

Abstract
The deleterious consequences of fatty acid (FA) and neutral lipid accumulation in nonadipose tissues, such as the heart, contribute to the pathogenesis of type 2 diabetes. To elucidate mechanisms of FA-induced cell death, or lipotoxicity, we generated Chinese hamster ovary (CHO) cell mutants resistant to palmitate-induced death and isolated a clone with disruption of eukaryotic elongation factor (eEF) 1A-1. eEF1A-1 involvement in lipotoxicity was confirmed in H9c2 cardiomyoblasts, in which small interfering RNA-mediated knockdown also conferred palmitate resistance. In wild-type CHO and H9c2 cells, palmitate increased reactive oxygen species and induced endoplasmic reticulum (ER) stress, changes accompanied by increased eEF1A-1 expression. Disruption of eEF1A-1 expression rendered these cells resistant to hydrogen peroxide- and ER stress-induced death, indicating that eEF1A-1 plays a critical role in the cell death response to these stressors downstream of lipid overload. Disruption of eEF1A-1 also resulted in actin cytoskeleton defects under basal conditions and in response to palmitate, suggesting that eEF1A-1 mediates lipotoxic cell death, secondary to oxidative and ER stress, by regulating cytoskeletal changes critical for this process. Furthermore, our observations of oxidative stress, ER stress, and induction of eEF1A-1 expression in a mouse model of lipotoxic cardiomyopathy implicate this cellular response in the pathophysiology of metabolic disease.
INTRODUCTION
The increased prevalence of obesity worldwide has contributed to the emergence of a disease cluster, the metabolic syndrome, that includes insulin resistance, type 2 diabetes, and cardiovascular disease. Elevated serum triglyceride and fatty acid (FA) levels associated with this syndrome contribute to lipid accumulation in many nonadipose tissues, including the heart. This inappropriate accumulation of excess lipid can lead to cellular dysfunction and cell death—a process called lipotoxicity (Unger, 2003).
Lipotoxic cardiomyocyte death has been proposed to play a central role in heart failure associated with diabetes and obesity in animal models and in humans. Although FAs are the principal source of energy for cardiomyocytes, high serum triglyceride and FA levels result in FA uptake by the heart that exceeds the anabolic and catabolic needs of the tissue. Triglyceride accumulation in cardiomyocytes of leptin- or leptin receptor-deficient obese diabetic animal models is associated with cardiomyocyte apoptosis (Zhou et al., 2000) and contractile dysfunction (Zhou et al., 2000; Aasum et al., 2002; Christoffersen et al., 2003), suggesting that lipotoxic cell death in the heart may be important in the genesis of diabetic cardiomyopathy. Recently, similar observations were reported in patients with metabolic syndrome and nonischemic heart failure (Sharma et al., 2004). Consistent with this apparent cardiac lipotoxicity, cardiomyocyte-specific increases in FA uptake in mice with cardiac-restricted overexpression of long-chain acyl-CoA synthetase 1 (ACS1), lipoprotein lipase, or fatty acid transport protein 1 are sufficient to cause cardiomyocyte abnormalities and/or death, leading to left ventricular dysfunction (Chiu et al., 2001, 2005; Yagyu et al., 2003).
A variety of tissue culture systems have been used to study mechanisms of lipotoxicity at the cellular level. These studies generally implicate saturated (i.e., palmitate) rather than unsaturated long-chain fatty acids as inducers of cell death. Potential mechanisms of palmitate-induced cell death include decreased cardiolipin synthesis (Ostrander et al., 2001), increased ceramide synthesis (Shimabukuro et al., 1998), and reactive oxygen species (ROS) generation (Shimabukuro et al., 1997; Listenberger et al., 2001). Decreased cardiolipin synthesis is associated with release of cytochrome c from the mitochondrial membrane into the cytosol, which may initiate apoptotic signals (Ostrander et al., 2001). Recent evidence suggests that ceramide and ROS, in contrast, trigger the release of calcium ions from the endoplasmic reticulum (ER) (Scorrano et al., 2003). Subsequent capture of these ions by mitochondria initiates apoptosis.
To further understand the mechanism of palmitate-induced lipotoxicity we used the ROSAβgeo retroviral promoter trap (Friedrich and Soriano, 1991) to perform a genetic screen in Chinese hamster ovary (CHO) cells. Here, we identify and characterize a mutant isolated from this screen on the basis of resistance to palmitate-induced cell death. We show that disruption of the gene encoding eukaryotic elongation factor (eEF) 1A-1 in this mutant as well as targeted disruption of eEF1A-1 using small interfering RNA (siRNA) confers resistance to palmitate-induced cell death. Furthermore, our studies support a central role for oxidative and ER stress during FA overload-induced death in cardiomyocytes, a mechanism that may be central to the pathogenesis of cardiomyopathy in metabolic diseases.
MATERIALS AND METHODS
Cell Culture and Chemicals
CHO-K1 cells (American Type Culture Collection, Manassas, VA) and CHO-derived cell lines were maintained in high glucose (4.5 mg/ml) DMEM and Ham's F-12 nutrient mixture (1:1), with 5% fetal bovine serum (FBS), 2 mM l-glutamine, 50 U/ml penicillin G sodium, 50 U/ml streptomycin sulfate, and 1 mM sodium pyruvate. H9c2 rat cardiomyoblasts (American Type Culture Collection) and H9c2-derived cell lines were maintained in high glucose DMEM with 10% FBS, and L-glutamine, penicillin, streptomycin, and sodium pyruvate at concentrations used in CHO cell growth medium. Wild-type H9c2 cells were differentiated to cardiomyocyte-like cells by incubation for 5-8 d in DMEM containing 1% FBS and 10 nM all-trans-retinoic acid, as described previously (Menard et al., 1999). For experiments, all CHO and H9c2 cell lines (90% confluent) were incubated in CHO cell growth medium (3.151 mg/ml glucose and 5% FBS) supplemented with palmitate (150-500 μM) (Nu-Chek Prep, Elysian, MN) complexed to bovine serum albumin (BSA) at a 2:1 M ratio (prepared as described previously; Listenberger et al., 2001). Staurosporine, actinomycin D, cycloheximide, and camptothecin were from Calbiochem (San Diego, CA); hydrogen peroxide (H2O2) was from Fisher Scientific (Pittsburgh, PA); and tunicamycin, thapsigargin, and α-tocopherol were from Sigma-Aldrich (St. Louis, MO).
Generation of Palmitate-resistant CHO Cell Mutants
Vesicular stomatitis virus G protein (VSV-G) pseudotyped murine retrovirus encoding the ROSAβgeo retroviral promoter trap (Friedrich and Soriano, 1991) was generated as described previously (Ory et al., 1996). CHO cells were transduced with retrovirus at a low multiplicity of infection to achieve an average of one integration per 10 genomes, as determined by Southern blotting analysis. CHO mutants were selected by growth in 0.5 mg/ml G418 (neomycin) for 8 d. Palmitate-resistant mutants were selected by growth for 2 d in media supplemented with 500 μM palmitate complexed to BSA at a 2:1 M ratio. Clones were isolated by plating at limiting dilution.
Identification of Trapped Gene
The endogenous gene disrupted by retroviral integration was identified by 5′ rapid amplification of cDNA ends (RACE) using primers corresponding to an oligonucleotide tag and ROSAβgeo sequences (SMART RACE cDNA amplification kit; Clontech, Mountain View, CA). The 5′RACE product was subsequently cloned and sequenced. CHO sequence located upstream of the retroviral insertion site was blasted against National Center of Biotechnology Information databases. Directed PCR assays with primer pairs to either wild-type CHO or wild type/retroviral fusion sequences were used to confirm that the sequence identified by 5′RACE comprised the gene disrupted by retroviral integration.
Northern Analysis
Total RNA was isolated using RNeasy reagents (QIAGEN, Valencia, CA). Northern blotting was performed as described previously (Schaffer and Lodish, 1994), using a 32P-labeled probe generated by SacII/EcoRI digestion of full-length hamster eEF1A-1. A murine actin probe was used as a loading control.
Cell Death, Caspase-3 Activity, and DNA Fragmentation
Cell death was assessed by membrane permeability to propidium iodide, as described previously (Listenberger et al., 2001). Briefly, cells incubated for 12-24 h with various treatments were harvested by trypsinization and stained with 1 μM propidium iodide. Percentage of propidium iodide-positive cells was determined by flow cytometry with quantification of 104 cells/sample. Apoptosis was assessed by quantitating caspase-3 activity and DNA fragmentation. Caspase-3 activity in cells incubated for 18 h with various inducers of apoptosis was determined using a fluorometric caspase-3 assay kit (Sigma-Aldrich). DNA cleavage in cells incubated for 24 h with various inducers of apoptosis was assessed using a fragment end labeling kit (FragEL; Calbiochem). Percentage of fragment end-labeled cells was determined by flow cytometry (5 × 104 cells/sample).
Targeted Knockdown of eEF1A-1 Expression
Full-length mouse eEF1A-1 cDNA sequence was used to design siRNA template oligonucleotides using a Web-based insert design tool (www.ambion.com/techlib/misc/psilencer_converter.html). Hairpin siRNA template oligonucleotide sequences predicted to reduce eEF1A-1 expression were used to generate pSilencer 2.1-U6 vectors (Ambion, Austin, TX) that were transfected into CHO and H9c2 cells. Transfected cells were selected by growth in 0.5 mg/ml hygromycin for 5 d, and clonal lines were isolated by plating at limiting dilution. Target sequences that conferred efficient knockdown of protein expression in these stable populations (as determined by immunoblotting) were 5′-TATGCCTTGGTTCAAGGGA-3′ and 5′-CGTGTCTGTCAAAGATGTC-3′, corresponding to nucleotides 696-714 and 1017-1035 of the eEF1A-1 coding sequence, respectively. For each parental cell line (CHO and H9c2), a single control siRNA-expressing clone in which eEF1A-1 expression was not reduced (designated siRNA1) and two clones in which eEF1A-1 expression was reduced to different extents (siRNA2 and siRNA3) were maintained for further study.
Detection of Reactive Oxygen Species
Relative cellular levels of ROS were determined as described previously (Listenberger et al., 2001), with minor modifications. CHO and H9c2 cells were incubated for 5 h with palmitate-supplemented media. Subsequently, cells were washed with phosphate-buffered saline (PBS) and incubated for 30 min with PBS containing 0.5 mM MgCl2, 0.92 mM CaCl2, and 10 μM 6-carboxy-2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA). Cells were harvested, resuspended in culture media, and mean fluorescence was determined by flow cytometry (104 cells/sample).
Total Cellular Protein Synthesis
Synthesis of total cellular proteins was measured in CHO and mutant cells after 30-min incubations with 5 μCi/ml of [35S]methionine in the absence or presence of cycloheximide (10 μg/ml). Cells were lysed in radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris, 150 mM sodium chloride, 5 mM EDTA, 1% Nonidet P-40, and 0.5% sodium deoxycholate), and proteins were precipitated with 50% trichloroacetic acid (TCA). Precipitates were collected on glass filters, and label incorporation was quantified by scintillation counting and normalized for total cellular protein.
Fatty Acid Uptake
Initial rates of long chain FA uptake were determined under conditions of low FA availability (0.6 μM fatty acid and 20 μM BSA) using a BODIPY-labeled palmitate analogue, 4, 4 difluoro-5-methyl-4-bora-3a,4a-diaza-3-indacenedodecanoic acid (Molecular Probes, Eugene, OR), as described previously (Schaffer and Lodish, 1994). A similar assay was performed under lipotoxic conditions using 495 μM palmitate (complexed to BSA at a molar ratio of 4:1) and 5 μM BODIPY-labeled analogue.
Microscopy
Cellular actin distribution was assessed by cytochemical staining. Wild-type and mutant CHO cells were plated on glass coverslips, fixed with 4% formaldehyde, and permeabilized with Triton X-100 before staining with rhodamine phalloidin (Molecular Probes). Cells were counterstained with 4,6-diamidino-2-phenylindole (DAPI) nucleic acid stain (Molecular Probes). Fluorescence microscopy was performed and digital images obtained using a Zeiss Axioskop2 microscope, equipped with an AxioCamMR5 camera.
Immunoblot Analyses
Whole cell lysates were prepared using RIPA buffer. eEF1A-1 and GRP78 from 10 μg of whole cell lysate protein were resolved by 10% SDS-PAGE and detected using mouse monoclonal (CBP-KK1; Upstate Biotechnology, Lake Placid, NY) and rabbit polyclonal (StressGen Biotechnologies, San Diego, CA) antibodies, respectively. CHOP-10 (GADD153) from 50 μg of whole cell lysate protein was resolved by 12% SDS-PAGE and detected using a rabbit polyclonal antibody from Sigma-Aldrich. Subcellular fractions containing soluble (monomeric) and insoluble (polymerized) actin were prepared by sequential extraction with cytoskeletal stabilizing buffer (150 mM potassium chloride, 20 mM PIPES, 10 mM imidazole, 1 mM magnesium chloride, 1 mM EGTA, and 0.2 mM dithiothreitol) containing 0.05% Triton X-100 (Sigma-Aldrich) and RIPA buffer. Actin from 5 μg of protein from each fraction was detected using a rabbit polyclonal antibody from Sigma-Aldrich. Proteins were visualized using appropriate horseradish peroxidase-conjugated secondary antibodies and chemiluminescence reagents (PerkinElmer Life and Analytical Sciences, Boston, MA). Bands were quantitated by densitometry. The efficiency and consistency of protein transfer to nitrocellulose membranes for all blots were determined by staining with Ponceau S.
Mice
Mice overexpressing human long-chain acyl-CoA synthetase in the heart (MHC-ACS O7) were generated previously (Chiu et al., 2001) and maintained on a standard chow diet with 5% fat. Animals were treated in accordance with approved Washington University Institutional Animal Care and Use Committee protocols. Animals were killed by cervical dislocation at 12 wk of age. For analyses of protein expression, ventricular tissue was harvested, snap frozen in liquid nitrogen, and tissue homogenates were prepared in RIPA buffer. Homogenates were precleared with protein G, and eEF1A-1 and GRP78 levels were determined by SDS-PAGE and immunoblotting, as described above. Catalase and protein disulfide isomerase (PDI) from 40 and 50 μg of tissue homogenates, respectively, were resolved by 10% SDS-PAGE and detected using rabbit polyclonal antibodies from Calbiochem and StressGen Biotechnologies, respectively. Blots were stripped and reprobed with a rabbit polyclonal actin antibody (Sigma-Aldrich).
RESULTS
Disruption by Insertional Mutagenesis or Targeted Knockdown of eEF1A-1 in CHO Cells Confers Resistance to Palmitate
To identify proteins critical for lipotoxic cell death, we performed a genetic screen in CHO cells. These cells are susceptible to palmitate-induced cell death (Listenberger et al., 2001) and are functionally hemizygous at many loci (Siminovitch, 1985), such that a single mutation is often sufficient to generate a recessive phenotype. VSV-G pseudotyped retrovirus encoding the ROSAβgeo promoter trap (Friedrich and Soriano, 1991) was titered and used at low multiplicity of infection to achieve an average of less than one retroviral integration per 10 cells. To isolate palmitate-resistant mutants, ROSAβgeo-trapped cells were selected in media containing 500 μM palmitate, conditions under which wild-type CHO cells were killed. Sequence obtained by 5′RACE of cDNA from a palmitate-resistant clone isolated through this screen corresponded to a portion of the 5′-untranslated region (UTR) of hamster eEF1A-1 (Figure 1A). Specific disruption of eEF1A-1 by retroviral insertion was confirmed in by directed PCR using primers for eEF1A-1, the closely related isoform eEF1A-2, and ROSAβgeo sequences (Figure 1B). A product was obtained only in wild-type CHO cDNA using forward and reverse primers for eEF1A-1, whereas products were obtained in both wild-type CHO and mutant cDNA using forward and reverse primers for eEF1A-2. A product was obtained only in mutant cDNA using forward primer for eEF1A-1 and reverse primer for ROSAβgeo. Thus, ROSAβgeo-mediated disruption occurred only in the eEF1A-1 gene, and only in the mutant cell line. Furthermore, eEF1A-1 mRNA and protein expression were reduced, as determined by Northern and immunoblotting (Figure 1, C and D). Residual bands detected by Northern and Western blotting in mutant cells are due to recognition of hamster eEF1A-2 by the Northern probe and monoclonal antibody, because eEF1A-1 and eEF1A-2 are 92% identical in amino acid sequence (Kahns et al., 1998).
Figure 1.

eEF1A-1 expression is disrupted in mutant CHO cells. (A) CHO sequence upstream of the retroviral insertion was obtained by 5′RACE of mutant cDNA (shaded) and corresponds to bases 1-41 of the 5′UTR of hamster eEF1A-1. Rat, mouse, and human sequences are aligned below. (B) Directed PCR for eEF1A-1 and eEF1A-2 expression in wild-type CHO and mutant cDNA. Control reactions contained no cDNA (lanes 5 and 6). Lanes 1, 3, and 5 include forward (F) and reverse (R) primers for either eEF1A-1 (top) or eEF1A-2 (bottom). Lanes 2, 4, and 6 include forward primers for either eEF1A-1 (top) or eEF1A-2 (bottom) and reverse primers for the retroviral sequence (ROSAβgeo). (C and D) eEF1A-1 expression in wild-type CHO and mutant cells was assessed by Northern blotting (C) and immunoblotting (D) using three independent RNA and cell lysate preparations.
To determine whether disruption of eEF1A-1 conferred selective resistance to palmitate, we assessed cell death and apoptosis in wild-type CHO and mutant cells in response to palmitate, staurosporine, actinomycin D, cycloheximide, and camptothecin. Mutant cells were markedly resistant to palmitate-induced cell death, but they were not resistant to the other inducers of apoptosis (Figure 2A). Moreover, mutant cells were selectively resistant to palmitate-induced apoptosis, as demonstrated by DNA fragmentation assays (Figure 2B) and caspase-3 activation (Figure 1S). Thus, the mutant cell line was not generally transformed or impaired in its ability to undergo apoptosis and cell death.
Figure 2.

Disruption by insertional mutagenesis or targeted knockdown of eEF1A-1 in CHO cells confers palmitate-resistance. (A) Wild-type and eEF1A-1 mutant CHO cells were incubated with 500 μM palmitate (Palm), 80 nM staurosporine (Staur), 2 μM actinomycin D (Act D), 20 μM cycloheximde (Cyclo), or 10 μM camptothecin (Camp) for 24 h. Cell death was determined by propidium iodide staining and flow cytometry. (B) Cells were incubated as described in A. Apoptosis was determined by FragEL and flow cytometry. (C) Basal eEF1A-1 protein levels in whole cell lysates from wild-type CHO, eEF1A-1 mutant CHO, and stable CHO-derived cell lines expressing control siRNA (siRNA1) or siRNA directed against eEF1A-1 (siRNA2 and siRNA3) were detected by immunoblotting and quantified by densitometry. Inset is a representative blot. (D) Cell lines from C were incubated for 12 h with 500 μM palmitate. Cell death was determined by propidium iodide staining and flow cytometry. All data expressed as mean ± SEM for five independent experiments, *p < 0.05.
To confirm the involvement of eEF1A-1 in palmitate-induced cell death, we generated stable CHO cell lines expressing siRNA directed against eEF1A-1. Although the knockdown of eEF1A-1 expression achieved in CHO-derived cell lines was modest (14-20% in siRNA2 and siRNA3) (Figure 2C), resistance to palmitate corresponded to the extent of knockdown of basal eEF1A-1 protein levels (Figure 2D). Thus, targeted knockdown of eEF1A-1 produces a palmitate-resistant phenotype similar to our CHO cell mutant generated by random insertional mutagenesis, providing independent confirmation of the involvement of this protein in palmitate-induced cell death.
Targeted Knockdown of eEF1A-1 in H9c2 Rat Cardiomyoblasts Confers Resistance to Palmitate
To extend these findings to a cell type affected by lipotoxicity in metabolic diseases, we investigated the role of eEF1A-1 in palmitate toxicity in H9c2 rat cardiomyoblasts (undifferentiated) and cardiomyocyte-like cultures (differentiated). H9c2 cardiomyoblasts generated ROS within 5 h (Figure 3A) and exhibited increased DNA fragmentation and cell death at 24 h (Figure 3, B and C) in response to increasing doses of palmitate. Similar trends were observed in differentiated H9c2 cells (Figure 2S). Thus, we generated stable H9c2 cardiomyoblast lines expressing siRNA directed against eEF1A-1 (Figure 3D). More robust reductions in eEF1A-1 expression were achieved in H9c2-derived cell lines (56-67% in siRNA2 and siRNA3) than in CHO-derived cell lines, and this greater degree of knockdown was accompanied by greater resistance to palmitate-induced death (Figure 3E). The extent of reduction in basal eEF1A-1 corresponded to the extent to which the slope of the palmitate dose-response relationship was reduced (Figure 3S). Together, data from CHO- and H9c2-derived cell lines with targeted knockdown of eEF1A-1 demonstrate a strong, positive correlation between basal eEF1A-1 expression and susceptibility to palmitate-induced cell death (Figure 3F).
Figure 3.

Targeted knockdown of eEF1A-1 expression confers palmitate-resistance in H9c2 rat cardiomyoblasts. (A) Myoblasts were incubated for 5 h with palmitate, followed by 30-min incubation with H2DCFDA. Mean DCF fluorescence, indicative of relative cellular ROS level, was measured by flow cytometry. (B) Cells were incubated for 24 h with palmitate. Apoptosis was determined by FragEL and flow cytometry. (C) Cells were incubated as described in B, and cell death was determined by propidium iodide staining and flow cytometry. (D) Basal eEF1A-1 protein levels in whole cell lysates from wild-type H9c2 myoblast and stable H9c2-derived cell lines expressing control siRNA (siRNA1) or siRNA directed against eEF1A-1 (siRNA2 and siRNA3) were detected by immunoblotting and quantified by densitometry. Inset is a representative blot. (E) Cell lines from D were incubated for 24 h with 500 μM palmitate. Cell death was determined by propidium iodide staining and flow cytometry. (F) Basal eEF1A-1 expression plotted against cell death in response to 500 μM palmitate for CHO-derived lines (wild type, mutant, and siRNA, closed symbols) and for H9c2-derived cell lines (wild type and siRNA, open symbols). All data expressed as mean ± SEM for five independent experiments, *p < 0.05.
eEF1A-1 Expression Is Rapidly Induced in Response to Palmitate and Oxidative Stress
Previous studies have demonstrated a role for eEF1A-1 in the apoptotic response to oxidative stress (Chen et al., 2000), and we reported previously that palmitate generates ROS in CHO cells, leading to apoptosis and cell death (Listenberger et al., 2001). Thus, we predicted that our mutant CHO cells might also be resistant to H2O2-induced cell death. eEF1A-1 null CHO cells exhibited significantly elevated basal ROS levels, which did not increase further after 5 h incubation with palmitate (Figure 4S). This likely reflects adaptive changes in the antioxidant and/or radical scavenging capacity of these cells. However, mutant cells were markedly resistant to cell death after 24-h incubation with 2.5 mM H2O2 (Figure 4A), consistent with previous data showing that H9c2 cells transfected with antisense eEF1A-1 cDNA are protected from oxidative stress-induced death (Chen et al., 2000). In addition, eEF1A-1 protein was rapidly induced and remained elevated in CHO and H9c2 cells in response to palmitate (Figure 4, B and D) or H2O2 (Figure 4C). However, the residual, nonspecific band detected in eEF1A-1 null mutant CHO cells decreased in response to palmitate (Figure 5S). These data are consistent with studies showing that under conditions of cell stress, eEF1A-1 protein increases posttranscriptionally to expedite apoptosis (Chen et al., 2000), whereas eEF1A-2, which promotes cell survival, decreases (Ruest et al., 2002). Although the magnitude of increase in eEF1A-1 expression in response to palmitate was modest (35%), eEF1A-1 is abundant in normal growing cells, making up 1-2% of total cellular protein (Condeelis, 1995), and small increases in relative eEF1A-1 protein levels constitute the production of large absolute quantities of this protein. Inhibition of eEF1A-1 expression using siRNA abrogated cell death in H9c2 myoblasts by preventing the normal, toxic induction of this protein in response to palmitate (Figure 6S). In summary, the induction of eEF1A-1 observed in response to palmitate or H2O2 is consistent with a role for this protein in mediating lipotoxic cell death secondary to oxidative stress.
Figure 4.

eEF1A-1 expression is rapidly induced in response to palmitate and oxidative stress. (A) Wild-type CHO and mutant cells were incubated for 24 h with 2.5 mM H2O2 and cell death was quantified by propidium iodide staining and flow cytometry. (B) Wild-type CHO cells were incubated with 500 μM palmitate, followed by detection of eEF1A-1 protein levels in whole cell lysates by immunoblotting. (C) Wild-type CHO cells were incubated with 2.5 mM H2O2, followed by detection of eEF1A-1 protein levels in whole cell lysates by immunoblotting. (D) H9c2 cardiomyoblasts were incubated as described in B, followed by detection of eEF1A-1 protein levels in whole cell lysates by immunoblotting. Insets are representative blots. All data expressed as mean ± SEM for five independent experiments, *p < 0.05.
eEF1A-1 Mediates Cell Death in Response to Palmitate-induced ROS Generation and ER Stress
Oxidative stress can lead to ER stress and cell death (Scorrano et al., 2003; Oyadomari and Mori, 2004; Rao et al., 2004), and ER stress has been implicated in the pathogenesis of metabolic diseases (Feng et al., 2003; Ozcan et al., 2004). Because palmitate can induce cell death through the generation of ROS, we investigated whether eEF1A-1 mediates lipotoxic cell death in response to ROS-induced ER stress in cardiomyoblasts. Treatment with palmitate for 5 h induced expression of the protein-folding chaperone glucose-regulated protein 78 (GRP78), and the transcription factor C/EBP-homologous protein CHOP-10, both markers of ER stress (Figure 5A). These changes occurred before cell death and concomitant with increases in cellular ROS and eEF1A-1 expression (Figure 5, A and B). Preincubation with 200 μM α-tocopherol prevented palmitate-induced cellular ROS accumulation (Figure 5B); blunted palmitate-induced increases in GRP78, CHOP-10, and eEF1A-1 protein (Figure 5A); and decreased cell death at 24 h (Figure 5C), suggesting that palmitate induces ER stress and cell death through the generation of ROS. In eEF1A-1 null CHO cells, GRP78 increased to the same extent as in wild-type CHO cells in response to palmitate (Figure 5D), indicating that the induction of an ER stress response was not impaired by disruption of eEF1A-1. However, knockdown of eEF1A-1 in H9c2 cardiomyoblasts (Figure 5E) and CHO cells (Figure 7S) conferred resistance to cell death by the established ER stress inducers tunicamycin and thapsigargin, consistent with a role for eEF1A-1 in mediating ER stress-induced cell death.
Figure 5.

Palmitate-induced ROS activate an ER stress response and induce eEF1A-1 expression and cell death in cardiomyoblasts. (A) Myoblasts were incubated for 1 h with ethanol (vehicle control) or 200 μM α-tocopherol, followed by 5 h with 500 μM palmitate. Expression of ER stress response proteins (GRP78 and CHOP-10) and eEF1A-1 were detected in whole cell lysates by immunoblotting. Cells incubated for 5 h with 2.5 μg/ml tunicamycin were included as a positive control. Relative densitometry values for the representative blots shown are given below each band. Open arrow indicates a nonspecific protein band. (B) Cells were incubated as described in A, followed by incubation for 30 min with H2DCFDA. Mean DCF fluorescence, indicative of relative cellular ROS level, was measured by flow cytometry. (C) Cells preincubated as described in A were incubated for 24 h with or without 500 μM palmitate. Cell death was determined by propidium iodide staining and flow cytometry. (D) Wild-type (closed symbols) and eEF1A-1 null mutant (open symbols) CHO cells were incubated with 500 μM palmitate. GRP78 protein levels in whole cell lysates were detected by immunoblotting. Insets are representative blots. (E) Wild-type H9c2 myoblast and stable H9c2-derived cell lines expressing control siRNA (siRNA1) or siRNA directed against eEF1A-1 (siRNA2 and siRNA3) were incubated for 48 h with either 2.5 μg/ml tunicamycin or 1 μM thapsigargin. Cell death was determined by propidium iodide staining and flow cytometry. For B-E, data expressed as mean ± SEM for at least three independent experiments, * p < 0.05.
Disruption of eEF1A-1 Prevents Palmitate-induced Changes in the Actin Cytoskeleton Preceding Cell Death
We next investigated the mechanism through which eEF1A-1 mediates palmitate-induced cell death. Because the canonical function of eEF1A-1 is to recruit aa-tRNA to the ribosome during peptide elongation (McKeehan and Hardesty, 1969), and the synthesis of new proteins is required for apoptosis, we quantified overall protein synthesis in eEF1A-1-deficient CHO cells. Mutant cells showed no change in rates of [35S]methionine incorporation into TCA-precipitable cellular proteins compared with wild-type cells (Figure 6A), suggesting that eEF1A-2 can compensate for eEF1A-1 in peptide elongation (Kahns et al., 1998) and that diminished overall protein synthesis is not the mechanism for palmitate resistance.
Figure 6.

Disruption of eEF1A-1 prevents palmitate-induced changes in the actin cytoskeleton preceding cell death. (A) To assess total protein synthesis, wild-type CHO and mutant cells were pulsed for 30 min with [35S]methionine, and TCA-precipitable proteins were collected. Radiolabel incorporation was quantified by scintillation counting and normalized to total cellular protein. (B) Initial uptake of BODIPY-labeled palmitate analogue was measured in wild-type CHO and mutant cells under conditions of either low (0.6 μM) or lipotoxic (500 μM) free FA availability. Mean fluorescence for each cell type was measured by flow cytometry. (C-F) Changes in actin cytoskeleton distribution (red) were assessed by rhodamine-phalloidin cytochemical staining and fluorescence microscopy of wild-type CHO cells under basal conditions (C) and after 5-h incubation with palmitate (D). Identical experiments were performed in eEF1A-1-deficient mutant CHO cells under basal conditions (E) and after 5-h palmitate treatment (F). Cells were counterstained with DAPI (blue). Bars, 20 μm. (G and H) Subcellular fractions containing soluble actin (G) and insoluble actin (H) were isolated from wild-type and mutant CHO cells under basal conditions and after treatment with palmitate for 5 h. Actin levels were assessed by immunoblotting and quantitated by densitometry. Insets are representative blots. Data expressed as mean ± SEM for four independent experiments, *p < 0.05.
eEF1A-1 also plays an important role in regulating the length and stability of cytoskeletal components, including actin filaments (Shiina et al., 1994; Murray et al., 1996). Thus, disruption of this protein might affect membrane events, such as FA uptake, or cytoskeletal rearrangements involved in the progression of cell death. eEF1A-1-deficient mutant CHO cells showed no defect in initial rates of FA uptake under conditions of low (physiological) FA availability or under conditions that simulated the lipotoxic levels of FA availability in our cell culture experiments (Figure 6B). However, under basal conditions, mutant cells contained 40% more insoluble (polymerized) actin than wild-type CHO cells (Figure 6, C, E, and H). Treatment with palmitate increased polymerized actin content of wild-type CHO cells by 17%. This effect on actin distribution is similar to changes observed in response to H2O2 before cell death (Dalle-Donne et al., 2001; Zhu et al., 2005). However, actin distribution remained unchanged in mutant cells (Figure 6, C-H). Our data are consistent with the induction of oxidative stress during FA overload and suggest that eEF1A-1 mediates the lipotoxic cell death response by regulating cytoskeletal changes during cell death.
eEF1A-1 and Markers of Oxidative and ER Stress Are Induced in a Mouse Model of Lipotoxic Cardiomyopathy
To determine whether oxidative stress, ER stress, and eEF1A-1 are involved in lipotoxic cardiomyocyte cell death in vivo, we assessed expression of the antioxidant enzyme catalase (Smith et al., 2005), the ER chaperones GRP78 and PDI, and eEF1A-1 in a mouse model of cardiac-specific lipotoxicity. We previously generated a transgenic mouse line with cardiac-restricted overexpression of ACS1 (MHC-ACS O7) (Chiu et al., 2001), a protein that catalyzes the esterification of long chain FFA to CoA, thereby facilitating uptake. These mice exhibit cardiomyocyte lipid accumulation leading to cardiomyocyte death, heart failure, and premature death. In ventricular tissue homogenates from 12-wk-old transgenic mice, catalase, GRP78, PDI, and eEF1A-1 protein levels were increased 2.6-, 2.5-, 6.2-, and 2.8-fold, respectively, compared with wild type (Figure 7, A and B). Total actin levels in wild-type versus transgenic ventricular tissue homogenates were not significantly different (Figure 7A). These data further support a model in which accumulation of excess lipid in the heart leads to oxidative and ER stress, followed by induction of eEF1A-1 that contributes to cardiomyocyte cell loss and cardiomyopathy.
Figure 7.
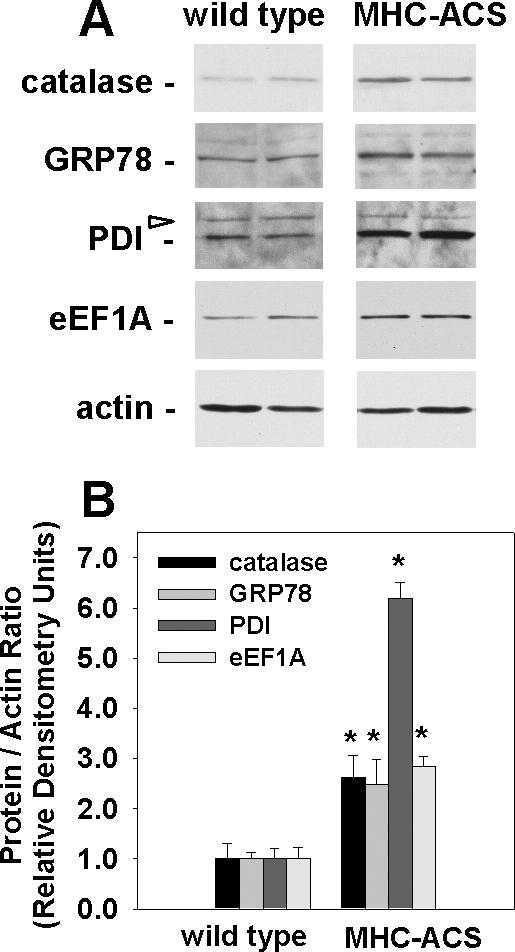
eEF1A-1 and markers of oxidative and ER stress are increased in lipotoxic cardiac tissue. (A) Representative immunoblotting of eEF1A-1, catalase, ER chaperone proteins (GRP78 and PDI), and actin in whole tissue homogenates from ventricles of 12-wk-old wild-type or MHC-ACS mice. Blots are representative of the four animals included in each group. Open arrow indicates nonspecific protein bands. (B) Densitometry of immunoblotting, as shown in A. Data expressed as mean of the protein to actin ratio ± SEM for four animals in each group, *p < 0.05.
DISCUSSION
Lipotoxicity has been proposed to play a role in the development of heart failure associated with diabetes and obesity. However, the molecular and cellular mechanisms of FA-induced cell death are not well understood. Here, we used the ROSAβgeo promoter trap in a novel mutagenic screen in CHO cells to identify eEF1A-1 as a key mediator of lipotoxic cell death. Our findings are consistent with a model in which eEF1A-1 mediates lipotoxicity secondary to oxidative and ER stress by regulating actin cytoskeletal changes during cell death. We extended our study to cardiomyoblasts and a murine model of lipotoxic cardiomyopathy, pointing to a role for this protein in the pathophysiology of metabolic heart disease.
eEF1A-1, originally identified as the factor required for GTP-dependent recruitment of aa-tRNA to the ribosome during peptide elongation (McKeehan and Hardesty, 1969), has since been shown to function in diverse cellular processes (Ejiri, 2002), including the cytotoxic response to oxidative stress in cardiomyoblasts (Chen et al., 2000). We show here that in both CHO and H9c2 cells, palmitate rapidly induces eEF1A-1 protein and that basal eEF1A-1 expression dictates sensitivity to palmitate-induced cell death. eEF1A-1 may mediate specific changes in protein translation required for palmitate-induced apoptosis and cell death. However, cellular levels of eEF1A-1 are not rate limiting for protein synthesis (Condeelis, 1995), and our eEF1A-1 null mutant CHO cells do not exhibit reduced total protein synthesis, suggesting that this protein mediates lipotoxic cell death through a mechanism independent of changes in overall rates of protein synthesis. In contrast, because eEF1A-1 plays an important role in remodeling microtubules and filamentous actin (Shiina et al., 1994; Murray et al., 1996) and because the cytoskeleton undergoes dramatic changes during apoptosis and cell death, eEF1A-1 may mediate cytoskeletal changes required to execute cell death programs in response to lipotoxic conditions. Our observation that disruption of eEF1A-1 results in actin cytoskeleton defects under basal conditions and in response to palmitate is consistent with a role for eEF1A-1 in regulating FA overload-induced changes in the actin cytoskeleton important for the progression of cell death. Although palmitate-induced death in primary ventricular cardiomyocytes is accompanied by degeneration of myofibrils (Dyntar et al., 2001), our observation of eEF1A-1-dependent palmitate-induced death in cell types devoid of myofibrils suggests that this response, in contrast to actin remodeling, is not central to the function of eEF1A-1 in palmitate-induced cell death.
ROS generation is likely a principal signal for cell death in response to lipotoxic FA concentrations. Our previous studies in CHO cells demonstrated that reactive intermediates are generated in response to palmitate supplementation (Listenberger et al., 2001, 2003). In the present study, we show that, in cardiomyoblasts, palmitate supplementation leads to ROS generation before evidence of cell death. Scavenging palmitate-induced ROS by preincubation with α-tocopherol, a known antioxidant, reduced cell death at 24 h after palmitate supplementation. ROS generation has been similarly implicated in lipotoxicity in cultured pancreatic β-cells (Shimabukuro et al., 1997), endothelial cells (Inoguchi et al., 2000), and skeletal muscle cells (Cabrero et al., 2002) as well as in animal models of lipotoxic injury to the liver (Browning and Horton, 2004) and heart (Finck et al., 2003). The source of ROS generated during lipotoxicity is controversial. Recent evidence, from a variety of cell types, suggests that processes other than mitochondrial β-oxidation, such as NADPH oxidase activity, may be involved (Inoguchi et al., 2003; Quagliaro et al., 2003; Cacicedo et al., 2005).
Because oxidative stress is closely linked to ER stress (Oyadomari and Mori, 2004), we hypothesized that eEF1A-1 mediates lipotoxic cell death in response ROS-induced ER stress in cardiomyocytes. Oxidative stress-induced apoptosis requires the release of ER calcium ions (Scorrano et al., 2003), and depletion of these stores can impair normal protein-folding functions, leading to ER stress (Rao et al., 2004; Rutkowski and Kaufman, 2004). The ER responds by activating specific pathways, including the unfolded protein response (UPR). Initially, the UPR involves inactivation of the translation initiation factor eIF2α, through its phosphorylation by an ER resident kinase (PERK), and increased expression of protein folding chaperones, such as GRP78 and PDI, in a combined attempt to reduce the frequency of translation initiation and relieve the ER of its burden of unfolded proteins. However, when ER function is severely impaired, apoptosis, mediated by the transcription factor CHOP-10, ensues (Rao et al., 2004). Here we demonstrate, in H9c2 cells, that palmitate increased cellular ROS and induced GRP78, CHOP-10, and eEF1A-1 expression at 5 h, resulting in cell death at 24 h. These effects were abrogated by preincubation with α-tocopherol. Our data suggest that eEF1A-1 mediates lipotoxic cell death secondary to ROS-induced ER stress (Figure 8). However, we cannot exclude the possibility that esterification of palmitate as it enters the cell may also directly induce ER stress, because in vitro evidence suggests that palmitoyl CoA may facilitate ER fission (Turner, 2004). This could lead to the release of ROS generated through oxidative protein folding (Harding et al., 2003), triggering apoptosis. Our data showing that disruption or knockdown of eEF1A-1 confers resistance to cell death by tunicamycin or thapsigargin suggests that eEF1A-1 acts downstream of induction of ER stress and thus is relatively distal in the pathway leading to cell death in the presence of palmitate. Together with the observation that ER stress is central to cholesterol-induced apoptosis in macrophages (Feng et al., 2003), our results are consistent with a general model in which perturbations of lipid metabolism can result in cell death mediated via the ER.
Figure 8.

Role of eEF1A-1 in palmitate-induced cell death. Under conditions of FA overload in nonadipose tissues, the cellular capacity to store FAs as triglycerides or to use FAs for energy is overwhelmed. This FFA overload can lead to the production of ROS, which can, in turn, induce ER stress. Prolonged or severe ER stress, which may occur in the presence of excess palmitate, can lead to further ROS accumulation, potentially amplifying the apoptotic/cell death response. Palmitoyl CoA, generated by esterification of palmitate as it enters the cell, may also induce ER stress directly, leading to the production and accumulation of ROS and subsequent apoptosis/cell death. In either of these scenarios, eEF1A-1 mediates actin cytoskeleton changes involved in the progression of the lipotoxic cell death response downstream of induction of oxidative and ER stress.
Oxidative and ER stress have recently been linked to the pathogenesis of several diseases, including insulin resistance and type 2 diabetes (Ozcan et al., 2004; Nakatani et al., 2005). Although glucotoxicity has been implicated in oxidative stress in diabetes (Brownlee, 2003), rodent models of poorly controlled diabetes are characterized by pleiotropic metabolic abnormalities, including high serum levels of both glucose and FAs. Our observations in cells and in MHC-ACS mice, a model of cardiac-specific lipotoxicity, indicate for the first time that increased FA uptake alone is associated with ER stress and cell death. Thus, elevated serum FAs in the metabolic syndrome and diabetes may play a central role in the pathogenesis of these disorders through the induction of oxidative and ER stress. Furthermore, a scheme in which excess FAs lead to oxidative and ER stress, followed by induction of eEF1A-1 that regulates cytoskeletal changes leading to cell death, may explain the increases in eEF1A-1 expression observed in skeletal muscle (Reynet and Kahn, 2001) and in renal cortex (Al-Maghrebi et al., 2004) from diabetic animals and humans. This cellular response may play a fundamental role in the genesis of complications of diabetes and the metabolic syndrome.
Supplementary Material
Acknowledgments
We thank Sarah Lewis and Jeff Harp for technical assistance. This work was supported by grants from the National Institutes of Health (DK064989 to J.E.S.) and the Washington University School of Medicine-Pharmacia Biomedical Program (to J.E.S.). N.M.B. is the recipient of a Research Fellowship from the Heart and Stroke Foundation of Canada.
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E05-08-0742) on November 30, 2005.
The online version of this article contains supplemental material at MBC Online (http://www.molbiolcell.org).
References
- Aasum, E., Belke, D. D., Severson, D. L., Riemersma, R. A., Cooper, M., Andreassen, M., and Larsen, T. S. (2002). Cardiac function and metabolism in Type 2 diabetic mice after treatment with BM 17.0744, a novel PPAR-alpha activator. Am. J. Physiol. 283, H949-H957. [DOI] [PubMed] [Google Scholar]
- Al-Maghrebi, M., Cojocel, C., and Thompson, M. (2004). Regulation of elongation factor-1 mRNA levels by vitamin E in diabetic rat kidneys. FASEB J. 18, C46. [DOI] [PubMed] [Google Scholar]
- Browning, J. D., and Horton, J. D. (2004). Molecular mediators of hepatic steatosis and liver injury. J. Clin. Investig. 114, 147-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brownlee, M. (2003). A radical explanation for glucose-induced beta cell dysfunction. J. Clin. Investig. 112, 1788-1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabrero, A., Alegret, M., Sanchez, R. M., Adzet, T., Laguna, J. C., and Carrera, M. V. (2002). Increased reactive oxygen species production down-regulates peroxisome proliferator-activated alpha pathway in C2C12 skeletal muscle cells. J. Biol. Chem. 277, 10100-10107. [DOI] [PubMed] [Google Scholar]
- Cacicedo, J. M., Benjachareowong, S., Chou, E., Ruderman, N. B., and Ido, Y. (2005). Palmitate-induced apoptosis in cultured bovine retinal pericytes: roles of NAD(P)H oxidase, oxidant stress, and ceramide. Diabetes 54, 1838-1845. [DOI] [PubMed] [Google Scholar]
- Chen, E., Proestou, G., Bourbeau, D., and Wang, E. (2000). Rapid up-regulation of peptide elongation factor EF-1alpha protein levels is an immediate early event during oxidative stress-induced apoptosis. Exp. Cell Res. 259, 140-148. [DOI] [PubMed] [Google Scholar]
- Chiu, H. C., et al. (2005). Transgenic expression of fatty acid transport protein 1 in the heart causes lipotoxic cardiomyopathy. Circ. Res. 96, 225-233. [DOI] [PubMed] [Google Scholar]
- Chiu, H. C., Kovacs, A., Ford, D. A., Hsu, F. F., Garcia, R., Herrero, P., Saffitz, J. E., and Schaffer, J. E. (2001). A novel mouse model of lipotoxic cardiomyopathy. J. Clin. Investig. 107, 813-822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christoffersen, C., Bollano, E., Lindegaard, M. L., Bartels, E. D., Goetze, J. P., Andersen, C. B., and Nielsen, L. B. (2003). Cardiac lipid accumulation associated with diastolic dysfunction in obese mice. Endocrinology 144, 3483-3490. [DOI] [PubMed] [Google Scholar]
- Condeelis, J. (1995). Elongation factor 1 alpha, translation and the cytoskeleton. Trends Biochem. Sci. 20, 169-170. [DOI] [PubMed] [Google Scholar]
- Dalle-Donne, I., Rossi, R., Milzani, A., Di Simplicio, P., and Colombo, R. (2001). The actin cytoskeleton response to oxidants: from small heat shock protein phosphorylation to changes in the redox state of actin itself. Free Radic. Biol. Med. 31, 1624-1632. [DOI] [PubMed] [Google Scholar]
- Dyntar, D., Eppenberger-Eberhardt, M., Maedler, K., Pruschy, M., Eppenberger, H. M., Spinas, G. A., and Donath, M. Y. (2001). Glucose and palmitic acid induce degeneration of myofibrils and modulate apoptosis in rat adult cardiomyocytes. Diabetes 50, 2105-2113. [DOI] [PubMed] [Google Scholar]
- Ejiri, S. (2002). Moonlighting functions of polypeptide elongation factor 1, from actin bundling to zinc finger protein R1-associated nuclear localization. Biosci. Biotechnol. Biochem. 66, 1-21. [DOI] [PubMed] [Google Scholar]
- Feng, B., et al. (2003). The endoplasmic reticulum is the site of cholesterol-induced cytotoxicity in macrophages. Nat. Cell Biol. 5, 781-792. [DOI] [PubMed] [Google Scholar]
- Finck, B. N., Han, X., Courtois, M., Aimond, F., Nerbonne, J. M., Kovacs, A., Gross, R. W., and Kelly, D. P. (2003). A critical role for PPARalpha-mediated lipotoxicity in the pathogenesis of diabetic cardiomyopathy: modulation by dietary fat content. Proc. Natl. Acad. Sci. USA 100, 1226-1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedrich, G., and Soriano, P. (1991). Promoter traps in embryonic stem cells: a genetic screen to identify and mutate developmental genes in mice. Genes Dev. 5, 1513-1523. [DOI] [PubMed] [Google Scholar]
- Harding, H. P., et al. (2003). An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 11, 619-633. [DOI] [PubMed] [Google Scholar]
- Inoguchi, T., et al. (2000). High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C (PKC)-dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes 49, 1939-1945. [DOI] [PubMed] [Google Scholar]
- Inoguchi, T., et al. (2003). A possible target of antioxidative therapy for diabetic vascular complications-vascular NAD(P)H oxidase. Curr. Med. Chem. 10, 1759-1764. [DOI] [PubMed] [Google Scholar]
- Kahns, S., Lund, A., Kristensen, P., Knudsen, C. R., Clark, B. F., Cavallius, J., and Merrick, W. C. (1998). The elongation factor 1 A-2 isoform from rabbit: cloning of the cDNA and characterization of the protein. Nucleic Acids Res. 26, 1884-1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Listenberger, L. L., Han, X., Lewis, S. E., Cases, S., Farese, R. V., Jr., Ory, D. S., and Schaffer, J. E. (2003). Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc. Natl. Acad. Sci. USA 100, 3077-3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Listenberger, L. L., Ory, D. S., and Schaffer, J. E. (2001). Palmitate-induced apoptosis can occur through a ceramide-independent pathway. J. Biol. Chem. 276, 14890-14895. [DOI] [PubMed] [Google Scholar]
- McKeehan, W. L., and Hardesty, B. (1969). Purification and partial characterization of the aminoacyl transfer ribonucleic acid binding enzyme from rabbit reticulocytes. J. Biol. Chem. 244, 4330-4339. [PubMed] [Google Scholar]
- Menard, C., Pupier, S., Mornet, D., Kitzmann, M., Nargeot, J., and Lory, P. (1999). Modulation of L-type calcium channel expression during retinoic acid-induced differentiation of H9C2 cardiac cells. J. Biol. Chem. 274, 29063-29070. [DOI] [PubMed] [Google Scholar]
- Murray, J. W., Edmonds, B. T., Liu, G., and Condeelis, J. (1996). Bundling of actin filaments by elongation factor 1 alpha inhibits polymerization at filament ends. J. Cell Biol. 135, 1309-1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatani, Y., et al. (2005). Involvement of endoplasmic reticulum stress in insulin resistance and diabetes. J. Biol. Chem. 280, 847-851. [DOI] [PubMed] [Google Scholar]
- Ory, D. S., Neugeboren, B. A., and Mulligan, R. C. (1996). A stable human-derived packaging cell line for production of high titer retrovirus/vesicular stomatitis virus G pseudotypes. Proc. Natl. Acad. Sci. USA 93, 11400-11406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrander, D. B., Sparagna, G. C., Amoscato, A. A., McMillin, J. B., and Dowhan, W. (2001). Decreased cardiolipin synthesis corresponds with cytochrome c release in palmitate-induced cardiomyocyte apoptosis. J. Biol. Chem. 276, 38061-38067. [DOI] [PubMed] [Google Scholar]
- Oyadomari, S., and Mori, M. (2004). Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 11, 381-389. [DOI] [PubMed] [Google Scholar]
- Ozcan, U., Cao, Q., Yilmaz, E., Lee, A. H., Iwakoshi, N. N., Ozdelen, E., Tuncman, G., Gorgun, C., Glimcher, L. H., and Hotamisligil, G. S. (2004). Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306, 457-461. [DOI] [PubMed] [Google Scholar]
- Quagliaro, L., Piconi, L., Assaloni, R., Martinelli, L., Motz, E., and Ceriello, A. (2003). Intermittent high glucose enhances apoptosis related to oxidative stress in human umbilical vein endothelial cells: the role of PKC and NAD(P)H-oxidase activation. Diabetes 52, 2795-2804. [DOI] [PubMed] [Google Scholar]
- Rao, R. V., Ellerby, H. M., and Bredesen, D. E. (2004). Coupling endoplasmic reticulum stress to the cell death program. Cell Death Differ. 11, 372-380. [DOI] [PubMed] [Google Scholar]
- Reynet, C., and Kahn, C. R. (2001). Unbalanced expression of the different subunits of elongation factor 1 in diabetic skeletal muscle. Proc. Natl. Acad. Sci. USA 98, 3422-3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruest, L. B., Marcotte, R., and Wang, E. (2002). Peptide elongation factor eEF1A-2/S1 expression in cultured differentiated myotubes and its protective effect against caspase-3-mediated apoptosis. J. Biol. Chem. 277, 5418-5425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutkowski, D. T., and Kaufman, R. J. (2004). A trip to the ER: coping with stress. Trends Cell Biol. 14, 20-28. [DOI] [PubMed] [Google Scholar]
- Schaffer, J. E., and Lodish, H. F. (1994). Expression cloning and characterization of a novel adipocyte long chain fatty acid transport protein. Cell 79, 427-436. [DOI] [PubMed] [Google Scholar]
- Scorrano, L., Oakes, S. A., Opferman, J. T., Cheng, E. H., Sorcinelli, M. D., Pozzan, T., and Korsmeyer, S. J. (2003). BAX and BAK regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science 300, 135-139. [DOI] [PubMed] [Google Scholar]
- Sharma, S., Adrogue, J. V., Golfman, L., Uray, I., Lemm, J., Youker, K., Noon, G. P., Frazier, O. H., and Taegtmeyer, H. (2004). Intramyocardial lipid accumulation in the failing human heart resembles the lipotoxic rat heart. FASEB J. 18, 1692-1700. [DOI] [PubMed] [Google Scholar]
- Shiina, N., Gotoh, Y., Kubomura, N., Iwamatsu, A., and Nishida, E. (1994). Microtubule severing by elongation factor 1 alpha. Science 266, 282-285. [DOI] [PubMed] [Google Scholar]
- Shimabukuro, M., Higa, M., Zhou, Y. T., Wang, M. Y., Newgard, C. B., and Unger, R. H. (1998). Lipoapoptosis in beta-cells of obese prediabetic fa/fa rats. Role of serine palmitoyltransferase overexpression. J. Biol. Chem. 273, 32487-32490. [DOI] [PubMed] [Google Scholar]
- Shimabukuro, M., Ohneda, M., Lee, Y., and Unger, R. H. (1997). Role of nitric oxide in obesity-induced beta cell disease. J. Clin. Investig. 100, 290-295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siminovitch, L. (1985). Mechanisms of genetic variation in Chinese hamster ovary cells. In: Molecular Cell Genetics, ed. M. M. Gottesman, New York: Wiley.
- Smith, H. M., Hamblin, M., and Hill, M. F. (2005). Greater propensity of diabetic myocardium for oxidative stress after myocardial infarction is associated with the development of heart failure. J. Mol. Cell Cardiol. 39, 657-665. [DOI] [PubMed] [Google Scholar]
- Turner, M. D. (2004). Fatty acyl CoA-mediated inhibition of endoplasmic reticulum assembly. Biochim. Biophys. Acta 1693, 1-4. [DOI] [PubMed] [Google Scholar]
- Unger, R. H. (2003). Lipid overload and overflow: metabolic trauma and the metabolic syndrome. Trends Endocrinol. Metab. 14, 398-403. [DOI] [PubMed] [Google Scholar]
- Yagyu, H., et al. (2003). Lipoprotein lipase (LpL) on the surface of cardiomyocytes increases lipid uptake and produces a cardiomyopathy. J. Clin. Invest. 111, 419-426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, Y. T., Grayburn, P., Karim, A., Shimabukuro, M., Higa, M., Baetens, D., Orci, L., and Unger, R. H. (2000). Lipotoxic heart disease in obese rats: implications for human obesity. Proc. Natl. Acad. Sci. USA 97, 1784-1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, D., Tan, K. S., Zhang, X., Sun, A. Y., Sun, G. Y., and Lee, J. C. (2005). Hydrogen peroxide alters membrane and cytoskeleton properties and increases intercellular connections in astrocytes. J. Cell Sci. 118, 3695-3703. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.