

Abstract
MCAK belongs to the Kinesin-13 family, whose members depolymerize microtubules rather than translocate along them. We defined the minimal functional unit of MCAK as the catalytic domain plus the class specific neck (MD-MCAK), which is consistent with previous reports. We used steady-state ATPase kinetics, microtubule depolymerization assays, and microtubule·MCAK cosedimentation assays to compare the activity of full-length MCAK, which is a dimer, with MD-MCAK, which is a monomer. Full-length MCAK exhibits higher ATPase activity, more efficient microtubule end binding, and reduced affinity for the tubulin heterodimer. Our studies suggest that MCAK dimerization is important for its catalytic cycle by promoting MCAK binding to microtubule ends, enhancing the ability of MCAK to recycle for multiple rounds of microtubule depolymerization, and preventing MCAK from being sequestered by tubulin heterodimers.
INTRODUCTION
Microtubules (MTs) are cytoskeletal polymers that serve two main cellular functions: they form tracks on which molecular motor proteins sort and deliver cellular components (Hirokawa and Takemura, 2004), and they are essential for the assembly of the mitotic spindle (Kline-Smith and Walczak, 2004). MTs are made of α/β tubulin heterodimers that assemble longitudinally to form protofilaments, 13 of which associate laterally to form the MT. MTs exhibit a behavior known as dynamic instability, in which populations of MTs coexist in states of growth and shrinkage and interconvert randomly between these two states (Desai and Mitchison, 1997; Nogales, 2000). Although solutions of purified tubulin exhibit dynamic instability, cellular factors have been shown to be important modulators of MT dynamics (Cassimeris and Spittle, 2001).
One important class of MT regulatory proteins is the Kinesin-13 family (Lawrence et al., 2004; Miki et al., 2005). Kinesin-13 family members, including MCAK (Kif2C), pKinI, and Kif2A, depolymerize MTs in vitro (Desai et al., 1999; Moores et al., 2002; Hunter et al., 2003) and regulate MT dynamics and chromosome segregation in cells (Maney et al., 1998; Maney et al., 2001; Kline-Smith and Walczak, 2002; Kline-Smith et al., 2004; Rogers et al., 2004). In addition to its important function in mitosis (Gaetz and Kapoor, 2004; Ganem and Compton, 2004), Kif2A is also required for proper neuronal outgrowth (Morfini et al., 1997; Homma et al., 2003).
MCAK is a homodimeric molecule that can depolymerize stabilized MTs as well as dynamic MTs in vitro (Desai et al., 1999; Hunter et al., 2003; Newton et al., 2004). It is composed of an N-terminal globular domain that functions in subcellular targeting (Maney et al., 1998; Wordeman et al., 1999; Walczak et al., 2002; Kline-Smith et al., 2004), a class-specific neck and catalytic core that are essential for MT depolymerization activity (Maney et al., 2001; Ovechkina et al., 2002; Ogawa et al., 2004), and a C-terminal tail that plays a role in dimerization and regulates ATPase activity (Maney et al., 2001; Moore and Wordeman, 2004). Although the native protein is dimeric, a monomeric form consisting of the neck and catalytic core is sufficient for MT depolymerization in vitro and in cells (Maney et al., 2001). It was originally proposed that a dimeric molecule would be necessary to push apart the lateral interactions of the protofilaments of the MT lattice, but this hypothesis seems not to be true because MCAK was shown to act on a single protofilament during depolymerization (Niederstrasser et al., 2002). The mechanistic significance for a two-headed molecule in vivo is therefore unknown.
The MT binding properties of Kinesin-13s are distinct from other kinesin superfamily members. Both MCAK and Kif2A bind to tubulin heterodimers and to the MT lattice, but they seem to exhibit a preference for the ends of MTs (Desai et al., 1999; Moore and Wordeman, 2004). Physiologically, this end binding activity may be important for the recently discovered tip-tracking activity of Kinesin-13s (Mennella et al., 2005; Moore et al., 2005). Mechanistically, this end binding is likely critical as the ATPase activity is preferentially stimulated by MT ends (Hunter et al., 2003; Moores et al., 2003). However, the ATPase activity is also stimulated in the presence of tubulin heterodimers (Moores et al., 2002, 2003; Hunter et al., 2003; Moore and Wordeman, 2004; Shipley et al., 2004).
Significant insight into how Kinesin-13 family members function, including a potential role for the class-specific neck, has been provided in the recently reported crystal structure of monomeric forms of Kif2C and pKinI (Ogawa et al., 2004; Shipley et al., 2004). Although the fold and placement of nucleotide in Kinesin-13s are conserved with those of other kinesins (Sack et al., 1999), the state of the ATP binding pocket is distinct, suggesting that Kinesin-13s may use their ATPase cycle differently than conventional kinesin. Interestingly, in silico modeling demonstrates that the Kinesin-13 structures fit better to curved protofilaments (Ogawa et al., 2004; Shipley et al., 2004), which are thought to be intermediates of MT depolymerization. In addition, the neck of Kif2C may associate laterally between the protofilaments of the MT, indicating that the neck may be crucial for MT destabilization (Ogawa et al., 2004).
In this study, we compare the activity of two Xenopus MCAK proteins to further probe the mechanistic cycle of MT depolymerization. We explored the catalytic differences between dimeric, full-length MCAK (FL-MCAK) and monomeric, minimal domain MCAK (MD-MCAK). Although the minimal domain of mammalian MCAK has been previously identified in cellular assays, it has never been fully characterized biochemically in comparison with full-length MCAK. Our data show that monomeric MD-MCAK exhibits significantly different properties compared with FL-MCAK. We propose that dimerization plays an important role in the catalytic cycle of MCAK-promoted MT depolymerization by enhancing the ability of MCAK to target to MT ends and by increasing the dissociation of the MCAK–tubulin heterodimer complex. The overall effect is to increase the free MCAK available for MT end binding to drive another cycle of ATP-promoted MT depolymerization.
MATERIALS AND METHODS
Cloning and Transfection of Deletion Constructs
MCAK deletion constructs were created by PCR amplification of Xenopus MCAK library clone pBS11B using sequence-specific primers and cloned back into pEGFPC1 using SacI and EcoRV restriction sites or KpnI and HindIII sites. Constructs for protein purification were cloned into pFastBac1 or pFastBac1-GFP using SacI and KpnI restriction sites. pFastBac1-GFP was created by PCR amplification of the enhanced green fluorescent protein (EGFP) sequence from pEGFPC1 and cloned into the BamHI and SacI sites of pFastBac1. All constructs were verified by DNA sequence analysis. Constructs were transfected into PtK2 cells and fixed and stained as described previously (Kline-Smith and Walczak, 2002). Cells were then categorized based on the intensity of green fluorescent protein (GFP) fluorescence. Cells expressing a moderate level of GFP fluorescence were scored without knowledge of the MCAK transfection construct, and the percentage of cells with destabilized MT arrays was determined for 40–100 cells per transfection as described previously (Kline-Smith and Walczak, 2002). Images were acquired using a Nikon E-600 with a 40× 1.0 Plan Apo objective and a MicroMax 1300 Y camera (Princeton Scientific Instruments, Monmouth Junction, NJ). The camera, shutters, and filters were controlled by MetaMorph (Molecular Devices, Sunnyvale, CA).
Protein Expression and Purification
FL-MCAK (MCAK amino acids 2–731) and MD-MCAK (MCAK amino acids 187–592) protein were expressed in either Sf9 or HighFive insect cells using the Bac-to-Bac baculovirus expression system (Invitrogen, Carlsbad, CA). Purification of FL-MCAK, GFL-MCAK (GFP-tagged FL-MCAK), and GMD-MCAK (GFP-tagged MD-MCAK) was as described previously (Desai et al., 1999). MD-MCAK was purified similarly to FL-MCAK, except MD-MCAK purified to near homogeneity with the first cation exchange column and did not require further purification. All protein concentrations are reported in terms of monomer and were determined using gel densitometry of Coomassie-stained gels relative to a BSA standard and quantified by NIH Image. Because the predicted molecular weight of MD-MCAK is roughly half that of the predicted molecular weight of FL-MCAK, the band intensities of equal molar concentrations of MD-MCAK and FL-MCAK on Coomassie-stained gels (Figures 2 and 6) are not equal. Multiple preparations of both FL-MCAK and MD-MCAK were used in all experiments.
Figure 2.

At high ratios of MCAK to MTs, FL-MCAK and MD-MCAK show similar affinities for MTs. (A) FL-MCAK or MD-MCAK (1.24 μM) was incubated in the absence of nucleotide with increasing concentrations of paclitaxel- and GMPCPP-stabilized MTs (0–8 μM) for 15 min at 22°C. FL-MCAK or MD-MCAK bound to MTs was separated from unbound enzyme by ultracentrifugation. Equal volumes of supernatant (S) and pellet (P) were run on SDS-PAGE gels and stained with Coomassie Brilliant Blue. (B) The amount of FL-MCAK or MD-MCAK in the supernatant and the pellet was quantified. The binding affinity curves were derived from Equation 3 and represent averaged data from at least three individual experiments. MT·E is the amount of FL- or MD-MCAK fractionating with MTs. (C) A summary of the binding affinities.
Figure 6.

MD-MCAK binds with higher affinity to tubulin heterodimer than FL-MCAK. (A) FL-MCAK (FL-M) or MD-MCAK (MD-M) at 500 nM was incubated in the absence of nucleotide with paclitaxel- and GMPCPP-stabilized MTs (3.25 μM tubulin) as a function of soluble GDP tubulin heterodimer (0–13 μM) for 15 min at 22°C followed by centrifugation. Equal volumes of the supernatant (S) and pellet (P) for each reaction were analyzed by SDS-PAGE followed by Colloidal Coomassie staining. Because of the high ratios of total tubulin to MCAK enzyme in each reaction, the regions of each gel containing FL-MCAK or MD-MCAK were equally contrast-enhanced relative to the tubulin portion of the gel. T, tubulin. B, FL-MCAK and MD-MCAK partitioning to the supernatant and the pellet were quantified. The fraction of MCAK in the pellet in the absence of additional GDP tubulin heterodimer (0:1) was considered 100%. The partitioning of MCAK to the pellet as a function of GDP tubulin heterodimer was normalized to the 0:1 control. The data represent mean ± SEM for three individual experiments.
Molecular Weight Determination
Molecular weights were determined using multiple preps of all proteins at equal concentrations. Stokes radii were determined using a Superose 6 10/300 GL column in 400 mM KCl, 20 mM PIPES, 1 mM MgCl2, 1 mM EGTA, 0.1 mM EDTA, 1 mM dithiothreitol (DTT), 10 μM MgATP, 0.1 μg/ml leupeptin, pepstatin A, chymostatin (LPC) using blue dextran, thyroglobulin, β-amylase, alcohol dehydrogenase, bovine serum albumin (BSA), carbonic anhydrase, and cytochrome c as standards. Protein peaks were detected by A280 readings, by Coomassie brilliant blue-stained gels, or by Western blot. Stokes radii were calculated as an average of Laurent and Killander and Porath plots (Porath and Flodin, 1959; Laurent and Killander, 1964). S values were determined using BSA, catalase, ovalbumin, carbonic anhydrase, and alcohol dehydrogenase as standards. Sucrose gradients were run in 100 mM KCl (or 400 mM KCl), 20 mM PIPES, 1 mM MgCl2, 1 mM EGTA, 0.1 mM EDTA, 1 mM DTT, 10 μM MgATP, 0.1 μg/ml LPC through a 5–20% sucrose gradient in a Beckman SW55 rotor for 15 h at 4°C. Fractions (250 μl) were collected manually, and protein peaks were detected by Western, Bradford reagent, or by GFP fluorescence with similar results. Molecular weights were calculated with Equation 1:
 |
1 |
M is the molecular weight, Rs is the Stokes radius, A is Avagadro's number, S is the sedimentation value, η is the solvent viscosity, ν is the calculated protein density from the amino acid composition, and ρ is the solvent density (Siegel and Monty, 1966).
Preparation of MT Substrates
Guanylyl-(α,β)-methylene-diphosphonate (GMPCPP)-stabilized MTs were polymerized from cycled bovine tubulin as described previously (Desai and Walczak, 2001). Briefly, tubulin was clarified with a high-speed spin for 5 min at 2°C in a TLA100 rotor (Beckman Coulter, Fullerton, CA) and then polymerized in the presence of 0.5 mM GMPCPP (Jena Bioscience USA, San Diego, CA), 1× BRB80 (80 mM PIPES, pH 6.8, 1 mM MgCl2, and 1 mM EGTA), 1 mM DTT for 30 min at 37°C. The MTs were then pelleted for 5 min in a TLA100 rotor at 37°C and resuspended in 1× BRB80, 1 mM DTT. GMPCPP- and paclitaxel-stabilized MTs were made in a similar manner except that paclitaxel was added to 10 μM at 20 min after the start of polymerization. After pelleting, MTs were resuspended in 1× BRB80, 1 mM DTT, 10 μM paclitaxel. To determine the average length of the MTs, MTs were polymerized in an identical manner using rhodamine-labeled tubulin, squashed onto coverslips, and visualized by fluorescence microscopy.
MT Depolymerization Assays
Depolymerization assays were performed using GMPCPP-stabilized MTs. For a direct comparison between fixed amounts of FL-MCAK and MD-MCAK, 50 nM enzyme was incubated at room temperature in 1.5× BRB80, 42 mM KCl, 2 mM MgATP, and 1.5 μM MTs. In assays to determine the EC50, FL- or MD-MCAK was titrated in a reaction (0–128 nM) that included 1 μM MTs. All reactions were incubated for 15 min at 22°C, and subsequently centrifuged in a Beckman TLA 100 rotor for 5 min at 90,000 rpm at 22°C. The pellet was resuspended in the original volume of 1× BRB80, and the supernatants and pellets were mixed with equal volumes of 2× Laemmli sample buffer (SB), boiled, and equal volumes were electrophoresed on a 10% SDS-PAGE gel. The gels were stained with Coomassie Brilliant Blue, scanned, and the fraction of soluble tubulin heterodimer was quantified by densitometry of the stained gel using NIH Image. In all cases, the amount of tubulin depolymerized in the absence of enzyme was subtracted as background so that the percentage of microtubules depolymerized is that from enzyme addition only. The data from at least three independent experiments were combined and fit to the four-parameter logistic equation or dose-response curve (Equation 2), and the EC50 was calculated for FL- and MD-MCAK using GraFit 5 software, where Response is the fraction of tubulin heterodimer in the supernatant, Amin is the baseline or background response, Amax is the maximal response, logEC50 is the log of the effective concentration that gives a 50% response, X is the log of the enzyme concentration, and H is the Hill slope.
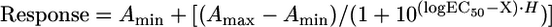 |
2 |
MCAK MT Cosedimentation Assays
The cosedimentation assays shown in Figure 2 were performed using GMPCPP- and paclitaxel-stabilized MTs (doubly stabilized) as described previously (Foster et al., 1998). Equal molar concentrations (1.24 μM) of purified protein (MD-MCAK or FL-MCAK) were incubated in 1.25× BRB80, 100 mM KCl, and increasing concentrations of doubly stabilized MTs (0.25–8 μM). The reactions were pelleted at 90,000 rpm in a Beckman TLA 100 rotor. The pellet of each reaction was resuspended in 1× BRB80 equal to the volume of its supernatant, and then the supernatant and pellet were diluted with equal amounts of 2× SB. Equal volumes of each sample were analyzed by SDS-PAGE and stained with Coomassie Blue. The concentration of MCAK partitioning to the supernatant and pellet was quantified by densitometry of the stained gel using NIH Image. The data from at least three independent experiments were fit to Equation 3 to determine the apparent Kd of FL- or MD-MCAK for MTs, where MT·E is the concentration of MCAK partitioning to the pellet with the MTs. MTt is the tubulin concentration as MTs, and E0 is the total MCAK concentration.
 |
3 |
Fluorescence-based MT Binding Assays
End binding assays were performed similarly to Desai et al. (1999). Briefly, 9 nM GFL-MCAK or GMD-MCAK was incubated with 400 nM GMPCPP-stabilized MTs in 1.5× BRB80, 100 mM KCl, plus or minus 5 mM nucleotide (MgAMPPNP or MgADP) for 15 min at 22°C (Figure 3). Reactions were fixed in 1% glutaraldehyde, diluted, and subsequently loaded onto 10% glycerol cushions in 1× BRB80. Reactions were sedimented for 45 min at 12,000 rpm at 20°C in a Beckman JS13.1 rotor onto poly-l-lysine-coated coverslips, which were processed for immunofluorescence to enhance the GFP signal. Anti-GFP antibodies were raised in rabbits to recombinant EGFP and affinity purified before use. All subsequent steps were carried out at room temperature. The coverslips were blocked in AbDil (2% BSA, 0.1% NaN3 in Tris-buffered saline-Triton X [20 mM Tris, 150 mM NaCl, pH 7.5 + 0.1% Triton X-100; TBS-TX]) for 30 min. All subsequent rinses between antibody incubations were performed using TBS-TX. Coverslips were incubated in primary anti-GFP antibody diluted to 2 μg/ml in AbDil for 30 min. They were washed in TBS-TX and incubated in 1/50 goat anti-rabbit fluorescein isothiocyanate (Jackson ImmunoResearch Laboratories, West Grove, PA). To visualize MTs, the coverslips were stained with 1/250 DM1α (Sigma-Aldrich) followed by 1/50 donkey anti-mouse Texas Red (Jackson ImmunoResearch Laboratories). Images were acquired from at least three independent experiments using a Micromax 1300 Y camera attached to a Nikon E-600 microscope with a 100× 1.3 Plan Fluor objective. To control for any nonspecific background binding of our anti-GFP antibody, we also performed the assay in the absence of enzyme. In no enzyme controls, >98% of the MTs had no staining. Within each independent experiment, photomicrographs were scaled similarly based on the average GFP fluorescence. MTs with binding events were then scored without knowledge of the sample identity. All binding events were compiled in Microsoft Excel and then analyzed using Student's t test.
Figure 3.

At substoichiometric ratios of MCAK to MTs, GFL-MCAK binds more effectively to MT ends than GMD-MCAK. GFL-MCAK or GMD-MCAK (9 nM) was mixed with 400 nM GMPCPP-stabilized MTs for 15 min at room temperature without additional nucleotide (A–C), in the presence of 5 mM MgAMPPNP (D–F), or in the presence 5 mM MgADP (G–I). Reactions were fixed, sedimented onto coverslips, and processed for immunofluorescence. MTs were scored for the percentage of binding events (A, B, D, E, G, and H) without knowledge of the sample identity. Asterisk (*) denotes a statistically significant difference between GFL-MCAK and GMD-MCAK with a p value < 0.05. Representative micrographs of GFL-MCAK (green) or GMD-MCAK (green) binding to MTs (red) without additional nucleotide (C), or in the presence of MgAMPPNP (F) or MgADP (I). Bar, 5 μm. The localization of GFL-MCAK or GMD-MCAK in fluorescence microscopy binding assays was quantified (J). The reported value is the average percentage of MTs with GFL-MCAK or GMD-MCAK localization ± SEM. n is the total number of MTs counted from five independent experiments.
Steady-State ATPase Assays
ATPase assays were performed using the Malachite green colorimetric assay to detect Pi (Pi) (Lanzetta et al., 1979). For these experiments, 50 nM FL- or MD-MCAK was incubated with 2 mM MgATP, 50 mM KCl, 1 mg/ml casein, 1.25× BRB80, and varying concentrations (0–20 μM) of tubulin heterodimer or doubly stabilized MTs for 30–60 min at 25°C. The rate of Pi release was linear over this time course. Data were collected and analyzed from at least three independent experiments, and the plots represent the average of these experiments. The microtubule concentration dependence data in Figure 5A were fit to the quadratic equation (Equation 4) because some of the microtubule concentrations used in the experiment were similar to the concentration of MCAK. The data in Figure 5, B and C, were fit to the Michaelis–Menten equation (Equation 5). For Figure 5C, the MgATP concentration was varied (0–1 mM) with doubly stabilized MTs held constant at 8 μM for FL-MCAK and 4 μM for MD-MCAK. The ATPase rates are reported per active site. Note that the MT concentration for FL-MCAK at 8 μM was subsaturating; therefore, the kcat obtained from the ATP concentration dependence experiment (Figure 5C) was somewhat lower than the kcat obtained from the MT concentration dependence (Figure 5A).
 |
4 |
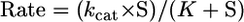 |
5 |
S is the substrate concentration (tubulin heterodimer or MgATP), kcat is the maximum rate constant of steady-state ATP turnover, E0 is the MCAK concentration, MTt is the microtubule concentration, K0.5,MT is the steady-state Km for MTs and represents the concentration needed to provide one-half the maximal velocity. K corresponds to the Km for tubulin (K0.5,Tubulin) or ATP (Km,ATP) and is the concentration of tubulin heterodimer or ATP, respectively, necessary to provide one-half the maximal velocity.
Figure 5.

MD-MCAK has lower ATPase activity than FL-MCAK. FL-MCAK, and MD-MCAK (50 nM) were assayed for steady-state ATPase activity in the presence of increasing concentrations of (A) paclitaxel- and GMPCPP-stabilized MTs (0–15 μM) or (B) tubulin heterodimer (0–20 μM) at 2 mM MgATP. The data in A and B were fit to Equations 4 (for MTs) and 5 (for tubulin heterodimer). (C) The steady-state kinetics for FL-MCAK or MD-MCAK (50 nM) were determined as a function of MgATP concentration at 8 μM MTs for FL-MCAK and 4 μM MTs for MD-MCAK with the fit of the data to equation 5. (D) Summary of the kinetic parameters derived from A to C.
Competition Binding Assays
To test the hypothesis that soluble tubulin heterodimer sequestered MD-MCAK, we designed a MT·MCAK cosedimentation assay using soluble tubulin heterodimer to compete with the binding sites on the MT polymer (Figure 6). In this experiment, 500 nM FL- or MD-MCAK was incubated with 3.25 μM doubly stabilized MTs, 1.25× BRB80, and 100 mM KCl in the presence of increasing concentrations of soluble tubulin heterodimer (0–13 μM) plus 250 μM GDP for 15 min. The GDP was added to decrease the ability of the added tubulin heterodimer to assemble into MT polymer. After centrifugation, the supernatant and pellet of each reaction were adjusted to equal volumes and electrophoresed on 10% SDS-PAGE gels. Colloidal Coomassie-stained gels from three independent experiments were quantified using Image J software.
The fraction of MCAK partitioning to the pellet was normalized to the amount of MCAK sedimenting with the MT pellet in the absence of additional soluble tubulin heterodimer (i.e., 0:1 reactions). The results are presented as a ratio of soluble tubulin heterodimer to MT polymer. A ratio of 1:1 represents 3.25 μM soluble tubulin heterodimer + 3.25 μM MTs; a ratio of 2:1 represents 6.5 μM soluble tubulin heterodimer + 3.25 μM MTs; and a ratio of 4:1 represents 13 μM soluble tubulin heterodimer + 3.25 μM MTs.
RESULTS
The Identification of the Minimal Domain of MCAK Necessary for MT Depolymerization
To initiate our biochemical investigations of what features of Xenopus MCAK are required for MT depolymerization activity, we first needed to determine the minimal domain of MCAK necessary to depolymerize MTs. We screened a series of GFP-fusion constructs in which MCAK sequences were deleted using a cellular-based transfection assay (Kline-Smith and Walczak, 2002) and found that the minimal domain of MCAK necessary for efficient depolymerization activity is contained within residues 187–592; we refer to these residues as the minimal domain of MCAK (MD-MCAK) (Figure S1 and Table S1). The MD-MCAK characterized here is similar in sequence to other MCAK monomers used for cellular and structural studies (Maney et al., 2001; Ogawa et al., 2004).
MD-MCAK Is Monomeric and Depolymerizes MTs In Vitro
To analyze the catalytic mechanism of MCAK, we expressed and purified FL-MCAK and MD-MCAK as well as GFP fusions of both proteins for biochemical assays (GFL-MCAK and GMD-MCAK) (Figure 1, A and B). We tested each pure protein in a sedimentation-based MT depolymerization assay and found that substoichiometric amounts of MD-MCAK depolymerized GMPCPP-stabilized MTs as well as FL-MCAK (Figure 1C), showing that both FL-MCAK and MD-MCAK (as well as the GFP fusion proteins) catalytically depolymerize MTs.
Figure 1.

FL-MCAK is dimeric and MD-MCAK is monomeric. (A) Schematic representation of the proteins used in this study. NT, N terminus; Neck, class-specific neck region; CD, catalytic domain; CT, C terminus. The dark gray box within the C terminus represents the putative coiled coil region. The numbers correspond to amino acid residues. (B) Equal amounts (0.5 μg) of purified recombinant proteins were run on SDS-PAGE gels and stained with Colloidal Coomassie Blue. Molecular mass markers in kilodaltons are indicated on the left. (C) GMPCPP-stabilized MTs (1.5 μM) were incubated with control buffer (No M) or equal molar amounts (50 nM) of FL-MCAK (FL-M), GFL-MCAK (GFL-M), MD-MCAK (MD-M), GMD-MCAK (GMD-M), or GFP alone for 30 min at 22°C. MTs were separated from soluble tubulin heterodimer by ultracentrifugation. Supernatant (S) and pellet (P) were analyzed by SDS-PAGE followed by Coomassie staining. The position of tubulin (T) is indicated on the right. (D) Purified proteins were analyzed by sucrose gradient sedimentation and gel filtration chromatography to determine estimated molecular weights using Equation 1. All values represent an average ± SEM for at least three experiments.
Because the C terminus of MCAK contains a weak coiled-coil domain and interacts with itself in yeast two-hybrid assays (our unpublished data), we performed hydrodynamic analysis of all proteins to determine their estimated molecular mass. We found that FL-MCAK and GFL-MCAK are dimeric but that MD-MCAK and GMD-MCAK are monomeric (Figure 1D). Because MD-MCAK is the smallest functional unit of MCAK that exhibited activity equal to that of FL-MCAK in our cellular assay, and because it is monomeric, we used it to probe the mechanistic differences between MT depolymerization induced by MD-MCAK in direct comparison with FL-MCAK.
FL-MCAK Binds with Greater Specificity to MT Ends Than MD-MCAK
We first measured the binding affinity of FL- or MD-MCAK for MTs using a cosedimentation assay in the absence of added nucleotides (Figure 2). In this experiment, the MTs were doubly stabilized with paclitaxel and GMPCPP to minimize destabilization of the MTs during the reaction. We found that both FL-MCAK and MD-MCAK exhibited similar microtubule affinities (0.84 μM for FL-MCAK versus 0.70 μM for MD-MCAK). In these cosedimentation assays, the concentration of MCAK (1.24 μM) to MT (0–8 μM tubulin) was much higher than in our depolymerization assays (Figures 1 and 4); therefore, the high-affinity end binding sites for MCAK on the MT are saturated, and MCAK is binding to the MT lattice as well. Thus, the experimentally determined Kd in this experiment is a composite constant reflecting the affinity for both the MT ends and the MT lattice. These results indicate that MCAK is capable of binding to the MT lattice, but the Kd observed is weaker than the 37–620 nM constants that have been reported for conventional kinesin (Kinesin-1), Ncd (Kinesin-14), and monomeric Eg5 (Kinesin-5), all of which translocate along MTs (Crevel et al., 1996; Foster et al., 1998; Cochran et al., 2005). Our next experiments were designed to evaluate the MT end binding behavior of MCAK.
Figure 4.

FL-MCAK depolymerizes MTs more efficiently than MD-MCAK. (A) GMPCPP-stabilized MTs (1 μM) were incubated with increasing concentrations (0–128 nM) of either FL-MCAK or MD-MCAK in the presence of saturating MgATP for 15 min at 22°C. Soluble tubulin heterodimer was separated from the remaining MTs by centrifugation. Equal volumes of supernatant (S) and pellet (P) were analyzed by SDS-PAGE followed by Coomassie staining, and the amount of depolymerization was then quantified using NIH Image. (B) The combined data derived from at least three separate experiments were fit to the dose-response curve (Equation 2). (C) Summary of the data derived from the graphs in B.
Previous work indicates that MCAK binds specifically to MT ends when MCAK is substoichiometric to the tubulin concentration (Desai et al., 1999; Hunter et al., 2003; Moore and Wordeman, 2004). We therefore wanted to determine whether MD-MCAK had similar end binding properties to FL-MCAK. To discriminate between the end binding and lattice binding properties of MCAK, we incubated substoichiometric concentrations of GFL-MCAK or GMD-MCAK with MTs in the presence of varying nucleotide conditions and then visualized the MTs by immunofluorescence microscopy (Figure 3) (Desai et al., 1999; Hunter et al., 2003; Moore and Wordeman, 2004).
In the absence of added nucleotide and at a molar ratio of one MCAK head to 44 tubulin heterodimers, GFL-MCAK was bound to 76 ± 5% of the MTs (Figure 3, A and J) and was highly enriched at the ends of MTs (Figure 3, B and C). GFL-MCAK bound exclusively to the ends of the majority of the MTs (67 ± 6%), whereas 25 ± 4% of the MTs had GFL-MCAK bound to both the lattice and MT ends. GFL-MCAK bound exclusively to the lattice of only 8 ± 2% of the MTs. Unlike GFL-MCAK, GMD-MCAK bound to only 52 ± 15% of the MTs, a 1.5-fold decrease in the percentage of MTs with enzyme bound compared with GFL-MCAK (Figure 3, A and J). Furthermore, in sharp contrast to GFL-MCAK, GMD-MCAK exhibited a great reduction in the preference for MT end binding in comparison with MT lattice binding (Figure 3, B and C). Overall, there was a significant decrease in the number of MTs with GMD-MCAK bound to the ends compared with GFL-MCAK, with a concurrent increase in the percentage of MTs with GMD-MCAK bound only to the lattice (Figure 3, B and C). These results suggest that GMD-MCAK has some preference for MT ends but that GFL-MCAK has a higher affinity for MT ends than does MD-MCAK.
Because GFL-MCAK clearly bound preferentially to the ends without added nucleotide, we wondered whether the addition of saturating concentrations of a nonhydrolyzable analogue of ATP (MgAMPPNP) could increase the affinity of GFL-MCAK for MT ends or enhance the binding of GMD-MCAK to the ends. Overall, the percentage of MTs bound by GFL-MCAK or GMD-MCAK did not differ from the experiments in which no additional nucleotide was added (Figure 3, A, D, and J). However, the distribution of binding events did change. MgAMPPNP addition significantly reduced the number of MTs with GFL-MCAK bound only to the ends by ∼1.5-fold (Figure 3, B, E, and J), suggesting that MgAMPPNP enhances lattice binding and suppresses end binding. However, the percentage of MTs with end binding was still greater than the percentage of MTs with lattice binding. For GMD-MCAK in the presence of MgAMPPNP, the percentage of MTs that remained unbound (48 ± 5%) was similar to what was seen without additional nucleotide (48 ± 7%). However, the percentage of MTs with only lattice binding increased 1.4-fold (Figure 3, B, E, and J). In general, there was a distinct shift (2-fold) toward more GMD-MCAK binding exclusively to the lattice versus exclusively to the end of the MT. Overall, our data suggest that AMPPNP promotes lattice binding of MD-MCAK and diminishes end binding of FL-MCAK.
To further address what effect the nucleotide state of MCAK has on MT binding properties, we determined whether the addition of MgADP altered MT binding. In the presence of saturating MgADP, both GFL-MCAK and GMD-MCAK behaved similarly (Figure 3, G, H, and J). The majority of MTs had no enzyme bound (88 ± 8% for GFL-MCAK and 88 ± 6% for GMD-MCAK), suggesting that the MCAK·ADP state is weakly bound to MTs. Of the few MTs that did have enzyme present (12% of all the MTs counted), most of GFL-MCAK and GMD-MCAK were bound to the lattice and not the MT ends. These data suggest that in the ADP state, MCAK does not bind well to the MT, and any binding that does occur is not MT end-specific.
In summary, our end binding analysis indicates that MD-MCAK does not bind to MT ends with the same high-affinity as FL-MCAK. Interestingly, we find that the greatest end binding of FL-MCAK occurs without the addition of nucleotide. An ATP-like state (MgAMPPNP) does not increase the end binding affinity of FL-MCAK and in fact increases the amount of enzyme on the lattice of the MT. However, the nucleotide state is less influential for the end binding affinity of MD-MCAK. These results suggest that the coupling of the ATP catalytic cycle to high-affinity MT end binding and presumably MT depolymerization may be differentially regulated by dimeric FL-MCAK in comparison with monomeric MD-MCAK.
FL-MCAK Depolymerizes MTs More Efficiently than MD-MCAK
Because MD-MCAK did not bind with high-affinity to MT ends, we expected it to be less efficient at inducing MT depolymerization because end binding is thought to be an essential part of the depolymerization mechanism (Desai et al., 1999; Hunter et al., 2003). However, our cellular and sedimentation-based depolymerization assays revealed no significant reduction in the activity of MD-MCAK compared with FL-MCAK; therefore, we needed an assay that was capable of reproducibly detecting subtle changes in depolymerization activity. Because we found that real-time MT depolymerization assays are highly variable and difficult to quantify accurately, we used a sedimentation assay in which we added increasing concentrations of enzyme to determine the effective concentration at which each enzyme gives 50% MT depolymerization (EC50; Figure 4). For each MCAK concentration analyzed, the fraction of MT polymer to soluble tubulin heterodimer at a fixed time point was quantified and compared with the log of the enzyme concentration (Figure 4B; Motulsky and Christopoulos, 2003). At high MCAK concentrations for both FL-MCAK and MD-MCAK, the MT polymer was almost completely converted to soluble tubulin heterodimer (Figure 4A); however, there was a clear difference in the concentration of enzyme at which there was 50% MT polymer and 50% soluble tubulin heterodimer. The fit of the data to the dose-response curve provided an EC50 for FL-MCAK of 5.6 nM and for MD-MCAK of 17 nM (Figure 4C). These data show that it takes nearly threefold more MD-MCAK than FL-MCAK to achieve the same molar quantity of soluble tubulin heterodimer relative to MT polymer.
FL-MCAK Has Higher ATPase Activity than MD-MCAK
To determine the underlying kinetic differences between FL-MCAK and MD-MCAK that may contribute to the observed differences in depolymerization activity, we measured the ATPase activity of each protein at concentrations substoichiometric to MT polymer. In these assays, we used doubly stabilized MTs, which are more resistant to depolymerization, to minimize MT substrate loss during the assay and to prevent accumulation of tubulin heterodimers in the assay, which can also stimulate ATPase activity (Moores et al., 2002, 2003; Hunter et al., 2003; Moore and Wordeman, 2004; Shipley et al., 2004; see below). Both FL-MCAK and MD-MCAK exhibited a low basal ATPase activity in the absence of MTs (0.03 ± 0.002 s-1), and exhibited MT-stimulated ATPase activity (Figure 5A); however, the ATPase kinetics differed greatly between the two proteins (Figure 5D).
We observed that the kcat,MT for the FL-MCAK was significantly higher than that of the monomeric MD-MCAK (0.73 versus 0.49 s-1, respectively; Figure 5A). These data indicate that each FL-MCAK head hydrolyzes ATP nearly twofold faster than the single head of MD-MCAK. However, what is most significant is the K0.5,MT, reflecting that dimeric FL-MCAK binds MTs much more weakly during ATP turnover than monomeric MD-MCAK (2.8 μM for FL-MCAK vs. 0.01 μM for MD-MCAK).
Both FL-MCAK and MD-MCAK also exhibited tubulin heterodimer-stimulated ATPase activity (Figure 5B). In comparison with the MT-stimulated ATPase activity, the overall magnitude of tubulin heterodimer-stimulated ATPase activity (kcat,Tubulin) was significantly lower, indicating that MTs are better stimulators of ATPase activity for both enzymes. In addition, for both FL-MCAK and MD-MCAK the K0.5,Tubulin indicates a weaker affinity for the tubulin heterodimer than for the MT polymer. This steady-state kinetic parameter indicates a dramatic difference in tubulin heterodimer affinity for FL-MCAK in comparison with MD-MCAK, which is relevant to the depolymerization mechanism. For FL-MCAK, the K0.5,Tubulin is 4.9 μM, suggesting that the full-length MCAK dimer is bound relatively weakly to the tubulin heterodimer. In contrast, the K0.5,Tubulin for MD-MCAK is 0.06 μM. This 60 nM constant indicates that the MCAK monomer is very tightly bound to the tubulin heterodimer.
We also explored MT-activated steady-state kinetics as a function of MgATP concentration for both FL-MCAK and MD-MCAK (Figure 5C). Although the Km,ATP constants reflect a difference in relative affinity for MgATP, the catalytic efficiency constants (kcat/Km,ATP) are similar: 0.008 μM-1 s-1 for FL-MCAK and 0.007 μM-1 s-1 for MD-MCAK. These results indicate that the differences in the ATP turnover are dictated by the differences in affinity for the MT polymer and the soluble tubulin heterodimer. We propose that the steady-state ATPase kinetics reflect the coupling of ATP turnover to MT depolymerization by both FL- and MD-MCAK. The reduction in ATPase activity of monomeric MD-MCAK and the apparent high-affinity for the tubulin heterodimer may account for some of the reduced efficiency seen in MT depolymerization assays.
MD-MCAK Has a Higher Affinity for Soluble Tubulin Heterodimer
We were surprised that the K0.5,Tubulin was much lower for MD-MCAK than for FL-MCAK, which suggested that MD-MCAK bound more tightly to tubulin heterodimer. However, our ATPase assays do not address the interaction of MCAK with MT polymer and tubulin heterodimer when both are present, a situation that is more physiologically relevant. To compare the relative binding of MCAK to MTs in the presence of tubulin heterodimer, we developed a MT cosedimentation competition assay in which FL- or MD-MCAK was incubated with a saturating concentration of doubly stabilized MTs in the presence of increasing concentrations of soluble tubulin heterodimer (Figure 6A). We quantified the amount of MCAK that partitioned to the supernatant (assumed to be associated with soluble tubulin heterodimer) or the pellet (associated with MTs). We found that as the tubulin heterodimer concentration was increased, MD-MCAK partitioned to the supernatant with the soluble tubulin heterodimer rather than in the pellet with the MT polymer (Figure 6A). At a 4:1 M ratio of soluble tubulin heterodimer to MTs, only 30 ± 0.05% of the MD-MCAK remained in the pellet compared with no tubulin heterodimer addition (Figure 6B). In contrast, as the concentration of tubulin heterodimer increased, the amount of FL-MCAK in the pellet remained relatively constant. These data are consistent with the hypothesis that MD-MCAK has a higher affinity for tubulin heterodimer than FL-MCAK. We propose that the high-affinity of MD-MCAK for tubulin heterodimer acts to stabilize the MCAK–tubulin heterodimer complex, thereby, slowing its dissociation for rebinding to the MT end for another cycle of ATP-promoted MT depolymerization.
DISCUSSION
A major question in the Kinesin-13 field is aimed at understanding the differences between MT-depolymerizing kinesins and MT-translocating kinesins. Our analysis extends previous work by showing that the cooperative interactions provided by the FL-MCAK are critical for effective depolymerization activity in vivo. The steady-state ATPase kinetics, the MT-end binding properties, and the affinity for soluble tubulin heterodimer revealed by our in vitro assays with MD-MCAK in comparison with FL-MCAK (Figures 3, 4, 5, 6) support this hypothesis.
MD-MCAK Exhibits Slower Kinetics Than FL-MCAK
Unlike most translocating kinesins, the ATPase activity of Kinesin-13 members is stimulated by MTs as well as by tubulin heterodimer (Moores et al., 2002, 2003; Hunter et al., 2003; Moore and Wordeman, 2004; Shipley et al., 2004). This difference in MT- and tubulin heterodimer-stimulated activity could be because Kinesin-13s recognize unique tubulin quaternary structures that are exposed only at the end of the MT or because they can bind directly to tubulin heterodimer (Desai et al., 1999). Interestingly, it was recently shown that Xklp1 (Kinesin-4 family) can translocate along MTs as well as regulate their dynamics (Bringmann et al., 2004). The ATPase activity of Xklp1 is also stimulated by tubulin heterodimer, suggesting the intriguing possibility that tubulin heterodimer-stimulated ATPase activity is a conserved feature of kinesins that regulate MT dynamics. However, Kar3 (kinesin-14 family), which influences MT dynamics in yeast (Meluh and Rose, 1990; Saunders et al., 1997; Troxell et al., 2001) and depolymerizes MTs in vitro (Endow et al., 1994; Sproul et al., 2005), exhibits only MT-stimulated ATPase activity and shows much less robust depolymerization activity compared with Kinesin-13 family members (Sproul et al., 2005). Thus, even within the kinesin superfamily, the mechanism of MT depolymerization has diverged and highlights the need to study the detailed catalytic mechanism of multiple members within a kinesin subfamily.
Our findings that the tubulin heterodimer-stimulated ATPase activity is lower than MT-stimulated ATPase activity for FL-MCAK are similar to what has been observed for mammalian MCAK (Hunter et al., 2003), yet curiously the tubulin heterodimer-stimulated ATPase activity is equal to MT-stimulated ATPase activity for the pKinI catalytic core (Moores et al., 2002, 2003; Shipley et al., 2004). This may be because of, in part, the type of MT substrate used in the assay: a recent report showed that MCAK exhibited MT-stimulated ATPase activity comparable with that of tubulin heterodimer when long paclitaxel-stabilized MTs were used as a substrate (Moore and Wordeman, 2004). Given that the ATPase activity of MCAK is highly stimulated by MT ends (Hunter et al., 2003; Moore and Wordeman, 2004), it is possible that the end structures of GMPCPP-stabilized MTs may be slightly different from paclitaxel-stabilized MTs. Alternatively, the differences in ATPase activity may simply be a reflection of the length of MTs that were used in the assay (Hunter et al., 2003; Moore and Wordeman, 2004) or a reflection of the differences between the expressed proteins (the pKinI construct used contained only the catalytic core).
Although the Kinesin-13s are stimulated by both MTs and tubulin heterodimer, the relative activities and apparent substrate affinities of FL-MCAK and MD-MCAK are very different. Our data suggest that although MD-MCAK still has a higher affinity for MTs than for tubulin heterodimer, it may be hampered in its depolymerization activity in part because its end binding ability is not as robust as FL-MCAK. For MD-MCAK, the K0.5,MT also may reflect both end and lattice affinities; therefore, the decreased ATPase activity measured may reflect a reduction of activity because of nonproductive lattice binding and compromised end binding.
Our data suggest that another difference between FL-MCAK and MD-MCAK is that a significant amount of MD-MCAK may remain tightly bound to tubulin heterodimer that is released during depolymerization. Consistent with this idea, MD-MCAK has a considerably higher affinity for soluble tubulin heterodimer than does FL-MCAK as assayed by binding competition assays. These results suggest that in the cellular transfection assay, MD-MCAK may be bound to free tubulin heterodimer and act as a tubulin-sequestering protein in addition to a microtubule-depolymerizing enzyme. In the future, it will be important to determine whether the ability of MCAK to sequester tubulin heterodimer is physiologically important.
Two Heads Are Better than One
Our data support a model in which dimerization of MCAK seems to be important for efficient MT depolymerization. The steady-state ATPase kinetics for FL-MCAK reveals a higher kcat in the presence of MTs and significantly weaker affinity for tubulin heterodimer in comparison with MD-MCAK. These results indicate that ATP turnover is coupled to the force production for MT depolymerization. We propose that the increased efficiency of MT destabilization by FL-MCAK results because of the cooperative interactions of the dimeric molecule that increase the MT end binding and decrease the affinity for the tubulin heterodimer. The mechanistic basis of the cooperativity, the pathway of communication, and whether the neck plays a role are questions to be addressed in the future.
Another model is that dimerization may contribute to processive depolymerization of stabilized MTs. One model of MCAK action is that the enzyme remains bound to the MT and continues to hydrolyze ATP while inducing tubulin heterodimer release from the MT end. This processivity seems to depend on the stability of the MT substrate (Hunter et al., 2003), which in vivo could depend on the number of stabilizing proteins present. Perhaps dimerization of MCAK plays a vital role in the establishment of processivity. For example, conventional kinesin as a monomer can walk along the MT, but it is not processive (Berliner et al., 1995; Hancock and Howard, 1998; Romberg et al., 1998; Inoue et al., 2001). If Kinesin-13s are processive depolymerizers, it is possible that monomeric Kinesin-13s are not processive depolymerizers in vitro, which could explain why MD-MCAK seems less efficient.
It is also possible that the differences between FL-MCAK and MD-MCAK are not a reflection solely of dimerization but are complicated by the presence of additional regulatory domains in the C terminus. Deletion of the C-terminal nine amino acids of mammalian MCAK increases depolymerization activity in vivo and also increases ATPase activity in vitro (Moore and Wordeman, 2004). The authors propose that MCAK binds weakly to the lattice but that ATPase activity is inhibited by the far C terminus until the enzyme reaches its high-affinity binding site at the end of the MT (Moore and Wordeman, 2004). From our transfection data, we found that truncation of 28 amino acids from the C terminus of Xenopus MCAK caused a small but significant decrease in depolymerization activity (Table S1) contrary to previous findings (Moore and Wordeman, 2004). Despite these differences, our monomeric MD-MCAK, which lacks the entire C terminus, shows similar MT depolymerization in vivo, decreased affinity for MT ends, and decreased ATPase activity in vitro compared with FL-MCAK. In addition, our preliminary data suggests that deletion of potential regulatory domains in either the N or C termini do not significantly influence depolymerization activity when assayed under similar conditions. However, it should be noted that the ionic strength used in the depolymerization assay buffers does affect the activity of several of the truncation mutants (our unpublished observations). Together, these results suggest that Kinesin-13s have more than one mechanism for the control of ATPase activity, which is another example of the complexity of this enzyme family.
MD-MCAK Binds Less Efficiently to MT Ends
One interesting finding from our studies is that FL-MCAK has a much stronger preference for binding to MT ends relative to MD-MCAK. MD-MCAK may be unable to recognize the end structure of the MT as well as FL-MCAK or it may be unable to reach the end by a nondirectional mechanism such as one-dimensional (1D) diffusion. It is also possible that other parts of the binding cycle, such as dissociation from the MT, may occur faster for MD-MCAK, and thus MD-MCAK may not remain on the MT end long enough to visualize its binding.
The nucleotide state of MCAK was also an important feature governing end binding. In contrast to previous findings (Desai et al., 1999; Moore and Wordeman, 2004), we found that although FL-MCAK still showed specificity for the MT ends in the presence of MgAMPPNP, the amount of FL-MCAK at the ends only was significantly decreased. Our results are curious considering that the 1D diffusion of MCAK, proposed to be necessary for end-targeting, requires the presence of added nucleotide (Hunter et al., 2003). However, we found that FL-MCAK bound most efficiently to MT ends without additional nucleotide, challenging this model. In comparison, studies involving the pKinI catalytic core have revealed that nucleotide state does not affect the affinity of pKinI binding to the MT lattice (Moores et al., 2003). Overall, these studies indicate that nucleotide state plays a role in regulating end binding and not lattice binding affinity for Kinesin-13s.
A Model for MCAK-induced Depolymerization
Given the difference in ATPase kinetics and end binding specificity, we propose the following model for how the two heads of MCAK may be used during MT depolymerization (Figure 7). FL-MCAK specifically binds to the end of the MT (Figure 7A). The binding of ATP may allow for stabilization of an already bent protofilament, or it may induce curvature of a protofilament that destabilizes the MT. Whereas several molecules of MCAK are likely necessary to depolymerize a stabilized MT, only one MCAK may be needed for the depolymerization of a dynamic MT. On end binding, ATP is hydrolyzed, tubulin heterodimer dissociates with bound MCAK, and then MCAK is released from the tubulin heterodimer and recycled for another round of depolymerization. MD-MCAK does not recognize or bind to the MT end as well as FL-MCAK (Figure 7B), thus the initial binding event is inefficient. Because MD-MCAK does not bind as well to the end, it takes more MD-MCAK than FL-MCAK for efficient depolymerization because the lattice serves as a sink for the binding of MD-MCAK molecules. Once the number of MD-MCAK molecules reaches a critical concentration at the end, depolymerization occurs. Because MD-MCAK has a higher affinity for tubulin heterodimer, it remains bound to the released tubulin heterodimer, which slows recycling and lowers the effective concentration of MD-MCAK available to initiate another round of depolymerization.
Figure 7.

Model for the role of dimerization of MCAK. The catalytic cycle is diagrammed at only one end of the MT, although MCAK depolymerizes MTs from both ends. (A) With FL-MCAK, a critical number of molecules (for clarity, only one is diagrammed) are necessary to induce ATP-promoted depolymerization at the end of the MT. The tubulin heterodimer·MCAK complex is released from the shortening MT. The affinity of FL-MCAK for the tubulin heterodimer is weak, resulting in release of MCAK with rapid rebinding to the MT end. (B) MD-MCAK also can depolymerize MTs, but it requires a greater number of molecules than FL-MCAK because of the lower binding specifically to the ends of the MT. The higher affinity of MD-MCAK for the tubulin heterodimer delays its release to the solution and therefore slows its overall recycling rate for MT end binding and subsequent MT depolymerization.
Supplementary Material
Acknowledgments
We thank S. Rosenfeld for stimulating discussions and ideas and E. Gan for help in cloning. This work was supported by grants from the American Heart Association (AHA), American Cancer Society (RSG-03-149-CSM), and National Institutes of Health (NIH) (GM-59618) to C.E.W.; by an NIH training grant (GM-007757) to K.M.H.; by a Walther Cancer Foundation postdoctoral fellowship to S.E.M.; by an AHA predoctoral fellowship to S.K.S.; and by a grant from NIH (GM-54141) and an NIH-National Institute of Arthritis & Musculoskeletal & Skin Diseases Career Development Award (K02-AR47841) to S.P.G. C.E.W. is a scholar of the Leukemia and Lymphoma Society.
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E05–08–0821) on November 16, 2005.
The online version of this article contains supplemental material at MBC Online (http://www.molbiolcell.org).
References
- Berliner, E., Young, E. C., Anderson, K., Mahtani, H. K., and Gelles, J. (1995). Failure of a single-headed kinesin to track parallel to microtubule protofilaments. Nature 373, 718-721. [DOI] [PubMed] [Google Scholar]
- Bringmann, H., Skiniotis, G., Spilker, A., Kandels-Lewis, S., Vernos, I., and Surrey, T. (2004). A kinesin-like motor inhibits microtubule dynamic instability. Science 303, 1519-1522. [DOI] [PubMed] [Google Scholar]
- Cassimeris, L., and Spittle, C. (2001). Regulation of microtubule-associated proteins. Int. Rev. Cytol. 210, 163-226. [DOI] [PubMed] [Google Scholar]
- Cochran, J. C., Gatial, J. E., 3rd, Kapoor, T. M., and Gilbert, S. P. (2005). Monastrol inhibition of the mitotic kinesin Eg5. J. Biol. Chem. 280, 12658-12667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crevel, I. M., Lockhart, A., and Cross, R. A. (1996). Weak and strong states of kinesin and ncd. J. Mol. Biol. 257, 66-76. [DOI] [PubMed] [Google Scholar]
- Desai, A., and Mitchison, T. J. (1997). Microtubule polymerization dynamics. Annu. Rev. Cell Dev. Biol. 13, 83-117. [DOI] [PubMed] [Google Scholar]
- Desai, A., Verma, S., Mitchison, T. J., and Walczak, C. E. (1999). Kin I kinesins are microtubule-destabilizing enzymes. Cell 96, 69-78. [DOI] [PubMed] [Google Scholar]
- Desai, A., and Walczak, C. E. (2001). Assays for microtubule-destabilizing kinesins. Methods Mol. Biol. 164, 109-121. [DOI] [PubMed] [Google Scholar]
- Endow, S. A., Kang, S. J., Satterwhite, L. L., Rose, M. D., Skeen, V. P., and Salmon, E. D. (1994). Yeast Kar3 is a minus-end microtubule motor protein that destabilizes microtubules preferentially at the minus ends. EMBO J. 13, 2708-2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster, K. A., Correia, J. J., and Gilbert, S. P. (1998). Equilibrium binding studies of non-claret disjunctional protein (Ncd) reveal cooperative interactions between the motor domains. J. Biol. Chem. 273, 35307-35318. [DOI] [PubMed] [Google Scholar]
- Gaetz, J., and Kapoor, T. M. (2004). Dynein/dynactin regulate metaphase spindle length by targeting depolymerizing activities to spindle poles. J. Cell Biol. 166, 465-471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganem, N. J., and Compton, D. A. (2004). The KinI kinesin Kif2a is required for bipolar spindle assembly through a functional relationship with MCAK. J. Cell Biol. 166, 473-478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock, W. O., and Howard, J. (1998). Processivity of the motor protein kinesin requires two heads. J. Cell Biol. 140, 1395-1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirokawa, N., and Takemura, R. (2004). Kinesin superfamily proteins and their various functions and dynamics. Exp. Cell Res. 301, 50-59. [DOI] [PubMed] [Google Scholar]
- Homma, N., Takei, Y., Nakata, T., Terada, S., Kikkawa, M., Noda, Y., and Hirokawa, N. (2003). Kinesin Superfamily Protein 2A (KIF2A) Functions in suppression of collateral branch extension. Cell 114, 229-239. [DOI] [PubMed] [Google Scholar]
- Hunter, A. W., Caplow, M., Coy, D. L., Hancock, W. O., Diez, S., Wordeman, L., and Howard, J. (2003). The Kin I kinesin MCAK is a microtubule depolymerase that forms an ATP-hydrolyzing complex at microtubule ends. Mol. Cell 11, 445-457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue, Y., Iwane, A. H., Miyai, T., Muto, E., and Yanagida, T. (2001). Motility of single one-headed kinesin molecules along microtubules. Biophys. J. 81, 2838-2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline-Smith, S. L., Khodjakov, A., Hergert, P., and Walczak, C. E. (2004). Depletion of centromeric MCAK leads to chromosome congression and segregation defects due to improper kinetochore attachments. Mol. Biol. Cell 15, 1146-1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline-Smith, S. L., and Walczak, C. E. (2002). The Microtubule-destabilizing kinesin XKCM1 regulates microtubule dynamic instability in cells. Mol. Biol. Cell 13, 2718-2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline-Smith, S. L., and Walczak, C. E. (2004). Mitotic spindle assembly and chromosome segregation: refocusing on microtubule dynamics. Mol. Cell 15, 317-327. [DOI] [PubMed] [Google Scholar]
- Lanzetta, P. A., Alvarez, L. J., Reinach, P. S., and Candia, O. A. (1979). An improved assay for nanomole amounts of inorganic phosphate. Anal. Biochem. 100, 95-97. [DOI] [PubMed] [Google Scholar]
- Laurent, T. C., and Killander, J. (1964). A theory of gel filtration and its experimental verification. J. Chromatogr. 14, 317-330. [Google Scholar]
- Lawrence, C. J., et al. (2004). A standardized kinesin nomenclature. J. Cell Biol. 167, 19-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maney, T., Hunter, A. W., Wagenbach, M., and Wordeman, L. (1998). Mitotic centromere-associated kinesin is important for anaphase chromosome segregation. J. Cell Biol. 142, 787-801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maney, T., Wagenbach, M., and Wordeman, L. (2001). Molecular dissection of the microtubule depolymerizing activity of mitotic centromere-associated kinesin. J. Biol. Chem. 276, 34753-34758. [DOI] [PubMed] [Google Scholar]
- Meluh, P. B., and Rose, M. D. (1990). KAR3, a kinesin-related gene required for yeast nuclear fusion. Cell 60, 1029-1041. [DOI] [PubMed] [Google Scholar]
- Mennella, V., Rogers, G. C., Rogers, S. L., Buster, D. W., Vale, R. D., and Sharp, D. J. (2005). Functionally distinct kinesin-13 family members cooperate to regulate microtubule dynamics during interphase. Nat. Cell Biol. 7, 235-245. [DOI] [PubMed] [Google Scholar]
- Miki, H., Okada, Y., and Hirokawa, N. (2005). Analysis of the kinesin superfamily: insights into structure and function. Trends Cell Biol. 15, 467-476. [DOI] [PubMed] [Google Scholar]
- Moore, A., and Wordeman, L. (2004). C-terminus of mitotic centromere-associated kinesin (MCAK) inhibits its lattice-stimulated ATPase activity. Biochem. J. 383, 227-235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore, A. T., Rankin, K. E., von Dassow, G., Peris, L., Wagenbach, M., Ovechkina, Y., Andrieux, A., Job, D., and Wordeman, L. (2005). MCAK associates with the tips of polymerizing microtubules. J. Cell Biol. 169, 391-397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moores, C. A., Hekmat-Nejad, M., Sakowicz, R., and Milligan, R. A. (2003). Regulation of KinI kinesin ATPase activity by binding to the microtubule lattice. J. Cell Biol. 163, 963-971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moores, C. A., Yu, M., Guo, J., Beraud, C., Sakowicz, R., and Milligan, R. A. (2002). A mechanism for microtubule depolymerization by KinI kinesins. Mol. Cell 9, 903-909. [DOI] [PubMed] [Google Scholar]
- Morfini, G., Quiroga, S., Rosa, A., Kosik, K., and Caceres, A. (1997). Suppression of KIF2 in PC12 cells alters the distribution of a growth cone nonsynaptic membrane receptor and inhibits neurite extension. J. Cell Biol. 138, 657-669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motulsky, H. J., and Christopoulos, A. (2003). Fitting Models to Biological Data using Linear and Nonlinear Regression. A Practical Guide to Curve Fitting. GraphPad Software Inc., San Diego.
- Newton, C. N., Wagenbach, M., Ovechkina, Y., Wordeman, L., and Wilson, L. (2004). MCAK, a Kin I kinesin, increases the catastrophe frequency of steady-state HeLa cell microtubules in an ATP-dependent manner in vitro. FEBS Lett. 572, 80-84. [DOI] [PubMed] [Google Scholar]
- Niederstrasser, H., Salehi-Had, H., Gan, E. C., Walczak, C., and Nogales, E. (2002). XKCM1 acts on a single protofilament and requires the C terminus of tubulin. J. Mol. Biol. 316, 817-828. [DOI] [PubMed] [Google Scholar]
- Nogales, E. (2000). Structural insights into microtubule function. Annu. Rev. Biochem. 69, 277-302. [DOI] [PubMed] [Google Scholar]
- Ogawa, T., Nitta, R., Okada, Y., and Hirokawa, N. (2004). A common mechanism for microtubule destabilizers-M type kinesins stabilize curling of the protofilament using the class-specific neck and loops. Cell 116, 591-602. [DOI] [PubMed] [Google Scholar]
- Ovechkina, Y., Wagenbach, M., and Wordeman, L. (2002). K-loop insertion restores microtubule depolymerizing activity of a “neckless” MCAK mutant. J. Cell Biol. 159, 557-562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porath, J., and Flodin, P. (1959). Gel filtration: a method for desalting and group separation. Nature 183, 1657-1659. [DOI] [PubMed] [Google Scholar]
- Rogers, G. C., Rogers, S. L., Schwimmer, T. A., Ems-McClung, S. C., Walczak, C. E., Vale, R. D., Scholey, J. M., and Sharp, D. J. (2004). Two mitotic kinesins cooperate to drive sister chromatid separation during anaphase. Nature 427, 364-370. [DOI] [PubMed] [Google Scholar]
- Romberg, L., Pierce, D. W., and Vale, R. D. (1998). Role of the kinesin neck region in processive microtubule-based motility. J. Cell Biol. 140, 1407-1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sack, S., Kull, F. J., and Mandelkow, E. (1999). Motor proteins of the kinesin family. Structures, variations, and nucleotide binding sites. Eur. J. Biochem. 262, 1-11. [DOI] [PubMed] [Google Scholar]
- Saunders, W., Hornack, D., Lengyel, V., and Deng, C. (1997). The Saccharomyces cerevisiae kinesin-related motor Kar3p acts at preanaphase spindle poles to limit the number and length of cytoplasmic microtubules. J. Cell Biol. 137, 417-431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shipley, K., Hekmat-Nejad, M., Turner, J., Moores, C., Anderson, R., Milligan, R., Sakowicz, R., and Fletterick, R. (2004). Structure of a kinesin microtubule depolymerization machine. EMBO J. 23, 1422-1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel, L. M., and Monty, K. J. (1966). Determination of molecular weights and frictional ratios of proteins in impure systems by use of gel filtration and density gradient centrifugation. Application to crude preparations of sulfite and hydroxylamine reductases. Biochim. Biophys. Acta 112, 346-362. [DOI] [PubMed] [Google Scholar]
- Sproul, L. R., Anderson, D. J., Mackey, A. T., Saunders, W. S., and Gilbert, S. P. (2005). Cik1 targets the minus-end Kinesin depolymerase kar3 to microtubule plus ends. Curr. Biol. 15, 1420-1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troxell, C. L., Sweezy, M. A., West, R. R., Reed, K. D., Carson, B. D., Pidoux, A. L., Cande, W. Z., and McIntosh, J. R. (2001). pkl1(+)and klp2(+): two kinesins of the Kar3 subfamily in fission yeast perform different functions in both mitosis and meiosis. Mol. Biol. Cell 12, 3476-3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walczak, C., Gan, E. C., Desai, A., Mitchison, T. J., and Kline-Smith, S. L. (2002). The microtubule-destabilizing kinesin, XKCM1 is required for chromosome positioning during spindle assembly. Curr. Biol. 12, 1885-1889. [DOI] [PubMed] [Google Scholar]
- Wordeman, L., Wagenbach, M., and Maney, T. (1999). Mutations in the ATP-binding domain affect the subcellular distribution of mitotic centromere-associated kinesin (MCAK). Cell Biol. Int. 23, 275-286. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.