarro, F., Kirchhausen, T., and Lieberman, J. (2005). Perforin triggers a plasma membrane-repair response that facilitates CTL induction of apoptosis. Immunity 23, 249. [DOI] [PubMed] [Google Scholar]
- Kolset, S. O., Prydz, K., and Pejler, G. (2004). Intracellular proteoglycans. Biochem. J. 379, 217-227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurschus, F. C., Bruno, R., Fellows, E., Falk, C. S., and Jenne, D. E. (2005). Membrane receptors are not required to deliver granzyme B during killer cell attack. Blood 105, 2049-2058. [DOI] [PubMed] [Google Scholar]
- Lord, S. J., Rajotte, R. V., Korbutt, G. S., and Bleackley, R. C. (2003). Granzyme B: a natural born killer. Immunol. Rev. 193, 31-38. [DOI] [PubMed] [Google Scholar]
- Los, M. et al. (1995). Requirement of an ICE/CED-3 protease for Fas/APO-1-mediated apoptosis. Nature 375, 81-83. [DOI] [PubMed] [Google Scholar]
- Lyon, M., Rushton, G., and Gallagher, J. T. (1994). The interaction of the transforming growth factor-βs with heparin/heparan sulfate is isoform-specific. J. Biol. Chem. 272, 18000-18006. [DOI] [PubMed] [Google Scholar]
- Merril, C. R., Dunau, M. L., and Goldman, D. (1981). A rapid sensitive silver stain for polypeptides in polyacrylamide gels. Analyt. Biochem. 110, 201-207. [DOI] [PubMed] [Google Scholar]
- Metkar, S. S., Wang, B., Aguilar-Santelises, M., Raja, S. M., Uhlin-Hansen, L., Podack, E., Trapani, J. A., and Froelich, C. J. (2002). Cytotoxic cell granule-mediated apoptosis: perforin delivers granzyme B-serglycin complexes into target cells without plasma membrane pore formation. Immunity 16, 417-428. [DOI] [PubMed] [Google Scholar]
- Min, H., and Cowman, M. K. (1986). Combined Alcian blue and silver staining of glycosaminoglycans in polyacrylamide gels: application to electrophoretic analysis of molecular weight distribution. Analyt. Biochem. 155, 275-285. [DOI] [PubMed] [Google Scholar]
- Motyka, B. et al. (2000). Mannose 6-phosphate/insulin-like growth factor II receptor is a death receptor for granzyme B during cytotoxic T cell-induced apoptosis. Cell 103, 491-500. [DOI] [PubMed] [Google Scholar]
- Odake, S., Kam, C.-M., Narasimhan, L., Poe, M., Blake, J. T., Krahenbuhl, O., Tschopp, J., and Powers, J. C. (1991). Human and murine cytotoxic T lymphocyte serine proteases: subsite mapping with peptide thioester substrates and inhibition of enzyme activity and cytolysis by isocoumarins. Biochemistry 30, 2217-2227. [DOI] [PubMed] [Google Scholar]
- Poe, M., Blake, J. T., Boulton, D. A., Gammon, M., Sigal, N. H., Wu, J. K., and Zweerink, H. J. (1991). Human cytotoxic lymphocyte granzyme B: its purification from granules and the characterization of substrate and inhibitor specificity. J. Biol. Chem. 266, 98-103. [PubMed] [Google Scholar]
- Raja, S. M. et al. (2005). A novel mechanism for protein delivery: granzyme B undergoes electrostatic exchange from serglycin to target cells. J. Biol. Chem. 280, 20752-20761. [DOI] [PubMed] [Google Scholar]
- Raja, S. M., Wang, B., Dantuluri, M., Desai, U. R., Demeler, B., Spiegel, K., Metkar, S. S., and Froelich, C. J. (2002). Cytotoxic cell granule-mediated apoptosis: characterization of the macromolecular complex of granzyme B with serglycin. J. Biol. Chem. 277, 49523-49530. [DOI] [PubMed] [Google Scholar]
- Roberts, D. L., Goping, I. S., and Bleackley, R. C. (2003). Mitochondria at the heart of the cytotoxic attack. Biochem. Biophys. Res. Comm. 304, 513-518. [DOI] [PubMed] [Google Scholar]
- Rouvier, E., Luciani, M.-F., and Golstein, P. (1993). Fas involvement in Ca2+-independent T cell-mediated cytotoxicity. J. Exp. Med. 177, 195-200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell, J. H., and Ley, T. J. (2002). Lymphocyte-mediated cytotoxicity. Annu. Rev. Immunol. 20, 323-370. [DOI] [PubMed] [Google Scholar]
- Shi, L., Keefe, D., Durand, E., Feng, H., Zhang, D., and Lieberman, J. (2005). Granzyme B binds to target cells mostly by charge and must be added at the same time as perforin to trigger apoptosis. J. Immunol. 174, 5456-5461. [DOI] [PubMed] [Google Scholar]
- Simon, M. M., Hausmann, M., Tran, T., Ebnet, K., Tschopp, J., ThaHla, R., and Müllbacher, A. (1997). In vitro- and ex vivo-derived cytolytic leukocytes from granzyme A × B double knockout mice are defective in granule-mediated apoptosis but not lysis of target cells. J. Exp. Med. 186, 1781-1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tewari, M., and Dixit, V. M. (1995). Fas- and tumor necrosis factor-induced apoptosis is inhibited by the poxvirus crmA gene product. J. Biol. Chem. 270, 3255-3260. [DOI] [PubMed] [Google Scholar]
- Thuy, L. P., and Nyhan, W. L. (1992). A new quantitative assay for glycosaminoglycans. Clin. Chim. Acta 212, 17-26. [DOI] [PubMed] [Google Scholar]
- Trapani, J. A., and Smyth, M. J. (2002). Functional significance of the perforin/granzyme cell death pathway. Nat. Rev. Immunol. 2, 735-747. [DOI] [PubMed] [Google Scholar]
- Trapani, J. A., and Sutton, V. R. (2003). Granzyme B: pro-apoptotic, antiviral and antitumor functions. Curr. Opin. Immunol. 15, 533-543. [DOI] [PubMed] [Google Scholar]
- Trapani, J. A., Sutton, V. R., Thia, K.Y.T., Li, Y. Q., Froelich, C. J., Jans, D. A., Sandrin, M. S., and Browne, K. A. (2003). A clathrin/dynamin- and mannose-6-phosphate receptor-independent pathway for granzyme B-induced cell death. J. Cell Biol. 160, 223-233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschopp, J., and Masson, D. (1987). Inhibition of the lytic activity of perforin (cytolysin) and of late complement components by proteoglycans. Mol. Immunol. 24, 907-913. [DOI] [PubMed] [Google Scholar]
- Veugelers, K., Motyka, B., Frantz, C., Shostak, I., Sawchuk, T., and Bleackley, R. C. (2004). The granzyme B-serglycin complex from cytotoxic granules requires dynamin for endocytosis. Blood 103, 3845-3853. [DOI] [PubMed] [Google Scholar]
- Watanabe, H., Grubb, J. H., and Sly, W. S. (1990). The overexpressed human 46-kDa mannose 6-phosphate receptor mediates endocytosis and sorting of b-glucuronidase. Proc. Natl. Acad. Sci. USA 87, 8036-8040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willingham, M. C., Pastan, I. H., Sahagian, G. G., Jourdian, G. W., and Neufeld, E. F. (1981). Morphologic study of the internalization of a lysosomal enzyme by the mannose 6-phosphate receptor in cultured Chinese hamster ovary cells. Proc. Natl. Acad. Sci. USA 78, 6967-6971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler, U., Pickett, T. M., and Hudig, D. (1996). Fractionation of perforin and granzymes by immobilized metal affinity chromatography (IMAC). J. Immunol. Methods 191, 11-20. [DOI] [PubMed] [Google Scholar]
- Yayon, A., Klagsbrun, M., Esko, J. D., Leder, P., and Ornitz, D. M. (1991). Cell surface, heparin-like molecules are required for binding of basic fibroblast growth factor to its high affinity receptor. Cell 64, 841-848. [DOI] [PubMed] [Google Scholar]
- Zioncheck, T. F. et al. (1995). Sulfated oligosaccharides promote hepatocyte growth factor association and govern its mitogenic activity. J. Biol. Chem. 270, 16871-16878. [DOI] [PubMed] [Google Scholar]