

Abstract
Multiply-charged peptide cations comprised of two polypeptide chains (designated A and B) bound via a disulfide linkage have been reacted with SO2−· in an electrodynamic ion trap mass spectrometer. These reactions proceed through both proton transfer (without dissociation) and electron transfer (with and without dissociation). Electron transfer reactions are shown to give rise to cleavage along the peptide backbone, loss of neutral molecules, and cleavage of the cystine bond. Disulfide bond cleavage is the preferred dissociation channel and both Chain A (or B)—S· and Chain A (or B)—SH fragment ions are observed, similar to those observed with electron capture dissociation (ECD) of disulfide-bound peptides. Electron transfer without dissociation produces [M + 2H]+· ions, which appear to be less kinetically stable than the proton transfer [M + H]+ product. When subjected to collision-induced dissociation (CID), the [M + 2H]+· ions fragment to give products that were also observed as dissociation products during the electron transfer reaction. However, not all dissociation channels noted in the electron transfer reaction were observed in the CID of the [M + 2H]+· ions. The charge state of the peptide has a significant effect on both the extent of electron transfer dissociation observed and the variety of dissociation products, with higher charge states giving more of each.
Tandem mass spectrometry currently plays a major role in the identification and characterization of proteins [1–3]. This application is enabled by the ability to form gaseous ions from peptides and proteins, typically via electrospray ionization [4, 5] or matrix-assisted laser desorption ionization [6, 7], the ability to form fragments from peptides and proteins of interest that reveal primary structure information, and the ability to measure and detect the fragments. Amino acid sequence information is usually sought for the identification of a protein. However, the identities and locations of post-translational modifications arising from, for example, phosphorylation, glycosylation, and disulfide bonding are also of interest for the complete characterization of the protein of interest. Unimolecular dissociation is the predominant chemical means for deriving primary structure information from a peptide or protein in the gas phase. The extent to which structural information can be derived from a post-translationally modified peptide or protein depends upon many factors including, for example, charge state and nature of the ion (e.g., protonated versus metal cationized), nature of the modification, and ion activation conditions. The nature of the modification itself can play a major role in directing dissociation chemistry. For this reason, it is of interest to explore various means for both forming and activating post-translationally modified ions. In this study, we focus on the behavior of polypeptide chains bound by a cystine bridge. The formation of such bridges takes place as a protein folds into its native conformation, and the bridges stabilize the three-dimensional structure of the protein [8–10].
CID is by far the most common means for deriving polypeptide structural information. Most tandem mass spectrometers currently applied to peptide cations employ multiple relatively low energy collisions as the means for parent ion excitation [11]. Under these conditions, CID induces amide backbone cleavage, producing b- and y-type ions [12, 13], which are useful in polypeptide characterization [14]. A complication of using low energy CID to study polypeptide ions generated from ESI arises when disulfide bonds are present. Competition between cleavage of amide bonds and disulfide linkage bonds can significantly affect the information content in the resulting product ion spectrum. For example, for multiply-charged polypeptides with one or more disulfide linkages, CID is frequently observed to lead to fragmentation that is generally limited to regions outside disulfide loops [15, 16], which can compromise protein identification. For singly charged species [17], however, and for negatively-charged species [18], cleavage at the disulfide linkage can be dominant. Characterization of disulfide-containing peptides usually requires chemical reduction of the S—S bonds, often followed by alkylation, prior to mass spectrometric analysis. Alternatively, cystine bonds may be broken using high energy CID [19] or matrix-assisted laser desorption ionization (MALDI) (in-source [20] or post-source [21]).
An alternative dissociation technique is electron capture dissociation (ECD), where low energy electrons are captured by multiply-protonated species [22, 23]. This overall process transforms the ion from an even-electron closed-shell system to an odd-electron hypervalent system while depositing the energy associated with electron capture into the ion. ECD of polypeptide ions typically produce c- and z- type (N—Cα bond cleavage) product ions. ECD tends to produce more extensive cleavage along the peptide backbone than CID, thereby yielding greater sequence coverage, and often preserves labile post-translational modifications such as phosphorylation and glycosylation [23] when CID does not. An interesting characteristic of ECD is that it has been shown to cleave polypeptide ions preferentially at disulfide bonds [24]. Such distinct fragmentation behaviors of polypeptide ions after capture of an electron make this process particularly interesting and, apparently, complementary to CID. ECD, however, has only been effected with analytically useful efficiency using Fourier transform ion resonance cyclotron (FTICR) mass spectrometers.
The ion/ion reaction analog to electron capture is electron transfer. Recently, ECD-like results have been obtained in electrodynamic ion traps resulting from ion/ion electron transfer reactions, in a process which has been termed electron transfer dissociation (ETD) [25–29]. For example, multiply-protonated peptides have been shown to fragment to yield c- and z- type products as a result of ion/ion reactions with suitable reagent anions. Because ECD has shown unique behavior with respect to disulfide linkages, it is desirable to determine how disulfide-linked polypeptide cations behave after electron transfer. Such a study can provide further observations to determine the degree to which electron capture and electron transfer are analogous. In this study, the effects of ion/ion electron transfer on disulfide bond-containing peptides are examined in a three-dimensional quadrupole ion trap mass spectrometer using SO2−· as the reagent anion. To simplify data interpretation, our attention was placed upon systems with two polypeptide chains bound by a single disulfide linkage. This motif avoids ambiguities that can arise for disulfide linked single-chain polypeptides because it does not require the cleavage of two bonds for detectable observation of a dissociation reaction.
Experimental
Pyridine, methanol, and glacial acetic acid were purchased from Mallinckrodt (Phillipsburg, NJ). Arg8-conopressin G was obtained from Bachem (King of Prussia, PA). Somatostatin was obtained from Anaspec (San Jose, CA). TPCK-treated trypsin and bovine α-lactalbumin were purchased from Sigma (St. Louis, MO). Sulfur dioxide was purchased from Scott Specialty Gases (Troy, MI). Tris(2-carboxyethyl)phosphine hydrochloride (TCEP·HCl) was obtained from Pierce (Rockford, IL). TPCK-treated trypsin was used to digest somatostatin, Arg8-conopressin G, and α-lactalbumin, using a previously described procedure [30]. Briefly, trypsin was added to aqueous peptide solutions and to α-lactalbumin in an ammonium bicarbonate (0.2 M) buffer. The solutions were incubated at 38 °C. The peptide solutions were diluted to 0.1 mg/mL in water/methanol/acetic acid 49.5/49.5/1 (vol/vol/vol) from which they were ionized. The α-lactalbumin solution was fractionated using reversed-phase HPLC as described previously [30]. After lyophilization, the samples were reconstituted to approximately 0.1 mg/mL in water/methanol/acetic acid 49.5/49.5/1 (vol/vol/vol) which were then used for nano-ESI. The three tryptic peptides studied are referred to as Peptide I, II, and III, and their sequences can be found in Table 1. For some experiments, the disulfide bond in Arg8-conopressin G was reduced with TCEP·HCl in a water/methanol/acetic acid solution as described previously [31]. The reduction products were used for nano-ESI directly from this solution without further purification.
Table 1.
Peptide Sequences
| Name | Sequence | Peptide digest sequence | Peptide label |
|---|---|---|---|
| Arg8- Conopressin |
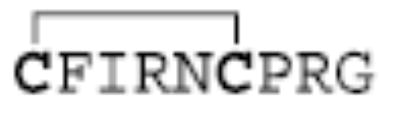
|

|
Peptide I |
| Somatostatin |
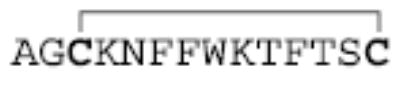
|

|
Peptide II |
| α-Lactalbumin |

|

|
Peptide III |
All experiments were carried out using a Hitachi (San Jose, CA) M-8000 3-DQ ion trap mass spectrometer, modified for ion/ion reactions, which has been described in detail elsewhere [32]. Anions were generated using atmospheric pressure glow discharge ionization and introduced into the mass spectrometer through a hole in the ring electrode. SO2 gas was leaked into the source to a pressure of approximately 530 mTorr. A software TTL trigger connected to a fast high voltage pulser (GRX-1.5K-E, Directed Energy Inc., Fort Collins, CO) was used to pulse the discharge.
Polypeptide cations were formed using nano-ESI [33, 34] with samples loaded into nanospray emitters pulled from borosilicate capillaries (1.5 mm o.d., 0.86 mm i.d.) using a P-87 Flaming/Brown micropipette puller (Sutter Instruments, Novato, CA). A stainless steel wire was inserted into the capillary and 1.2–2 kV were applied to the wire to induce ionization. In order to form lower charge states for some peptides, it was necessary to place a small dish of pyridine under the nanospray tip to allow ion/molecule proton transfer reactions to occur in the ion sampling region [35].
A typical experiment consisted of about 1 s of cation injection time. This was followed by an isolation step using the Hitachi’s filtered noise field (FNF) [36, 37] waveforms and by raising the amplitude of the radio frequency signal applied to the ring electrode of the ion trap to eject unwanted ions (~50 ms). SO2−· anions were then injected for 200 to 300 ms, during which time reactions could occur, and an AC signal was applied to the endcaps of the ion trap to eject any SO3− ions (formed from SO2−· via ion/molecule reactions in both the source and the ion trap). Because low RF levels are required to trap SO2−· ions, continual injection of anions is necessary during the reaction time to trap high mass positive ions efficiently. This process is known as “trapping by proxy” [38]. Following the reaction time, the remaining anions are ejected by raising the RF level of the trap, and cations are subsequently analyzed by resonance ejection. For some experiments, subsequent isolation and activation steps are performed prior to mass analysis. Isolation steps were performed as described above, and a software TTL trigger connected to an auxiliary Agilent (Palo Alto, CA) 33120A arbitrary waveform generator was used to resonantly excite ions of interest for approximately 300 ms. Spectra shown here are averaged over approximately 5 min (~250 scans).
Results and Discussion
Electron Transfer with Dissociation
When multiply-charged disulfide linked polypeptides are reacted with SO2−· anions, electron transfer and proton transfer are the two main pathways through which reaction can occur. Extensive previous work has shown that proton transfer reactions involving multiply-protonated peptides generally do not lead to fragmentation of the species during reaction [39, 40]. Unlike proton transfer, electron transfer is likely to lead to fragmentation [25, 41]. Figure 1 shows the reaction of SO2−· anions with +3 ions of Peptide I (generated by digestion of Arg8-conopressin G). The sequence CFIR is referred to herein as the A-chain and NCPR as the B-chain, for ease of discussion. To note peptide backbone cleavages we use the notation ABmn to specify that the ion contains the full A-chain linked to the B-chain, which is cleaved so as to produce the mn sequence ion. Alternately, BAmn would indicate the full B-chain linked to the A-chain, which is cleaved so as to produce the mn sequence ion. The main reaction products observed here are the +2 and +1 peptide species. These are expected to be largely comprised of proton transfer products, although an electron transfer without dissociation is also likely to make up part of the observed abundance (see below). The major dissociation products observed during the reaction, the A-chain and B-chain ions, arise from cleavage of the S—S disulfide bond. There is also evidence for some cleavage of the peptide backbone to yield c- and z-type ions, as shown by the presence of the signal labeled as ABc3+/BAc3+ (because both chains end in arginine residues, the masses of these ions are identical and they are thus indistinguishable). ECD of polypeptides has led to the observation of small neutral losses of NH3 and portions of the arginine side chain [42], and these also are noted here, as well as the loss of SH2 (or possibly, loss of two NH3 molecules). The loss of SH2 is somewhat surprising, as the loss of a sulfur would be expected to lead to the separation of the two peptide chains. The observed loss of only SH2 might be explained by either a rearrangement to form a monosulfide bond between the two peptide chains, or by the existence of noncovalent bonding between the chains that might hold them together after disulfide bond cleavage [43]. The dearth of backbone cleavages and the relatively abundant chain ions observed in the reaction product ions of Figure 1 indicate that the disulfide bonds are cleaved preferentially to the backbone bonds upon electron transfer in this reaction. Previous work with ECD has also shown a preference for cleavage of the disulfide bond [24].
Figure 1.
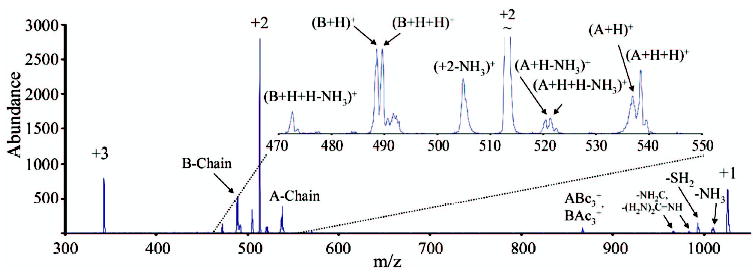
Product spectrum from the reaction of Peptide I [M + 3H]3+ with SO2−·.
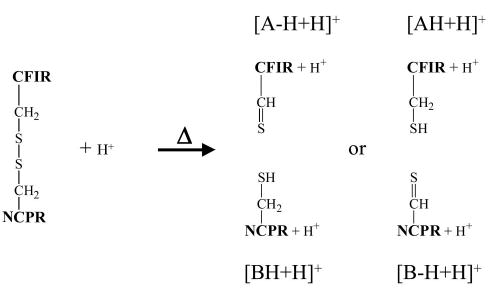
In the ECD work, it was reported that disulfide-bound peptide chains cleaved to yield an odd electron Chain-S· product, and an even electron Chain-SH product [24]. The work with ECD also reports that the even electron Chain-SH product tends to come from the more highly charged chain, presumably due to polarization of the S—S bond. It has been proposed that this occurs when a hydrogen atom (H·), generated by electron capture at a protonation site, attacks one of the sulfur atoms, leading to cleavage of the disulfide bond. The preference for cleavage of disulfide bonds is then explained by the greater hydrogen affinity of the disulfide bond [24], in comparison with the peptide backbone. Some other studies have indicated that this may not be the only mechanism by which ECD occurs at disulfide linkages, and that direct dissociative electron attachment may also be a possibility [44]. It is of interest to determine if the cleavage observed for electron transfer during reaction with SO2−· yields the same products as those produced by ECD. The inset in Figure 1 shows an expanded view of the region that includes the two peptide chains. It can be seen that both the A-chain and B-chain product ions are doublets, with the peaks separated by 1 Da. This indicates that, for each chain, there are two products, differing in mass by one hydrogen. These can be explained as the Chain-S· product and the Chain-SH product for each chain. Scheme 1 summarizes the various ion types and their designations according to the labeling in Figure 1. It is assumed here that, given the presence of an arginine residue at each of the C-termini, each chain retains one charge after a single electron transfer reaction that leads to dissociation. Clearly, for this reaction both products can be generated for each chain. As both chains would be expected to be singly charged, no particular preference for the location of the hydrogen atom would be predicted on the basis of the ECD observation mentioned above.
Scheme 1.

The dissociation reactions of these product ions were further explored with MS3 experiments using CID. The results of these MS3 experiments on the A-chain ions produced in the reaction discussed above are shown in Figure 2. Figure 2a shows the results from the collisional activation of the lower mass ion, which is expected to be the odd-electron Chain A-S· product. The major product corresponds to the loss of 46 Da, which is likely to be loss of SCH2 from the cysteine side chain. In addition, there is also a small signal corresponding to a loss of 34 Da, expected to be loss of SH2 from the cysteine side chain. Figure 2b shows the activation of the higher mass ion, which is expected to be the even electron Chain A-SH product. The most abundant product is the loss of NH3/H2O. In addition, there are small signals corresponding to the loss of SH2 and SCH2, as well as several sequence ions corresponding to cleavage of backbone amide bonds. These include the b3+, y2+, y1+, as well as y2*+ ions (the asterisk denotes sequential loss of an NH3 molecule from the y2+ ion). For comparison, the disulfide bond of Peptide I was reduced in solution with TCEP, which reduces the disulfide bond and yields both chains as even electron, Chain-SH products. The resulting A-chain +1 ion was isolated from electrospray and subjected to CID. The results for this experiment are shown in Figure 2c. The results are almost entirely the same as those observed for the higher mass reaction product ion in Figure 2b, save for the minor SCH2 loss product observed in Figure 2b. It is believed that the SCH2 loss is a result of a small amount of the lower mass ion not being removed by the isolation prior to CID. This result indicates that the protonated Chain A-SH species formed via electron transfer dissociation and via digestion in solution followed by electrospray ionization cannot be distinguished with ion trap CID.
Figure 2.
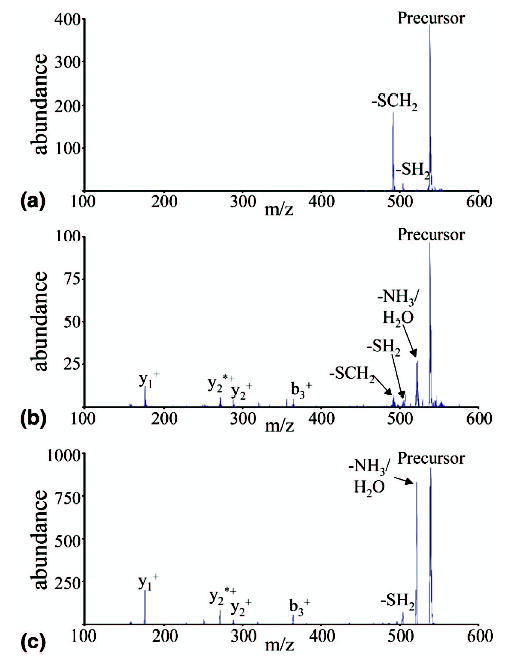
(a) MS3 of Peptide I A-Chain [A + H]+. (b) MS3 of Peptide I A-Chain [AH + H]+. (c) MS2 of A-Chain formed from solution-phase reduction of disulfide bond in Peptide I.
Analogous MS3 experiments were conducted on the B-chain product ions shown in Figure 1 (data not shown). The results were very similar to those observed for the A-chain. The lower mass ion, expected to be Chain B-S·, yielded loss of 46 Da (SCH2) as the major product, as well as a new product, loss of 44 Da, in similar abundance, and loss of 34 Da (SH2) as a minor product. The origin of the 44 Da loss is not entirely clear but loss of CO2 from the C-terminus is a likely source. The higher mass ion, expected to be Chain B-SH, again gave loss of NH3/H2O as the major product as well as some sequence ions arising from cleavage of backbone amide bonds, and a small SH2 loss. Once again, the CID spectrum of the chain generated by reduction of the disulfide bond in solution was collected for comparison, and found to be nearly identical to that of the nominal Chain B-SH product ion formed as a result of the electron transfer reaction. The CID behavior of the first generation product ions is consistent with the observations described by Wee et al. for the CID of tripeptide radical cations [45]. This work showed that GCR+·, when subjected to CID, fragments to yield loss of SCH2 and SH· as the main products, as well as a very minor y1+ ion. They suggest that loss of SCH2 occurs when the radical site is located on the sulfur atom, while SH· loss occurs when the radical site is located on the Cα of the cysteine residue. At least initially, the predominant radical site would be expected to be the sulfur atom for the reaction products studied here, which is consistent with SCH2 loss constituting the main dissociation process as is observed in the MS3 experiments of the Chain-S· ions. Furthermore, they also report for other tripeptides that a loss of CO2 (44 Da) can occur when the radical site is located on the C-terminus, which might account for the observation of this ion in the B-chain MS3 experiments. Taken collectively, the data described above are all consistent with cleavage of the disulfide bond of the two-chain peptide to yield either odd- or even-electron products, apparently entirely analogous with electron capture.
Electron Transfer Without Dissociation
While it is clear from Figure 1 that some electron transfer reactions lead to dissociation of the resulting charge-reduced product ions, it is of interest to examine the ion/ion reaction products that do not lead to fragmentation. We examined, in particular, the +1 intact polypeptide ions that resulted from the ion/ion reactions. These products can be comprised of a mixture of species formed exclusively by proton transfer reactions, a mixture of both electron transfer and proton transfer, and exclusively by electron transfer reactions. However, given that SO2−· reacts primarily via proton transfer [40] and that at least some of the ions that undergo dissociation as a result of electron transfer are not available for a second electron transfer step, the relative contribution to the intact +1 ion population from successive electron transfer reactions is expected to be very small. If an ion that had undergone one electron transfer is present as a +1 ion (i.e., it had either undergone a proton transfer prior to electron transfer or after electron transfer), it is expected to be an [M + 2H]+· ion. An ion that underwent two successive proton transfers to reach +1 is an [M + H]+ ion. These products differ in mass by 1 Da. However, under the conditions used in this study, the ion trap was not able to determine these relative contributions of these products on the basis of mass measurement alone because of the overlap of the more abundant [M + H]+ ions’ isotopic distribution upon that of the [M + 2H]+· ions. Nevertheless, CID experiments can be used to probe for evidence of [M + 2H]+· if, as was the case for the MS3 experiments discussed above, odd electron species fragment differently from the even electron products upon activation.
Figure 3a shows the CID product ion spectrum of the +1 peptide ions formed in the reaction of +3 Peptide I ions with SO2−·. Figure 3b shows the CID of exclusively [M + H]+ ions made via ESI for comparison. (The latter species was formed using pyridine placed under the spray to increase the singly charged ion signal relative to that of higher charge states [see the Experimental section for details]). As would be expected, the spectra are largely similar, as most of the +1 ions sampled in the experiment leading to Figure 3a are expected to be [M + H]+ ions. The most abundant products in both cases are loss of NH3/H2O and formation of the (ABb3+H2O)+/(BAb3+H2O)+ ions (this occurs via a rearrangement that is common when arginine is the C-terminal amino acid, as is true for both chains here [46]). In addition, a variety of ions arising from separation of the two peptide chains, at either the S—S or the C—S bonds are apparent in both spectra. Such cleavages are known to occur for disulfide linked singly protonated species [17]. The ABc3+ and BAc3+ ions observed in the reaction leading to Figure 1 differ in mass from the (ABb3+H2O)+/(BAb3+H2O)+ ions by only 1 Da, and might also be present in these data but would likely be obscured by the more abundant (ABb3+H2O)+/(BAb3+H2O)+ ions. However, there are also some key differences between the two spectra. For example, Figure 3a shows a prominent SH2 loss product that is almost completely absent from Figure 3b. Other significant differences are apparent in the mass-to-charge region that encompasses the single chain ions, as shown in the insets of Figure 3. The ions observed in Figure 3b can be accounted for exclusively by heterolytic cleavage of the S—S and C—S bonds. When the C—S bonds cleave, the side taking both sulfurs takes a hydrogen from the now sulfurless side, yielding thiocysteine and dehydroalanine structures, respectively. This type of heterolytic cleavage has been reported previously for activation of a disulfide linked two-chain peptide in the negative ion mode [18]. It is also interesting to note that the C—S bonds appear to cleave more readily than the S—S bond, and that B-chain ions are more prevalent, which suggests that the B-chain is likely to be slightly more basic than the A-chain.
Figure 3.
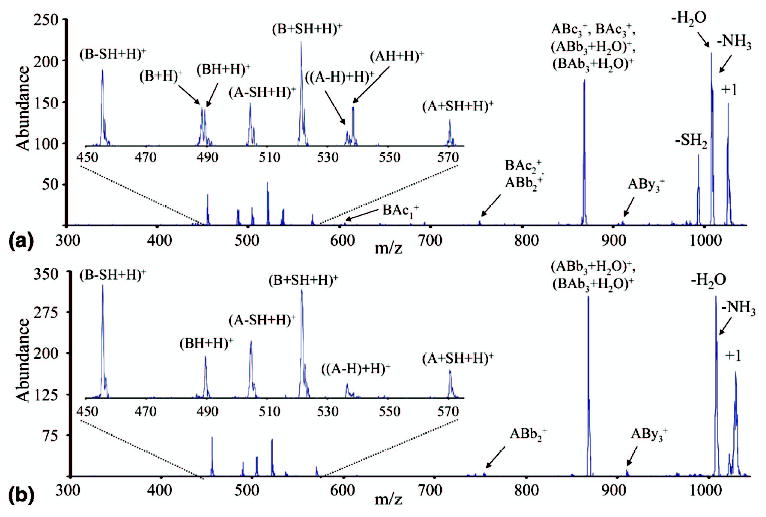
(a) CID of the singly charged peptide species formed from reaction of Peptide I [M + 3H]3+ with SO2−·. (b) CID of Peptide I [M + H]+ formed from electrospray.
Based on the electron transfer dissociation results of Figure 1, cleavage of C—S bonds is not expected to arise from [M + 2H]+·. ions. However, cleavage of the S—S bond of the disulfide linkage is expected to occur from both the [M + H]+ and [M + 2H]+· ions. While Scheme 1 shows products expected from [M + 3H]2+· ions, the same nominal products can be formed by fragmentation of [M + 2H]+·. That is, the [A + H]+ and [BH + H]+ ions (radical site retained on the A-chain) and the [AH + H]+ and [B + H]+ ions (radical site retained on the B-chain) can all, in principle, be formed. Scheme 2 summarizes the single chain ions that can be formed via heterolytic cleavage of the S—S bond from the [M + H]+ ion. These include the [A − H + H]+ and [BH + H]+ ions (hydrogen transferred to the B-chain), and [AH + H]+ and [B − H + H)+ ions (hydrogen transferred to the A-chain). Figure 3b shows predominantly the [A − H+H]+ and [BH + H]+ ions and little or no evidence for [AH + H]+ and [B − H+H]+ ions, thereby indicating a strong preference for hydrogen transfer from the A-chain to the B-chain in the heterolytic cleavage of the [M + H]+ ion. When the inset in Figure 3a is examined all the same ions are present, but there are two new products evident. These are the [B + H]+ ion and the [AH + H]+ ion, the pair of products expected to arise from radical site retention on the B-chain. These ions are expected to arise from [M + 2H]+· present in the +1 ion population subjected to collisional activation because essentially no [AH + H]+ ions were formed via collisional activation of the [M + H]+ ion (Figure 3b) and because no [B + H]+ ions are expected to be readily formed from the [M + H]+ ion (and, indeed, no such ions were observed in Figure 3b). (Similar differences between CID of the +1 intact peptide ions made in the reaction of +3 Peptide II with SO2−· and [M + H]+ ions formed directly via electrospray (data not shown) have been observed, suggesting that this is a general observation, not a phenomenon specific to Peptide I.) Interestingly, there appears to be little evidence for cleavage of the disulfide bond of the [M + 2H]+· ion with retention of the radical site on the A-chain. The expected products are either not observed, as is the case for [A + H]+ ions, or they can be accounted for by heterolytic cleavage of the [M + H]+ ions in the activated population, as is the case for the [BH + H]+ ions. In contrast, the ETD results from the [M + 3H]3+ ion (see Figure 1) show products with radical retention of either chain in roughly comparable abundance. The latter observation suggests that the dissociation step occurred in the reaction of the +3 ion to the +2 ion. However, given that the activation conditions and time-frames for the experiments are different, no firm conclusions can be drawn. In any case, neither the CID of the [M + H]+ nor the [M + 2H]+· ions shows comparable contributions from the two possible S—S cleavage channels available to each ion.
Scheme 2.
Kinetic stability is another possible characteristic upon which the distinction between [M + H]+ and [M + 2H]+· ions might be made. Based on discussions in the ECD literature, it might be expected that the [M + 2H]+· are more readily fragmented than the [M + H]+ ions. Two hypotheses can be proposed to account for how electron transfer can occur without leading to discrete fragments. In the first, a covalent bond is broken, but the fragments remain bound via noncovalent interactions [43]. In the second, a bond may be significantly weakened by the electron transfer [47], but the dissociation rate is sufficiently slow relative to cooling rates in the ion trap to allow for at least some of the electron transfer products to be stabilized. The latter interpretation assumes that the covalent bonds in the electron transfer products are sufficiently strong to survive under the normal ion trap storage conditions. For either hypothesis, it would be expected that the [M + 2H]+· ions would require less energy to induce dissociation. For this reason, the dependence of the product ion abundances on the activation amplitude was investigated. In Figure 4 the m/z 450-575 region is shown for the CID of the +1 intact peptide ions formed in the reaction of +3 peptide ions with SO2−· at two different activation amplitudes, 687 mV (Figure 4a) and 1030 mV (Figure 4b). It can be seen that with the lower activation amplitude (Figure 4a), the [B + H]+ and [A − H + H]+ ions, which are the two ions unique to the [M + 2H]+· ion, are clearly present, while the [BH + H]+ and [(A − H + H)+ ions are largely absent. Cleavage products from C—S bonds of the [M + H]+ are present at this amplitude but at lower abundance relative to those of the [B + H]+ and [AH + H]+ ions compared with the case in Figure 4b. In Figure 4b, the higher amplitude activation, all of the ions are present. In addition, the SH2 loss product shows a similar abundance to the NH3/H2O loss product in the lower activation amplitude experiment, while it is significantly smaller than the NH3/H2O loss product in the higher activation amplitude experiment (data not shown). The increase in the relative contributions of the major electron transfer products, the [B + H]+ and [AH + H]+ ions as well as the SH2 loss product, with decreasing activation amplitude, are consistent with the [M + 2H]+· ions being less kinetically stable than the [M + H]+ ions.
Figure 4.
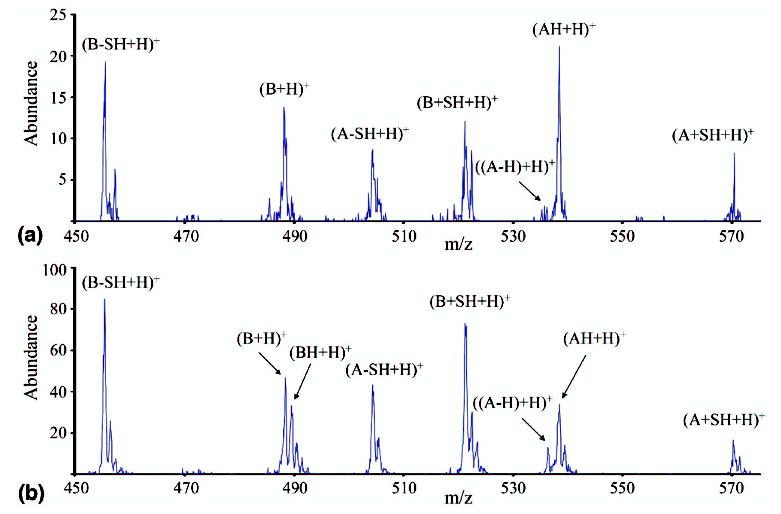
CID of the singly charged peptide species formed from reaction of Peptide I [M + 3H]3+ with SO2−· at (a) 687 mV and (b) 1030 mV.
Another means for identifying the existence of two different ion populations of the same or similar m/z is with a “burn-up” or sequential activation experiment [48]. If two different populations exist, and they differ in dissociation rate under a given set of activation conditions, low amplitude activation can be used to deplete the faster dissociation population preferentially. If the remaining precursor ion is then reisolated and activated again, the relative abundances of the product ions should change, with the products that come from the population with a lower threshold being depleted. When such an experiment was employed with the +1 intact peptide ions made in the reaction of +3 Peptide I ions with SO2−·, the burn-up experiment resulted in a significant depletion of the [B + H]+, [AH + H]+, and SH2 loss ions (data not shown), which supports the conclusion drawn from Figure 4 that the [M + 2H]+··ions are less kinetically stable than [M + H]+ ions.
The results of all these CID experiments, taken together, provide definitive evidence that electron transfer occurs to some of these disulfide linked polypeptide ions without formation of discrete fragments. Upon subsequent activation, they fragment to give primarily loss of SH2, formation of a [B + H]+ ion, and formation of a [AH + H]+ ion. All of these ions are also observed directly as products in the reaction of +3 Peptide I ions with SO2−·. Furthermore, the surviving [M + 2H]+· ions are more readily fragmented than the [M + H]+ ions. It is interesting to speculate about what insight these results provide into the nature of the [M + 2H]+· ions, with respect to the two hypotheses mentioned above. In the ECD cases for which covalent bond cleavage with stabilization by noncovalent bonds has been proposed [43], the ions in question are typically much larger whole protein ions in which there are likely to be a substantial number of noncovalent interactions. The relatively small size of the fragments involved here would give rise to relatively few noncovalent interactions between them. The H2S fragment, for example, is not expected to engage in strong noncovalent interactions. The simpler explanation of these data is that the covalent bonds are intact in the surviving [M + 2H]+··ions and that they are cleaved by CID. However, as these data are only indirect, no definitive conclusions regarding the nature of bonding in the surviving [M + 2H]+· ions can be drawn.
Effect of Cation Charge State on Extent of Dissociation and Reaction Channel Diversity
Previously it has been shown that the cation charge state plays a major role in determining both the extent of electron transfer and the dissociation products observed resulting from reactions with SO2−· [41]. Reactions of +3 peptides yielded much greater abundances of c- and z-type ions, as well as a much greater variety of products, than +2 peptides, typically allowing complete sequencing of the peptide. It is of interest, therefore, to determine if charge state plays an important role in the dissociation of peptide chains linked by a disulfide bond, in which most of the electron transfer dissociation might be expected to result in cleavage of the disulfide bond rather than the peptide backbone. Peptide I and Peptide II form both the +3 and +2 charge states directly via nano-ESI, and results are similar to those discussed in this section for Peptide III. Nano-ESI of Peptide III (made from digestion of α-lactalbumin) forms the +4 and +3 charge states directly, and when pyridine is placed under the nanoelectrospray assembly (see the Experimental section for details), the +2 charge state can also be formed. Again, for ease of discussion, the sequence CEVFR is referred to as the A-chain, and LDQWLCEKL referred to as the B-chain (see Table 1), and backbone cleavages are denoted using the ABmn or BAmn notation described previously as well. The results of reacting each of these charge states with SO2−· anions are summarized in Figure 5a, b, and c. As Figure 5c shows, +2 disulfide linked peptides show very little dissociation as a result of reaction with SO2−· anions. The only products seen here are small SH2 and NH3/H2O losses. The cleavage of the disulfide bond to give the separate peptide chains is not observed, nor are any ions resulting from cleavage of the peptide backbone. The reaction of the +3 Peptide III ions in Figure 5b shows substantially more dissociation products. As with the reaction of +3 Peptide I ions shown in Figure 1, the main dissociation products result from cleavage of the S—S disulfide bond. There are also neutral losses as well as a substantial number of ions which indicate cleavage of the N—Cα peptide backbone bonds to give c- and z-type sequence ions. Peptide III is notable for giving a much wider range of c- and z-type ions than either Peptides I or II. In this case, 9 of the 12 possible N—Cα bonds are broken. Most of the dissociation is expected to occur in the reaction of the +3 ions with SO2−· anions, as the results in Figure 5c, as well as the earlier work with nondisulfide-bonded peptides, show that little electron transfer induced dissociation occurs in the reactions of SO2−· anions with +2 peptide ions. Figure 5a shows the reaction of +4 Peptide III ions with SO2−· anions, and displays still more dissociation products than the results for +3 ions in Figure 5b. Once again, the main dissociation products result from cleavage of the S—S bond. An even wider range of c- and z-type ions are observed, with 11 of 12 possible N—Cα bond cleavages being represented by fragment ions. This increase in dissociation variety and abundance likely arises from the integration of fragmentation that takes place in the +4 → +3 and +3 → +2 steps, with most of the fragments likely arising from the +4 ion. Not surprisingly, there is a greater abundance of +2 c-and z-type ions (marked with a asterisk) observed in Figure 5a than in Figure 5b. For reasons discussed above, it is unlikely that products that arise from sequential electron transfer reactions make major contributions to the data. No major products are observed in Figure 5a that indicate two steps of electron transfer dissociation, although such species may well contribute to the small signals in the baseline.
Figure 5.
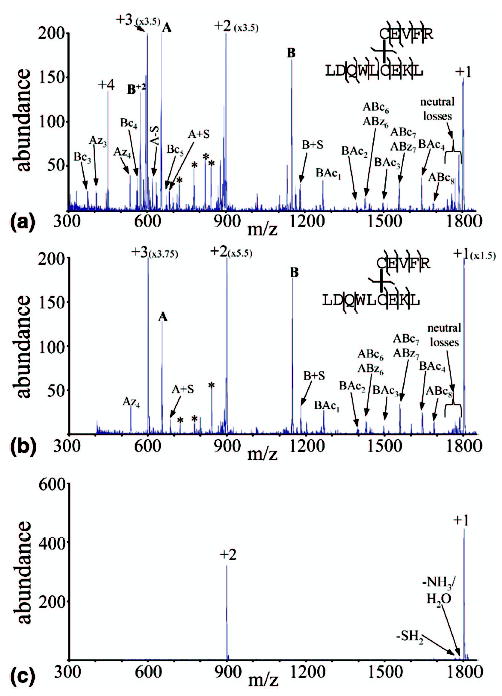
(a) Product spectrum from the reaction of Peptide III [M + 4H]4+ with SO2−·. (b) Product spectrum from the reaction of Peptide III [M + 3H]3+ with SO2−·. (c) Product spectrum from the reaction of Peptide III [M + 2H]2+ with SO2−·. An asterisk is used to denote doubly charged sequence ions for which the singly charged species is already identified.
The charge state dependence noted here for this disulfide linked polypeptide ion, as well as the others examined, is qualitatively consistent with observations made with unmodified polypeptide cations. The trend is consistent both with reaction exothermicity and with the expected kinetic stabilities of the products. That is, the ion/ion reaction of the more highly charged cation is inherently more exothermic than the reaction of the same peptide at a lower charge state. Furthermore, the kinetic stabilities of the first generation ion/ion reaction products differ, as the products experience greater electrostatic repulsion with increase in parent ion charge state. With regard to reaction exothermicity, it was reported in a recent work that the ionization energies of polypeptide cations show an average increase of about 1.1 eV/charge [49]. Trends in ionization energies (electron removal) are expected to be mirrored in recombination energies (electron capture). The reaction exothermicity, ΔHrxn, for the general reaction:
| (1) |
| (2) |
where EA(N) is the electron affinity of N and RE(MHnn+) is the recombination energy of the cation. So, as the charge state of the cation increases, the increase in recombination energy will lead to a more exothermic reaction. If the energy partitioned into the polypeptide ion as a result of the ion/ion reaction correlates with reaction exothermicity, the rates of the various dissociation channels would be expected to increase with ion/ion reaction exothermicity. Fewer first generation products would therefore be expected to be stabilized either by collisions or emission. The kinetic stabilities of the products also play a major role in determining dissociation rates. To the extent that electrostatic repulsion reduces dissociation barriers, it can be expected that product ion dissociation rates will generally increase with charge. Both of these factors suggest that higher charge state reactants should be expected to lead to a greater extent of dissociation and a more varied range of dissociation channels, as has been observed.
Conclusions
Proton transfer and electron transfer are competitive processes in the ion/ion reactions of multiply-charged disulfide-bound peptide cations with SO2−· anions. Proton transfer, as has been noted previously for other types of polypeptide ions, proceeds without dissociation of the charge-reduced product ions. Electron transfer has been shown to lead preferentially, although not exclusively, to cleavage of the S—S disulfide bond, producing Chain-S· and Chain-SH product ions, similar to observations made for ECD of disulfide-bound peptides. Cleavage of the peptide backbone to produce c-and z-type ions also occurs upon electron transfer, as do the losses of various small molecules. It has also been shown that at least some of the initially formed electron transfer products survive and are observed as intact polypeptide species. The resulting [M + 2H]+· ions appear to be less kinetically stable than [M + H]+ ions, and fragment upon CID to produce many of the same products observed for dissociation induced spontaneously via electron transfer. The range of electron transfer induced dissociation products observed, as well as the overall amount of electron transfer induced dissociation observed, appears to depend strongly on the charge state of the cationic reagent, with increasing charge leading to more abundant dissociation products as well as more widely varied dissociation products.
Collectively, these findings provide useful insights into the dissociation chemistries of disulfide linked polypeptide cations. In those cases in which even-electron protonated peptides show significant disulfide bond cleavage, C—S bond dissociation dominates over S—S bond dissociation. Electron transfer product ions show the opposite trend, with disulfide bond dissociation preferred. Furthermore, the odd-electron hypervalent species show preference for cleavage of the disulfide linkage, whereas multiply-protonated peptides tend not to show disulfide bond cleavage until the charge is sequestered by strongly basic sites. The latter characteristic makes problematic the extraction of structural information within loops defined by intra-chain disulfide bonds via CID of multiply-protonated polypeptides. In general, reduction, usually followed by alkylation, of disulfide linkages is required prior to mass spectrometry. Electron transfer ion/ion reactions may prove to be useful in cleaving disulfide linkages in the gas phase to allow for subsequent interrogation via CID or any other activation method.
Acknowledgments
This research was sponsored by the National Institutes of Health under grant GM 45372 and the U.S. Department of Energy under award no. DE-FG02-00ER15105.
References
- 1.Jennings KR. The Changing Impact of the Collision-Induced Decomposition of Ions on Mass Spectrometry. Int J Mass Spectrom. 2000;200:479–493. [Google Scholar]
- 2.Aebersold R, Goodlett DR. Mass Spectrometry in Proteomics. Chem Rev. 2001;101:269–295. doi: 10.1021/cr990076h. [DOI] [PubMed] [Google Scholar]
- 3.Griffin TJ, Aebersold R. Advances in Proteome Analysis by Mass Spectrometry. J Biol Chem. 2001;276:45497–45500. doi: 10.1074/jbc.R100014200. [DOI] [PubMed] [Google Scholar]
- 4.Fenn JB, Mann M, Meng CK, Wong SF, Whitehouse CM. Electrospray Ionization for Mass Spectrometry of Large Biomolecules. Mass Spectrom Rev. 1990;9:37–70. doi: 10.1126/science.2675315. [DOI] [PubMed] [Google Scholar]
- 5.Fenn JB, Mann M, Meng CK, Wong SF, Whitehouse CM. Electrospray Ionization for Mass Spectrometry of Large Biomolecules. Science. 1989;246:64–71. doi: 10.1126/science.2675315. [DOI] [PubMed] [Google Scholar]
- 6.Hillenkamp F, Karas M, Beavis RC, Chait BT. Matrix-Assisted Laser Desorption/Ionization Mass Spectrometry of Biopolymers. Anal Chem. 1991;63:1193A–1203A. doi: 10.1021/ac00024a002. [DOI] [PubMed] [Google Scholar]
- 7.Karas M, Hillenkamp F. Laser Desorption Ionization of Proteins with Molecular Masses Exceeding 10,000 Daltons. Anal Chem. 1988;60:2299–2301. doi: 10.1021/ac00171a028. [DOI] [PubMed] [Google Scholar]
- 8.Matsumura M, Signor G, Matthews BW. Substantial Increase of Protein Stability by Multiple Disulphide Bonds. Nature. 1989;342:291–293. doi: 10.1038/342291a0. [DOI] [PubMed] [Google Scholar]
- 9.Creighton TE. Disulphide Bonds and Protein Stability. BioEssays. 1988;8:57–63. doi: 10.1002/bies.950080204. [DOI] [PubMed] [Google Scholar]
- 10.Thornton JM. Disulphide Bridges in Globular Proteins. J Mol Biol. 1981;151:261–287. doi: 10.1016/0022-2836(81)90515-5. [DOI] [PubMed] [Google Scholar]
- 11.McLuckey SA, Goeringer DE. Slow Heating Methods in Tandem Mass Spectrometry. J Mass Spectrom. 1997;32:461–474. [Google Scholar]
- 12.Roepstorff P, Fohlman J. Proposal for a Common Nomenclature for Sequence Ions in Mass-Spectra of Peptides. Biomed Mass Spectrom. 1984;11:601. doi: 10.1002/bms.1200111109. [DOI] [PubMed] [Google Scholar]
- 13.Biemann K. Contributions of Mass-Spectrometry to Peptide and Protein Structure. Biomed Environ Mass Spectrom. 1988;16:99–111. doi: 10.1002/bms.1200160119. [DOI] [PubMed] [Google Scholar]
- 14.Hunt DF, Yates JR, III, Shabanowitz J, Winston S, Hauer CH. Protein Sequencing by Tandem Mass Spectrometry. Proc Natl Acad Sci USA. 1986;83:6233–6237. doi: 10.1073/pnas.83.17.6233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hogan JM, McLuckey SA. Charge State Dependent Collision-Induced Dissociation of Native and Reduced Porcine Elastase. J Mass Spectrom. 2003;38:245–256. doi: 10.1002/jms.458. [DOI] [PubMed] [Google Scholar]
- 16.Loo JA, Edmonds CG, Udseth HR, Smith RD. Effect of Reducing Disulfide-Containing Proteins on Electrospray Ionization Mass Spectra. Anal Chem. 1990;62:693–698. doi: 10.1021/ac00206a009. [DOI] [PubMed] [Google Scholar]
- 17.Wells JM, Stephenson JL, Jr, McLuckey SA. Charge Dependence of Positive Insulin Ion Decompositions. Int J Mass Spectrom. 2000;203:A1–A9. [Google Scholar]
- 18.Chrisman PA, McLuckey SA. Dissociations of Disulfide-Linked Gaseous Polypeptide/Protein Anions: Ion Chemistry with Implications for Protein Identification and Characterization. J Proteome Res. 2002;1:549–557. doi: 10.1021/pr025561z. [DOI] [PubMed] [Google Scholar]
- 19.Bean MF, Carr SA. Characterization of Disulfide Bond Position in Proteins and Sequence Analysis of Cystine-Bridged Peptides by Tandem Mass Spectrometry. Anal Biochem. 1992;201:216–226. doi: 10.1016/0003-2697(92)90331-z. [DOI] [PubMed] [Google Scholar]
- 20.Patterson SD, Katta V. Prompt Fragmentation of Disulfide-Linked Peptides during Matrix-Assisted Laser Desorption Ionization Mass Spectrometry. Anal Chem. 1994;66:3727–3732. doi: 10.1021/ac00093a030. [DOI] [PubMed] [Google Scholar]
- 21.Zhou J, Ens W, Poppe-Schriemer N, Standing KG, Westmore JB. Cleavage of Interchain Disulfide Bonds Following Matrix-Assisted Laser Desorption. Int J Mass Spectrom Ion Processes. 1993;126:115–122. [Google Scholar]
- 22.Zubarev RA, Kelleher NL, McLafferty FW. Electron Capture Dissociation of Multiply Charged Protein Cations. A Nonergodic Process. J Am Chem Soc. 1998;120:3265–3266. [Google Scholar]
- 23.Zubarev RA. Reactions of Polypeptide Ions with Electrons in the Gas Phase. Mass Spectrom Rev. 2003;22:57–77. doi: 10.1002/mas.10042. [DOI] [PubMed] [Google Scholar]
- 24.Zubarev RA, Kruger NA, Fridriksson EK, Lewis MA, Horn DM, Carpenter BK, McLafferty FW. Electron Capture Dissociation of Gaseous Multiply-Charged Proteins is Favored at Disulfide Bonds and Other Sites of High Hydrogen Atom Affinity. J Am Chem Soc. 1999;121:2857–2862. [Google Scholar]
- 25.Coon JJ, Syka JEP, Schwartz JC, Shabanowitz J, Hunt DF. Anion Dependence in the Partitioning Between Proton and Electron Transfer in Ion/Ion Reactions. Int J Mass Spectrom. 2004;236:33–42. [Google Scholar]
- 26.Coon, J. J.; Syka, J. E. P.; Schroeder, M. J.; Shabanowitz, J.; Hunt, D. F. Proceedings of the 52nd ASMS Conference on Mass Spectrometry; Nashville, TN, May, 2004.
- 27.Syka JEP, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF. Peptide and Protein Sequence Analysis by Electron Transfer Dissociation Mass Spectrometry. Proc Natl Acad Sci USA. 2004;101:9528–9533. doi: 10.1073/pnas.0402700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schroeder, M. J.; Coon, J. J.; Syka, J. E. P.; Shabanowitz, J.; Hunt, D. F. Proceedings of the 52nd ASMS Conference on Mass Spectrometry; Nashville, TN, May, 2004.
- 29.Syka, J. E. P.; Coon, J. J.; Schwartz, J. C.; Shabanowitz, J.; Hunt, D. F. Proceedings of the 52nd ASMS Conference on Mass Spectrometry; Nashville, TN, May, 2004.
- 30.Hogan JM, Pitteri SJ, McLuckey SA. Phosphorylation Site Identification via Ion Trap Tandem Mass Spectrometry of Whole Protein and Peptide Ions: Bovine α-Crystallin A Chain. Anal Chem. 2003;75:6509–6516. doi: 10.1021/ac034410s. [DOI] [PubMed] [Google Scholar]
- 31.Newton KA, Pitteri SJ, Laskowski M, Jr, McLuckey SA. Effects of Single Amino Acid Substitution on the Collision-Induced Dissociation of Intact Protein Ions: Turkey Ovomucoid Third Domain. J Proteome Res. 2004;3:1033–1041. doi: 10.1021/pr049910w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reid GE, Wells JM, Badman ER, McLuckey SA. Performance of a Quadrupole Ion Trap Mass Spectrometer Adapted for Ion/Ion Reaction Studies. Int J Mass Spectrom. 2003;222:243–258. [Google Scholar]
- 33.Van Berkel GJ, Asano KG, Schnier PD. Electrochemical Processes in a Wire-in-a-Capillary Bulk-Loaded, Nano-Electrospray Emitter. J Am Soc Mass Spectrom. 2001;12:853–862. doi: 10.1016/S1044-0305(01)00264-1. [DOI] [PubMed] [Google Scholar]
- 34.Kelleher NL, Senko MW, Siegel MM, McLafferty FW. Unit Resolution Mass Spectra of 112 kDa Molecules with 3 Da Accuracy. J Am Soc Mass Spectrom. 1997;8:380–383. [Google Scholar]
- 35.Shelimov KB, Jarrold MF. Conformations, Unfolding, and Refolding of Apomyoglobin in Vacuum: An Activation Barrier for Gas-Phase Protein Folding. J Am Chem Soc. 1997;119:2987–2994. [Google Scholar]
- 36.Goeringer DE, Asano KG, McLuckey SA, Hoekman D, Stiller SE. Filtered Noise Field Signals for Mass-Selective Accumulation of Externally Formed Ions in a Quadrupole Ion Trap. Anal Chem. 1994;66:313–318. [Google Scholar]
- 37.Kelley, P. E. Mass Spectrometry Method Using Notch Filter; U.S. Patent 5134286, 1992.
- 38.McLuckey SA, Wu J, Bundy JL, Stephenson JL, Jr, Hurst GB. Oligonucleotide Mixture Analysis via Electrospray and Ion/Ion Reactions in a Quadrupole Ion Trap. Anal Chem. 2002;74:976–984. doi: 10.1021/ac011015y. [DOI] [PubMed] [Google Scholar]
- 39.Pitteri, S. J., McLuckey, S. A. Recent Developments in the Ion/Ion Chemistry of High-Mass Multiply Charged Ions. Mass Spectrom. Rev., in press. [DOI] [PubMed]
- 40.Stephenson JL, Jr, McLuckey SA. Adaptation of the Paul Trap for Study of the Reaction of Multiply Charged Cations with Singly Charged Anions. Int J Mass Spectrom Ion Processes. 1997;162:89–106. [Google Scholar]
- 41.Pitteri, S. J.; Chrisman, P. A.; Hogan, J. M.; McLuckey, S. A. Electron Transfer Ion/Ion Reactions in a Three-Dimensional Quadrupole Ion Trap: Reactions of Doubly and Triply Protonated Peptides with SO2−· Anal. Chem., in press. [DOI] [PMC free article] [PubMed]
- 42.Cooper HJ, Hudgins RR, Håkansson K, Marshall AG. Characterization of Amino Acid Side Chain Losses in Electron Capture Dissociation. J Am Soc Mass Spectrom. 2002;13:241–249. doi: 10.1016/S1044-0305(01)00357-9. [DOI] [PubMed] [Google Scholar]
- 43.Breuker K, Oh H, Horn DM, Cerda BA, McLafferty FW. Detailed Unfolding and Folding of Gaseous Ubiquitin Ions Characterized by Electron Capture Dissociation. J Am Chem Soc. 2002;124:6407–6420. doi: 10.1021/ja012267j. [DOI] [PubMed] [Google Scholar]
- 44.Sawicka A, Skurski P, Hudgins RR, Simons J. Model Calculations Relevant to Disulfide Bond Cleavage via Electron Capture Influenced by Positively Charged Groups. J Phys Chem B. 2003;107:13505–13511. [Google Scholar]
- 45.Wee S, O’Hair RAJ, McFadyen WD. Comparing the Gas-Phase Fragmentation Reactions of Protonated and Radical Cations of the Tripeptides GXR. Int J Mass Spectrom. 2004;234:101–122. [Google Scholar]
- 46.Ballard KD, Gaskell SJ. Intramolecular [18O] Isotopic Exchange in the Gas Phase Observed during the Tandem Mass Spectrometric Analysis of Peptides. J Am Chem Soc. 1992;114:64–71. [Google Scholar]
- 47.Tureĉek F. N—Cα Bond Dissociation Energies and Kinetics in Amide and Peptide Radicals. Is the Dissociation a Nonergodic Process? J Am Chem Soc. 2003;125:5954–5963. doi: 10.1021/ja021323t. [DOI] [PubMed] [Google Scholar]
- 48.Chrisman PA, Newton KA, Reid GE, Wells JM, McLuckey SA. Loss of Charged Versus Neutral Heme from Gaseous Holomyoglobin Ions. Rapid Commun Mass Spectrom. 2001;15:2334–2340. doi: 10.1002/rcm.512. [DOI] [PubMed] [Google Scholar]
- 49.Budnik BA, Tsybin YO, Håkansson P, Zubarev RA. Ionization Energies of Multiply Protonated Polypeptides Obtained by Tandem Ionization in Fourier Transform Mass Spectrometers. J Mass Spectrom. 2002;37:1141–1144. doi: 10.1002/jms.376. [DOI] [PubMed] [Google Scholar]