

Abstract
Dengue fever is a mosquito-borne viral disease prevalent mainly in tropical countries. As the clinical manifestations of dengue are not very unique, laboratory diagnosis is crucial in identifying cases of dengue infection. Detection of dengue infection based on the identification of antidengue antibodies has emerged as a practical and reliable means of diagnosing dengue fever. We recently developed a customized recombinant dengue multiepitope protein (r-DME-G) that can specifically detect the immunoglobulin G (IgG) class of antidengue antibodies in patient sera. Using this strategy, we have now created another dengue multiepitope protein, r-DME-M, with specificity for the IgM class of antidengue antibodies. A synthetic gene encoding the r-DME-M protein was expressed as a maltose-binding protein fusion in Escherichia coli. The recombinant protein was purified in a single affinity chromatographic step to obtain yields of ∼15 mg purified protein/liter of culture. The purified protein was used to develop an in-house IgM enzyme-linked immunosorbent assay (ELISA) and tested using a panel of 172 patient sera characterized using the commercially available Dengue Duo rapid strip test from PanBio, Australia. The IgM ELISA results showed that the r-DME-M protein not only recognized all IgM+ samples identified by the PanBio test but also identified samples missed by the latter test. We also successfully adapted the r-DME-M protein to a rapid strip test format. This approach of creating customized antigens coupled to overexpression in E. coli and simple purification offers a promising alternative option to dengue diagnosis with the potential to circumvent the drawbacks of the whole virus antigen-based commercial kits.
Infection with dengue (DEN) viruses, of which there are four closely related but antigenically distinct serotypes (DEN-1, -2, -3 and -4), can cause a whole spectrum of illness ranging from the relatively mild dengue fever (DF) to potentially fatal dengue hemorrhagic fever (DHF) and dengue shock syndrome (DSS). The disease is transmitted to humans by mosquitoes and represents a major, rapidly expanding global public health problem in more than 100 nations worldwide. Annually, about 100 million people are infected, of which about 500,000, mostly children, are affected by DHF/DSS. The World Health Organization estimates that about 2.5 billion people are at risk of dengue infection (7-9). The at-risk population is predicted to double in the coming years (10). The high case fatality rates associated with DHF/DSS can be significantly minimized by supportive care and symptomatic treatment through fluid replacement. The key to survival therefore depends on early, definitive diagnosis of dengue infection (17, 21). In this context, the detection of dengue infections through identification of antidengue antibodies in the serum has emerged as a very reliable diagnostic approach (7).
The immunological status of the infected individual determines if the antidengue antibody response is primary or secondary (19). While individuals previously unexposed to flaviviral infection mount a primary antibody response, those who have experienced prior flavivirus infection manifest a secondary antibody response. In primary infection, anti-dengue immunoglobulin M (IgM) antibodies appear as early as 3 to 5 days after onset of illness, peak at about 2 weeks postinfection, and wane thereafter. IgG antibodies appear shortly afterwards and persist for several years. In contrast, secondary infection is characterized by the appearance of high-titer antidengue IgG antibodies, which appear either before or along with IgM antibodies. While the kinetics of IgM production is similar in primary and secondary infections, IgM antibody titers tend to be significantly lower in the latter instance. This difference in the pattern of antibody response has provided the basis for the serological detection of IgM and IgG antidengue antibodies to differentiate between primary and secondary dengue infections (19).
In recent years, several dengue diagnostic kits, in a multiplicity of formats, have become commercially available (6). These are used primarily to screen travelers returning from countries where dengue is endemic. Some of these kits can detect both antidengue IgM and IgG antibodies simultaneously and distinguish between primary and secondary dengue infections. Many of the diagnostic tests use whole virus antigen, produced in tissue culture or suckling mouse brain, for antidengue antibody detection. The use of such material, while presenting a potential health hazard through exposure to infectious virus, is also expensive, as production costs associated with virus cultivation are generally high. Further, the whole virus antigens manifest cross-reactivity towards antibodies against other flaviviruses as well as unrelated infectious agents, resulting in false positivity (6). One way to address these concerns is to replace the whole virus antigens with customized recombinant antigens, consisting of carefully chosen epitopes. Immunodominant, linear dengue viral epitopes, potentially suitable for constructing multiepitope proteins, have been mapped extensively on the major structural protein, the envelope (E) protein (12, 15, 20), and the nonstructural (NS) protein NS1 (3, 4, 22, 23). The majority of these epitopes specifically recognize IgG antibodies.
We recently reported a novel multiepitope approach that entails splicing together unique dengue-specific epitopes that do not cross-react with antibodies to non-dengue flavivirus antibodies and overexpressing the resultant recombinant protein in an Escherichia coli host (1). The multiepitope approach, which eliminates the whole virus antigen (thus contributing to safety), permits a high epitope density (sensitivity), careful choice of unique dengue-specific epitopes (specificity), and E. coli-based overexpression (cost-effective antigen production). Using this approach we created a recombinant dengue multiepitope protein, r-DME-G, by fusing together several IgG-specific dengue virus-encoded epitopes. Further, we demonstrated that this protein could be used in an indirect enzyme-linked immunosorbent assay (ELISA) to specifically detect an antidengue IgG class of antibodies in patient sera (1).
We have now used this approach to develop a new recombinant dengue multiepitope protein, designed to detect IgM-specific antidengue antibodies. A thorough examination of the available literature revealed a single report by Huang et al. (11) that described an IgM-specific dengue epitope that appeared potentially useful for the purpose of creating an IgM-specific dengue multiepitope protein. These investigators examined potential B-cell epitope-containing peptides of DEN-2 NS1 for their reactivity to IgM and IgG classes of antibodies in dengue and Japanese encephalitis (JE) patient sera. They identified a single peptide, D2 NS1-P1, which reacted with the IgM class of antibodies in sera of dengue, but not JE, patients. This peptide, corresponding to amino acid (aa) residues 1 to 15 of DEN-2 NS1, defined a linear, immunodominant IgM-specific epitope which was reactive towards patient sera from all four dengue serotypes (11). We used this epitope as the basis for designing the recombinant dengue multiepitope protein, r-DME-M, with built-in specificity for the IgM class of antidengue antibodies. In this paper, we describe the design of the r-DME-M protein, its expression, and purification. Further, we present data demonstrating its utility as a diagnostic reagent in the detection of antidengue IgM antibodies. Finally, we describe the design of a simple strip test using the r-DME-M protein that could prove useful in identifying dengue infections under field conditions where adequate diagnostic facilities may be lacking.
MATERIALS AND METHODS
Materials.
Escherichia coli host strain DH5α was purchased from Invitrogen. The plasmid pQE60 (Ampr), Ni-nitrilotriacetic acid (NTA) superflow resin and anti-His monoclonal antibody (catalog no. 34660) were from QIAGEN, Germany. The plasmid pMALc2x was purchased from New England Biolabs. The secondary antibody-enzyme conjugates (anti-mouse IgG-alkaline phosphatase [AP] and anti-human IgM-horseradish peroxidase [HRPO]) and the AP substrate 5-bromo-4-chloro-3-indolyl phosphate-nitroblue tetrazolium (BCIP/NBT) were from Calbiochem. The HRPO substrate 3,3′,5,5′-tetramethylbenzidine (TMB) was from Kirkegaard & Perry Laboratories.
Construction of the r-DME-M gene expression vector.
A synthetic gene, codon-optimized for E. coli expression, was first generated by ligation (GeneArt, Germany) of oligonucleotides encoding four linear immunodominant epitopes from NS1 proteins of DEN serotypes 1 to 4. These epitopes were 15 aa residues long with adjacent epitopes joined together by tetraglycyl linkers. The resultant 0.24-kb gene, the r-DME-M gene, was ligated into the BamHI and BglII sites of the bacterial expression vector pQE-60 to generate the plasmid pQ-r-DME-M. In this plasmid, the r-DME-M gene was inserted in frame with the ATG codon (at the 5′ end) and the six-His tag-encoding sequence (at the 3′ end) provided by the pQE-60 vector. Recombinant clones were selected on ampicillin plates and subjected to direct colony PCR screening, using vector-specific primers, to identify recombinants harboring the synthetic r-DME-M gene. Orientation of the insert was verified by restriction analysis of plasmid minipreps. The r-DME-M gene plus the six-His tag-encoding sequence were retrieved from the pQ-rDME-M construct as a 0.26-kb BamHI/HindIII restriction fragment and ligated in frame with the malE gene of plasmid pMALc2x. In this vector, the malE gene, which encodes the maltose-binding protein (MBP), is driven by the isopropylthiogalactoside (IPTG)-inducible Ptac promoter. The ligated product was transformed into E. coli DH5α cells and selected on ampicillin plates. The resultant DH5α transformants were screened by restriction analysis. Positive clones were further subjected to expression screening (see below) to identify clones capable of efficiently expressing the r-MBP-DME-M fusion protein.
Expression screening.
Expression screening was done using test tube cultures, essentially as described earlier (1). Several of the positive DH5α clones, chosen based on restriction analysis, were inoculated into 3-ml test tube cultures and allowed to grow at 37°C in a shaker at 200 rpm. When the cultures were in logarithmic growth phase (corresponding to an optical density at 600 nm [OD600] of ∼0.6), they were induced with 0.4 mM IPTG for ∼4 h. After induction, equivalent numbers of cells from the different cultures (normalized on the basis of OD600 values) were lysed in sample buffer and analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (14). Noninduced controls were analyzed in parallel. One clone that expressed maximal levels of the recombinant protein was chosen for further work. Plasmid DNA isolated from this clone was verified by restriction analysis.
Purification of r-DME-M protein.
A preculture was set up by inoculating 20 ml LB medium containing ampicillin (100 μg/ml) with 10 μl glycerol stock of a DH5α clone (chosen above) harboring the r-MBP-DME-M gene. The culture was grown overnight in a shaker at 37°C, at 200 rpm, and inoculated into 1 liter LB (containing 100 μg ampicillin/ml), in a 4-liter Haffkine flask, which was placed in the shaker at 37°C for about 2 to 3 h at 125 rpm. When the OD600 of the culture reached ∼0.6 (a small aliquot of the noninduced culture was set aside for subsequent SDS-PAGE analysis), it was induced by the addition of IPTG to a final concentration of 0.4 mM. Induction was allowed to proceed for 4 h before harvesting the cells. Aliquots of the induced and noninduced cell cultures were analyzed by SDS-PAGE prior to initiating purification.
The induced culture was centrifuged in a Sorvall GS3 rotor at 6,000 rpm for 15 min at 4°C. About 1 g of induced cell pellet (corresponding to 250 ml E. coli culture) was lysed by stirring in 10 ml lysis buffer (8 M urea-100 mM sodium phosphate-10 mM Tris-HCl-10 mM imidazole [pH 8.0]), at room temperature (RT) for 1 h. The lysate was clarified by centrifugation (10,000 rpm in a Sorvall GSA rotor at RT for 30 min). The resultant supernatant was mixed with 1 ml Ni-NTA Superflow resin that had been preequilibrated with the lysis buffer. This suspension was gently rocked for 1 h at RT and then packed into a column. After collecting the flowthrough, the column was washed extensively with buffer 1 (lysis buffer containing 20 mM imidazole) and eluted with buffer 2 (lysis buffer containing 250 mM imidazole). Fractions of 3 ml were collected and analyzed by SDS-PAGE. Peak fractions were pooled together, mixed with gentamicin to a final concentration of 50 μg/ml, flash-frozen in liquid nitrogen, and stored at −80°C until use.
Western blot analysis.
The procedure adopted was identical to that reported earlier by us for r-DME-G protein (1). The purified r-DME-M protein was run on a 15% denaturing gel (SDS-PAGE), along with appropriate controls and prestained protein markers, and transferred electrophoretically to a nitrocellulose membrane. The membrane was blocked with 1% polyvinyl pyrrolidone in 1× phosphate-buffered saline, pH 7.2 (PBS) for 2 h at RT. It was then washed three times with 1× PBS containing 0.1% Tween 20 (1× PBS plus 0.1% T) and incubated with a commercially available murine penta-His monoclonal antibody (MAb; 1:2,000 dilution) for 90 min at RT. The membrane was washed again, as above, and incubated with anti-mouse IgG-AP conjugate (1:5,000 dilution) for a further 90 min at RT. The blot was washed and developed by incubation in BCIP/NBT substrate solution for 15 min at RT.
In-house ELISA for the detection of antidengue IgM antibodies.
Again, the IgM ELISA protocol we used was essentially similar to the one developed earlier for the r-DME-G (1), with the only differences being the nature of the coating antigen and the specificity of the secondary antibody conjugate. Briefly, the purified r-DME-M protein (diluted to 10 μg/ml in 0.1 M carbonate buffer, pH 9.5) was used to coat 96-well microtiter plates (100 μl/well) at 4°C overnight. The coated wells were washed once with 1 PBS plus 0.1% T and blocked with 5% skim milk powder (SMP) in 1× PBS (1× PBS plus 5% SMP) for 4 h at 37°C. The wells were washed once again with 1× PBS plus 0.1% T and then incubated for 15 min at 37°C with 100 μl dengue patient sera (at a 1:10 dilution in 1× PBS plus 5% SMP containing 1% sodium deoxycholate [SDC]). Wells were washed using 1× PBS plus 0.1% T plus 1% SDC and incubated with murine anti-human IgM-HRPO conjugate (at a 1:7,500 dilution). The wells were washed once again as above and incubated with 100 μl of TMB substrate for 15 min at 37°C. A peroxidase reaction was terminated with 100 μl 1 M H2SO4, and the absorbance was read at 450 nm. Control experiments in which we either omitted the addition of dengue patient serum or used r-MBP as the coating antigen (instead of the r-MBP-DME-M fusion protein) were run in parallel to correct for any nonspecific ELISA reactivity. Sera were designated as positive or negative for the presence of antidengue IgM antibodies using a cutoff absorbance of 0.25 (450 nm). This cutoff value was obtained from the mean ELISA absorbance of 59 “normal” sera (that tested negative for both IgM and IgG antibodies using the PanBio test) plus twice the standard deviation.
Design of an in-house rapid strip test.
For this test, we prepared gold-labeled r-DME-M, test sample, and the test strip as follows. Purified r-DME-M was conjugated to 40-nm gold colloid using a standardized protocol of British Biocell International, Cardiff, United Kingdom. The test sample was prepared by preincubating the patient sera (30 μl) with gold-conjugated r-DME-M (15 μl) in a final volume of 50 μl made up with 1× PBS plus 0.1% T. The test strip (available from a local vendor), which was a ∼12-cm strip of nitrocellulose membrane mounted on a plastic support and provided with an absorbent pad at one end (the top end while performing the test), was prepared by placing two horizontal streaks across its mid-section. One streak was of the purified r-DME-M protein, while the other was of murine anti-r-DME-M polyclonal serum. The relative positions of these two streaks on the strip are indicated below in Fig. 5A. To perform the test, the lower, nonpadded end of the strip was dipped into a microtiter well containing 50 μl of the test sample prepared as described above. About ∼10 min later, when the sample reached the absorbent pad (which prevents the sample from running down the strip), the strip was washed in 1× PBS plus 0.1% T and read by visual inspection while still wet. Two pink lines at both the test and control positions indicated a positive test, while a single pink line at the control position alone indicated a negative test.
FIG. 5.
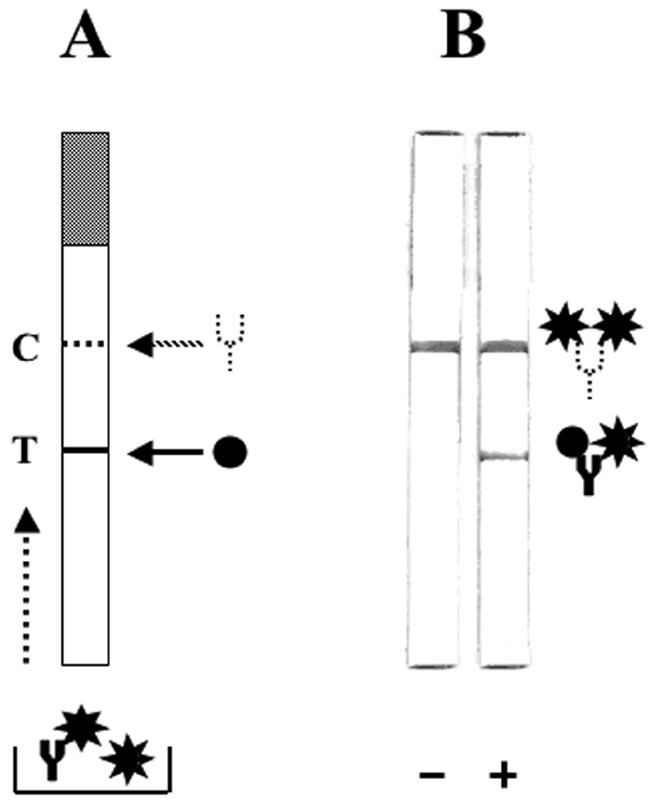
Design and use of the rapid strip assay to detect antidengue antibodies using the r-DME-M protein. (A) Schematic representation of the strip test template. The strip consists of a nitrocellulose membrane mounted on a plastic support. The shaded portion at the top represents the absorbent pad. The test line (T, indicated by the solid line) denotes the position on the strip at which the r-DME-M protein (indicated by the black circle) is immobilized as a thin line. The control line (C, indicated by the dashed line) denotes the position on the strip at which murine anti-r-DME-M polyclonal serum (indicated by the dotted Y) is coated as a thin line. Shown below is the sample receptacle containing the test serum mixed with gold-labeled r-DME-M (indicated by the black star). Antidengue IgM antibody (indicated by the solid Y) if present will form a complex with the gold-labeled r-DME-M in the sample well. The dotted arrow at the left side of the strip template indicates the direction of sample migration during the test. (B) Depiction of the actual results of the strip test performed with sera that lack (−) or contain (+) antidengue IgM antibodies. A schematic representation of the complexes formed at the T and C lines is shown to the right.
RESULTS
Design of a novel IgM-specific dengue multiepitope protein (r-DME-M).
In order to create a multiepitope protein that could be useful in early detection of dengue infection, we focused on an immunodominant epitope of the DEN-2 NS1 protein. This epitope, corresponding to the N-terminal 15 aa residues, was shown to display predominantly IgM specificity and was reactive towards sera from patients infected with all four DEN virus serotypes (11). A sequence alignment of this peptide with the corresponding regions from the NS1 proteins of DEN-1, DEN-3, and DEN-4, depicted in Fig. 1A, revealed a high degree of homology among these four NS1 peptides. We designed a synthetic gene based on these four epitopes. Adjacent epitopes were linked by a string of four glycyl residues. A synthetic gene encoding this protein, codon optimized for expression in E. coli, was created and cloned into the bacterial expression vector pQE60. The insert was designed to be in frame with the initiator codon and a carboxy-terminal six-histidine tag-encoding sequence provided by the vector. The synthetic gene, designated rDME-M (recombinant dengue multiepitope, IgM specific), is predicted to encode a ∼9-kDa recombinant protein. The aa sequence of the r-DME-M protein is shown in Fig. 1B. A computer modeling analysis of the structure of the r-DME-M protein, depicted in Fig. 1C, suggested that the proposed design of the multiepitope protein would be consistent with a structure that permits easy accessibility of all the constituent epitopes.
FIG. 1.

Design of the r-DME-M protein. (A) Multiple sequence alignment (ClustalW) of the amino-terminal 15 aa residues (single letter code) of the NS1 proteins of DEN serotypes 1 to 4 (D1 to D4). Conserved residues are shown in gray boxes. (B) Complete amino acid sequence of the r-DME-M protein assembled from the peptides indicated in panel A. The epitope amino acids are shown in normal font, and the tetraglycyl linkers are shown in bold font. The amino-terminal residues 1 to 4 and the carboxy-terminal eight residues, inclusive of the six-His tag, shown in italics, are encoded by plasmid-derived sequences. (C) A computer-generated representation of the r-DME-M protein in solution. The protein aa sequence was submitted to the 3D-PSSM web server and visualized using ViewerLite software. Regions in red are α-helical, and those in white are unstructured.
We transformed the pQ-r-DME-M construct into E. coli host strain SG13009 containing the pREP4 plasmid, which encodes the lacI repressor (required for regulated recombinant gene expression) and the kanamycin marker. Expression screening of several of the resultant double recombinants failed to identify a single clone capable of expressing the r-DME-M gene (data not shown). Given that we had codon optimized our gene for E. coli expression and had successfully used this vector system and cloning strategy earlier (1), the observed lack of expression is puzzling. Based on information in the literature that recombinant proteins of 100 to 200 aa residues are more likely to be expressed efficiently in E. coli (5), it is probable that the failure to express the r-DME-M protein (88 aa residues) is a reflection of its relatively smaller size.
Expression of the r-DME-M protein as an MBP fusion.
As attempts to express the r-DME-M protein in E. coli using the pQE expression plasmid were unsuccessful, we explored the possibility of expressing it as an MBP fusion. To this end, we retrieved the r-DME-M gene as a 0.26-kb BamHI/HindIII fragment (inclusive of the 3′ six-histidine tag-encoding sequence) from the pQ-r-DME-M plasmid and fused it in frame with the MBP-encoding malE gene in the vector pMALc2x. This expression vector is depicted in Fig. 2A. In this vector, the MBP-r-DME-M fusion gene is under the transcriptional control of the IPTG-inducible Ptac promoter. This vector was introduced into the E. coli host DH5α, and transformants selected in the presence of ampicillin were analyzed by expression screening. In this experiment, IPTG-induced cells were directly lysed in Laemmli sample buffer and analyzed by SDS-PAGE. The MBP fusion strategy resulted in successful expression. Figure 2B depicts the induction profile of a typical clone. Induction of the MBP-rDME-M gene resulted in the appearance of a new ∼52-kDa band, consistent with the predicted size of the MBP-r-DME-M fusion protein (MBP component, ∼43 kDa; r-DME-M component, ∼9 kDa). When induced cells were lysed by sonication in a native buffer, separated into supernatant and pellet fractions as described before (13), and analyzed by SDS-PAGE, the protein was found to be present partially in the soluble fraction (∼30%) with the rest in the insoluble fraction (compare lanes 2 and 3 in Fig. 2C). For the sake of convenience, the ∼52-kDa MBP-r-DME-M fusion protein is referred to as the r-DME-M protein in the rest of this paper.
FIG. 2.

Expression of r-DME-M protein in E. coli as an MBP fusion. (A) Map of the expression plasmid. The synthetic r-DME-M gene with a 3′ six-His tag-encoding sequence (rDME) was inserted into the BamHI and HindIII sites of plasmid pMALc2x in frame with the MBP-encoding gene (Mal E). Abbreviations: Ptac, E. coli Ptac promoter; TT, transcriptional terminator; Lac I, lac repressor; Ori and Ampr, plasmid replication origin and ampicillin resistance marker, respectively. (B) Coomassie-stained denaturing gel showing the polypeptide profiles of E. coli harboring the plasmid in panel A, before (lane 3) and after (lane 2) IPTG induction (I, induced; U, noninduced). (C) Localization of the r-DME-M protein in induced cell lysates. An aliquot of the induced cell lysate prepared by sonication in a native buffer was separated into supernatant (S) and pellet (P) fractions and analyzed by SDS-PAGE followed by Coomassie staining. In both gels shown in panels B and C, protein molecular mass markers were run in lane 1; their sizes in kDa are shown to the left. The arrows at the right indicate the position of the rDME-M fusion protein.
Purification of the r-DME-M protein.
As the r-DME-M protein was only partially soluble with a major proportion in the insoluble fraction, we proceeded to purify it under denaturing conditions to obtain maximal yields. For purification, we opted to take advantage of the C-terminal six-His tag, rather than the N-terminal MBP tag, mainly because the Ni-NTA affinity purification had already been optimized in our laboratory for several recombinant six-His-tagged proteins (1, 13). It should be mentioned here that the r-DME-M protein was predesigned to carry the six-His tag, and the MBP fusion strategy was adopted mainly to promote expression in E. coli, rather than to provide an affinity handle for purification. Fractions collected during different steps of the purification were analyzed by SDS-PAGE as shown in Fig. 3A. Under the experimental conditions employed, almost all of the induced protein bound to the column, as evident from a comparison of the polypeptide profile of the initial lysate loaded on the affinity matrix (lane 2) with that of the flowthrough material (lane 3). An extensive wash in the presence of 20 mM imidazole progressively removed most of the nonspecific contaminants. This can be seen from a comparison of lanes 4 (initial wash fraction) and 5 (final wash fraction). Elution of the bound proteins using 250 mM imidazole resulted in the emergence of highly purified recombinant protein from the column (lanes 6 to 8). We estimate that a >95% purity was achieved on the basis of comparison of the protein profiles of the eluted material and the crude lysate (lane 2). Starting from ∼1 g induced cell pellet (equivalent to 250 ml of E. coli culture), we obtained ∼3.7 mg purified recombinant protein (pooled fractions in lanes 6 to 8). This corresponds to a recovery of 69%, as the crude lysate was estimated to contain ∼5.4 mg of the recombinant protein, based on densitometric analysis. A summary of the purification is presented in Table 1.
FIG. 3.

Purification and characterization of the r-DME-M protein. (A) SDS-PAGE analysis of Ni-NTA affinity column fractions obtained during the purification of r-DME-M protein. Samples analyzed in the gel are load (L, lane 2), flowthrough (F, lane 3), wash (W1 and W2, lanes 4 and 5), and eluates (E1 to E3, lanes 6 to 8). Protein molecular mass markers (M) were run in lane 1; their sizes in kDa are shown to the left. The arrow at the right indicates the position of the r-DME-M protein. (B) Western blot analysis of the purified r-DME-M protein using penta-His MAb. An aliquot of the purified recombinant protein was run in lane 2. Negative (a protein without His tag; BSA) and positive (rED2-III, a protein with a six-His tag [13]) controls were run in lanes 3 and 4, respectively. Protein molecular mass markers were run in lane 1; their sizes in kDa are shown to the left. The top and bottom arrows at the right indicate the positions of the r-DME-M protein and the rD2E-III protein, respectively. Abbreviations: P, prestained protein markers; R, r-DME-M; B, BSA; E, rED2-III. (C) Antibodies induced by the r-DME-M protein interact specifically with rNS1. Serial 10-fold dilutions of murine sera obtained from animals immunized with PBS (black circles), r-DME-G (open traingles), r-DME-M (open squares), or DEN-2 rNS1 (black squares) were tested in an ELISA using DEN-2 rNS1 as the capture antigen.
TABLE 1.
Summary of r-DME-M purification from 1 liter of induced culture
| Purification step | Total protein (mg)a | Purity (%)b | Recovery (%)c |
|---|---|---|---|
| Crude lysated | 272 | 0 | 100 |
| Ni-NTA affinity | 14.8 | 95 | 69 |
| Chromatography |
Protein content was determined by the BioRad method, using BSA as standard.
Purity was assessed by SDS-PAGE analysis.
The amount of r-DME-M protein in the crude lysate (which was estimated to be ∼5.4 mg by densitometric analysis) was designated as 100%.
The purification was done using a 250-ml culture (wet cell weight of ∼1 g).
Next, we tested the Ni-NTA affinity-purified protein in a Western blot assay. An aliquot of pooled purified protein was probed with a commercially available murine penta-His MAb specific to the engineered six-His tag at the carboxy-terminal end of the recombinant protein. In this blot, we also included a previously described six-His-tagged recombinant protein (13) as a positive control. As a negative control (a protein lacking the tag), we used bovine serum albumin (BSA). It is evident from the data shown in Fig. 3B that the penta-His MAb, which specifically recognizes the six-His-tagged positive control protein (compare lanes 3 and 4), also recognized the ∼52-kDa recombinant protein purified as described in the experiment shown in Fig. 3A. To further characterize the r-DME-M protein, we raised murine polyclonal antibodies against it and tested these in an ELISA using recombinant NS1 as the capture antigen. The rationale for this experiment was that the NS1 epitope-derived r-DME-M protein should elicit antibodies capable of recognizing the NS1 protein. This was borne out by data depicted in Fig. 3C. The data show that the antibodies elicited by the r-DME-M protein were as good as the antibodies elicited by the r-NS1 protein. Interestingly, sera from mice immunized with the r-DME-G protein, designed to be IgG specific, did not show any ELISA reactivity in this assay.
Dengue patient sera recognize the r-DME-M protein.
Having obtained purified r-DME-M protein, we next proceeded to ascertain if it would be reactive towards antidengue antibodies in patient sera. For this, we obtained a large panel (n = 172) of suspected dengue patient sera from regions in Sri Lanka where dengue is endemic. All samples were tested for the presence of infectious virus and viral RNA. We then analyzed the presence of antidengue antibodies using the commercially available Dengue Duo rapid strip test (PanBio Pty Ltd., Australia), which detects polyclonal dengue virus-specific antibodies of both IgM and IgG classes (2). The presence of antidengue IgG antibodies in all these sera was also tested independently using our r-DME-G protein described earlier (1). We next proceeded to evaluate the purified r-DME-M protein as a diagnostic reagent to detect antidengue IgM antibodies using this well-characterized serum panel. To this end, we adapted the in-house ELISA protocol developed previously for the detection of antidengue antibodies of the IgG class (1). In this assay, the rDME-M protein was used to capture the IgM class of antidengue antibodies from patient sera and revealed using murine anti-human IgM-HRPO conjugate.
The data pertaining to the evaluation of the r-DME-M protein are summarized in Table 2. Based on the data obtained, we could categorize the samples broadly into a virus-positive group (I) and a virus-negative group (II). With regard to the serology, both groups were heterogenous, as reflected by their division into subgroups (group I is comprised of subgroups 1 to 3; group II is comprised of subgroups 4 to 8). Table 3 presents serum-wise data for group I samples (n = 22). All these sera contained viral RNA, and infectious virus could be isolated from all but one sample of this group. A total of six samples (all five of subgroup 2 plus the single one from subgroup 3) were found to contain IgM antibodies. Out of these six, the PanBio strip test identified three samples (two from subgroup 2 and the single subgroup 3 sample) as IgM+. With regard to IgG, only one sample (subgroup 3) was found to be positive; this was corroborated by the PanBio test. The remaining samples (subgroup 1; n = 16) were negative for both IgM and IgG. Once again, these results were borne out by the PanBio test.
TABLE 2.
Evaluation of patient sera using the r-DME proteins
| Groupa | IgM/IgG statusb | Total no. of samples | No. positive by:
|
|
|---|---|---|---|---|
| r-DMEc | PanBiod | |||
| Virus-positive sera | ||||
| 1 | IgM−/IgG− | 16 | 16 | 16 |
| 2 | IgM+/IgG− | 5 | 5 | 2 |
| 3 | IgM+/IgG+ | 1 | 1 | 1 |
| Virus-negative sera | ||||
| 4 | IgM+/IgG+ | 12 | 12 | 12 |
| 5 | IgM+/IgG− | 37 | 37 | 37 |
| 6 | IgM−/IgG+ | 21 | 21 | 21 |
| 7 | IgM−/IgG− | 59 | 59 | 59 |
| 8 | ?e | 21 | ? | ? |
Groups 1 to 3 tested positive for virus; groups 4 to 8 tested negative for virus.
Presence and absence of IgM or IgG is indicated by + and −, respectively.
The data were obtained using rDME-M and rDME-G (1) separately as capture antigens in ELISAs to detect IgM and IgG, respectively; samples were designated as + or − using a cutoff value of 0.25 (representing the mean absorbance of sera belonging to subgroup 7 plus twice the standard deviation).
Data were generated using the Dengue Duo IgM & IgG Rapid Strip test purchased from PanBio Pty., Australia.
?, borderline samples, with results from r-DME protein-based in-house ELISAs and the commercial PanBio tests not in agreement with each other.
TABLE 3.
Single serum profiles of dengue virus-positive sera
| Sample no. and ID | Identity based on:
|
IgM/IgG statusa
|
||
|---|---|---|---|---|
| RT-PCRf | Virus isolation | r-DMEb | PanBioc | |
| 1, D176 | DEN-2 | DEN-2 | IgM−/IgG− | IgM−/IgG− |
| 2, D192 | DEN-2 | DEN-2 | IgM−/IgG− | IgM−/IgG− |
| 3, D211 | DEN-2 | DEN-2 | IgM−/IgG− | IgM−/IgG− |
| 4, D223 | DEN-2 | DEN-2 | IgM−/IgG− | IgM−/IgG− |
| 5, D225 | DEN-2 | DEN-2 | IgM−/IgG− | IgM−/IgG− |
| 6, D226 | DEN-2 | DEN-2 | IgM−/IgG− | IgM−/IgG− |
| 7, D086 | DEN-2 | DEN-2 | IgM−/IgG− | IgM−/IgG− |
| 8, D168 | DEN-2 | DEN-2 | IgM−/IgG− | IgM−/IgG− |
| 9, D173 | DEN-2 | DEN-2 | IgM−/IgG− | IgM−/IgG− |
| 10, D174 | DEN-2 | DEN-2 | IgM−/IgG− | IgM−/IgG− |
| 11, D039 | DEN-3 | DEN-3 | IgM−/IgG− | IgM−/IgG− |
| 12, D019 | DEN-2 | DEN-2 | IgM−/IgG− | IgM−/IgG− |
| 13, D119 | DEN-2 | DEN-2 | IgM−/IgG− | IgM−/IgG− |
| 14, D092 | DEN-3 | DEN-3 | IgM−/IgG− | IgM−/IgG− |
| 15, D022 | DEN-2 | DEN-2 | IgM−/IgG− | IgM−/IgG− |
| 16, D030 | DEN-2 | DEN-2 | IgM−/IgG− | IgM−/IgG− |
| 17,d 0075 | DEN-2 | DEN-2 | IgM+/IgG− | IgM−/IgG− |
| 18,d D065 | DEN-2 | DEN-2 | IgM+/IgG− | IgM−/IgG− |
| 19, D016 | DEN-2 | DEN-2 | IgM+/IgG− | IgM+/IgG− |
| 20, D027 | DEN-2 | DEN-2 | IgM+/IgG− | IgM+/IgG− |
| 21, D011 | DEN-2 | —e | IgM+/IgG− | IgM+/IgG− |
| 22,d D068 | DEN-2 | DEN-2 | IgM+/IgG+ | IgM−/IgG+ |
Presence and absence of IgM or IgG are indicated by + and − superscripts, respectively.
The data were obtained using rDME-M and rDME-G (1) separately as capture antigens in ELISAs to detect IgM and IgG, respectively; samples were designated as + or − using a cutoff value of 0.25 (representing the mean absorbance of sera belonging to subgroup 7 [Table 2] plus twice the standard deviation).
Data were generated using Dengue Duo IgM & IgG Rapid Strip test purchased from PanBio Pty., Australia.
Virus-positive samples containing IgM antibodies (IgM+ in bold) picked up by the r-DME-M-based ELISA but missed by the PanBio test.
—, virus could not be isolated successfully from this sample.
RT-PCR, reverse transcription-PCR.
The samples comprising group II (n = 150) were all found to be virus−/RNA−. Of the 129 samples, represented collectively by subgroups 4 to 7, we could detect the presence of IgM in 49 samples (12 samples of subgroup 4 plus 37 samples of subgroup 5) and IgG in 33 samples (12 samples of subgroup 4 plus 21 samples of subgroup 6), using the r-DME-G-based IgG ELISA (1). Samples in subgroup 7 were all IgM−/IgG− and are presumably “normal,” as all of them tested negative in the virus isolation and reverse transcription-PCR assays as well. With regard to the PanBio test, 19 samples of group I (16 from subgroup 1, 2 from subgroup 2, and the single sample from subgroup 3) and all samples of subgroups 4 to 7 (n = 129) yielded results that were identical to those obtained using the r-DME-based IgM and IgG ELISAs.
For samples in subgroup 8 (n = 21), the results of the r-DME-based ELISAs did not agree with those of the PanBio test. It must be pointed out that neither virus nor viral RNA could be detected in all of these “indeterminate” samples. With regard to IgM status, a closer examination of the ELISA data revealed that the PanBio and the r-DME-M-based ELISA results were not consistent with each other for 14 of these 21 indeterminate samples. Similarly, the remaining sera were inconsistent with respect to IgG status. Of the 14 indeterminate samples above, seven sera, which were identified as IgM+ by the PanBio test, were not picked up by the r-DME-M-based IgM ELISA. Likewise, the remaining seven, which tested positive using the r-DME-M protein, scored negative in the PanBio test. An inspection of the ELISA absorbances of these 14 sera revealed that all of them manifested borderline reactivity towards the r-DME-M protein, as depicted in Fig. 4. For comparison, this figure also includes the three IgM+ samples of subgroup 2, which were missed by the PanBio test. Similarly, the discrepancies in IgG status using the PanBio test and our previously developed r-DME-G-based ELISA could be traced to borderline samples (data not shown). The overall comparative analysis of our data with the PanBio results suggests that there is an excellent agreement between the r-DME-based ELISA and the PanBio tests, with ∼86% (148 out of 172) of the samples analyzed giving identical results. Further, the r-DME-M-based IgM ELISA picked up three additional samples (group I, subgroup 2) which the PanBio test failed to identify. The fact that these three samples were virus+/RNA+ suggests that the r-DME-M-based IgM ELISA test is more effective in early diagnosis of dengue infection.
FIG. 4.

Analysis of indeterminate sera with widely ranging r-DME-M reactivities. The scatter diagram displays the reactivities (absorbance at 450 nm) of sera manifesting weak and strong reactivities towards the r-DME-M protein used as the capture antigen in the in-house IgM ELISA. The IgM status of the sera determined by the PanBio strip test (PanBio) or the in-house IgM ELISA (r-DME-M) is shown in the inset; the subgroup, with reference to Table 2, to which the serum belongs is shown in parentheses. The dotted line indicates the cutoff absorbance (0.25 at 450 nm) used to designate samples as either positive or negative.
The r-DME-M protein as a dengue diagnostic reagent.
We next sought to devise a rapid test that might be useful in identifying the presence of antidengue antibodies in patient sera under field conditions, that is, without having to use any laboratory facilities. Accordingly, a rapid strip test, shown in Fig. 5A, was designed. Across this strip we coated r-DME-M protein as a streak. We placed a second streak of murine polyclonal anti-r-DME-M sera. To perform the test, we mixed the patient serum with gold-labeled r-DME-M protein in a microtiter well, dipped the lower end of the strip into this mixture, and allowed sufficient time for the sample to rise to the level of the absorbent pad by capillary action. Shown in Fig. 5B are the results of typical negative and positive tests. In the negative (-) sample that did not contain any antidengue antibodies (IgM−/IgG−, using the PanBio test), a single pale pink line appeared (Fig. 5A). This represents free gold-labeled r-DME-M protein captured by the murine polyclonal antibodies coated at the control line. In the case of the dengue antibody-positive (either IgM+/IgG+ or IgM+/IgG−, using the PanBio test) serum sample, however, a second pale pink line appeared at the test line. This represents the gold-labeled r-DME-M protein-antidengue antibody complex captured by unlabeled r-DME-M coated at the test line. The results of the strip test showed that the r-DME-M protein could indeed be adapted to a rapid test format for the detection of dengue-specific antibodies.
DISCUSSION
Dengue diagnosis based on antibody identification has emerged as the most practical approach (7). Most methods of antibody detection rely on the use of whole dengue virus antigens. As mentioned earlier, the serodiagnosis of dengue based on antibody detection is complicated by the existence of cross-reactive antigenic determinants shared by members of the Flaviviridae family (6, 7). Apart from compromising sensitivity and specificity, the preparation of whole virus antigen is associated with biohazard risk and high production costs. While replacing the whole virus antigen with a recombinantly produced viral protein can eliminate the safety risk, it may not necessarily address the cross-reactivity and cost considerations. This was underscored by a recent report which showed that a mixture of recombinant E proteins (of all four DEN serotypes) could effectively replace the whole virus antigen in a commercial test but failed to eliminate cross-reactivity with antibodies to other flaviviruses (2). Further, this test also did not effectively address the cost factor, as it used four recombinant proteins produced using an insect cell culture system. Thus, there is a need for development of diagnostic tests that incorporate not only a high degree of sensitivity and specificity of detection but also safety and cost-effectiveness as well. The use of a customized, recombinant antigen that can be overexpressed in E. coli may offer an alternative that effectively addresses the concerns mentioned above. Therefore, the major objective of this study was to design an IgM-specific recombinant multiepitope protein and evaluate its potential in the detection of antidengue IgM antibodies. To this end, we applied our recently developed strategy of creating customized antigens to develop a multiepitope protein, r-DME-M, designed to detect IgM-specific antidengue antibodies to aid in early diagnosis of primary dengue infections. This strategy depends on fusing together linear, immunodominant epitopes using flexible linkers.
A search for immunodominant IgM-specific dengue epitopes revealed a single linear epitope which mapped to the amino-terminal 15 residues of the DEN-2 NS1 protein (11). Presumably, most IgM-specific epitopes may be conformational, and this may perhaps explain the paucity of information regarding linear IgM-specific dengue epitopes. However, in an effort to include additional IgM-specific epitopes in designing the r-DME-M protein, we compared the amino-terminal sequences of the NS1 proteins of the remaining three DEN serotypes with that of the DEN-2 NS1 protein. This revealed a high degree of sequence homology among all four DEN serotypes, prompting the notion that the amino termini of the NS1 proteins of all four DEN serotypes represent potential IgM-specific epitopes. Thus, we created the synthetic gene, r-DME-M, encoding all four of these NS1 epitopes, in which adjacent epitopes were joined using flexible tetraglycyl linkers. Glycine was used as the linker residue, as it can provide flexibility owing to its lack of a β-carbon (18). Computer modeling analyses showed that the r-DME-M protein adopts a structure in which all the epitopes are freely accessible, suggesting that each of them would be able to collectively contribute to the overall IgM specificity of the molecule. We engineered a six-His tag-encoding sequence at the 3′ end of the r-DME-M gene to facilitate one-step affinity purification of the expressed protein. We failed to express this gene using the pQE60 vector. This is presumably a reflection of the small size (88 aa residues) of the protein it encodes (5). This notion appears consistent with our observation that this same gene could be expressed successfully when fused to the MBP-encoding malE gene. Analysis of the Ni-NTA-purified r-DME-M (MBP fusion) protein by SDS-PAGE showed that a high degree of purity, >95%, had been achieved in a single step. Our data show that starting from a 1-liter culture of induced cells, ∼15 mg of purified protein, representing a ∼69% recovery from the crude lysate, can be obtained. Purified r-DME-M protein could be detected in a Western blot assay using anti-His MAb, consistent with its ability to bind Ni-NTA matrix.
Since one of the major objectives of this study was to investigate the potential of the r-DME-M protein as a diagnostic antigen for the early detection of dengue infections, we tested the ability of the r-DME-M protein to detect antidengue IgM antibodies in an in-house ELISA. Using our recombinant protein as the capture antigen and dengue patient serum as the test sample, captured antidengue IgM antibodies were detected using murine anti-human IgM-HRPO conjugate in conjunction with TMB substrate. We tested a panel of 172 serum samples drawn from patients in Sri Lanka suspected of dengue infection. Prior to using this panel for the evaluation of r-DME-M, we characterized the samples for the presence and type of dengue virus infection (by virus isolation and RT-PCR) and antidengue antibodies (using the commercially available PanBio test). We also determined the presence of antidengue IgG antibodies in these sera using our IgG-specific r-DME-G protein developed recently (1). We then evaluated the utility of the r-DME-M protein developed in this study to detect the presence of antidengue antibodies of the IgM class in this serum panel. This analysis showed that 80 (subgroups 6 and 7) (Table 2) out of the 172 sera which were IgM− based on the PanBio test displayed no significant reactivity towards the r-DME-M protein in our in-house ELISA test. All these sera were virus negative. As 21 (corresponding to subgroup 6) (Table 2) out of these 80 sera were IgG+ (based on both the PanBio test as well as their reactivity to the IgG-specific r-DME-G protein), we interpret the remaining 59 (subgroup 7 in Table 2) sera to be “normal.” The reactivities of these 59 samples were used to compute the cutoff ELISA absorbance value (equivalent to an OD450 of 0.25) to designate the remaining samples as either positive or negative. Based on this cutoff, 55 sera (corresponding to subgroups 2 to 5) were found to be IgM+. All but 3 out of these 55 sera could be corroborated as IgM+ using the PanBio test. We believe that these three sera (subgroup 2 in Table 2) which manifested significant reactivity towards the r-DME-M protein (Fig. 4) are indeed IgM+ despite their not being scored positive for IgM antibodies in the PanBio test. Our conclusion stems from the fact that our in-house ELISA relies on an IgM-specific NS1 epitope-based r-DME-M antigen while the PanBio test utilizes a cocktail of E proteins to detect antidengue IgM antibodies. Our preliminary data suggest that the NS1-based r-DME-M protein offers better sensitivity in the early detection of IgM antibodies. The observation that these sera were also virus positive lends credence to this notion. Subgroup 1 sera (n = 16), which were also virus positive, however, tested negative for both IgM and IgG in both the PanBio as well as the r-DME-M (or r-DME-G) protein-based assays. These presumably represent samples from patients still in the viremic phase. Results for 21 sera (subgroup 8) were not consistent between the PanBio test on the one hand and the r-DME-M (or r-DME-G) protein-based assay on the other. These were borderline samples whose ELISA reactivities were close to the cutoff. It is likely that the borderline sera that tested positive using the r-DME-M protein contained predominantly anti-NS1 antibodies and represent “early” samples that were missed by the PanBio test designed to detect anti-E antibodies. A two-copy r-DME-M protein yielded essentially the same results (data not shown).
Interestingly, of the 55 IgM-positive sera samples referred to above, 13 sera (corresponding to subgroups 3 and 4 in Table 2) were also positive for IgG antibodies, while the remaining were IgG negative. Given that we have used anti-human IgM-HRPO to detect the antidengue IgM antibodies, this observation suggests that the r-DME-M protein is capable of specifically interacting with the IgM antibodies and that this interaction is not affected by the presence of antidengue IgG antibodies. This notion is further strengthened by the observed lack of reactivity of the IgG+ subgroup 6 sera (n = 21) towards the r-DME-M protein (Table 2). The overall comparative analysis of our data with the PanBio results suggests that there is an excellent agreement between the r-DME-protein-based ELISAs and the PanBio test, with ∼86% (148 out of 172) of the samples analyzed giving identical results. With reference to our in-house NS1 epitope-derived r-DME-M-based IgM ELISA, the E protein-based PanBio test was found to be ∼95% sensitive in detecting antidengue IgM antibodies.
The standard IgM antibody capture ELISA routinely used to detect antidengue IgM antibodies typically uses anti-human IgM to capture serum IgM, which in turn is detected using a mixture of all four DEN whole-virus antigens in conjunction with an antidengue antibody conjugate (16). This entails the production of two different antibodies and all four DEN antigens. The PanBio test is based on a similar antibody capture format, with some minor variations. In contrast, the in-house ELISA we have described uses just a single customized recombinant antigen designed to directly bind antidengue IgM antibodies in conjunction with a secondary antibody conjugate. In this context, it is noteworthy that the in-house ELISA results are in complete agreement with the PanBio test results.
Having shown that the r-DME-M protein does indeed have the capacity to detect antidengue IgM antibodies, we have used it to devise a simple, rapid strip test that may be of use in a field setting. This test consists of incubating the patient serum with gold-labeled r-DME-M protein and capturing the gold-labeled antigen-antidengue antibody complex using nonlabeled r-DME-M immobilized on a nitrocellulose strip. The captured complex is visualized as a pink line on the test strip. In conjunction with our previously developed r-DME-G protein, this strip test can be modified to enable simultaneous detection of both IgM and IgG antibodies. Further evaluation of the r-DME-M protein must await the availability of more patient sera. We would also need to test our recombinant protein against antibodies to other infectious pathogens (such as those causing Japanese encephalitis, yellow fever, malaria, and typhoid) to evaluate its specificity. The D2 NS1-P1 peptide, on which the r-DME-M protein is based, has been shown to be uniquely specific for sera from dengue patients, showing no reactivity towards JE patient sera (11). While it is very likely that this specificity is preserved in the r-DME-M protein, it needs to be tested experimentally. We have been unable to address this issue at the present time due to the nonavailability of JE and yellow fever patient sera. It is also necessary to identify additional linear, immunodominant IgM-specific epitopes. This will enable the design of improved versions of r-DME-M protein with enhanced diagnostic potential. In conclusion, our approach to creating recombinant antigens with tailored specificity to either the IgM (this study) or IgG (1) class of antibodies coupled to E. coli-based overexpression and simple one-step purification could be a precursor to developing alternate diagnostic options that may eliminate the concerns associated with currently available tests based on whole-virus antigens. This would be a strategy not restricted to the detection of dengue infections alone.
Acknowledgments
This work has been supported by institutional core funds and a grant from the Ministry of Defense, Government of India. R.A. is a Senior Research Fellow supported by the Indian Council of Scientific and Industrial Research.
REFERENCES
- 1.AnandaRao, R., S. Swaminathan, S. Fernando, A. M. Jana, and N. Khanna. 2005. A custom-designed recombinant multiepitope protein as a dengue diagnostic reagent. Protein Expr. Purif. 41:136-141. [DOI] [PubMed] [Google Scholar]
- 2.Cuzzubbo, A. J., T. P. Endy, A. Nisalak, S. Kalyanarooj, D. W. Vaughn, S. A. Ogata, D. E. Clements, and P. L. Devine. 2001. Use of recombinant envelope proteins for serological diagnosis of dengue virus infection in an immunochromatographic assay. Clin. Diagn. Lab. Immunol. 8:1150-1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Falconar, A. K. I., P. R. Young, and M. A. Miles. 1994. Precise location of sequential dengue virus subcomplex and complex B cell epitopes on the nonstructural-1 glycoprotein. Arch. Virol. 137:315-326. [DOI] [PubMed] [Google Scholar]
- 4.Garcia, G., D. W. Vaughn, and R. M. del Angel. 1997. Recognition of synthetic oligopeptides from nonstructural proteins NS1 and NS3 of dengue-4 virus by sera from dengue virus-infected children. Am. J. Trop. Med. Hyg. 56:466-470. [DOI] [PubMed] [Google Scholar]
- 5.Goeddel, D. V. 1990. Systems for heterologous gene expression. Methods Enzymol. 185:3-7. [DOI] [PubMed] [Google Scholar]
- 6.Groen, J., P. Koraka, J. Velzing, C. Copra, and A. D. M. E. Osterhaus. 2000. Evaluation of six immunoassays for detection of dengue virus-specific immunoglobulin M and G antibodies. Clin. Diagn. Lab. Immunol. 7:867-871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gubler, D. J. 1998. Dengue and dengue hemorrhagic fever. Clin. Microbiol. Rev. 11:480-496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gubler, D. J. 2002. Epidemic dengue/dengue hemorrhagic fever as a public health, social and economic problem in the 21st century. Trends Microbiol. 10:100-103. [DOI] [PubMed] [Google Scholar]
- 9.Gubler, D. J. 2004. Cities spawn epidemic dengue viruses. Nat. Med. 10:129-130. [DOI] [PubMed] [Google Scholar]
- 10.Hales, S., N. de Wet, J. Maindonald, and A. Woodward. 2002. Potential effect of population and climate changes on global distribution of dengue fever: an empirical model. Lancet 360:830-834. [DOI] [PubMed] [Google Scholar]
- 11.Huang, J. H., J. J. Wey, Y. C. Sun, C. Chin, L. J. Chien, and Y. C. Wu. 1999. Antibody responses to an immunodominant nonstructural 1 synthetic peptide in patients with dengue fever and dengue hemorrhagic fever. J. Med. Virol. 57:1-8. [PubMed] [Google Scholar]
- 12.Innis, B. L., V. Thirawuth, and C. Hemachudha. 1989. Identification of continuous epitopes of the envelope glycoprotein of dengue type 2 virus. Am. J. Trop. Med. Hyg. 40:676-687. [DOI] [PubMed] [Google Scholar]
- 13.Jaiswal, S., N. Khanna, and S. Swaminathan. 2004. High-level expression and one-step purification of recombinant dengue virus type 2 envelope domain III protein in Escherichia coli. Protein Expr. Purif. 33:80-91. [DOI] [PubMed] [Google Scholar]
- 14.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680-685. [DOI] [PubMed] [Google Scholar]
- 15.Megret, F., J. P. Hugnot, A. Falconar, M. K. Gentry, D. M. Morens, J. M. Murray, J. J. Schlesinger, P. J. Wright, P. Young, M. H. V. van Regenmortel, and V. Deubel. 1992. Use of recombinant fusion proteins and monoclonal antibodies to define linear and discontinuous antigenic sites on the dengue envelope glycoprotein. Virology 187:480-491. [DOI] [PubMed] [Google Scholar]
- 16.Nawa, M., K.-I. Yamada, T. Takasaki, T. Akatsuka, and I. Kurane. 2000. Serotype-cross-reactive immunoglobulin M responses in dengue virus infections determined by enzyme-linked immunosorbent assay. Clin. Diagn. Lab. Immunol. 7:774-777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nimmannitya, S. 1997. Dengue hemorrhagic fever: diagnosis and management, p. 133-145. In D. J. Gubler and G. Kuno (ed.), Dengue and dengue hemorrhagic fever. CAB International, Wallingford, United Kingdom.
- 18.Robinson, C. R., and R. T. Sauer. 1998. Optimizing the stability of single-chain proteins by linker length and composition mutagenesis. Proc. Natl. Acad. Sci. USA 95:5929-5934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shu, P. Y., and J. H. Huang. 2004. Current advances in dengue diagnosis. Clin. Diagn. Lab. Immunol. 11:642-650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Trirawatanapong, T., B. Chandran, R. Putnak, and R. Padmanabhan. 1992. Mapping of a region of dengue virus type-2 glycoprotein required for binding by a neutralizing monoclonal antibody. Gene 116:139-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.World Health Organization. 1997. Dengue haemorrhagic fever: diagnosis, treatment, prevention and control, 2nd ed. World Health Organization, Geneva, Switzerland.
- 22.Wu, H.-C., Y.-L. Huang, T.-T. Chao, J.-T. Jan, J.-L. Huang, H.-Y. Chiang, C.-C. King, and M.-F. Shaio. 2001. Identification of B-cell epitope of dengue virus type 1 and its application in diagnosis of patients. J. Clin. Microbiol. 39:977-982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu, H.-C., M.-Y. Jung, C.-Y. Chiu, T.-T. Chao, S.-C. Lai, J.-T. Jan, and M.-F. Shaio. 2003. Identification of a dengue virus type 2 (DEN-2) serotype-specific B-cell epitope and detection of DEN-2-immunized animal serum samples using an epitope-based peptide antigen. J. Gen. Virol. 84:2771-2779. [DOI] [PubMed] [Google Scholar]